Microbiome-Gut-Brain Axis and Toll-Like Receptors in Parkinson’s Disease


Abstract
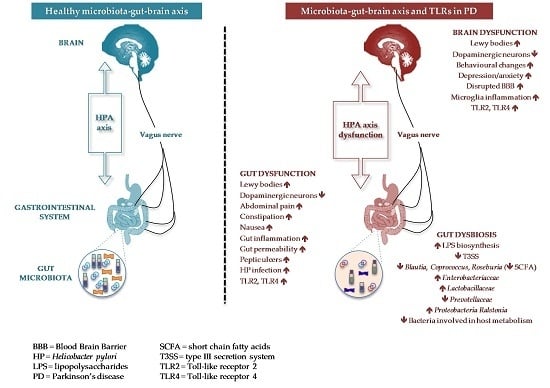
1. Introduction
2. Microbiota-Gut-Brain Axis and Host Health
3. The Gastrointestinal System in Parkinson’s Disease
4. The Gut Microbiota in Parkinson’s Disease
5. Toll-Like Receptors
6. TLRs in Parkinson’s Disease
7. Microbiota-Gut-Brain-Axis and Toll-Like Receptor Signaling: Potential Implications in Parkinson’s Disease
8. Potential Therapeutic Strategies
8.1. Dietary Supplements
8.2. Probiotics
8.3. Prebiotics
8.4. Antibiotics
8.5. TLRs Modulators
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Tysnes, O.B.; Storstein, A. Epidemiology of Parkinson’s disease. J. Neural. Transm. (Vienna) 2017, 124, 901–905. [Google Scholar] [PubMed]
- Nair, A.T.; Ramachandran, V.; Joghee, N.M.; Antony, S.; Ramalingam, G. Gut microbiota dysfunction as reliable non-invasive early diagnostic biomarkers in the pathophysiology of Parkinson’s disease: A critical review. J. Neurogastroenterol. Motil. 2018, 24, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Dorsey, E.R.; Constantinescu, R.; Thompson, J.P.; Biglan, K.M.; Holloway, R.G.; Kieburtz, K.; Marshall, F.J.; Ravina, B.M.; Schifitto, G.; Siderowf, A.; et al. Projected number of people with parkinson disease in the most populous nations, 2005 through 2030. Neurology 2007, 68, 384–386. [Google Scholar] [CrossRef] [PubMed]
- Beach, T.G.; Adler, C.H.; Sue, L.I.; Vedders, L.; Lue, L.; White Iii, C.L.; Akiyama, H.; Caviness, J.N.; Shill, H.A.; Sabbagh, M.N.; et al. Multi-organ distribution of phosphorylated alpha-synuclein histopathology in subjects with lewy body disorders. Acta Neuropathol. 2010, 119, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Sastre, M.; Bohl, J.R.; de Vos, R.A.; Del Tredici, K. Parkinson’s disease: Lesions in dorsal horn layer i, involvement of parasympathetic and sympathetic pre- and postganglionic neurons. Acta Neuropathol. 2007, 113, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Fasano, A.; Visanji, N.P.; Liu, L.W.; Lang, A.E.; Pfeiffer, R.F. Gastrointestinal dysfunction in Parkinson’s disease. Lancet Neurol. 2015, 14, 625–639. [Google Scholar] [CrossRef]
- Poewe, W.; Antonini, A.; Zijlmans, J.C.; Burkhard, P.R.; Vingerhoets, F. Levodopa in the treatment of Parkinson’s disease: An old drug still going strong. Clin. Interv. Aging 2010, 5, 229–238. [Google Scholar] [PubMed]
- Shah, E.; Rezaie, A.; Riddle, M.; Pimentel, M. Psychological disorders in gastrointestinal disease: Epiphenomenon, cause or consequence? Ann. Gastroenterol. 2014, 27, 224–230. [Google Scholar] [PubMed]
- Fadgyas-Stanculete, M.; Buga, A.M.; Popa-Wagner, A.; Dumitrascu, D.L. The relationship between irritable bowel syndrome and psychiatric disorders: From molecular changes to clinical manifestations. J. Mol. Psychiatry 2014, 2, 4. [Google Scholar] [CrossRef] [PubMed]
- Dembinski, A.; Warzecha, Z.; Ceranowicz, P.; Pawlik, M.; Dembinski, M.; Kabat, K.; Konturek, S.J.; Kownacki, P.; Hladki, W.; Pawlik, W.W. Influence of central and peripheral administration of pancreatic polypeptide on gastric mucosa growth. J. Physiol. Pharmacol. 2004, 55, 223–237. [Google Scholar] [PubMed]
- Ceranowicz, P.; Cieszkowski, J.; Warzecha, Z.; Dembinski, A. Experimental models of acute pancreatitis. Postepy Hig. Med. Dosw. (Online) 2015, 69, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Ceranowicz, P.; Cieszkowski, J.; Warzecha, Z.; Kusnierz-Cabala, B.; Dembinski, A. The beginnings of pancreatology as a field of experimental and clinical medicine. Biomed. Res. Int. 2015, 2015, 128095. [Google Scholar] [CrossRef] [PubMed]
- Dumnicka, P.; Maduzia, D.; Ceranowicz, P.; Olszanecki, R.; Drozdz, R.; Kusnierz-Cabala, B. The interplay between inflammation, coagulation and endothelial injury in the early phase of acute pancreatitis: Clinical implications. Int. J. Mol. Sci. 2017, 18, 354. [Google Scholar] [CrossRef] [PubMed]
- Moloney, R.D.; O’Mahony, S.M.; Dinan, T.G.; Cryan, J.F. Stress-induced visceral pain: Toward animal models of irritable-bowel syndrome and associated comorbidities. Front. Psychiatry 2015, 6, 15. [Google Scholar] [CrossRef] [PubMed]
- Felice, V.D.; Quigley, E.M.; Sullivan, A.M.; O’Keeffe, G.W.; O’Mahony, S.M. Microbiota-gut-brain signalling in Parkinson’s disease: Implications for non-motor symptoms. Parkinsonism Relat. Disord. 2016, 27, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Straub, R.H.; Wiest, R.; Strauch, U.G.; Harle, P.; Scholmerich, J. The role of the sympathetic nervous system in intestinal inflammation. Gut 2006, 55, 1640–1649. [Google Scholar] [CrossRef] [PubMed]
