Molecular Mechanisms of Oligodendrocyte Regeneration in White Matter-Related Diseases


Abstract
1. Introduction
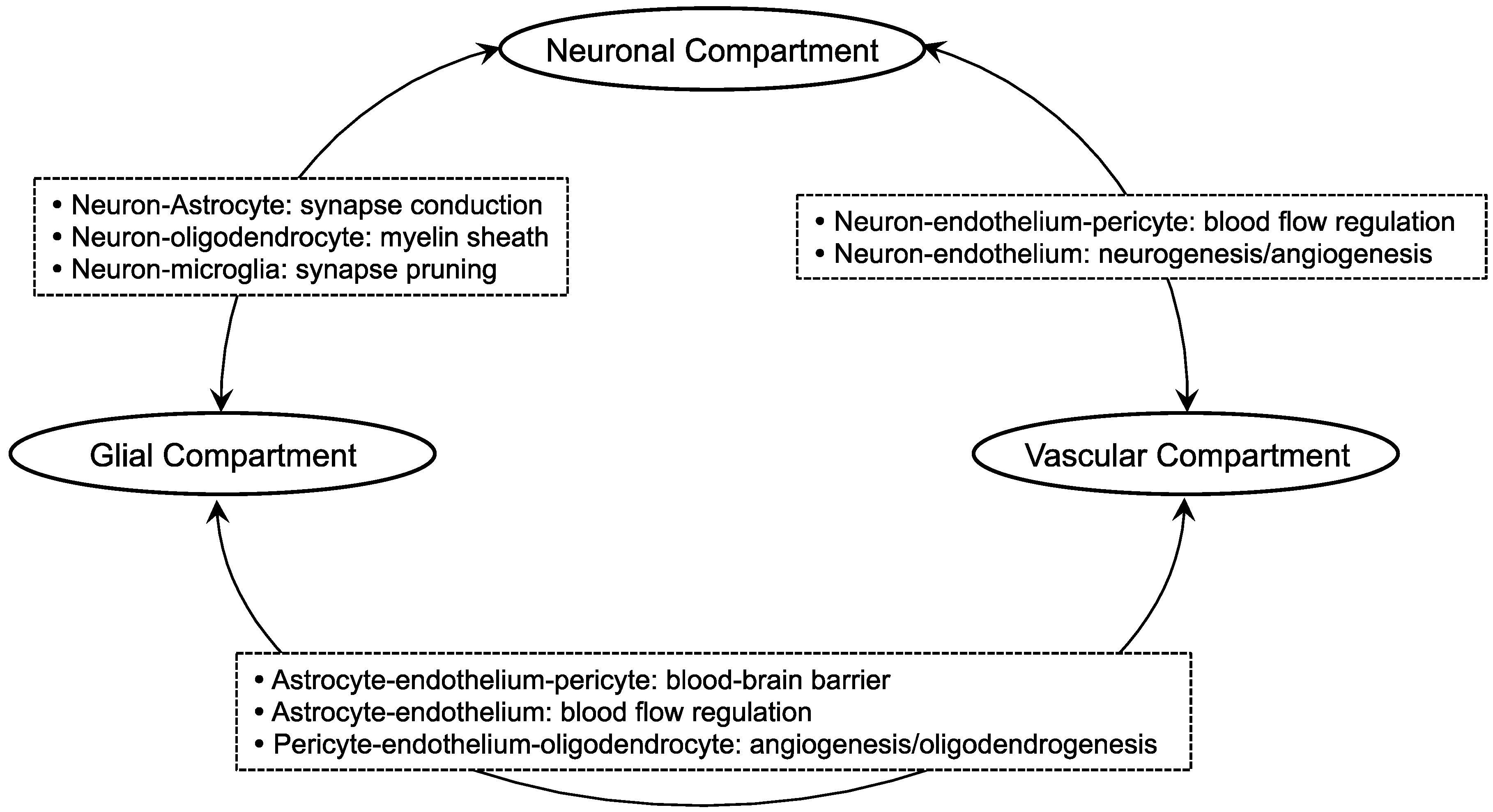
2. Oligodendrocyte-Related Cell-Cell Interaction in White Matter
3. Molecular Mechanisms of Oligodendrocyte Repair/Damage in Neurodegenerative Diseases
3.1. Vascular Cognitive Impairment Syndrome (VCI)
3.2. Alzheimer’s Disease (AD)
3.3. Multiple Sclerosis (MS)
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Del Zoppo, G.J.; Mabuchi, T. Cerebral microvessel responses to focal ischemia. J. Cereb. Blood Flow Metab. 2003, 23, 879–894. [Google Scholar] [CrossRef] [PubMed]
- Lo, E.H.; Dalkara, T.; Moskowitz, M.A. Mechanisms, challenges and opportunities in stroke. Nat. Rev. Neurosci. 2003, 4, 399–415. [Google Scholar] [CrossRef] [PubMed]
- Maki, T.; Hayakawa, K.; Pham, L.D.; Xing, C.; Lo, E.H.; Arai, K. Biphasic mechanisms of neurovascular unit injury and protection in CNS diseases. CNS Neurol. Disord. Drug Targets 2013, 12, 302–315. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C. Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat. Rev. Neurosci. 2004, 5, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 2008, 57, 178–201. [Google Scholar] [CrossRef] [PubMed]
- Garbuzova-Davis, S.; Rodrigues, M.C.; Hernandez-Ontiveros, D.G.; Louis, M.K.; Willing, A.E.; Borlongan, C.V.; Sanberg, P.R. Amyotrophic lateral sclerosis: A neurovascular disease. Brain Res. 2011, 1398, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, A.; Majed, H.; Layfield, R.; Compston, A.; Chandran, S. Oligodendrocytes promote neuronal survival and axonal length by distinct intracellular mechanisms: A novel role for oligodendrocyte-derived glial cell line-derived neurotrophic factor. J. Neurosci. 2003, 23, 4967–4974. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Sdrulla, A.D.; diSibio, G.; Bush, G.; Nofziger, D.; Hicks, C.; Weinmaster, G.; Barres, B.A. Notch receptor activation inhibits oligodendrocyte differentiation. Neuron 1998, 21, 63–75. [Google Scholar] [CrossRef]
- Doyle, S.; Hansen, D.B.; Vella, J.; Bond, P.; Harper, G.; Zammit, C.; Valentino, M.; Fern, R. Vesicular glutamate release from central axons contributes to myelin damage. Nat. Commun. 2018, 9, 1032. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C.; Nedergaard, M. Glial regulation of the cerebral microvasculature. Nat. Neurosci. 2007, 10, 1369–1376. [Google Scholar] [CrossRef] [PubMed]
- Butt, A.M.; Ibrahim, M.; Ruge, F.M.; Berry, M. Biochemical subtypes of oligodendrocyte in the anterior medullary velum of the rat as revealed by the monoclonal antibody Rip. Glia 1995, 14, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Orthmann-Murphy, J.L.; Abrams, C.K.; Scherer, S.S. Gap junctions couple astrocytes and oligodendrocytes. J. Mol. Neurosci. 2008, 35, 101–116. [Google Scholar] [CrossRef] [PubMed]
- Arai, K.; Lo, E.H. Astrocytes protect oligodendrocyte precursor cells via MEK/ERK and PI3K/Akt signaling. J. Neurosci. Res. 2010, 88, 758–763. [Google Scholar] [CrossRef] [PubMed]
- Franklin, R.J.; Crang, A.J.; Blakemore, W.F. Transplanted type-1 astrocytes facilitate repair of demyelinating lesions by host oligodendrocytes in adult rat spinal cord. J. Neurocytol. 1991, 20, 420–430. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, S.P.; Rome, L.H. Stimulation of in vitro myelin synthesis by microglia. Glia 1994, 11, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Nicholas, R.S.; Stevens, S.; Wing, M.G.; Compston, D.A. Microglia-derived IGF-2 prevents TNFα induced death of mature oligodendrocytes in vitro. J. Neuroimmunol. 2002, 124, 36–44. [Google Scholar] [CrossRef]
- Nicholas, R.S.; Wing, M.G.; Compston, A. Nonactivated microglia promote oligodendrocyte precursor survival and maturation through the transcription factor NF-κB. Eur. J. Neurosci. 2001, 13, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Shi, Y.; Jiang, X.; Leak, R.K.; Hu, X.; Wu, Y.; Pu, H.; Li, W.W.; Tang, B.; Wang, Y.; et al. HDAC inhibition prevents white matter injury by modulating microglia/macrophage polarization through the GSK3β/PTEN/Akt axis. Proc. Natl. Acad. Sci. USA 2015, 112, 2853–2858. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.H.; Maki, T.; Maeda, M.; Miyamoto, N.; Liang, A.C.; Hayakawa, K.; Pham, L.D.; Suwa, F.; Taguchi, A.; Matsuyama, T.; et al. Oligodendrocyte precursor cells support blood-brain barrier integrity via TGF-β signaling. PLoS ONE 2014, 9, e103174. [Google Scholar] [CrossRef] [PubMed]
