Molecular Genetics of Frontotemporal Dementia Elucidated by Drosophila Models—Defects in Endosomal–Lysosomal Pathway


Abstract
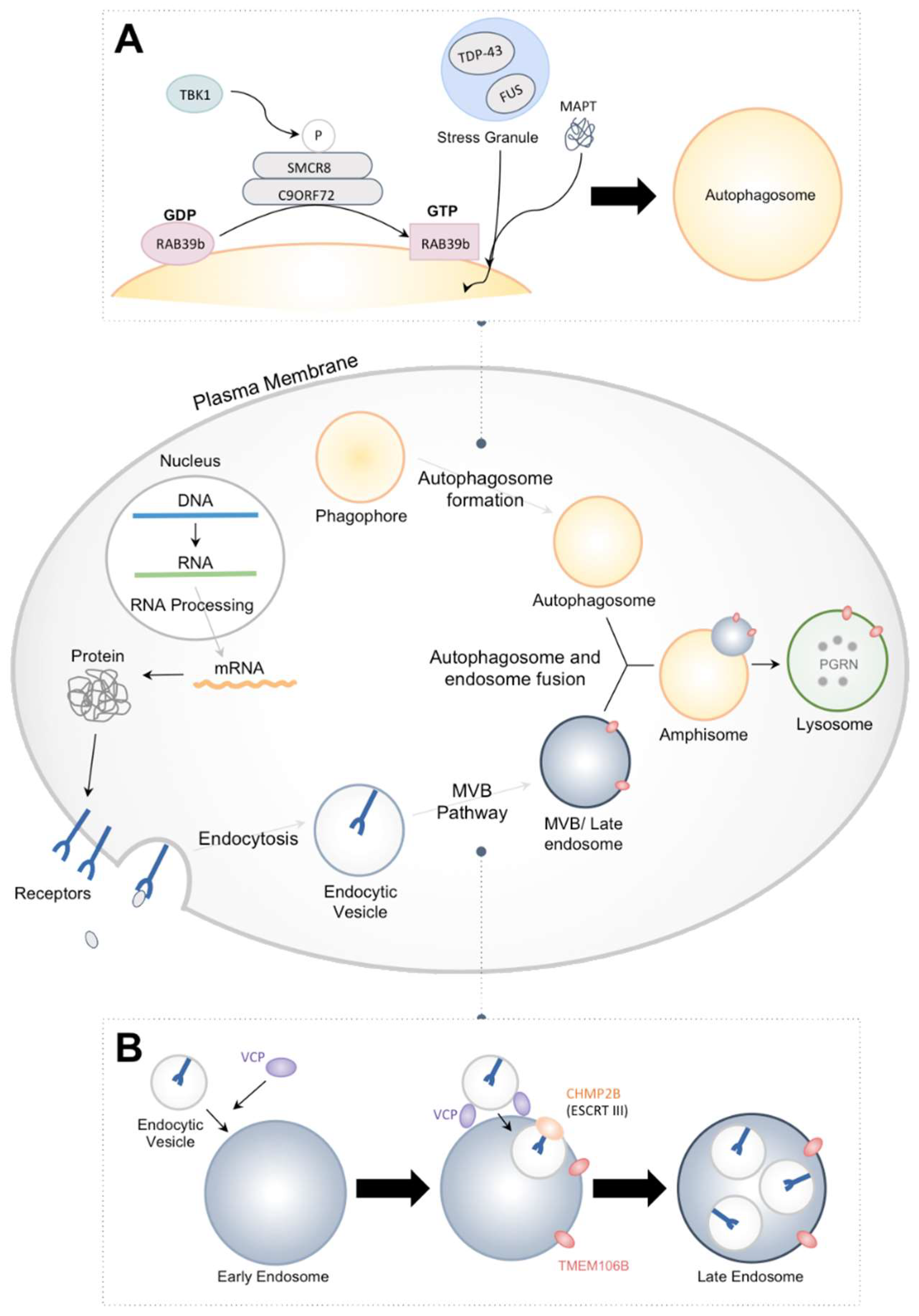
1. Introduction
2. Mutations Associated with FTD
3. FTD Associated with Mutant CHMP2B
3.1. Structure and Function of CHMP2B
3.2. Defects Caused by CHMP2Bintron5
4. Future Perspectives
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Olszewska, D.A.; Lonergan, R.; Fallon, E.M.; Lynch, T. Genetics of frontotemporal dementia. Curr. Neurol. Neuroscie. Rep. 2016, 16, 107. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.-D.; Chen, X.-C. Clinic, neuropathology and molecular genetics of frontotemporal dementia: A mini-review. Transl. Neurodegener. 2013, 2, 8. [Google Scholar] [CrossRef] [PubMed]
- Young, J.J.; Lavakumar, M.; Tampi, D.; Balachandran, S.; Tampi, R.R. Frontotemporal dementia: Latest evidence and clinical implications. Ther. Adv. Psychopharmacol. 2018, 8, 33–48. [Google Scholar] [CrossRef] [PubMed]
- Krasniak, C.S.; Ahmad, S.T. The role of CHMP2B(Intron5) in autophagy and frontotemporal dementia. Brain Res. 2016, 1649, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Lindquist, S.G.; Brændgaard, H.; Svenstrup, K.; Isaacs, A.M.; Nielsen, J.E. on behalf of the FReJA Consortium. Frontotemporal dementia linked to chromosome 3 (FTD-3)—Current concepts and the detection of a previously unknown branch of the danish FTD-3 family. Eur. J. Neurol. 2008, 15, 667–670. [Google Scholar] [CrossRef] [PubMed]
- Belly, A.; Bodon, G.; Blot, B.; Bouron, A.; Sadoul, R.; Goldberg, Y. CHMP2B mutants linked to frontotemporal dementia impair maturation of dendritic spines. J. Cell Sci. 2010, 123, 2943–2954. [Google Scholar] [CrossRef] [PubMed]
- Skibinski, G.; Parkinson, N.J.; Brown, J.M.; Chakrabarti, L.; Lloyd, S.L.; Hummerich, H.; Nielsen, J.E.; Hodges, J.R.; Spillantini, M.G.; Thusgaard, T.; et al. Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat. Genet. 2005, 37, 806–808. [Google Scholar] [CrossRef] [PubMed]
- Rainero, I.; Rubino, E.; Michelerio, A.; D’Agata, F.; Gentile, S.; Pinessi, L. Recent advances in the molecular genetics of frontotemporal lobar degeneration. Funct. Neurol. 2017, 32, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, D.; Parlato, R. C9ORF72-associated neurodegeneration in ALS-FTD: Breaking new ground in ribosomal RNA and nucleolar dysfunction. Cell Tissue Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Renton, A.E.; Majounie, E.; Waite, A.; Simon-Sanchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Almeida, S.; Gao, F.B. Lost & found: C9ORF72 and the autophagy pathway in ALS/FTD. EMBO J. 2016, 35, 1251–1253. [Google Scholar] [PubMed]
- Mizielinska, S.; Grönke, S.; Niccoli, T.; Ridler, C.E.; Clayton, E.L.; Devoy, A.; Moens, T.; Norona, F.E.; Woollacott, I.O.C.; Pietrzyk, J.; et al. C9ORF72 repeat expansions cause neurodegeneration in drosophila through arginine-rich proteins. Science (N. Y.) 2014, 345, 1192–1194. [Google Scholar] [CrossRef] [PubMed]