- Costantini, T.W.; Baird, A. Lost your nerve? Modulating the parasympathetic nervous system to treat inflammatory bowel disease. J. Physiol. 2016, 594, 4097–4098. [Google Scholar] [CrossRef] [PubMed]
- Warzecha, Z.; Dembinski, A.; Ceranowicz, P.; Konturek, P.C.; Stachura, J.; Tomaszewska, R.; Konturek, S.J. Calcitonin gene-related peptide can attenuate or augment pancreatic damage in caerulein-induced pancreatitis in rats. J. Physiol. Pharmacol. 1999, 50, 49–62. [Google Scholar] [PubMed]
- Warzecha, Z.; Dembinski, A.; Ceranowicz, P.; Stachura, J.; Tomaszewska, R.; Konturek, S.J. Effect of sensory nerves and cgrp on the development of caerulein-induced pancreatitis and pancreatic recovery. J. Physiol. Pharmacol. 2001, 52, 679–704. [Google Scholar] [PubMed]
- Dembinski, A.; Warzecha, Z.; Ceranowicz, P.; Jaworek, J.; Sendur, R.; Knafel, A.; Dembinski, M.; Bilski, J.; Pawlik, W.W.; Tomaszewska, R.; et al. Stimulation of sensory nerves and cgrp attenuate pancreatic damage in ischemia/reperfusion induced pancreatitis. Med. Sci. Monit. 2003, 9, BR418–BR425. [Google Scholar] [PubMed]
- Dembinski, A.; Warzecha, Z.; Ceranowicz, P.; Konturek, S.J. The role of capsaicin-sensitive sensory neurons and nitric oxide in regulation of gastric mucosal growth. J. Physiol. Pharmacol. 1995, 46, 351–362. [Google Scholar] [PubMed]
- Dembinski, A.; Warzecha, Z.; Konturek, P.J.; Ceranowicz, P.; Konturek, S.J. Influence of capsaicin-sensitive afferent neurons and nitric oxide (no) on cerulein-induced pancreatitis in rats. Int. J. Pancreatol. 1996, 19, 179–189. [Google Scholar] [PubMed]
- Warzecha, Z.; Dembinski, A.; Jaworek, J.; Ceranowicz, P.; Szlachcic, A.; Walocha, J.; Konturek, S.J. Role of sensory nerves in pancreatic secretion and caerulein-induced pancreatitis. J. Physiol. Pharmacol. 1997, 48, 43–58. [Google Scholar] [PubMed]
- Warzecha, Z.; Dembinski, A.; Ceranowicz, P.; Dembinski, M.; Cieszkowski, J.; Kownacki, P.; Konturek, P.C. Role of sensory nerves in gastroprotective effect of anandamide in rats. J. Physiol. Pharmacol. 2011, 62, 207–217. [Google Scholar] [PubMed]
- Ramos, L.R.; Sachar, D.B.; DiMaio, C.J.; Colombel, J.F.; Torres, J. Inflammatory bowel disease and pancreatitis: A review. J. Crohn’s Colitis 2016, 10, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Dembinski, A.; Warzecha, Z.; Ceranowicz, P.; Dembinski, M.; Cieszkowski, J.; Pawlik, W.W.; Konturek, S.J.; Tomaszewska, R.; Hladki, W.; Konturek, P.C. Cannabinoids in acute gastric damage and pancreatitis. J. Physiol. Pharmacol. 2006, 57 (Suppl. S5), 137–154. [Google Scholar] [PubMed]
- Dembinski, A.; Warzecha, Z.; Ceranowicz, P.; Warzecha, A.M.; Pawlik, W.W.; Dembinski, M.; Rembiasz, K.; Sendur, P.; Kusnierz-Cabala, B.; Tomaszewska, R.; et al. Dual, time-dependent deleterious and protective effect of anandamide on the course of cerulein-induced acute pancreatitis. Role of sensory nerves. Eur. J. Pharmacol. 2008, 591, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Endres, K.; Schafer, K.H. Influence of commensal microbiota on the enteric nervous system and its role in neurodegenerative diseases. J. Innate Immun. 2018, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kubinak, J.L.; Round, J.L. Toll-like receptors promote mutually beneficial commensal-host interactions. PLoS Pathog. 2012, 8, e1002785. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D. Gut microbiota—At the intersection of everything? Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 321–322. [Google Scholar] [CrossRef] [PubMed]
- Carabotti, M.; Scirocco, A.; Maselli, M.A.; Severi, C. The gut-brain axis: Interactions between enteric microbiota, central and enteric nervous systems. Ann. Gastroenterol. 2015, 28, 203–209. [Google Scholar] [PubMed]
- Sender, R.; Fuchs, S.; Milo, R. Are we really vastly outnumbered? Revisiting the ratio of bacterial to host cells in humans. Cell 2016, 164, 337–340. [Google Scholar] [CrossRef] [PubMed]