- Maki, T.; Maeda, M.; Uemura, M.; Lo, E.K.; Terasaki, Y.; Liang, A.C.; Shindo, A.; Choi, Y.K.; Taguchi, A.; Matsuyama, T.; et al. Potential interactions between pericytes and oligodendrocyte precursor cells in perivascular regions of cerebral white matter. Neurosci. Lett. 2015, 597, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Dimou, L.; Simon, C.; Kirchhoff, F.; Takebayashi, H.; Gotz, M. Progeny of Olig2-expressing progenitors in the gray and white matter of the adult mouse cerebral cortex. J. Neurosci. 2008, 28, 10434–10442. [Google Scholar] [CrossRef] [PubMed]
- Young, K.M.; Psachoulia, K.; Tripathi, R.B.; Dunn, S.J.; Cossell, L.; Attwell, D.; Tohyama, K.; Richardson, W.D. Oligodendrocyte dynamics in the healthy adult CNS: Evidence for myelin remodeling. Neuron 2013, 77, 873–885. [Google Scholar] [CrossRef] [PubMed]
- Maki, T.; Liang, A.C.; Miyamoto, N.; Lo, E.H.; Arai, K. Mechanisms of oligodendrocyte regeneration from ventricular-subventricular zone-derived progenitor cells in white matter diseases. Front. Cell. Neurosci. 2013, 7, 275. [Google Scholar] [CrossRef] [PubMed]
- Hughes, E.G.; Kang, S.H.; Fukaya, M.; Bergles, D.E. Oligodendrocyte progenitors balance growth with self-repulsion to achieve homeostasis in the adult brain. Nat. Neurosci. 2013, 16, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Menn, B.; Garcia-Verdugo, J.M.; Yaschine, C.; Gonzalez-Perez, O.; Rowitch, D.; Alvarez-Buylla, A. Origin of oligodendrocytes in the subventricular zone of the adult brain. J. Neurosci. 2006, 26, 7907–7918. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Perez, O.; Romero-Rodriguez, R.; Soriano-Navarro, M.; Garcia-Verdugo, J.M.; Alvarez-Buylla, A. Epidermal growth factor induces the progeny of subventricular zone type B cells to migrate and differentiate into oligodendrocytes. Stem Cells 2009, 27, 2032–2043. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Perez, O.; Alvarez-Buylla, A. Oligodendrogenesis in the subventricular zone and the role of epidermal growth factor. Brain Res. Rev. 2011, 67, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Marinovich, A.; Barres, B.A. Purification and characterization of adult oligodendrocyte precursor cells from the rat optic nerve. J. Neurosci. 1998, 18, 4627–4636. [Google Scholar] [CrossRef] [PubMed]
- McTigue, D.M.; Wei, P.; Stokes, B.T. Proliferation of NG2-positive cells and altered oligodendrocyte numbers in the contused rat spinal cord. J. Neurosci. 2001, 21, 3392–3400. [Google Scholar] [CrossRef] [PubMed]
- Moyon, S.; Dubessy, A.L.; Aigrot, M.S.; Trotter, M.; Huang, J.K.; Dauphinot, L.; Potier, M.C.; Kerninon, C.; Melik Parsadaniantz, S.; Franklin, R.J.; et al. Demyelination causes adult CNS progenitors to revert to an immature state and express immune cues that support their migration. J. Neurosci. 2015, 35, 4–20. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Goderie, S.K.; Jin, L.; Karanth, N.; Sun, Y.; Abramova, N.; Vincent, P.; Pumiglia, K.; Temple, S. Endothelial cells stimulate self-renewal and expand neurogenesis of neural stem cells. Science 2004, 304, 1338–1340. [Google Scholar] [CrossRef] [PubMed]
- Snapyan, M.; Lemasson, M.; Brill, M.S.; Blais, M.; Massouh, M.; Ninkovic, J.; Gravel, C.; Berthod, F.; Gotz, M.; Barker, P.A.; et al. Vasculature guides migrating neuronal precursors in the adult mammalian forebrain via brain-derived neurotrophic factor signaling. J. Neurosci. 2009, 29, 4172–4188. [Google Scholar] [CrossRef] [PubMed]
- Chintawar, S.; Cayrol, R.; Antel, J.; Pandolfo, M.; Prat, A. Blood-brain barrier promotes differentiation of human fetal neural precursor cells. Stem Cells 2009, 27, 838–846. [Google Scholar] [CrossRef] [PubMed]