- Sellier, C.; Campanari, M.L.; Julie Corbier, C.; Gaucherot, A.; Kolb-Cheynel, I.; Oulad-Abdelghani, M.; Ruffenach, F.; Page, A.; Ciura, S.; Kabashi, E.; et al. Loss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin-2 to induce motor neuron dysfunction and cell death. EMBO J. 2016, 35, 1276–1297. [Google Scholar] [CrossRef] [PubMed]
- Farg, M.A.; Sundaramoorthy, V.; Sultana, J.M.; Yang, S.; Atkinson, R.A.; Levina, V.; Halloran, M.A.; Gleeson, P.A.; Blair, I.P.; Soo, K.Y.; et al. C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum. Mol. Genet. 2014, 23, 3579–3595. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.; Mackenzie, I.R.; Pickering-Brown, S.M.; Gass, J.; Rademakers, R.; Lindholm, C.; Snowden, J.; Adamson, J.; Sadovnick, A.D.; Rollinson, S.; et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 2006, 442, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Cruts, M.; Gijselinck, I.; van der Zee, J.; Engelborghs, S.; Wils, H.; Pirici, D.; Rademakers, R.; Vandenberghe, R.; Dermaut, B.; Martin, J.J.; et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 2006, 442, 920–924. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, P.M.; Zhou, X.; Hu, F. Autophagy-lysosome dysfunction in amyotrophic lateral sclerosis and frontotemporal lobar degeneration. In Lysosomes—Associated Diseases and Methods to Study Their Function; Sharma, P.D., Ed.; InTech: Rijeka, Croatia, 2017; Chapter 4. [Google Scholar]
- Boxer, A.L.; Gold, M.; Huey, E.; Gao, F.-B.; Burton, E.A.; Chow, T.; Kao, A.; Leavitt, B.; Lamb, B.; Grether, M.; et al. Frontotemporal degeneration, the next therapeutic frontier: Molecules and animal models for FTD drug development (part 1 of 2 articles). Alzheimer Dement. J. Alzheimer Assoc. 2013, 9, 176–188. [Google Scholar] [CrossRef] [PubMed]
- Cenik, B.; Sephton, C.F.; Kutluk Cenik, B.; Herz, J.; Yu, G. Progranulin: A proteolytically processed protein at the crossroads of inflammation and neurodegeneration. J. Biol. Chem. 2012, 287, 32298–32306. [Google Scholar] [CrossRef] [PubMed]
- Rademakers, R.; Cruts, M.; Van Broeckhoven, C. The role of tau (MAPT) in frontotemporal dementia and related tauopathies. Hum. Mutat. 2004, 24, 277–295. [Google Scholar] [CrossRef] [PubMed]
- Shulman, J.M.; Feany, M.B. Genetic modifiers of tauopathy in drosophila. Genetics 2003, 165, 1233–1242. [Google Scholar] [PubMed]
- Jun, M.-H.; Han, J.-H.; Lee, Y.-K.; Jang, D.-J.; Kaang, B.-K.; Lee, J.-A. TMEM106B, a frontotemporal lobar dementia (FTLD) modifier, associates with FTD-3-linked CHMP2B, a complex of ESCRT-III. Mol. Brain 2015, 8, 85. [Google Scholar] [CrossRef] [PubMed]
- Rostgaard, N.; Roos, P.; Budtz-Jørgensen, E.; Johannsen, P.; Waldemar, G.; Nørremølle, A.; Lindquist, S.G.; Gydesen, S.; Brown, J.M.; Collinge, J. TMEM106B and apoe polymorphisms in CHMP2B-mediated frontotemporal dementia (FTD-3). Neurobiol. Aging 2017, 59, 221.e1–221.e7. [Google Scholar] [CrossRef] [PubMed]
- Wang, I.F.; Wu, L.S.; Chang, H.Y.; Shen, C.K. TDP-43, the signature protein of FTLD-U, is a neuronal activity-responsive factor. J. Neurochem. 2008, 105, 797–806. [Google Scholar] [CrossRef] [PubMed]
- Lagier-Tourenne, C.; Polymenidou, M.; Cleveland, D.W. TDP-43 and FUS/TLS: Emerging roles in RNA processing and neurodegeneration. Hum. Mol. Genet. 2010, 19, R46–R64. [Google Scholar] [CrossRef] [PubMed]