- Costea, P.I.; Hildebrand, F.; Arumugam, M.; Backhed, F.; Blaser, M.J.; Bushman, F.D.; de Vos, W.M.; Ehrlich, S.D.; Fraser, C.M.; Hattori, M.; et al. Enterotypes in the landscape of gut microbial community composition. Nat. Microbiol. 2018, 3, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Eckburg, P.B.; Bik, E.M.; Bernstein, C.N.; Purdom, E.; Dethlefsen, L.; Sargent, M.; Gill, S.R.; Nelson, K.E.; Relman, D.A. Diversity of the human intestinal microbial flora. Science 2005, 308, 1635–1638. [Google Scholar] [CrossRef] [PubMed]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Ubeda, C.; Pamer, E.G. Antibiotics, microbiota, and immune defense. Trends Immunol. 2012, 33, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.; Kolodziejczyk, A.A.; Thaiss, C.A.; Elinav, E. Dysbiosis and the immune system. Nat. Rev. Immunol. 2017, 17, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Dembinski, A.; Warzecha, Z.; Ceranowicz, P.; Dembinski, M.; Cieszkowski, J.; Gosiewski, T.; Bulanda, M.; Kusnierz-Cabala, B.; Galazka, K.; Konturek, P.C. Synergic interaction of rifaximin and mutaflor (escherichia coli nissle 1917) in the treatment of acetic acid-induced colitis in rats. Gastroenterol. Res. Pract. 2016, 2016, 3126280. [Google Scholar] [CrossRef] [PubMed]
- Jaworek, J.; Tudek, B.; Kowalczyk, P.; Kot, M.; Szklarczyk, J.; Leja-Szpak, A.; Pierzchalski, P.; Bonior, J.; Dembinski, A.; Ceranowicz, P.; et al. Effect of endotoxemia in suckling rats on pancreatic integrity and exocrine function in adults: A review report. Gastroenterol. Res. Pract. 2018, 2018, 6915059. [Google Scholar] [CrossRef] [PubMed]
- Cryan, J.F.; Dinan, T.G. Mind-altering microorganisms: The impact of the gut microbiota on brain and behaviour. Nat. Rev. Neurosci. 2012, 13, 701–712. [Google Scholar] [CrossRef] [PubMed]
- Foster, J.A.; McVey Neufeld, K.A. Gut-brain axis: How the microbiome influences anxiety and depression. Trends Neurosci. 2013, 36, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Mazzoli, R.; Pessione, E. The neuro-endocrinological role of microbial glutamate and gaba signaling. Front. Microbiol. 2016, 7, 1934. [Google Scholar] [CrossRef] [PubMed]
- Lyte, M. Probiotics function mechanistically as delivery vehicles for neuroactive compounds: Microbial endocrinology in the design and use of probiotics. Bioessays 2011, 33, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Sivamaruthi, B.S.; Madhumita, R.; Balamurugan, K.; Rajan, K.E. Cronobacter sakazakii infection alters serotonin transporter and improved fear memory retention in the rat. Front. Pharmacol. 2015, 6, 188. [Google Scholar] [CrossRef] [PubMed]
- Ceranowicz, P.; Warzecha, Z.; Dembinski, A. Peptidyl hormones of endocrine cells origin in the gut—Their discovery and physiological relevance. J. Physiol. Pharmacol. 2015, 66, 11–27. [Google Scholar] [PubMed]
- Bellono, N.W.; Bayrer, J.R.; Leitch, D.B.; Castro, J.; Zhang, C.; O’Donnell, T.A.; Brierley, S.M.; Ingraham, H.A.; Julius, D. Enterochromaffin cells are gut chemosensors that couple to sensory neural pathways. Cell 2017, 170, 185–198.e116. [Google Scholar] [CrossRef] [PubMed]
- O’Mahony, S.M.; Clarke, G.; Borre, Y.E.; Dinan, T.G.; Cryan, J.F. Serotonin, tryptophan metabolism and the brain-gut-microbiome axis. Behav. Brain Res. 2015, 277, 32–48. [Google Scholar] [CrossRef] [PubMed]
- Alam, R.; Abdolmaleky, H.M.; Zhou, J.R. Microbiome, inflammation, epigenetic alterations, and mental diseases. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2017, 174, 651–660. [Google Scholar] [CrossRef] [PubMed]
- Galland, L. The gut microbiome and the brain. J. Med. Food 2014, 17, 1261–1272. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.J.; Chiu, I.M. Bacterial signaling to the nervous system through toxins and metabolites. J. Mol. Biol. 2017, 429, 587–605. [Google Scholar] [CrossRef] [PubMed]
- Collins, S.M.; Surette, M.; Bercik, P. The interplay between the intestinal microbiota and the brain. Nat. Rev. Microbiol. 2012, 10, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Mulak, A.; Bonaz, B. Brain-gut-microbiota axis in Parkinson’s disease. World J. Gastroenterol. 2015, 21, 10609–10620. [Google Scholar] [CrossRef] [PubMed]
- Tomik, J.; Tomik, B.; Gajec, S.; Ceranowicz, P.; Pihut, M.; Olszanecki, R.; Strek, P.; Skladzien, J. The balloon-based manometry evaluation of swallowing in patients with amyotrophic lateral sclerosis. Int. J. Mol. Sci. 2017, 18, 707. [Google Scholar] [CrossRef] [PubMed]