- Plane, J.M.; Andjelkovic, A.V.; Keep, R.F.; Parent, J.M. Intact and injured endothelial cells differentially modulate postnatal murine forebrain neural stem cells. Neurobiol. Dis. 2010, 37, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Arai, K.; Lo, E.H. An oligovascular niche: Cerebral endothelial cells promote the survival and proliferation of oligodendrocyte precursor cells. J. Neurosci. 2009, 29, 4351–4355. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, K.; Seo, J.; Pham, L.-D.; Miyamoto, N.; Som, A.; Guo, S.; Kim, K.-W.; Lo, E.; Arai, K. Cerebral endothelial derived vascular endothelial growth factor promotes the migration but not the proliferation of oligodendrocyte precursor cells in vitro. Neurosci. Lett. 2012, 513, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Kim, W.J.; Lok, J.; Lee, S.R.; Besancon, E.; Luo, B.H.; Stins, M.F.; Wang, X.; Dedhar, S.; Lo, E.H. Neuroprotection via matrix-trophic coupling between cerebral endothelial cells and neurons. Proc. Natl. Acad. Sci. USA 2008, 105, 7582–7587. [Google Scholar] [CrossRef] [PubMed]
- Montagne, A.; Nikolakopoulou, A.M.; Zhao, Z.; Sagare, A.P.; Si, G.; Lazic, D.; Barnes, S.R.; Daianu, M.; Ramanathan, A.; Go, A.; et al. Pericyte degeneration causes white matter dysfunction in the mouse central nervous system. Nat. Med. 2018, 24, 326–337. [Google Scholar] [CrossRef] [PubMed]
- Maki, T.; Choi, Y.K.; Miyamoto, N.; Shindo, A.; Liang, A.C.; Ahn, B.J.; Mandeville, E.T.; Kaji, S.; Itoh, K.; Seo, J.H.; et al. A-Kinase Anchor Protein 12 Is Required for Oligodendrocyte Differentiation in Adult White Matter. Stem Cells 2018. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C.; Anrather, J. The immunology of stroke: From mechanisms to translation. Nat. Med. 2011, 17, 796–808. [Google Scholar] [CrossRef] [PubMed]
- Erkinjuntti, T.; Inzitari, D.; Pantoni, L.; Wallin, A.; Scheltens, P.; Rockwood, K.; Desmond, D.W. Limitations of clinical criteria for the diagnosis of vascular dementia in clinical trials. Is a focus on subcortical vascular dementia a solution? Ann. N.Y. Acad. Sci. 2000, 903, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Erkinjuntti, T.; Inzitari, D.; Pantoni, L.; Wallin, A.; Scheltens, P.; Rockwood, K.; Roman, G.C.; Chui, H.; Desmond, D.W. Research criteria for subcortical vascular dementia in clinical trials. J. Neural Transm. Suppl. 2000, 59, 23–30. [Google Scholar] [PubMed]
- Roman, G.C.; Erkinjuntti, T.; Wallin, A.; Pantoni, L.; Chui, H.C. Subcortical ischaemic vascular dementia. Lancet Neurol. 2002, 1, 426–436. [Google Scholar] [CrossRef]
- Prins, N.D.; Scheltens, P. White matter hyperintensities, cognitive impairment and dementia: An update. Nat. Rev. Neurol. 2015, 11, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Roh, J.H.; Lee, J.H. Recent updates on subcortical ischemic vascular dementia. J. Stroke 2014, 16, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Ekonomou, A.; Ballard, C.G.; Pathmanaban, O.N.; Perry, R.H.; Perry, E.K.; Kalaria, R.N.; Minger, S.L. Increased neural progenitors in vascular dementia. Neurobiol. Aging 2011, 32, 2152–2161. [Google Scholar] [CrossRef] [PubMed]
- Baltan, S. Ischemic injury to white matter: An age-dependent process. Neuroscientist 2009, 15, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, M.; Sako, K.; Yura, S.; Yonemasu, Y. Cerebral blood flow and histopathological changes following permanent bilateral carotid artery ligation in Wistar rats. Exp. Brain Res. 1992, 89, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Kurumatani, T.; Kudo, T.; Ikura, Y.; Takeda, M. White matter changes in the gerbil brain under chronic cerebral hypoperfusion. Stroke 1998, 29, 1058–1062. [Google Scholar] [CrossRef] [PubMed]
- Shibata, M.; Ohtani, R.; Ihara, M.; Tomimoto, H. White matter lesions and glial activation in a novel mouse model of chronic cerebral hypoperfusion. Stroke 2004, 35, 2598–2603. [Google Scholar] [CrossRef] [PubMed]
- Venkat, P.; Chopp, M.; Chen, J. Models and mechanisms of vascular dementia. Exp. Neurol. 2015, 272, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Ihara, M.; Taguchi, A.; Maki, T.; Washida, K.; Tomimoto, H. A mouse model of chronic cerebral hypoperfusion characterizing features of vascular cognitive impairment. Methods Mol. Biol. 2014, 1135, 95–102. [Google Scholar] [PubMed]
