Identification of Bacteriophage Virion Proteins Using Multinomial Naïve Bayes with g-Gap Feature Tree




Abstract
1. Introduction
2. Results
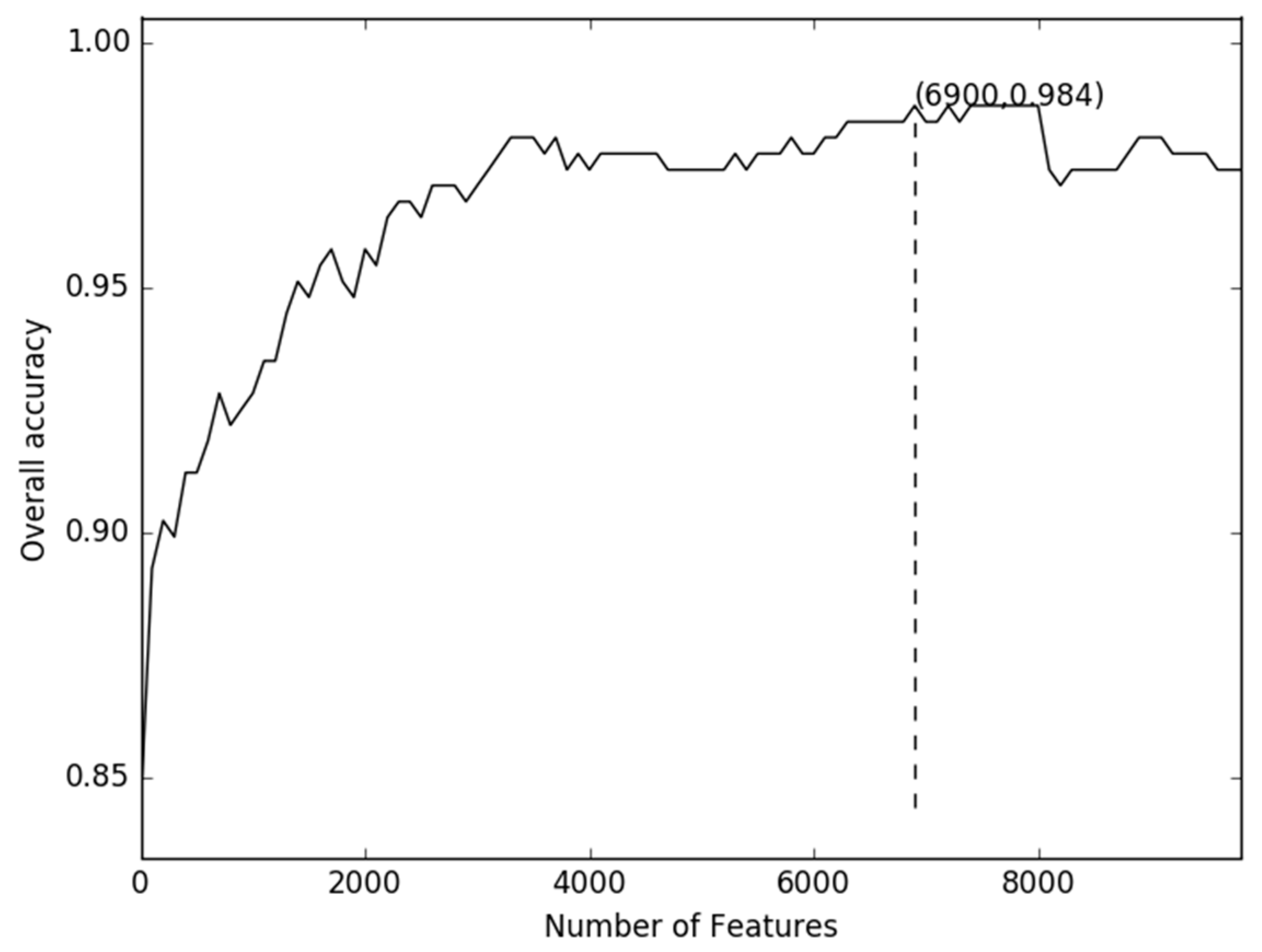
2.1. Comparison of Discrete Feature Vector in Different Dimensions
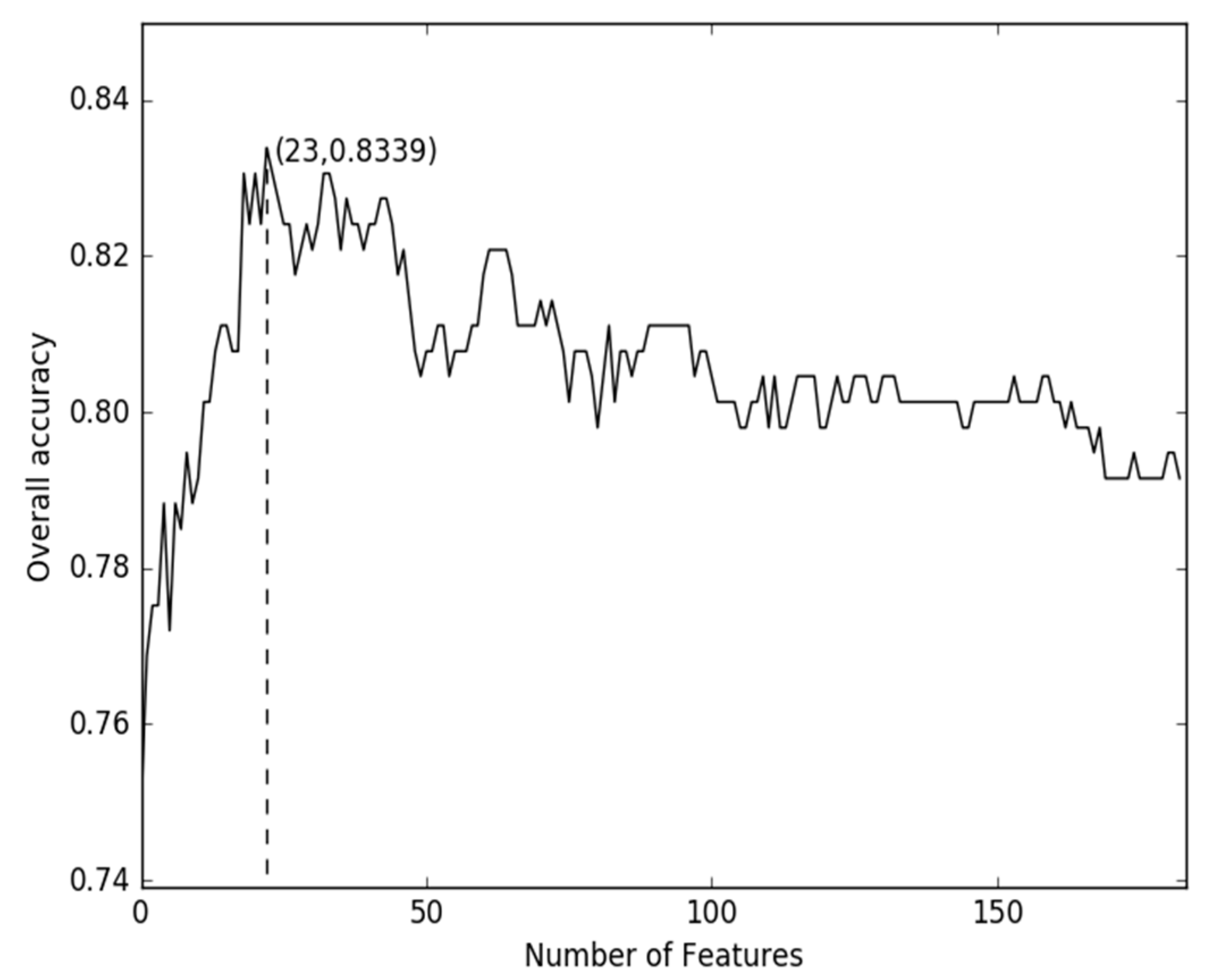
2.2. Comparison of g-Gap Features with Others
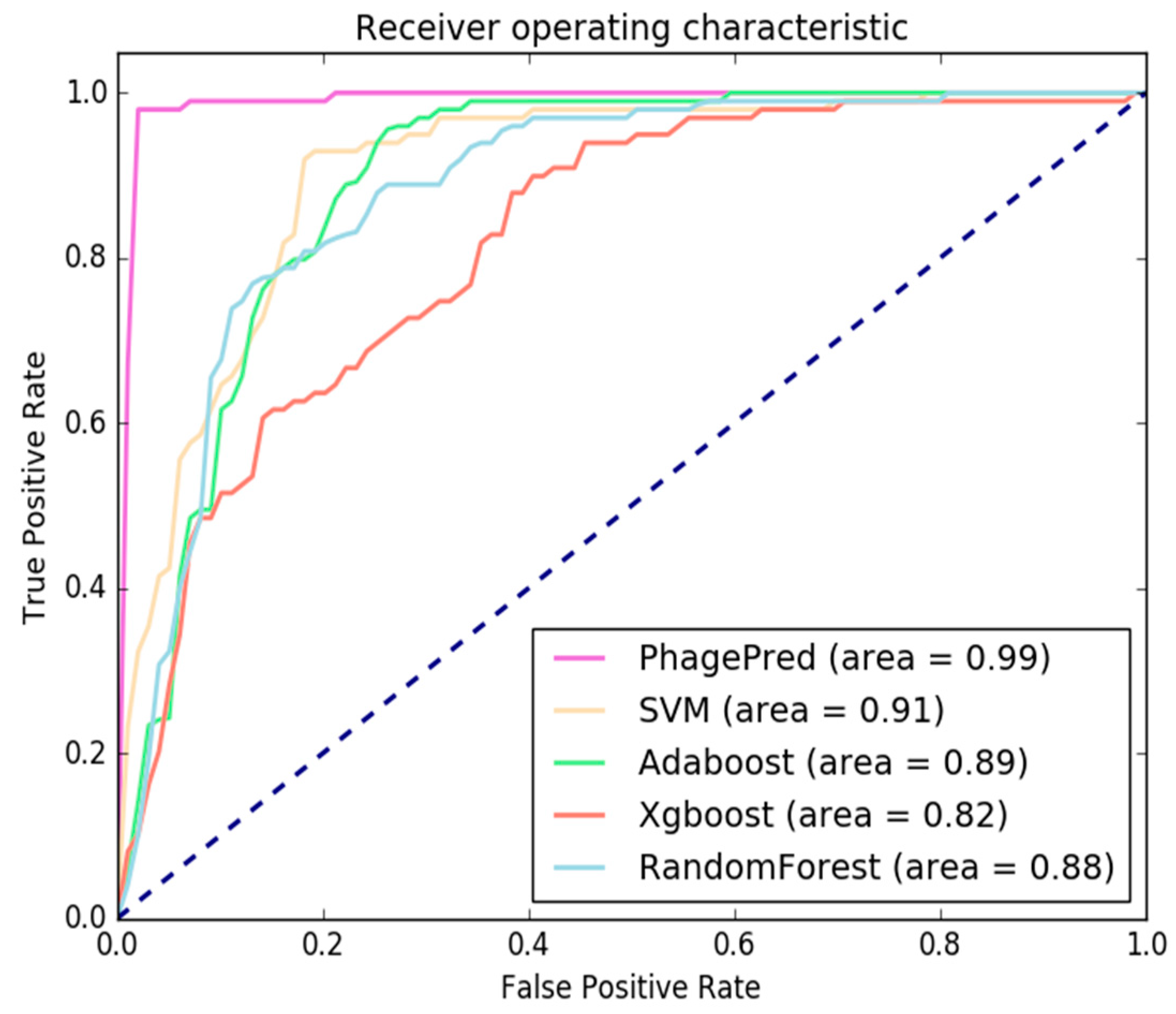
2.3. Comparison with Different Classifiers
2.4. Comparison with Existing Methods
3. Discussion
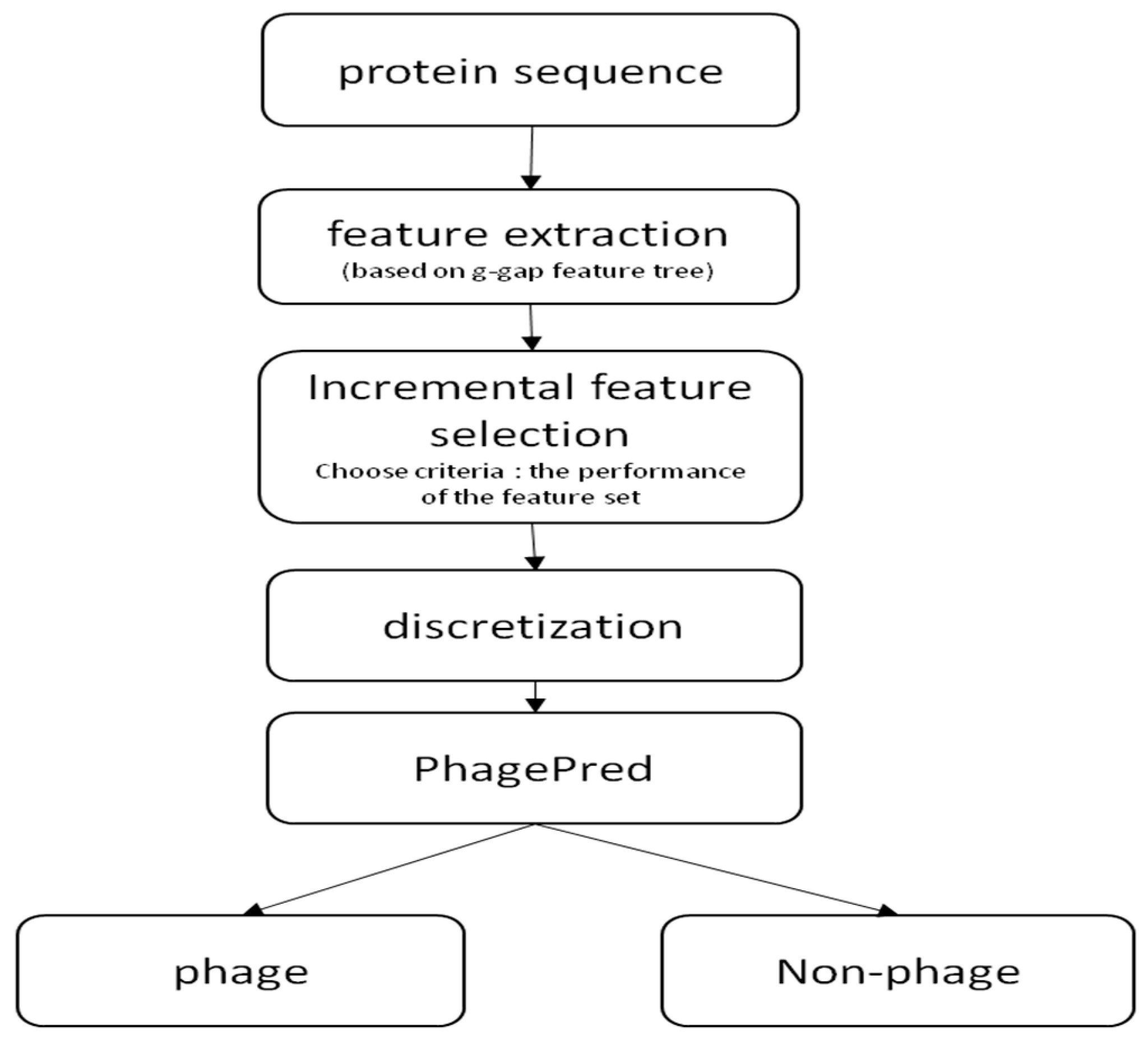
4. Materials and Methods
4.1. Benchmark Dataset
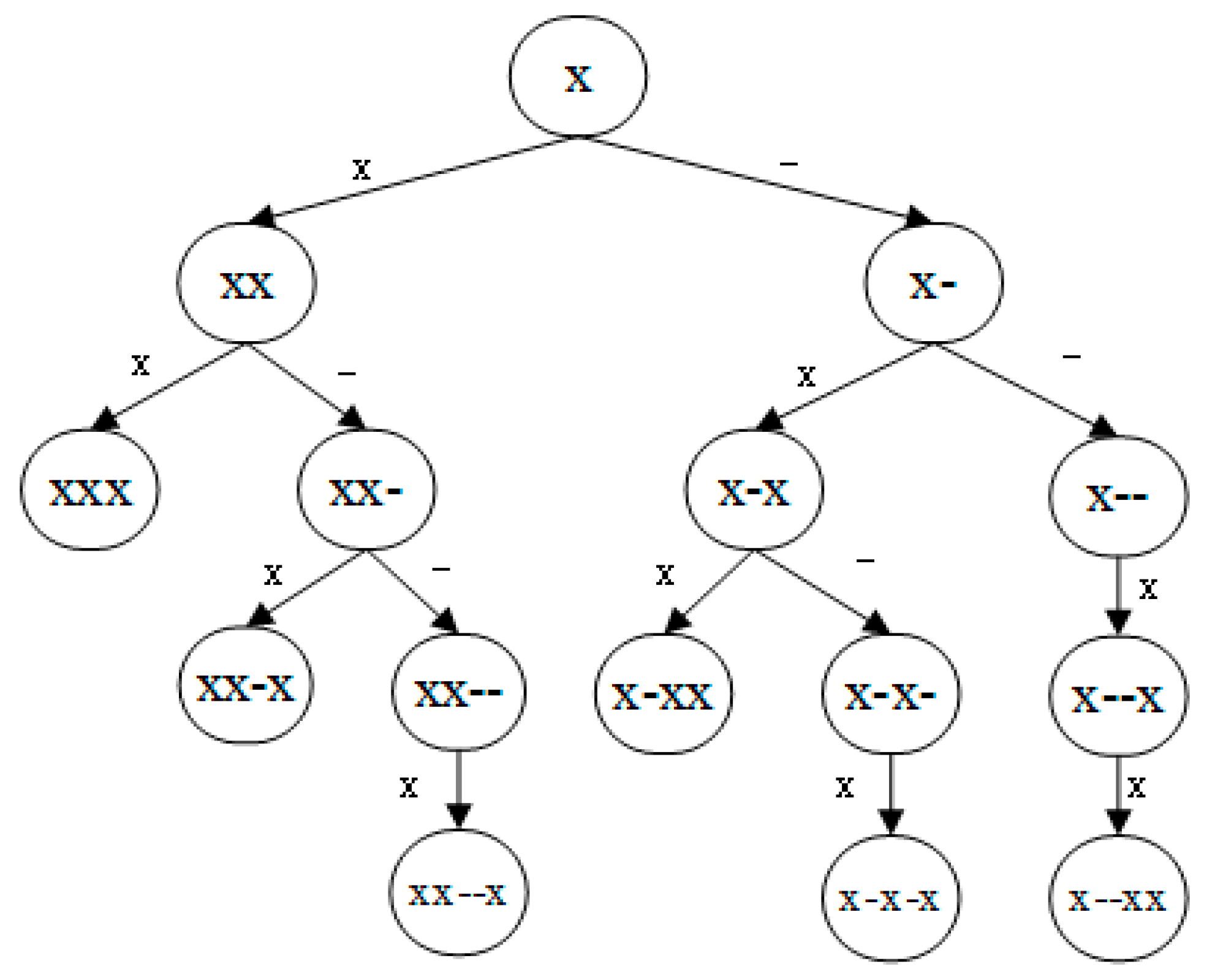
4.2. g-Gap Feature Tree
4.3. Discrete Feature Vector and Classifier
4.3.1. Transformation
4.3.2. ANOVA
4.3.3. Discretization
4.3.4. Multinomial Naïve Bayes
4.4. Evaluation Measurements
5. Web Server
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Denton, C.; Crosby, R.J. Bacteriophages: Biology, Applications and Role in Health and Disease; Nova Biomedical Press: Waltham, MA, USA, 2013. [Google Scholar]
- Wommack, K.E.; Colwell, R.R. Virioplankton: Viruses in aquatic ecosystems. Microbiol. Mol. Biol. Rev. 2000, 64, 69–114. [Google Scholar] [CrossRef] [PubMed]
- Wagner, P.L.; Waldor, M.K. Bacteriophage control of bacterial virulence. Infect. Immun. 2002, 70, 3985–3993. [Google Scholar] [CrossRef] [PubMed]
- Drulis-Kawa, Z.; Majkowska-Skrobek, G.; Maciejewska, B.; Delattre, A.S.; Lavigne, R. Learning from bacteriophages-advantages and limitations of bacteriophage and bacteriophage-encoded protein applications. Curr. Protein Pept. Sci. 2002, 13, 699–722. [Google Scholar] [CrossRef]