- Warzecha, Z.; Dembinski, A.; Ceranowicz, P.; Dembinski, M.; Sendur, R.; Pawlik, W.W.; Konturek, S.J. Deleterious effect of helicobacter pylori infection on the course of acute pancreatitis in rats. Pancreatology 2002, 2, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, C.; Colucci, R.; Antonioli, L.; Barocelli, E.; Ballabeni, V.; Bernardini, N.; Blandizzi, C.; de Jonge, W.J.; Fornai, M. Intestinal dysfunction in Parkinson’s disease: Lessons learned from translational studies and experimental models. Neurogastroenterol. Motil. 2016, 28, 1781–1791. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; de Vos, R.A.; Bohl, J.; Del Tredici, K. Gastric alpha-synuclein immunoreactive inclusions in meissner’s and auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci. Lett. 2006, 396, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Svensson, E.; Horvath-Puho, E.; Thomsen, R.W.; Djurhuus, J.C.; Pedersen, L.; Borghammer, P.; Sorensen, H.T. Vagotomy and subsequent risk of Parkinson’s disease. Ann. Neurol. 2015, 78, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Pan-Montojo, F.; Schwarz, M.; Winkler, C.; Arnhold, M.; O’Sullivan, G.A.; Pal, A.; Said, J.; Marsico, G.; Verbavatz, J.M.; Rodrigo-Angulo, M.; et al. Environmental toxins trigger pd-like progression via increased alpha-synuclein release from enteric neurons in mice. Sci. Rep. 2012, 2, 898. [Google Scholar] [CrossRef] [PubMed]
- Forsyth, C.B.; Shannon, K.M.; Kordower, J.H.; Voigt, R.M.; Shaikh, M.; Jaglin, J.A.; Estes, J.D.; Dodiya, H.B.; Keshavarzian, A. Increased intestinal permeability correlates with sigmoid mucosa alpha-synuclein staining and endotoxin exposure markers in early Parkinson’s disease. PLoS ONE 2011, 6, e28032. [Google Scholar] [CrossRef] [PubMed]
- Keshavarzian, A.; Green, S.J.; Engen, P.A.; Voigt, R.M.; Naqib, A.; Forsyth, C.B.; Mutlu, E.; Shannon, K.M. Colonic bacterial composition in Parkinson’s disease. Mov. Disord. 2015, 30, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Erickson, M.A. The blood-brain barrier and immune function and dysfunction. Neurobiol. Dis. 2010, 37, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Scheperjans, F.; Aho, V.; Pereira, P.A.; Koskinen, K.; Paulin, L.; Pekkonen, E.; Haapaniemi, E.; Kaakkola, S.; Eerola-Rautio, J.; Pohja, M.; et al. Gut microbiota are related to Parkinson’s disease and clinical phenotype. Mov. Disord. 2015, 30, 350–358. [Google Scholar] [CrossRef] [PubMed]
- De Vadder, F.; Kovatcheva-Datchary, P.; Goncalves, D.; Vinera, J.; Zitoun, C.; Duchampt, A.; Backhed, F.; Mithieux, G. Microbiota-generated metabolites promote metabolic benefits via gut-brain neural circuits. Cell 2014, 156, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Andrews, Z.B.; Erion, D.; Beiler, R.; Liu, Z.W.; Abizaid, A.; Zigman, J.; Elsworth, J.D.; Savitt, J.M.; DiMarchi, R.; Tschoep, M.; et al. Ghrelin promotes and protects nigrostriatal dopamine function via a ucp2-dependent mitochondrial mechanism. J. Neurosci. 2009, 29, 14057–14065. [Google Scholar] [CrossRef] [PubMed]
- Unger, M.M.; Moller, J.C.; Mankel, K.; Eggert, K.M.; Bohne, K.; Bodden, M.; Stiasny-Kolster, K.; Kann, P.H.; Mayer, G.; Tebbe, J.J.; et al. Postprandial ghrelin response is reduced in patients with Parkinson’s disease and idiopathic rem sleep behaviour disorder: A peripheral biomarker for early Parkinson’s disease? J. Neurol. 2011, 258, 982–990. [Google Scholar] [CrossRef] [PubMed]
- Sampson, T.R.; Debelius, J.W.; Thron, T.; Janssen, S.; Shastri, G.G.; Ilhan, Z.E.; Challis, C.; Schretter, C.E.; Rocha, S.; Gradinaru, V.; et al. Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell 2016, 167, 1469–1480.e1412. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, T.H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 2009, 22, 240–273. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R.; Janeway, C., Jr. Innate immunity. N. Engl. J. Med. 2000, 343, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, C.; Hudson, K.L.; Anderson, K.V. The toll gene of drosophila, required for dorsal-ventral embryonic polarity, appears to encode a transmembrane protein. Cell 1988, 52, 269–279. [Google Scholar] [CrossRef]
- Brun, P.; Giron, M.C.; Qesari, M.; Porzionato, A.; Caputi, V.; Zoppellaro, C.; Banzato, S.; Grillo, A.R.; Spagnol, L.; De Caro, R.; et al. Toll-like receptor 2 regulates intestinal inflammation by controlling integrity of the enteric nervous system. Gastroenterology 2013, 145, 1323–1333. [Google Scholar] [CrossRef] [PubMed]
- Brun, P.; Gobbo, S.; Caputi, V.; Spagnol, L.; Schirato, G.; Pasqualin, M.; Levorato, E.; Palu, G.; Giron, M.C.; Castagliuolo, I. Toll like receptor-2 regulates production of glial-derived neurotrophic factors in murine intestinal smooth muscle cells. Mol. Cell. Neurosci. 2015, 68, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Okun, E.; Griffioen, K.J.; Mattson, M.P. Toll-like receptor signaling in neural plasticity and disease. Trends Neurosci. 2011, 34, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Prehaud, C.; Megret, F.; Lafage, M.; Lafon, M. Virus infection switches tlr-3-positive human neurons to become strong producers of beta interferon. J. Virol. 2005, 79, 12893–12904. [Google Scholar] [CrossRef] [PubMed]