- Edaravone Acute Infarction Study Group. Effect of a novel free radical scavenger, edaravone (MCI-186), on acute brain infarction. Randomized, placebo-controlled, double-blind study at multicenters. Cerebrovasc. Dis. 2003, 15, 222–229. [Google Scholar]
- Lapchak, P.A. A critical assessment of edaravone acute ischemic stroke efficacy trials: Is edaravone an effective neuroprotective therapy? Expert Opin. Pharmacother. 2010, 11, 1753–1763. [Google Scholar] [CrossRef] [PubMed]
- Morihara, R.; Kono, S.; Sato, K.; Hishikawa, N.; Ohta, Y.; Yamashita, T.; Deguchi, K.; Manabe, Y.; Takao, Y.; Kashihara, K.; et al. Thrombolysis with Low-Dose Tissue Plasminogen Activator 3–4.5 h After Acute Ischemic Stroke in Five Hospital Groups in Japan. Transl. Stroke Res. 2016, 7, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Nagase, M.; Yamamoto, Y.; Miyazaki, Y.; Yoshino, H. Increased oxidative stress in patients with amyotrophic lateral sclerosis and the effect of edaravone administration. Redox Rep. 2016, 21, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Itoyama, Y.; Sobue, G.; Tsuji, S.; Aoki, M.; Doyu, M.; Hamada, C.; Kondo, K.; Yoneoka, T.; Akimoto, M.; et al. Confirmatory double-blind, parallel-group, placebo-controlled study of efficacy and safety of edaravone (MCI-186) in amyotrophic lateral sclerosis patients. Amyotroph. Lateral Scler. Frontotemporal Degener. 2014, 15, 610–617. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, N.; Maki, T.; Pham, L.D.; Hayakawa, K.; Seo, J.H.; Mandeville, E.T.; Mandeville, J.B.; Kim, K.W.; Lo, E.H.; Arai, K. Oxidative stress interferes with white matter renewal after prolonged cerebral hypoperfusion in mice. Stroke 2013, 44, 3516–3521. [Google Scholar] [CrossRef] [PubMed]
- Takase, H.; Liang, A.C.; Miyamoto, N.; Hamanaka, G.; Ohtomo, R.; Maki, T.; Pham, L.D.; Lok, J.; Lo, E.H.; Arai, K. Protective effects of a radical scavenger edaravone on oligodendrocyte precursor cells against oxidative stress. Neurosci. Lett. 2018, 668, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, N.; Pham, L.D.; Maki, T.; Liang, A.C.; Arai, K. A radical scavenger edaravone inhibits matrix metalloproteinase-9 upregulation and blood-brain barrier breakdown in a mouse model of prolonged cerebral hypoperfusion. Neurosci. Lett. 2014, 573, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, N.; Pham, L.D.; Hayakawa, K.; Matsuzaki, T.; Seo, J.H.; Magnain, C.; Ayata, C.; Kim, K.W.; Boas, D.; Lo, E.H.; et al. Age-related decline in oligodendrogenesis retards white matter repair in mice. Stroke 2013, 44, 2573–2578. [Google Scholar] [CrossRef] [PubMed]
- Ohtomo, R.; Bannai, T.; Ohtomo, G.; Shindo, A.; Tomimoto, H.; Tsuji, S.; Iwata, A. Cilostazol alleviates white matter degeneration caused by chronic cerebral hypoperfusion in mice: Implication of its mechanism from gene expression analysis. Neurosci. Lett. 2018, 662, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Torta, F.; Arai, K.; Wenk, M.R.; Herr, D.R.; Wong, P.T.; Lai, M.K. Sphingosine kinase inhibition ameliorates chronic hypoperfusion-induced white matter lesions. Neurochem. Int. 2016, 94, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Higaki, A.; Mogi, M.; Iwanami, J.; Min, L.J.; Bai, H.Y.; Shan, B.S.; Kukida, M.; Yamauchi, T.; Tsukuda, K.; Kan-No, H.; et al. Beneficial Effect of Mas Receptor Deficiency on Vascular Cognitive Impairment in the Presence of Angiotensin II Type 2 Receptor. J. Am. Heart Assoc. 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Fu, P.; Yu, Z.; Xie, M.; Wang, W.; Luo, X. NKCC1 Inhibition Attenuates Chronic Cerebral Hypoperfusion-Induced White Matter Lesions by Enhancing Progenitor Cells of Oligodendrocyte Proliferation. J. Mol. Neurosci. 2018, 64, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Miyanohara, J.; Kakae, M.; Nagayasu, K.; Nakagawa, T.; Mori, Y.; Arai, K.; Shirakawa, H.; Kaneko, S. TRPM2 Channel Aggravates CNS Inflammation and Cognitive Impairment via Activation of Microglia in Chronic Cerebral Hypoperfusion. J. Neurosci. 2018, 38, 3520–3533. [Google Scholar] [CrossRef] [PubMed]