- Geser, F.; Martinez-Lage, M.; Kwong, L.K.; Lee, V.M.; Trojanowski, J.Q. Amyotrophic lateral sclerosis, frontotemporal dementia and beyond: The TDP-43 diseases. J. Neurol. 2009, 256, 1205–1214. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Miguel, L.; Frebourg, T.; Campion, D.; Lecourtois, M. Both cytoplasmic and nuclear accumulations of the protein are neurotoxic in drosophila models of TDP-43 proteinopathies. Neurobiol. Dis. 2011, 41, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ray, P.; Rao, E.J.; Shi, C.; Guo, W.; Chen, X.; Woodruff, E.A., 3rd; Fushimi, K.; Wu, J.Y. A drosophila model for TDP-43 proteinopathy. Proc. Natl. Acad. Sci. USA 2010, 107, 3169–3174. [Google Scholar] [CrossRef] [PubMed]
- Lanson, N.A., Jr.; Maltare, A.; King, H.; Smith, R.; Kim, J.H.; Taylor, J.P.; Lloyd, T.E.; Pandey, U.B. A drosophila model of FUS-related neurodegeneration reveals genetic interaction between fus and TDP-43. Hum. Mol. Genet. 2011, 20, 2510–2523. [Google Scholar] [CrossRef] [PubMed]
- Rumpf, S.; Bagley, J.A.; Thompson-Peer, K.L.; Zhu, S.; Gorczyca, D.; Beckstead, R.B.; Jan, L.Y.; Jan, Y.N. Drosophila valosin-containing protein is required for dendrite pruning through a regulatory role in mRNA metabolism. Proc. Natl. Acad. Sci. USA 2014, 111, 7331–7336. [Google Scholar] [CrossRef] [PubMed]
- Ritson, G.P.; Custer, S.K.; Freibaum, B.D.; Guinto, J.B.; Geffel, D.; Moore, J.; Tang, W.; Winton, M.J.; Neumann, M.; Trojanowski, J.Q.; et al. TDP-43 mediates degeneration in a novel drosophila model of disease caused by mutations in VCP/p97. J. Neurosci. 2010, 30, 7729–7739. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-W.; Brent, J.R.; Tomlinson, A.; Shneider, N.A.; McCabe, B.D. The ALS-associated proteins FUS and TDP-43 function together to affect drosophila locomotion and life span. J. Clin. Investig. 2011, 121, 4118–4126. [Google Scholar] [CrossRef] [PubMed]
- Azuma, Y.; Tokuda, T.; Shimamura, M.; Kyotani, A.; Sasayama, H.; Yoshida, T.; Mizuta, I.; Mizuno, T.; Nakagawa, M.; Fujikake, N.; et al. Identification of TER94, drosophila VCP, as a strong modulator of motor neuron degeneration induced by knockdown of CAZ, drosophila FUS. Hum. Mol. Genet. 2014, 23, 3467–3480. [Google Scholar] [CrossRef] [PubMed]
- Freischmidt, A.; Wieland, T.; Richter, B.; Ruf, W.; Schaeffer, V.; Muller, K.; Marroquin, N.; Nordin, F.; Hubers, A.; Weydt, P.; et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat. Neurosci. 2015, 18, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Sieben, A.; Van Mossevelde, S.; Wauters, E.; Engelborghs, S.; van der Zee, J.; Van Langenhove, T.; Santens, P.; Praet, M.; Boon, P.; Miatton, M.; et al. Extended FTLD pedigree segregating a belgian GRN-null mutation: Neuropathological heterogeneity in one family. Alzheimer Res. Ther. 2018, 10, 7. [Google Scholar] [CrossRef] [PubMed]
- Cui, R.; Tuo, M.; Li, P.; Zhou, C. Association between TBK1 mutations and risk of amyotrophic lateral sclerosis/frontotemporal dementia spectrum: A meta-analysis. Neurol. Sci. 2018, 39, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Oakes, J.A.; Davies, M.C.; Collins, M.O. TBK1: A new player in als linking autophagy and neuroinflammation. Mol. Brain 2017, 10, 5. [Google Scholar] [CrossRef] [PubMed]