- Schaechter, M. Desk Encyclopedia of Microbiology; Academic Press: Cambridge, MA, USA, 2010. [Google Scholar]
- Hanlon, G.W. Bacteriophages: An appraisal of their role in the treatment of bacterial infections. Int. J. Antimicrob. Agents 2007, 30, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, H.W. 5500 phages examined in the electron microscope. Arch. Virol. 2007, 152, 227–243. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Halgamuge, S.K.; Kells, C.I.; Tang, S.L. Gene function prediction based on genomic context clustering and discriminative learning: An application to bacteriophages. BMC Bioinf. 2007, 8, S6. [Google Scholar] [CrossRef] [PubMed]
- Seguritan, V.; Alves, N., Jr.; Arnoult, M.; Raymond, A.; Lorimer, D.; Burgin, A.B., Jr.; Salamon, P.; Segall, A.M. Artificial neural networks trained to detect viral and bacteriophage structural proteins. PloS Comput. Biol. 2012, 8, e1002657. [Google Scholar] [CrossRef] [PubMed]
- Feng, P.M.; Ding, H.; Chen, W.; Lin, H. Naïve bayes classifier with feature selection to identify bacteriophage virion proteins. Comput. Math. Methods Med. 2013, 2013, 530696. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Feng, P.M.; Chen, W.; Lin, H. Identification of bacteriophage virion proteins by the anova feature selection and analysis. Mol. Biosyst. 2014, 10, 2229–2235. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, C.; Gao, R.; Yang, R. An ensemble method to distinguish bacteriophage virion from non-virion proteins based on protein sequence characteristics. Int. J. Mol. Sci. 2015, 16, 21734–21758. [Google Scholar] [CrossRef] [PubMed]