- Barajon, I.; Serrao, G.; Arnaboldi, F.; Opizzi, E.; Ripamonti, G.; Balsari, A.; Rumio, C. Toll-like receptors 3, 4, and 7 are expressed in the enteric nervous system and dorsal root ganglia. J. Histochem. Cytochem. 2009, 57, 1013–1023. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Ho, D.H.; Suk, J.E.; You, S.; Michael, S.; Kang, J.; Joong Lee, S.; Masliah, E.; Hwang, D.; Lee, H.J.; et al. Neuron-released oligomeric alpha-synuclein is an endogenous agonist of tlr2 for paracrine activation of microglia. Nat. Commun. 2013, 4, 1562. [Google Scholar] [CrossRef] [PubMed]
- Daniele, S.G.; Beraud, D.; Davenport, C.; Cheng, K.; Yin, H.; Maguire-Zeiss, K.A. Activation of myd88-dependent tlr1/2 signaling by misfolded alpha-synuclein, a protein linked to neurodegenerative disorders. Sci. Signal. 2015, 8, ra45. [Google Scholar] [CrossRef] [PubMed]
- Beraud, D.; Maguire-Zeiss, K.A. Misfolded alpha-synuclein and toll-like receptors: Therapeutic targets for Parkinson’s disease. Parkinsonism. Relat. Disord. 2012, 18 (Suppl. S1), S17–S20. [Google Scholar] [CrossRef]
- Fellner, L.; Irschick, R.; Schanda, K.; Reindl, M.; Klimaschewski, L.; Poewe, W.; Wenning, G.K.; Stefanova, N. Toll-like receptor 4 is required for alpha-synuclein dependent activation of microglia and astroglia. Glia 2013, 61, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Noelker, C.; Morel, L.; Lescot, T.; Osterloh, A.; Alvarez-Fischer, D.; Breloer, M.; Henze, C.; Depboylu, C.; Skrzydelski, D.; Michel, P.P.; et al. Toll like receptor 4 mediates cell death in a mouse mptp model of parkinson disease. Sci. Rep. 2013, 3, 1393. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Jiang, W.; Liu, L.; Wang, X.; Ding, C.; Tian, Z.; Zhou, R. Dopamine controls systemic inflammation through inhibition of nlrp3 inflammasome. Cell 2015, 160, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Codolo, G.; Plotegher, N.; Pozzobon, T.; Brucale, M.; Tessari, I.; Bubacco, L.; de Bernard, M. Triggering of inflammasome by aggregated alpha-synuclein, an inflammatory response in synucleinopathies. PLoS ONE 2013, 8, e55375. [Google Scholar] [CrossRef] [PubMed]
- Labzin, L.I.; Heneka, M.T.; Latz, E. Innate immunity and neurodegeneration. Annu. Rev. Med. 2018, 69, 437–449. [Google Scholar] [CrossRef] [PubMed]
- Rietdijk, C.D.; van Wezel, R.J.; Garssen, J.; Kraneveld, A.D. Neuronal toll-like receptors and neuro-immunity in Parkinson’s disease, Alzheimer’s disease and stroke. Neuroimmunol. Neuroinflamm. 2016, 3, 27–37. [Google Scholar] [CrossRef]
- Drouin-Ouellet, J.; Cicchetti, F. Inflammation and neurodegeneration: The story ‘retolled’. Trends Pharmacol. Sci. 2012, 33, 542–551. [Google Scholar] [CrossRef] [PubMed]
- Mosher, K.I.; Wyss-Coray, T. Microglial dysfunction in brain aging and Alzheimer’s disease. Biochem. Pharmacol. 2014, 88, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Yanez, A.; Hassanzadeh-Kiabi, N.; Ng, M.Y.; Megias, J.; Subramanian, A.; Liu, G.Y.; Underhill, D.M.; Gil, M.L.; Goodridge, H.S. Detection of a tlr2 agonist by hematopoietic stem and progenitor cells impacts the function of the macrophages they produce. Eur. J. Immunol. 2013, 43, 2114–2125. [Google Scholar] [CrossRef] [PubMed]
- Houser, M.C.; Tansey, M.G. The gut-brain axis: Is intestinal inflammation a silent driver of Parkinson’s disease pathogenesis? NPJ Parkinsons Dis. 2017, 3, 3. [Google Scholar] [CrossRef] [PubMed]
- Stefanova, N.; Fellner, L.; Reindl, M.; Masliah, E.; Poewe, W.; Wenning, G.K. Toll-like receptor 4 promotes alpha-synuclein clearance and survival of nigral dopaminergic neurons. Am. J. Pathol. 2011, 179, 954–963. [Google Scholar] [CrossRef] [PubMed]
- Friedland, R.P. Mechanisms of molecular mimicry involving the microbiota in neurodegeneration. J. Alzheimers Dis. 2015, 45, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Caputi, V.; Marsilio, I.; Filpa, V.; Cerantola, S.; Orso, G.; Bistoletti, M.; Paccagnella, N.; De Martin, S.; Montopoli, M.; Dall’Acqua, S.; et al. Antibiotic-induced dysbiosis of the microbiota impairs gut neuromuscular function in juvenile mice. Br. J. Pharmacol. 2017, 174, 3623–3639. [Google Scholar] [CrossRef] [PubMed]
- Latorre, E.; Layunta, E.; Grasa, L.; Castro, M.; Pardo, J.; Gomollon, F.; Alcalde, A.I.; Mesonero, J.E. Intestinal serotonin transporter inhibition by toll-like receptor 2 activation. A feedback modulation. PLoS ONE 2016, 11, e0169303. [Google Scholar] [CrossRef] [PubMed]