- West, R.J.; Lu, Y.; Marie, B.; Gao, F.-B.; Sweeney, S.T. RAB8, POSH, and TAK1 regulate synaptic growth in a drosophila model of frontotemporal dementia. J. Cell Biol. 2015, 208, 931. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, O.; Teis, D. The escrt machinery. Curr. Biol. 2012, 22, R116–R120. [Google Scholar] [CrossRef] [PubMed]
- Henne, W.M.; Stenmark, H.; Emr, S.D. Molecular mechanisms of the membrane sculpting escrt pathway. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar]
- Katzmann, D.J.; Odorizzi, G.; Emr, S.D. Receptor downregulation and multivesicular-body sorting. Nat. Rev. Mol. Cell Biol. 2002, 3, 893–905. [Google Scholar] [CrossRef] [PubMed]
- Morita, E.; Sundquist, W.I. Retrovirus budding. Ann. Rev. Cell Dev. Biol. 2004, 20, 395–425. [Google Scholar] [CrossRef] [PubMed]
- Carlton, J.G.; Martin-Serrano, J. Parallels between cytokinesis and retroviral budding: A role for the escrt machinery. Science 2007, 316, 1908–1912. [Google Scholar] [CrossRef] [PubMed]
- Christ, L.; Raiborg, C.; Wenzel, E.M.; Campsteijn, C.; Stenmark, H. Cellular functions and molecular mechanisms of the ESCRT membrane-scission machinery. Trends Biochem. Sci. 2017, 42, 42–56. [Google Scholar] [CrossRef] [PubMed]
- Frankel, E.B.; Audhya, A. ESCRT-dependent cargo sorting at multivesicular endosomes. Semin. Cell Dev. Biol. 2018, 74, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Wollert, T.; Hurley, J.H. Molecular mechanism of multivesicular body biogenesis by ESCRT complexes. Nature 2010, 464, 864–869. [Google Scholar] [CrossRef] [PubMed]
- Hurley, J.H. The escrt complexes. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 463–487. [Google Scholar] [CrossRef] [PubMed]
- Henne, W.M.; Buchkovich, N.J.; Emr, S.D. The ESCRT pathway. Dev. Cell 2011, 21, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Pineda-Molina, E.; Belrhali, H.; Piefer, A.J.; Akula, I.; Bates, P.; Weissenhorn, W. The crystal structure of the C-terminal domain of Vps28 reveals a conserved surface required for Vps20 recruitment. Traffic 2006, 7, 1007–1016. [Google Scholar] [CrossRef] [PubMed]
- Im, Y.J.; Hurley, J.H. Integrated structural model and membrane targeting mechanism of the human ESCRT-II complex. Dev. Cell 2008, 14, 902–913. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Stjepanovic, G.; Shen, Q.; Martin, A.; Hurley, J.H. Vps4 disassembles an ESCRT-III filament by global unfolding and processive translocation. Nat. Struct. Mol. Biol. 2015, 22, 492–498. [Google Scholar] [CrossRef] [PubMed]
- McCullough, J.; Colf, L.A.; Sundquist, W.I. Membrane fission reactions of the mammalian ESCRT pathway. Ann. Rev. Biochem. 2013, 82, 663–692. [Google Scholar] [CrossRef] [PubMed]
- Adell, M.A.Y.; Migliano, S.M.; Teis, D. ESCRT-III and Vps4: A dynamic multipurpose tool for membrane budding and scission. FEBS J. 2016, 283, 3288–3302. [Google Scholar] [CrossRef] [PubMed]
- Truebestein, L.; Leonard, T.A. Coiled-coils: The long and short of it. Bioessays 2016, 38, 903–916. [Google Scholar] [CrossRef] [PubMed]