- Manavalan, B.; Shin, T.H.; Lee, G. PVP-SVM: Sequence-based prediction of Phage Virion proteins using a support vector machine. Front. Microbiol. 2018, 9, 476. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Guo, S.H.; Deng, E.Z.; Yuan, L.F.; Guo, F.B.; Huang, J.; Rao, N.N.; Chen, W.; Lin, H. Prediction of Golgi-resident protein types by using feature selection technique. Chemom. Intell. Lab. Syst. 2013, 124, 9–13. [Google Scholar] [CrossRef]
- Lin, H.; Chen, W.; Ding, H. Acalpred: A sequence-based tool for discriminating between acidic and alkaline enzymes. PLoS ONE 2013, 8, e75726. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Tang, J.; Zou, Q. SkipCPP-Pred: An Improved and Promising Sequence-based Predictor for Predicting Cell-Penetrating Peptides. BMC Genom. 2017, 18, 742. [Google Scholar] [CrossRef] [PubMed]
- Caragea, C.; Silvescu, A.; Mitra, P. Protein sequence classification using feature hashing. Proteome Sci. 2012, 10, 14. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Liu, F.; Wang, X.; Chen, J.; Fang, L.; Chou, K.C. Pse-in-One: A web server for generating various modes of pseudo components of DNA, RNA, and protein sequences. Nucleic Acids Res. 2015, 43, W65–W71. [Google Scholar] [CrossRef] [PubMed]
- Magrane, M. UniProt Knowledgebase: A hub of integrated protein data. Database 2011, 2011, bar009. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Godzik, A. Fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- Feng, P.; Yang, H.; Ding, H.; Lin, H.; Chen, W.; Chou, K.C. iDNA6mA-PseKNC: Identifying DNA N6-methyladenosine sites by incorporating nucleotide physicochemical properties into PseKNC. Genomics 2018. [Google Scholar] [CrossRef] [PubMed]