- Caputi, V.; Marsilio, I.; Cerantola, S.; Roozfarakh, M.; Lante, I.; Galuppini, F.; Rugge, M.; Napoli, E.; Giulivi, C.; Orso, G.; et al. Toll-like receptor 4 modulates small intestine neuromuscular function through nitrergic and purinergic pathways. Front. Pharmacol. 2017, 8, 350. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Jiang, E.; Weng, H.R. Activation of toll like receptor 4 attenuates gaba synthesis and postsynaptic gaba receptor activities in the spinal dorsal horn via releasing interleukin-1 beta. J. Neuroinflamm. 2015, 12, 222. [Google Scholar] [CrossRef] [PubMed]
- Erny, D.; Hrabe de Angelis, A.L.; Jaitin, D.; Wieghofer, P.; Staszewski, O.; David, E.; Keren-Shaul, H.; Mahlakoiv, T.; Jakobshagen, K.; Buch, T.; et al. Host microbiota constantly control maturation and function of microglia in the cns. Nat. Neurosci. 2015, 18, 965–977. [Google Scholar] [CrossRef] [PubMed]
- Larraufie, P.; Dore, J.; Lapaque, N.; Blottiere, H.M. Tlr ligands and butyrate increase pyy expression through two distinct but inter-regulated pathways. Cell. Microbiol. 2017, 19. [Google Scholar] [CrossRef] [PubMed]
- Clemente, J.C.; Ursell, L.K.; Parfrey, L.W.; Knight, R. The impact of the gut microbiota on human health: An integrative view. Cell 2012, 148, 1258–1270. [Google Scholar] [CrossRef] [PubMed]
- Maslowski, K.M.; Mackay, C.R. Diet, gut microbiota and immune responses. Nat. Immunol. 2011, 12, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Perez-Pardo, P.; Dodiya, H.B.; Broersen, L.M.; Douna, H.; van Wijk, N.; Lopes da Silva, S.; Garssen, J.; Keshavarzian, A.; Kraneveld, A.D. Gut-brain and brain-gut axis in Parkinson’s disease models: Effects of a uridine and fish oil diet. Nutr. Neurosci. 2017, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Perez-Pardo, P.; Kliest, T.; Dodiya, H.B.; Broersen, L.M.; Garssen, J.; Keshavarzian, A.; Kraneveld, A.D. The gut-brain axis in Parkinson’s disease: Possibilities for food-based therapies. Eur. J. Pharmacol. 2017, 817, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.L.; James-Kracke, M.; Sun, G.Y.; Sun, A.Y. Oxidative and inflammatory pathways in Parkinson’s disease. Neurochem. Res. 2009, 34, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Afshordel, S.; Hagl, S.; Werner, D.; Rohner, N.; Kogel, D.; Bazan, N.G.; Eckert, G.P. Omega-3 polyunsaturated fatty acids improve mitochondrial dysfunction in brain aging—Impact of bcl-2 and npd-1 like metabolites. Prostaglandins Leukot. Essent. Fatty Acids 2015, 92, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Bazan, N.G.; Molina, M.F.; Gordon, W.C. Docosahexaenoic acid signalolipidomics in nutrition: Significance in aging, neuroinflammation, macular degeneration, alzheimer’s, and other neurodegenerative diseases. Ann. Rev. Nutr. 2011, 31, 321–351. [Google Scholar] [CrossRef] [PubMed]
- Coulombe, K.; Kerdiles, O.; Tremblay, C.; Emond, V.; Lebel, M.; Boulianne, A.S.; Plourde, M.; Cicchetti, F.; Calon, F. Impact of dha intake in a mouse model of synucleinopathy. Exp. Neurol. 2018, 301, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Hwang, D.H.; Kim, J.A.; Lee, J.Y. Mechanisms for the activation of toll-like receptor 2/4 by saturated fatty acids and inhibition by docosahexaenoic acid. Eur. J. Pharmacol. 2016, 785, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Zhao, L.; Hwang, D.H. Modulation of pattern recognition receptor-mediated inflammation and risk of chronic diseases by dietary fatty acids. Nutr. Rev. 2010, 68, 38–61. [Google Scholar] [CrossRef] [PubMed]
- Beamer, C.A.; Shepherd, D.M. Inhibition of tlr ligand- and interferon gamma-induced murine microglial activation by panax notoginseng. J. Neuroimmune Pharmacol. 2012, 7, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Haddadi, R.; Nayebi, A.M.; Eyvari Brooshghalan, S. Silymarin prevents apoptosis through inhibiting the bax/caspase-3 expression and suppresses toll like receptor-4 pathway in the snc of 6-ohda intoxicated rats. Biomed. Pharmacother. 2018, 104, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.; Guarner, F.; Reid, G.; Gibson, G.R.; Merenstein, D.J.; Pot, B.; Morelli, L.; Canani, R.B.; Flint, H.J.; Salminen, S.; et al. Expert consensus document. The international scientific association for probiotics and prebiotics consensus statement on the scope and appropriate use of the term probiotic. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Derrien, M.; van Hylckama Vlieg, J.E. Fate, activity, and impact of ingested bacteria within the human gut microbiota. Trends Microbiol. 2015, 23, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Sanders, M.E.; Benson, A.; Lebeer, S.; Merenstein, D.J.; Klaenhammer, T.R. Shared mechanisms among probiotic taxa: Implications for general probiotic claims. Curr. Opin. Biotechnol. 2018, 49, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Wallace, C.J.K.; Milev, R. The effects of probiotics on depressive symptoms in humans: A systematic review. Ann. Gen. Psychiatry 2017, 16, 14. [Google Scholar] [CrossRef] [PubMed]