- Yatomi, Y.; Tanaka, R.; Shimada, Y.; Yamashiro, K.; Liu, M.; Mitome-Mishima, Y.; Miyamoto, N.; Ueno, Y.; Urabe, T.; Hattori, N. Type 2 diabetes reduces the proliferation and survival of oligodendrocyte progenitor cells in ishchemic white matter lesions. Neuroscience 2015, 289, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, N.; Maki, T.; Shindo, A.; Liang, A.C.; Maeda, M.; Egawa, N.; Itoh, K.; Lo, E.K.; Lok, J.; Ihara, M.; et al. Astrocytes Promote Oligodendrogenesis after White Matter Damage via Brain-Derived Neurotrophic Factor. J. Neurosci. 2015, 35, 14002–14008. [Google Scholar] [CrossRef] [PubMed]
- Uemura, M.T.; Ihara, M.; Maki, T.; Nakagomi, T.; Kaji, S.; Uemura, K.; Matsuyama, T.; Kalaria, R.N.; Kinoshita, A.; Takahashi, R. Pericyte-derived bone morphogenetic protein 4 underlies white matter damage after chronic hypoperfusion. Brain Pathol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Maki, T.; Morancho, A.; Martinez-San Segundo, P.; Hayakawa, K.; Takase, H.; Liang, A.C.; Gabriel-Salazar, M.; Medina-Gutierrez, E.; Washida, K.; Montaner, J.; et al. Endothelial Progenitor Cell Secretome and Oligovascular Repair in a Mouse Model of Prolonged Cerebral Hypoperfusion. Stroke 2018, 49, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Kimura-Ohba, S.; Thompson, J.; Rosenberg, G.A. Rodent Models of Vascular Cognitive Impairment. Transl. Stroke Res. 2016, 7, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Brickman, A.M.; Provenzano, F.A.; Muraskin, J.; Manly, J.J.; Blum, S.; Apa, Z.; Stern, Y.; Brown, T.R.; Luchsinger, J.A.; Mayeux, R. Regional white matter hyperintensity volume, not hippocampal atrophy, predicts incident Alzheimer disease in the community. Arch. Neurol. 2012, 69, 1621–1627. [Google Scholar] [CrossRef] [PubMed]
- Brickman, A.M. Contemplating Alzheimer’s disease and the contribution of white matter hyperintensities. Curr. Neurol. Neurosci. Rep. 2013, 13, 415. [Google Scholar] [CrossRef] [PubMed]
- Brickman, A.M.; Zahodne, L.B.; Guzman, V.A.; Narkhede, A.; Meier, I.B.; Griffith, E.Y.; Provenzano, F.A.; Schupf, N.; Manly, J.J.; Stern, Y.; et al. Reconsidering harbingers of dementia: Progression of parietal lobe white matter hyperintensities predicts Alzheimer’s disease incidence. Neurobiol. Aging 2015, 36, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Tosto, G.; Zimmerman, M.E.; Carmichael, O.T.; Brickman, A.M. Predicting aggressive decline in mild cognitive impairment: The importance of white matter hyperintensities. JAMA Neurol. 2014, 71, 872–877. [Google Scholar] [CrossRef] [PubMed]
- Marner, L.; Nyengaard, J.R.; Tang, Y.; Pakkenberg, B. Marked loss of myelinated nerve fibers in the human brain with age. J. Comp. Neurol. 2003, 462, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Bartzokis, G. Alzheimer’s disease as homeostatic responses to age-related myelin breakdown. Neurobiol. Aging 2011, 32, 1341–1371. [Google Scholar] [CrossRef] [PubMed]
- Brown, W.R.; Moody, D.M.; Thore, C.R.; Anstrom, J.A.; Challa, V.R. Microvascular changes in the white mater in dementia. J. Neurol. Sci. 2009, 283, 28–31. [Google Scholar] [CrossRef] [PubMed]
- McAleese, K.E.; Walker, L.; Graham, S.; Moya, E.L.J.; Johnson, M.; Erskine, D.; Colloby, S.J.; Dey, M.; Martin-Ruiz, C.; Taylor, J.P.; et al. Parietal white matter lesions in Alzheimer's disease are associated with cortical neurodegenerative pathology, but not with small vessel disease. Acta Neuropathol. 2017, 134, 459–473. [Google Scholar] [CrossRef] [PubMed]
- Raj, D.; Yin, Z.; Breur, M.; Doorduin, J.; Holtman, I.R.; Olah, M.; Mantingh-Otter, I.J.; van Dam, D.; de Deyn, P.P.; den Dunnen, W.; et al. Increased White Matter Inflammation in Aging- and Alzheimer’s Disease Brain. Front. Mol. Neurosci. 2017, 10, 206. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R., Jr.; Wiste, H.J.; Weigand, S.D.; Therneau, T.M.; Knopman, D.S.; Lowe, V.; Vemuri, P.; Mielke, M.M.; Roberts, R.O.; Machulda, M.M.; et al. Age-specific and sex-specific prevalence of cerebral β-amyloidosis, tauopathy, and neurodegeneration in cognitively unimpaired individuals aged 50–95 years: A cross-sectional study. Lancet Neurol. 2017, 16, 435–444. [Google Scholar] [CrossRef]