- Dubchak, I.; Muchnik, I.; Holbrook, S.R.; Kim, S.H. Prediction of protein folding class using global description of amino acid sequence. Proc. Natl. Acad. Sci. USA 1995, 92, 8700–8704. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Liu, W.X.; He, J.; Liu, X.H.; Ding, H.; Chen, W. Predicting cancerlectins by the optimal g-gap dipeptides. Sci. Rep. 2015, 5, 16964. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Gallego, S.; García, S.; Mouriño-Talín, H.; Martínez-Rego, D.; Bolón-Canedo, V.; Alonso-Betanzos, A.; Benitez, J.M.; Herrera, F. Data discretization: Taxonomy and big data challenge. Wiley Interdiscip. Rev. 2016, 6, 5–21. [Google Scholar] [CrossRef]
- Lewis, D.D. Naive (Bayes) at forty: The independence assumption in information retrieval. In European Conference on Machine Learning 1998; Springer: Berlin/Heidelberg, Germany, 1998; pp. 4–15. [Google Scholar]
- Miguel-Hurtado, O.; Guest, R.; Stevenage, S.V.; Neil, G.J.; Black, S. Comparing Machine Learning Classifiers and Linear/Logistic Regression to Explore the Relationship between Hand Dimensions and Demographic Characteristics. PLoS ONE 2016, 11, e0165521. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.R.; Sun, B.Q.; Xiao, X.; Xu, Z.C.; Chou, K.C. iHyd-PseCp: Identify hydroxyproline and hydroxylysine in proteins by incorporating sequence-coupled effects into general PseAAC. Oncotarget 2016, 7, 44310. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.Y.; Shi, S.P.; Qiu, J.D.; Liu, M.C. Using support vector machines to identify protein phosphorylation sites in viruses. J. Mol. Graph. Model. 2015, 56, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Liu, Z.; Xiao, X.; Liu, B.; Chou, K.C. iCar-PseCp: Identify carbonylation sites in proteins by Monte Carlo sampling and incorporating sequence coupled effects into general PseAAC. Oncotarget 2016, 7, 34558–34570. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.C.; Shen, H.B. Recent progress in protein subcellular location prediction. Anal. Biochem. 2007, 370, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Fang, L.; Liu, F.; Wang, X.; Chou, K.C. iMiRNA-PseDPC: MicroRNA precursor identification with a pseudo distance-pair composition approach. J. Biomol. Struct. Dynam. 2016, 1, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Hou, T.; Zheng, G.; Zhang, P.; Jia, J.; Li, J.; Xie, L.; Wei, C.; Li, Y. LAceP: Lysine acetylation site prediction using logistic regression classifiers. PLoS ONE 2014, 9, e89575. [Google Scholar] [CrossRef] [PubMed]
- Kibriya, A.M.; Frank, E.; Pfahringer, B.; Holmes, G. Multinomial naive bayes for text categorization revisited. In Australasian Joint Conference on Artificial Intelligence; Springer: Berlin/Heidelberg, Germany, 2014; pp. 488–499. [Google Scholar]
- Tang, H.; Zhao, Y.W.; Zou, P.; Zhang, C.M.; Chen, R.; Huang, P.; Lin, H. HBPred: A tool to identify growth hormone-binding proteins. Int. J. Biol. Sci. 2018, 14, 957–964. [Google Scholar] [CrossRef]