- Obita, T.; Saksena, S.; Ghazi-Tabatabai, S.; Gill, D.J.; Perisic, O.; Emr, S.D.; Williams, R.L. Structural basis for selective recognition of ESCRT-III by the AAA ATpase Vps4. Nature 2007, 449, 735–739. [Google Scholar] [CrossRef] [PubMed]
- Stuchell-Brereton, M.D.; Skalicky, J.J.; Kieffer, C.; Karren, M.A.; Ghaffarian, S.; Sundquist, W.I. ESCRT-III recognition by Vps4 ATpases. Nature 2007, 449, 740–744. [Google Scholar] [CrossRef] [PubMed]
- Bodon, G.; Chassefeyre, R.; Pernet-Gallay, K.; Martinelli, N.; Effantin, G.; Hulsik, D.L.; Belly, A.; Goldberg, Y.; Chatellard-Causse, C.; Blot, B. Charged multivesicular body protein 2B (CHMP2B) of the endosomal sorting complex required for transport-III (ESCRT-III) polymerizes into helical structures deforming the plasma membrane. J. Biol. Chem. 2011, 286, 40276–40286. [Google Scholar] [CrossRef] [PubMed]
- Shim, S.; Kimpler, L.A.; Hanson, P.I. Structure/function analysis of four core ESCRT-III proteins reveals common regulatory role for extreme C-terminal domain. Traffic 2007, 8, 1068–1079. [Google Scholar] [CrossRef] [PubMed]
- Hanson, P.I.; Roth, R.; Lin, Y.; Heuser, J.E. Plasma membrane deformation by circular arrays of ESCRT-III protein filaments. J. Cell Biol. 2008, 180, 389–402. [Google Scholar] [CrossRef] [PubMed]
- Lata, S.; Schoehn, G.; Jain, A.; Pires, R.; Piehler, J.; Gőttlinger, H.G.; Weissenhorn, W. Helical structures of ESCRT-III are disassembled by Vps4. Science (N. Y.) 2008, 321, 1354–1357. [Google Scholar] [CrossRef] [PubMed]
- Bajorek, M.; Schubert, H.L.; McCullough, J.; Langelier, C.; Eckert, D.M.; Stubblefield, W.-M.B.; Uter, N.T.; Myszka, D.G.; Hill, C.P.; Sundquist, W.I. Structural basis for ESCRT-III protein autoinhibition. Nat. Struct. Mol. Biol. 2009, 16, 754–762. [Google Scholar] [CrossRef] [PubMed]
- Buchkovich, N.J.; Henne, W.M.; Tang, S.; Emr, S.D. Essential N-terminal insertion motif anchors the ESCRT-III filament during MVB vesicle formation. Dev. Cell 2013, 27, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Malerød, L.; Stuffers, S.; Brech, A.; Stenmark, H. Vps22/EAP30 in ESCRT-II mediates endosomal sorting of growth factor and chemokine receptors destined for lysosomal degradation. Traffic 2007, 8, 1617–1629. [Google Scholar] [CrossRef] [PubMed]
- Bache, K.G.; Stuffers, S.; Malerød, L.; Slagsvold, T.; Raiborg, C.; Lechardeur, D.; Wälchli, S.; Lukacs, G.L.; Brech, A.; Stenmark, H. The ESCRT-III subunit hVps24 is required for degradation but not silencing of the epidermal growth factor receptor. Mol. Biol. Cell 2006, 17, 2513–2523. [Google Scholar] [CrossRef] [PubMed]
- Doyotte, A.; Russell, M.R.G.; Hopkins, C.R.; Woodman, P.G. Depletion of TSG101 forms a mammalian ‘class E′ compartment: A multicisternal early endosome with multiple sorting defects. J. Cell Sci. 2005, 118, 3003–3017. [Google Scholar] [CrossRef] [PubMed]
- Verma, R.; Marchese, A. The endosomal sorting complex required for transport pathway mediates chemokine receptor CXCR4-promoted lysosomal degradation of the mammalian target of rapamycin antagonist deptor. J. Biol. Chem. 2015, 290, 6810–6824. [Google Scholar] [CrossRef] [PubMed]