- Abildgaard, A.; Elfving, B.; Hokland, M.; Wegener, G.; Lund, S. Probiotic treatment reduces depressive-like behaviour in rats independently of diet. Psychoneuroendocrinology 2017, 79, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.F.; Shen, Y.Q. Dysbiosis of gut microbiota and microbial metabolites in Parkinson’s disease. Ageing Res. Rev. 2018, 45, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Bravo, J.A.; Forsythe, P.; Chew, M.V.; Escaravage, E.; Savignac, H.M.; Dinan, T.G.; Bienenstock, J.; Cryan, J.F. Ingestion of lactobacillus strain regulates emotional behavior and central gaba receptor expression in a mouse via the vagus nerve. Proc. Natl. Acad. Sci. USA 2011, 108, 16050–16055. [Google Scholar] [CrossRef] [PubMed]
- Barichella, M.; Pacchetti, C.; Bolliri, C.; Cassani, E.; Iorio, L.; Pusani, C.; Pinelli, G.; Privitera, G.; Cesari, I.; Faierman, S.A.; et al. Probiotics and prebiotic fiber for constipation associated with parkinson disease: An rct. Neurology 2016, 87, 1274–1280. [Google Scholar] [CrossRef] [PubMed]
- Cassani, E.; Privitera, G.; Pezzoli, G.; Pusani, C.; Madio, C.; Iorio, L.; Barichella, M. Use of probiotics for the treatment of constipation in Parkinson’s disease patients. Minerva Gastroenterol. Dietol. 2011, 57, 117–121. [Google Scholar] [PubMed]
- Ojetti, V.; Ianiro, G.; Tortora, A.; D’Angelo, G.; Di Rienzo, T.A.; Bibbo, S.; Migneco, A.; Gasbarrini, A. The effect of lactobacillus reuteri supplementation in adults with chronic functional constipation: A randomized, double-blind, placebo-controlled trial. J. Gastrointest. Liver Dis. JGLD 2014, 23, 387–391. [Google Scholar]
- Gibson, G.R.; Hutkins, R.; Sanders, M.E.; Prescott, S.L.; Reimer, R.A.; Salminen, S.J.; Scott, K.; Stanton, C.; Swanson, K.S.; Cani, P.D.; et al. Expert consensus document: The international scientific association for probiotics and prebiotics (isapp) consensus statement on the definition and scope of prebiotics. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, H.E.; Piazza, B.R.; Forsyth, C.B.; Keshavarzian, A. Nutrition and gastrointestinal health as modulators of Parkinson’s disease. In Pharma-Nutrition: An Overview; Folkerts, G., Garssen, J., Eds.; Springer International Publishing: Cham, Germany, 2014; pp. 213–242. [Google Scholar]
- Savignac, H.M.; Corona, G.; Mills, H.; Chen, L.; Spencer, J.P.; Tzortzis, G.; Burnet, P.W. Prebiotic feeding elevates central brain derived neurotrophic factor, n-methyl-d-aspartate receptor subunits and d-serine. Neurochem. Int. 2013, 63, 756–764. [Google Scholar] [CrossRef] [PubMed]
- Barboza, J.L.; Okun, M.S.; Moshiree, B. The treatment of gastroparesis, constipation and small intestinal bacterial overgrowth syndrome in patients with Parkinson’s disease. Expert Opin. Pharmacother. 2015, 16, 2449–2464. [Google Scholar] [CrossRef] [PubMed]
- Parashar, A.; Udayabanu, M. Gut microbiota: Implications in Parkinson’s disease. Parkinsonism Relat. Disord. 2017, 38, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Villena, J.; Kitazawa, H. Modulation of intestinal tlr4-inflammatory signaling pathways by probiotic microorganisms: Lessons learned from lactobacillus jensenii tl2937. Front. Immunol. 2014, 4, 512. [Google Scholar] [CrossRef] [PubMed]
- Barochia, A.; Solomon, S.; Cui, X.; Natanson, C.; Eichacker, P.Q. Eritoran tetrasodium (e5564) treatment for sepsis: Review of preclinical and clinical studies. Expert Opin. Drug Metab. Toxicol. 2011, 7, 479–494. [Google Scholar] [CrossRef] [PubMed]
- Schwenkgrub, J.; Zaremba, M.; Joniec-Maciejak, I.; Cudna, A.; Mirowska-Guzel, D.; Kurkowska-Jastrzebska, I. The phosphodiesterase inhibitor, ibudilast, attenuates neuroinflammation in the mptp model of Parkinson’s disease. PLoS ONE 2017, 12, e0182019. [Google Scholar] [CrossRef] [PubMed]
- Pant, K.; Yadav, A.K.; Gupta, P.; Islam, R.; Saraya, A.; Venugopal, S.K. Butyrate induces ros-mediated apoptosis by modulating mir-22/sirt-1 pathway in hepatic cancer cells. Redox Biol. 2017, 12, 340–349. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Chen, W.D.; Wang, Y.D. Gut microbiota: An integral moderator in health and disease. Front. Microbiol. 2018, 9, 151. [Google Scholar] [CrossRef] [PubMed]