- Yang, H.; Qiu, W.R.; Liu, G.; Guo, F.B.; Chen, W.; Chou, K.C.; Lin, H. iRSpot-Pse6NC: Identifying recombination spots in Saccharomyces cerevisiae by incorporating hexamer composition into general PseKNC. Int. J. Biol. Sci. 2018, 14, 883–891. [Google Scholar] [CrossRef]
- Cao, R.; Bhattacharya, D.; Hou, J.; Cheng, J. DeepQA: Improving the estimation of single protein model quality with deep belief networks. BMC Bioinf. 2016, 17, 495. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Yang, H.; Feng, P.M.; Ding, H.; Lin, H. iDNA4mC: Identifying DNA N4-methylcytosine sites based on nucleotide chemical properties. Bioinformatics 2017, 33, 3518–3523. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Freitas, C.; Chan, L.; Sun, M.; Jiang, H.; Chen, Z. ProLanGO: Protein Function Prediction Using Neural Machine Translation Based on a Recurrent Neural Network. Molecules 2017, 22, 1732. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.W.; Su, Z.D.; Yang, W.; Lin, H.; Chen, W.; Tang, H. IonchanPred 2.0: A tool to predict ion channels and their types. Int. J. Mol. Sci. 2017, 18, 1838. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; QAdhikari, B.; Bhattacharya, D.; Sun, M.; Hou, J.; Cheng, J. Acon: Single model quality assessment using protein structural and contact information with machine learning techniques. Bioinformatics 2017, 33, 586–588. [Google Scholar] [PubMed]
- Chen, W.; Feng, P.; Yang, H.; Ding, H.; Lin, H.; Chou, K.C. iRNA-3typeA: Identifying Three Types of Modification at RNA’s Adenosine Sites. Mol. Ther. Nucleic Acids 2018, 11, 468–474. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Tang, H.; Chen, X.X.; Zhang, C.J.; Zhu, P.P.; Ding, H.; Chen, W.; Lin, H. Identification of secretory proteins in mycobacterium tuberculosis using pseudo amino acid composition. Biomed. Res. Int. 2016, 2016, 5413903. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.X.; Tang, H.; Li, W.C.; Wu, H.; Chen, W.; Ding, H.; Lin, H. Identification of Bacterial Cell Wall Lyases via Pseudo Amino Acid Composition. Biomed. Res. Int. 2016, 2016, 1654623. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.; Yang, Q.; Zou, Q. NucPosPred: Predicting species-specific genomic nucleosome positioning via four different modes of general PseKNC. J. Theor. Biol. 2018, 450, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Tan, P.; Wang, L.; Jin, N.; Li, Y.; Zhang, L.; Yang, H.; Hu, Z.; Zhang, L.; Hu, C.; et al. RNALocate: A resource for RNA Subcellular Localizations. Nucleic Acids Res. 2017, 45, D135–D138. [Google Scholar] [PubMed]
- Jiang, J.; Xing, F.; Zeng, X.; Zou, Q. RicyerDB: A Database For Collecting Rice Yield-related Genes with Biological Analysis. Int. J. Biol. Sci. 2018, 14, 965–970. [Google Scholar] [CrossRef]
- Liang, Z.Y.; Lai, H.Y.; Yang, H.; Zhang, C.J.; Yang, H.; Wei, H.H.; Chen, X.X.; Zhao, Y.W.; Su, Z.D.; Li, W.C.; et al. Pro54DB: A database for experimentally verified sigma-54 promoters. Bioinformatics 2017, 33, 467–469. [Google Scholar] [CrossRef] [PubMed]
- Zou, Q.; Li, X.; Jiang, W.; Lin, Z.; Lin, G.; Chen, K. Survey of MapReduce Frame Operation in Bioinformatics. Brief. Bioinf. 2014, 15, 637–647. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Classifier | Sn (%) | Sp (%) | Acc (%) | MCC (%) |