- Lobert, V.H.; Brech, A.; Pedersen, N.M.; Wesche, J.; Oppelt, A.; Malerød, L.; Stenmark, H. Ubiquitination of α5β1 integrin controls fibroblast migration through lysosomal degradation of fibronectin-integrin complexes. Dev. Cell 2010, 19, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Mamińska, A.; Bartosik, A.; Banach-Orłowska, M.; Pilecka, I.; Jastrzębski, K.; Zdżalik-Bielecka, D.; Castanon, I.; Poulain, M.; Neyen, C.; Wolińska-Nizioł, L.; et al. Escrt proteins restrict constitutive NF-κb signaling by trafficking cytokine receptors. Sci. Signal. 2016, 9, ra8. [Google Scholar] [CrossRef] [PubMed]
- Husebye, H.; Halaas, Ø.; Stenmark, H.; Tunheim, G.; Sandanger, Ø.; Bogen, B.; Brech, A.; Latz, E.; Espevik, T. Endocytic pathways regulate toll-like receptor 4 signaling and link innate and adaptive immunity. EMBO J. 2006, 25, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Le Borgne, R. Regulation of notch signalling by endocytosis and endosomal sorting. Curr. Opin. Cell Biol. 2006, 18, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Szymanska, E.; Budick-Harmelin, N.; Miaczynska, M. Endosomal “sort” of signaling control: The role of escrt machinery in regulation of receptor-mediated signaling pathways. Semin. Cell Dev. Biol. 2018, 74, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Thompson, B.J.; Mathieu, J.; Sung, H.-H.; Loeser, E.; Rørth, P.; Cohen, S.M. Tumor suppressor properties of the ESCRT-II complex component Vps25 in drosophila. Dev. Cell 2005, 9, 711–720. [Google Scholar] [CrossRef] [PubMed]
- Rodahl, L.M.; Haglund, K.; Sem-Jacobsen, C.; Wendler, F.; Vincent, J.-P.; Lindmo, K.; Rusten, T.E.; Stenmark, H. Disruption of Vps4 and JNK function in drosophila causes tumour growth. PLoS ONE 2009, 4, e4354. [Google Scholar] [CrossRef] [PubMed]
- Moberg, K.H.; Schelble, S.; Burdick, S.K.; Hariharan, I.K. Mutations in erupted, the drosophila ortholog of mammalian tumor susceptibility gene 101, elicit non-cell-autonomous overgrowth. Dev. Cell 2005, 9, 699–710. [Google Scholar] [CrossRef] [PubMed]
- Herz, H.M.; Bergmann, A. Genetic analysis of escrt function in drosophila: A tumour model for human TSG101. Biochem. Soc. Trans. 2009, 37, 204–207. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-A.; Gao, F.-B. Escrt, autophagy, and frontotemporal dementia. BMB Rep. 2008, 41, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, C.; Legouis, R.; Culetto, E. ESCRT and Autophagies: Endosomal Functions and Beyond. Semin. Cell Dev. Biol. 2018, 74, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zhang, Z.; Sun, D.; Sweeney, S.T.; Gao, F.-B. Syntaxin 13, a genetic modifier of mutant CHMP2B in frontotemporal dementia, is required for autophagosome maturation. Mol. Cell 2013, 52, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Sadoul, R.; Laporte, M.H.; Chassefeyre, R.; Chi, K.I.; Goldberg, Y.; Chatellard, C.; Hemming, F.J.; Fraboulet, S. The role of escrt during development and functioning of the nervous system. Semin. Cell Dev. Biol. 2018, 74, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Chassefeyre, R.; Martínez-Hernández, J.; Bertaso, F.; Bouquier, N.; Blot, B.; Laporte, M.; Fraboulet, S.; Couté, Y.; Devoy, A.; Isaacs, A.M. Regulation of postsynaptic function by the dementia-related ESCRT-III subunit CHMP2B. J. Neurosci. 2015, 35, 3155–3173. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.T.; Sweeney, S.T.; Lee, J.A.; Sweeney, N.T.; Gao, F.B. Genetic screen identifies SERPIN5 as a regulator of the toll pathway and CHMP2B toxicity associated with frontotemporal dementia. Proc. Natl. Acad. Sci. USA 2009, 106, 12168–12173. [Google Scholar] [CrossRef] [PubMed]