- Skoog, I.; Kern, S.; Zetterberg, H.; Ostling, S.; Borjesson-Hanson, A.; Guo, X.; Blennow, K. Low Cerebrospinal Fluid Aβ42 and Aβ40 are Related to White Matter Lesions in Cognitively Normal Elderly. J. Alzheimer’s Dis. 2018, 62, 1877–1886. [Google Scholar] [CrossRef] [PubMed]
- Van Leijsen, E.M.C.; Kuiperij, H.B.; Kersten, I.; Bergkamp, M.I.; van Uden, I.W.M.; Vanderstichele, H.; Stoops, E.; Claassen, J.; van Dijk, E.J.; de Leeuw, F.E.; et al. Plasma Aβ (Amyloid-β) Levels and Severity and Progression of Small Vessel Disease. Stroke 2018, 49, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Behrendt, G.; Baer, K.; Buffo, A.; Curtis, M.A.; Faull, R.L.; Rees, M.I.; Gotz, M.; Dimou, L. Dynamic changes in myelin aberrations and oligodendrocyte generation in chronic amyloidosis in mice and men. Glia 2013, 61, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Gagyi, E.; Kormos, B.; Castellanos, K.J.; Valyi-Nagy, K.; Korneff, D.; LoPresti, P.; Woltjer, R.; Valyi-Nagy, T. Decreased oligodendrocyte nuclear diameter in Alzheimer's disease and Lewy body dementia. Brain Pathol. 2012, 22, 803–810. [Google Scholar] [CrossRef] [PubMed]
- Fernando, M.S.; Simpson, J.E.; Matthews, F.; Brayne, C.; Lewis, C.E.; Barber, R.; Kalaria, R.N.; Forster, G.; Esteves, F.; Wharton, S.B.; et al. White matter lesions in an unselected cohort of the elderly: Molecular pathology suggests origin from chronic hypoperfusion injury. Stroke 2006, 37, 1391–1398. [Google Scholar] [CrossRef] [PubMed]
- Pak, K.; Chan, S.L.; Mattson, M.P. Presenilin-1 mutation sensitizes oligodendrocytes to glutamate and amyloid toxicities, and exacerbates white matter damage and memory impairment in mice. Neuromol. Med. 2003, 3, 53–64. [Google Scholar] [CrossRef]
- Desai, M.K.; Mastrangelo, M.A.; Ryan, D.A.; Sudol, K.L.; Narrow, W.C.; Bowers, W.J. Early oligodendrocyte/myelin pathology in Alzheimer's disease mice constitutes a novel therapeutic target. Am. J. Pathol. 2010, 177, 1422–1435. [Google Scholar] [CrossRef] [PubMed]
- Collins-Praino, L.E.; Francis, Y.I.; Griffith, E.Y.; Wiegman, A.F.; Urbach, J.; Lawton, A.; Honig, L.S.; Cortes, E.; Vonsattel, J.P.; Canoll, P.D.; et al. Soluble amyloid β levels are elevated in the white matter of Alzheimer’s patients, independent of cortical plaque severity. Acta Neuropathol. Commun. 2014, 2, 83. [Google Scholar] [CrossRef] [PubMed]
- Desai, M.K.; Guercio, B.J.; Narrow, W.C.; Bowers, W.J. An Alzheimer’s disease-relevant presenilin-1 mutation augments amyloid-β-induced oligodendrocyte dysfunction. Glia 2011, 59, 627–640. [Google Scholar] [CrossRef] [PubMed]
- Jantaratnotai, N.; Ryu, J.K.; Kim, S.U.; McLarnon, J.G. Amyloid β peptide-induced corpus callosum damage and glial activation in vivo. Neuroreport 2003, 14, 1429–1433. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.T.; Xu, J.; Lee, J.M.; Ku, G.; Han, X.; Yang, D.I.; Chen, S.; Hsu, C.Y. Amyloid-β peptide induces oligodendrocyte death by activating the neutral sphingomyelinase-ceramide pathway. J. Cell Biol. 2004, 164, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Chen, S.; Ahmed, S.H.; Chen, H.; Ku, G.; Goldberg, M.P.; Hsu, C.Y. Amyloid-β peptides are cytotoxic to oligodendrocytes. J. Neurosci. 2001, 21, RC118. [Google Scholar] [CrossRef] [PubMed]
- Stephen, R.; Hongisto, K.; Solomon, A.; Lonnroos, E. Physical Activity and Alzheimer’s Disease: A Systematic Review. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2017, 72, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Nigam, S.M.; Xu, S.; Kritikou, J.S.; Marosi, K.; Brodin, L.; Mattson, M.P. Exercise and BDNF reduce Aβ production by enhancing α-secretase processing of APP. J. Neurochem. 2017, 142, 286–296. [Google Scholar] [CrossRef] [PubMed]
- Gratuze, M.; Julien, J.; Morin, F.; Marette, A.; Planel, E. Differential effects of voluntary treadmill exercise and caloric restriction on tau pathogenesis in a mouse model of Alzheimer's disease-like tau pathology fed with Western diet. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2017, 79, 452–461. [Google Scholar] [CrossRef] [PubMed]