|---|---|---|---|---|
| xgboost | 52.52 | 81.25 | 71.98 | 46.05 |
| Random Forest | 25.25 | 97.60 | 74.26 | 38.67 |
| Adaboost + CART | 52.53 | 88.94 | 77.20 | 41.03 |
| SVM | 73.74 | 90.87 | 85.34 | 65.92 |
| PhagePred | 96.97 | 99.04 | 98.37 | 96.27 |
| Classifier | Sn (%) | Sp (%) | Acc (%) | MCC (%) |
|---|---|---|---|---|
| Naïve bayes | 75.76 | 80.77 | 79.15 | 54.59 |
| SVM | 75.76 | 89.42 | 85.02 | 65.53 |
| PVP-SVM | 73.73 | 93.27 | 86.97 | 69.50 |
| PhagePred | 96.97 | 98.56 | 98.05 | 96.27 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, Y.; Gao, H.; Lin, H.; Liu, Z.; Tang, L.; Li, S. Identification of Bacteriophage Virion Proteins Using Multinomial Naïve Bayes with g-Gap Feature Tree. Int. J. Mol. Sci. 2018, 19, 1779. https://doi.org/10.3390/ijms19061779
Pan Y, Gao H, Lin H, Liu Z, Tang L, Li S. Identification of Bacteriophage Virion Proteins Using Multinomial Naïve Bayes with g-Gap Feature Tree. International Journal of Molecular Sciences. 2018; 19(6):1779. https://doi.org/10.3390/ijms19061779
Chicago/Turabian StylePan, Yanyuan, Hui Gao, Hao Lin, Zhen Liu, Lixia Tang, and Songtao Li. 2018. "Identification of Bacteriophage Virion Proteins Using Multinomial Naïve Bayes with g-Gap Feature Tree" International Journal of Molecular Sciences 19, no. 6: 1779. https://doi.org/10.3390/ijms19061779
APA StylePan, Y., Gao, H., Lin, H., Liu, Z., Tang, L., & Li, S. (2018). Identification of Bacteriophage Virion Proteins Using Multinomial Naïve Bayes with g-Gap Feature Tree. International Journal of Molecular Sciences, 19(6), 1779. https://doi.org/10.3390/ijms19061779