- Cheruiyot, A.; Lee, J.-A.; Gao, F.-B.; Ahmad, S.T. Expression of mutant CHMP2B, an ESCRT-III component involved in frontotemporal dementia, causes eye deformities due to notch misregulation in drosophila. FASEB J. 2014, 28, 667–675. [Google Scholar] [CrossRef] [PubMed]
- West, R.J.; Ugbode, C.; Gao, F.-B.; Sweeney, S.T. The pro-apoptotic JNK scaffold POSH/SH3RF1 mediates CHMP2Bintron5-associated toxicity in animal models of frontotemporal dementia. Hum. Mol. Genet. 2018, 27, 1382–1395. [Google Scholar] [CrossRef] [PubMed]
- Filimonenko, M.; Stuffers, S.; Raiborg, C.; Yamamoto, A.; Malerød, L.; Fisher, E.M.C.; Isaacs, A.; Brech, A.; Stenmark, H.; Simonsen, A. Functional multivesicular bodies are required for autophagic clearance of protein aggregates associated with neurodegenerative disease. J. Cell Biol. 2007, 179, 485–500. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-A.; Beigneux, A.; Ahmad, S.T.; Young, S.G.; Gao, F.-B. ESCRT-III dysfunction causes autophagosome accumulation and neurodegeneration. Curr. Biol. 2007, 17, 1561–1567. [Google Scholar] [CrossRef] [PubMed]
- Vernay, A.; Therreau, L.; Blot, B.; Risson, V.; Dirrig-Grosch, S.; Waegaert, R.; Lequeu, T.; Sellal, F.; Schaeffer, L.; Sadoul, R. A transgenic mouse expressing CHMP2Bintron5 mutant in neurons develops histological and behavioural features of amyotrophic lateral sclerosis and frontotemporal dementia. Hum. Mol. Genet. 2016, 25, 3341–3360. [Google Scholar] [CrossRef] [PubMed]
- Ghazi-Noori, S.; Froud, K.E.; Mizielinska, S.; Powell, C.; Smidak, M.; Fernandez de Marco, M.; O’Malley, C.; Farmer, M.; Parkinson, N.; Fisher, E.M.C.; et al. Progressive neuronal inclusion formation and axonal degeneration in CHMP2B mutant transgenic mice. Brain 2012, 135, 819–832. [Google Scholar] [CrossRef] [PubMed]
- Urwin, H.; Authier, A.; Nielsen, J.E.; Metcalf, D.; Powell, C.; Froud, K.; Malcolm, D.S.; Holm, I.; Johannsen, P.; Brown, J. Disruption of endocytic trafficking in frontotemporal dementia with CHMP2B mutations. Hum. Mol. Genet. 2010, 19, 2228–2238. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-A.; Liu, L.; Javier, R.; Kreitzer, A.C.; Delaloy, C.; Gao, F.-B. ESCRT-III subunits Snf7-1 and Snf7-2 differentially regulate transmembrane cargos in hESC-derived human neurons. Mol. Brain 2011, 4, 37. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Schmid, B.; Nielsen, T.T.; Nielsen, J.E.; Clausen, C.; Hyttel, P.; Holst, B.; Freude, K.K. Generation of a human induced pluripotent stem cell line via CRISPR-CAS9 mediated integration of a site-specific homozygous mutation in CHMP2B. Stem Cell Res. 2016, 17, 151–153. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Schmid, B.; Nikolaisen, N.K.; Rasmussen, M.A.; Aldana, B.I.; Agger, M.; Calloe, K.; Stummann, T.C.; Larsen, H.M.; Nielsen, T.T. Patient IPSC-derived neurons for disease modeling of frontotemporal dementia with mutation in CHMP2B. Stem Cell Rep. 2017, 8, 648–658. [Google Scholar] [CrossRef] [PubMed]