- Baek, S.S.; Kim, S.H. Treadmill exercise ameliorates symptoms of Alzheimer disease through suppressing microglial activation-induced apoptosis in rats. J. Exerc. Rehabil. 2016, 12, 526–534. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chao, F.L.; Zhou, C.N.; Jiang, L.; Zhang, L.; Chen, L.M.; Luo, Y.M.; Xiao, Q.; Tang, Y. Effects of exercise on capillaries in the white matter of transgenic AD mice. Oncotarget 2017, 8, 65860–65875. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Chao, F.L.; Luo, Y.M.; Xiao, Q.; Jiang, L.; Zhou, C.N.; Zhang, Y.; Lv, F.L.; He, Q.; Ma, J.; et al. Exercise Prevents Cognitive Function Decline and Demyelination in the White Matter of APP/PS1 Transgenic AD Mice. Curr. Alzheimer Res. 2017, 14, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Arai, K.; Lo, E. Oligovascular signaling in white matter stroke. Biol. Pharm. Bull. 2009, 32, 1639–1683. [Google Scholar] [CrossRef] [PubMed]
- Steinman, L. Immunology of relapse and remission in multiple sclerosis. Annu. Rev. Immunol. 2014, 32, 257–281. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H.; Bruck, W.; Lucchinetti, C.; Rodriguez, M. Remyelination in multiple sclerosis. Mult. Scler. 1997, 3, 133–136. [Google Scholar] [CrossRef] [PubMed]
- Patrikios, P.; Stadelmann, C.; Kutzelnigg, A.; Rauschka, H.; Schmidbauer, M.; Laursen, H.; Sorensen, P.S.; Bruck, W.; Lucchinetti, C.; Lassmann, H. Remyelination is extensive in a subset of multiple sclerosis patients. Brain J. Neurol. 2006, 129, 3165–3172. [Google Scholar] [CrossRef] [PubMed]
- Prineas, J.W.; Barnard, R.O.; Kwon, E.E.; Sharer, L.R.; Cho, E.S. Multiple sclerosis: Remyelination of nascent lesions. Ann. Neurol. 1993, 33, 137–151. [Google Scholar] [CrossRef] [PubMed]
- Prineas, J.W.; Connell, F. Remyelination in multiple sclerosis. Ann. Neurol. 1979, 5, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Ludwin, S.K.; Maitland, M. Long-term remyelination fails to reconstitute normal thickness of central myelin sheaths. J. Neurol. Sci. 1984, 64, 193–198. [Google Scholar] [CrossRef]
- Blakemore, W.F.; Murray, J.A. Quantitative examination of internodal length of remyelinated nerve fibres in the central nervous system. J. Neurol. Sci. 1981, 49, 273–284. [Google Scholar] [CrossRef]
- Ozawa, K.; Suchanek, G.; Breitschopf, H.; Bruck, W.; Budka, H.; Jellinger, K.; Lassmann, H. Patterns of oligodendroglia pathology in multiple sclerosis. Brain J. Neurol. 1994, 117, 1311–1322. [Google Scholar] [CrossRef]
- Sospedra, M.; Martin, R. Immunology of multiple sclerosis. Annu. Rev. Immunol. 2005, 23, 683–747. [Google Scholar] [CrossRef] [PubMed]
- Nait-Oumesmar, B.; Picard-Riera, N.; Kerninon, C.; Decker, L.; Seilhean, D.; Hoglinger, G.U.; Hirsch, E.C.; Reynolds, R.; Baron-van Evercooren, A. Activation of the subventricular zone in multiple sclerosis: Evidence for early glial progenitors. Proc. Natl. Acad. Sci. USA 2007, 104, 4694–4699. [Google Scholar] [CrossRef] [PubMed]
- Kotter, M.R.; Stadelmann, C.; Hartung, H.P. Enhancing remyelination in disease—Can we wrap it up? Brain J. Neurol. 2011, 134, 1882–1900. [Google Scholar] [CrossRef] [PubMed]
- de la Pena, I.; Pabon, M.; Acosta, S.; Sanberg, P.R.; Tajiri, N.; Kaneko, Y.; Borlongan, C.V. Oligodendrocytes engineered with migratory proteins as effective graft source for cell transplantation in multiple sclerosis. Cell Med. 2014, 6, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Picard-Riera, N.; Decker, L.; Delarasse, C.; Goude, K.; Nait-Oumesmar, B.; Liblau, R.; Pham-Dinh, D.; Baron-van Evercooren, A. Experimental autoimmune encephalomyelitis mobilizes neural progenitors from the subventricular zone to undergo oligodendrogenesis in adult mice. Proc. Natl. Acad. Sci. USA 2002, 99, 13211–13216. [Google Scholar] [CrossRef] [PubMed]
- Kuhlmann, T.; Miron, V.; Cui, Q.; Wegner, C.; Antel, J.; Bruck, W. Differentiation block of oligodendroglial progenitor cells as a cause for remyelination failure in chronic multiple sclerosis. Brain J. Neurol. 2008, 131, 1749–1758. [Google Scholar] [CrossRef] [PubMed]