- Moloney, G.M.; O’Leary, O.F.; Salvo-Romero, E.; Desbonnet, L.; Shanahan, F.; Dinan, T.G.; Clarke, G.; Cryan, J.F. Microbial regulation of hippocampal mirna expression: Implications for transcription of kynurenine pathway enzymes. Behav. Brain Res. 2017, 334, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Paschon, V.; Takada, S.H.; Ikebara, J.M.; Sousa, E.; Raeisossadati, R.; Ulrich, H.; Kihara, A.H. Interplay between exosomes, micrornas and toll-like receptors in brain disorders. Mol. Neurobiol. 2016, 53, 2016–2028. [Google Scholar] [CrossRef] [PubMed]
- Tang, R.; Lin, Y.M.; Liu, H.X.; Wang, E.S. Neuroprotective effect of docosahexaenoic acid in rat traumatic brain injury model via regulation of tlr4/nf-kappa b signaling pathway. Int. J. Biochem. Cell Biol. 2018, 99, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Kumar, H.; Kawai, T.; Akira, S. Toll-like receptors and innate immunity. Biochem. Biophys. Res. Commun. 2009, 388, 621–625. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, A.; Akbari, P.; Difilippo, E.; Schols, H.A.; Ulfman, L.H.; Schoterman, M.H.; Garssen, J.; Fink-Gremmels, J.; Braber, S. The piglet as a model for studying dietary components in infant diets: Effects of galacto-oligosaccharides on intestinal functions. Br. J. Nutr. 2016, 115, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Riviere, A.; Selak, M.; Lantin, D.; Leroy, F.; De Vuyst, L. Bifidobacteria and butyrate-producing colon bacteria: Importance and strategies for their stimulation in the human gut. Front. Microbiol. 2016, 7, 979. [Google Scholar] [CrossRef] [PubMed]

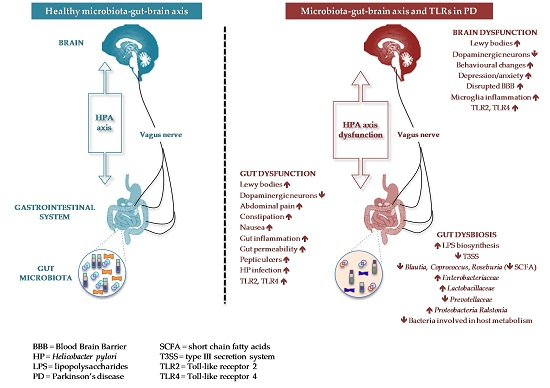{kind=link}
| Modulator | Influence on Microbiota-Gut-Brain Axis | Potential TLRs Target | References |
|---|---|---|---|
| Dietary supplements | |||
| Docosahexaenoic acid (DHA) | Reduction of oxidative stress by improving neuronal mitochondrial dysfunction | TLR2, TLR4 | [104,108,134] |
| Panax Notoginseng (NotoG) | Suppression of microglial activation and reduction of IL-6 and TNF-α release | TLR4 | [110] |
| Sylimarin | Antioxidant and neuroprotective effects (salvaging of free radicals) | TLR4 | [111] |
| Probiotics | |||
| L. rhamnosus (JB-1) | Modulation of GABAA and GABAB receptors in the brain | TLR1, TLR2, TLR6 | [118,135] |
| Lactobacillus casei Shirota | Decrease of visceral pain and bloating; improvement of stool consistency | TLR1, TLR2, TLR6 | [120,135] |
| Lactobacillus reuteri | Improvement of bowel movement and increase of the frequency of evacuation | TLR1, TLR2, TLR6 | [121,135] |
| Prebiotics | |||
| Fructo oligosaccharides (FOS) | Increase of BDNF expression in the hippocampus | TLR2, TLR4 | [73,124,135] |
| Galacto oligosaccharides (GOS) | Improvement of villus surface area in the small intestine | TLR1-TLR13 | [135,136] |
| Short-chain fatty acids (SCFA)-Butyrate | Maintenance of colonic epithelium integrity | TLR1, TLR2, TLR6 | [135,137] |
| Antibiotics | |||
| Rifaximin | Treatment of small intestinal overgrowth | TLR4 | [125] |
| Mynocicline | Neuroprotective effect on nigrostriatal dopaminergic neurons | TLR4 | [126] |
| TLRs modulators | |||
| Eritoran tetrasodium | Inhibition of LPS-induced proinflammatory cytokine release | TLR4 | [128] |
| Ibudilast | Improvement of anti-inflammatory cytokine release, modulation of glial cells activity and induction of GDNF expression | TLR4 | [129] |
| MiR-22 | Induced by butyrate-producing commensal bacteria | TLR1, TLR2, TLR6 | [130,135] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caputi, V.; Giron, M.C. Microbiome-Gut-Brain Axis and Toll-Like Receptors in Parkinson’s Disease. Int. J. Mol. Sci. 2018, 19, 1689. https://doi.org/10.3390/ijms19061689
Caputi V, Giron MC. Microbiome-Gut-Brain Axis and Toll-Like Receptors in Parkinson’s Disease. International Journal of Molecular Sciences. 2018; 19(6):1689. https://doi.org/10.3390/ijms19061689
Chicago/Turabian StyleCaputi, Valentina, and Maria Cecilia Giron. 2018. "Microbiome-Gut-Brain Axis and Toll-Like Receptors in Parkinson’s Disease" International Journal of Molecular Sciences 19, no. 6: 1689. https://doi.org/10.3390/ijms19061689
APA StyleCaputi, V., & Giron, M. C. (2018). Microbiome-Gut-Brain Axis and Toll-Like Receptors in Parkinson’s Disease. International Journal of Molecular Sciences, 19(6), 1689. https://doi.org/10.3390/ijms19061689