- Clayton, E.L.; Mizielinska, S.; Edgar, J.R.; Nielsen, T.T.; Marshall, S.; Norona, F.E.; Robbins, M.; Damirji, H.; Holm, I.E.; Johannsen, P.; et al. Frontotemporal dementia caused by CHMP2B mutation is characterised by neuronal lysosomal storage pathology. Acta Neuropathol. 2015, 130, 511–523. [Google Scholar] [CrossRef] [PubMed]
- Gascon, E.; Lynch, K.; Ruan, H.; Almeida, S.; Verheyden, J.; Seeley, W.W.; Dickson, D.W.; Petrucelli, L.; Sun, D.; Jiao, J.; et al. Alterations in microRNA-124 and AMPA receptors contribute to social behavioral deficits in frontotemporal dementia. Nat. Med. 2014, 20, 1444–1451. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-T.; Lee, Y.-C.; Soong, B.-W. What we have learned from the next-generation sequencing: Contributions to the genetic diagnoses and understanding of pathomechanisms of neurodegenerative diseases. J. Neurogenet. 2015, 29, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Gene Name | C9ORF72 | MAPT | CHMP2B | FUS | VCP | TARDBP (TDP-43) | PGRN | TBK1 | TMEM106B |
|---|---|---|---|---|---|---|---|---|---|
| Full Name | Chromosome 9 open reading frame 72 | Microtubule—associated protein tau | Chromatin modifying protein 2B | Fused in sarcoma | Valosin containing protein | Transactive DNA-binding protein | Progranulin | TANK-binding kinase 1 | Transmembrane protein 106B |
| Location | 9p21.2 | 17q21.32 | 3p11.2 | 16q11.22 | 9p13.3 | 1p36.22 | 17q21.32 | 12q14.2 | 7q21.3 |
| Incidence Rate | 25% | Familial: 10–20% Sporadic: 0–3% | Rare | Rare | 1.6% | Rare (<20 cases) | Familial: 5–20% Sporadic: 1–5% | 1.1% | Unknown |
| Normal Function |
|
|
|
|
|
|
|
|
|
| Deficits Caused |
|
|
|
|
|
|
|
|
|
| Drosophila Model | Yes 13 | Yes 22 | Yes 41, 81, 84–86 | Yes 32 | Yes 34 | Yes 32 | No | No | No |
| Drosophila Homolog | None | tau/ CG45110 | CG4618 | caz | ter94 | TBPH | N/A | N/A | N/A |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vandal, S.E.; Zheng, X.; Ahmad, S.T. Molecular Genetics of Frontotemporal Dementia Elucidated by Drosophila Models—Defects in Endosomal–Lysosomal Pathway. Int. J. Mol. Sci. 2018, 19, 1714. https://doi.org/10.3390/ijms19061714
Vandal SE, Zheng X, Ahmad ST. Molecular Genetics of Frontotemporal Dementia Elucidated by Drosophila Models—Defects in Endosomal–Lysosomal Pathway. International Journal of Molecular Sciences. 2018; 19(6):1714. https://doi.org/10.3390/ijms19061714
Chicago/Turabian StyleVandal, Sarah E., Xiaoyue Zheng, and S. Tariq Ahmad. 2018. "Molecular Genetics of Frontotemporal Dementia Elucidated by Drosophila Models—Defects in Endosomal–Lysosomal Pathway" International Journal of Molecular Sciences 19, no. 6: 1714. https://doi.org/10.3390/ijms19061714
APA StyleVandal, S. E., Zheng, X., & Ahmad, S. T. (2018). Molecular Genetics of Frontotemporal Dementia Elucidated by Drosophila Models—Defects in Endosomal–Lysosomal Pathway. International Journal of Molecular Sciences, 19(6), 1714. https://doi.org/10.3390/ijms19061714