- Wolswijk, G. Chronic stage multiple sclerosis lesions contain a relatively quiescent population of oligodendrocyte precursor cells. J. Neurosci. Off. J. Soc. Neurosci. 1998, 18, 601–609. [Google Scholar] [CrossRef]
- Fischer, M.T.; Sharma, R.; Lim, J.L.; Haider, L.; Frischer, J.M.; Drexhage, J.; Mahad, D.; Bradl, M.; van Horssen, J.; Lassmann, H. NADPH oxidase expression in active multiple sclerosis lesions in relation to oxidative tissue damage and mitochondrial injury. Brain J. Neurol. 2012, 135, 886–899. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ramenaden, E.R.; Peng, J.; Koito, H.; Volpe, J.J.; Rosenberg, P.A. Tumor necrosis factor α mediates lipopolysaccharide-induced microglial toxicity to developing oligodendrocytes when astrocytes are present. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 5321–5330. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhang, H.; Pu, H.; Wang, G.; Li, W.; Leak, R.K.; Chen, J.; Liou, A.K.; Hu, X. n-3 PUFA supplementation benefits microglial responses to myelin pathology. Sci. Rep. 2014, 4, 7458. [Google Scholar] [CrossRef] [PubMed]
- Miron, V.E.; Boyd, A.; Zhao, J.W.; Yuen, T.J.; Ruckh, J.M.; Shadrach, J.L.; van Wijngaarden, P.; Wagers, A.J.; Williams, A.; Franklin, R.J.M.; et al. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat. Neurosci. 2013, 16, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- Rawji, K.S.; Yong, V.W. The benefits and detriments of macrophages/microglia in models of multiple sclerosis. Clin. Dev. Immunol. 2013, 2013, 948976. [Google Scholar] [CrossRef] [PubMed]
- Lalive, P.H.; Paglinawan, R.; Biollaz, G.; Kappos, E.A.; Leone, D.P.; Malipiero, U.; Relvas, J.B.; Moransard, M.; Suter, T.; Fontana, A. TGF-β-treated microglia induce oligodendrocyte precursor cell chemotaxis through the HGF-c-Met pathway. Eur. J. Immunol. 2005, 35, 727–737. [Google Scholar] [CrossRef] [PubMed]
- Butovsky, O.; Ziv, Y.; Schwartz, A.; Landa, G.; Talpalar, A.E.; Pluchino, S.; Martino, G.; Schwartz, M. Microglia activated by IL-4 or IFN-γ differentially induce neurogenesis and oligodendrogenesis from adult stem/progenitor cells. Mol. Cell. Neurosci. 2006, 31, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Hammond, T.R.; Gadea, A.; Dupree, J.; Kerninon, C.; Nait-Oumesmar, B.; Aguirre, A.; Gallo, V. Astrocyte-derived endothelin-1 inhibits remyelination through notch activation. Neuron 2014, 81, 588–602. [Google Scholar] [CrossRef] [PubMed]
- Nash, B.; Ioannidou, K.; Barnett, S.C. Astrocyte phenotypes and their relationship to myelination. J. Anat. 2011, 219, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Domingues, H.S.; Portugal, C.C.; Socodato, R.; Relvas, J.B. Oligodendrocyte, Astrocyte, and Microglia Crosstalk in Myelin Development, Damage, and Repair. Front. Cell Dev. Biol. 2016, 4, 71. [Google Scholar] [PubMed]



{kind=link}
{kind=link}
| Parameter | SHR-SP (Rat) | BCAO (Rat) | BCAS (Gerbil) | BCAS (Mouse) |
|---|---|---|---|---|
| Operation/surgery | No surgery | Ligation only | Coil placement | Coil placement |
| Cerebral blood flow (CBF) decline (%) | 50–70% | ~70% | ~70% | |
| White matter legion | ~20 weeks | ~1 week | over 8 weeks | ~2 weeks |
| Cognitive dysfunction | ~4 weeks | ~2 weeks | ~4 weeks |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ohtomo, R.; Iwata, A.; Arai, K. Molecular Mechanisms of Oligodendrocyte Regeneration in White Matter-Related Diseases. Int. J. Mol. Sci. 2018, 19, 1743. https://doi.org/10.3390/ijms19061743
Ohtomo R, Iwata A, Arai K. Molecular Mechanisms of Oligodendrocyte Regeneration in White Matter-Related Diseases. International Journal of Molecular Sciences. 2018; 19(6):1743. https://doi.org/10.3390/ijms19061743
Chicago/Turabian StyleOhtomo, Ryo, Atsushi Iwata, and Ken Arai. 2018. "Molecular Mechanisms of Oligodendrocyte Regeneration in White Matter-Related Diseases" International Journal of Molecular Sciences 19, no. 6: 1743. https://doi.org/10.3390/ijms19061743
APA StyleOhtomo, R., Iwata, A., & Arai, K. (2018). Molecular Mechanisms of Oligodendrocyte Regeneration in White Matter-Related Diseases. International Journal of Molecular Sciences, 19(6), 1743. https://doi.org/10.3390/ijms19061743