The Role of Endoplasmic Reticulum Stress-Glycogen Synthase Kinase-3 Signaling in Atherogenesis


Abstract

{kind=link}
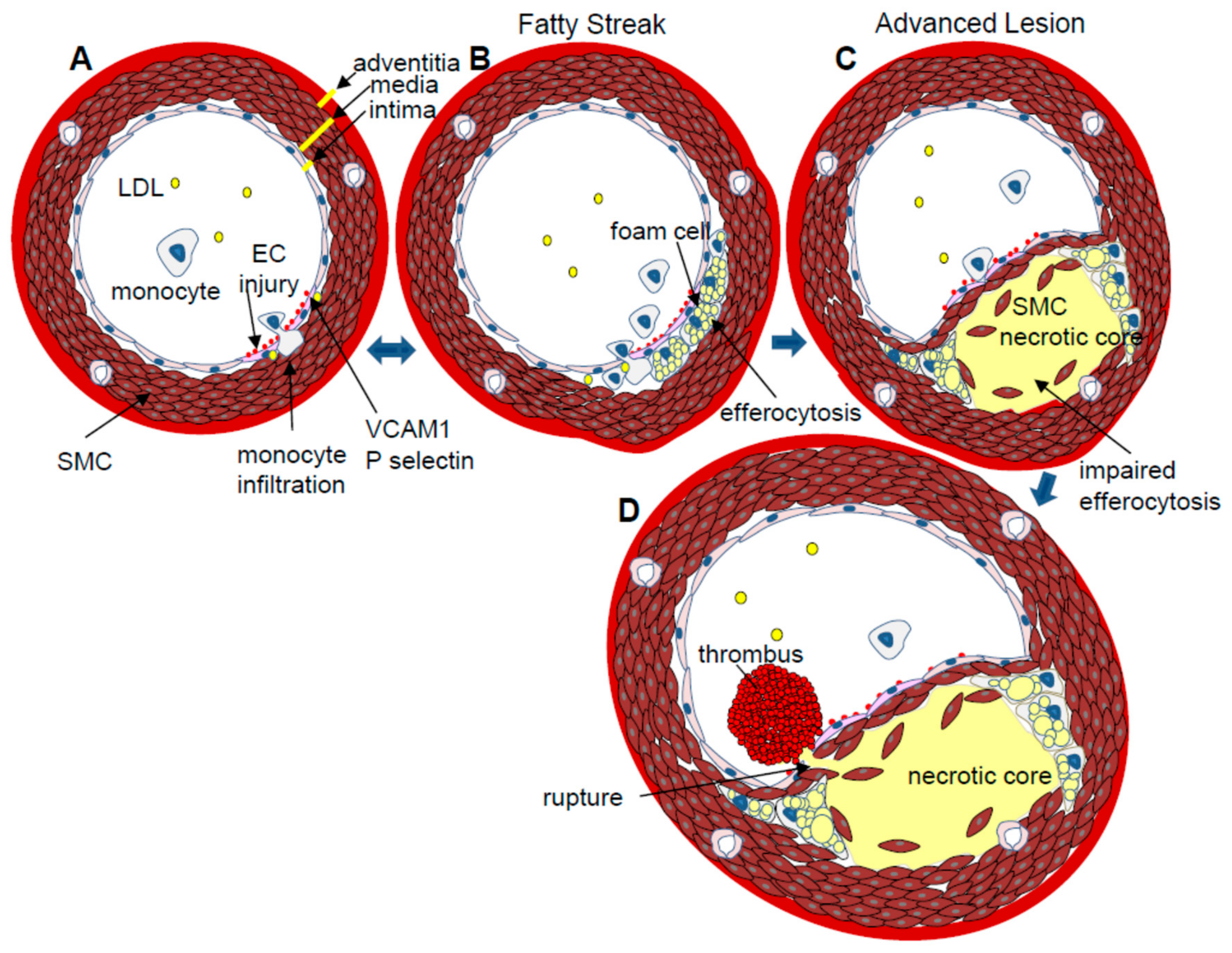{kind=link}
{kind=link}
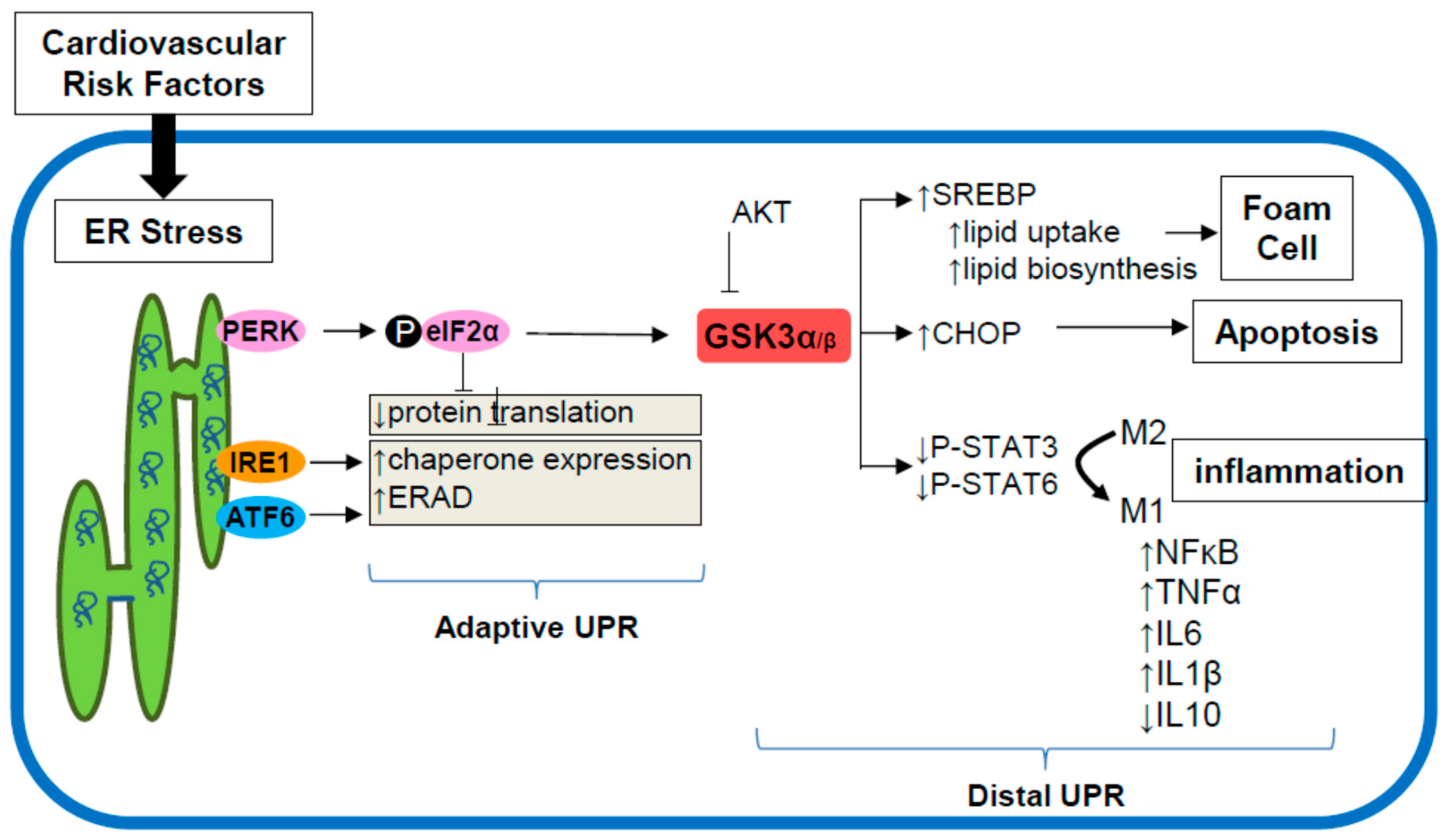{kind=link}
1. Introduction
2. Atherosclerosis
3. Molecular Mechanisms that Promote Atherosclerosis
4. The Endoplasmic Reticulum (ER) and ER Stress
5. ER Stress and Atherosclerosis
6. Glycogen Synthase Kinase (GSK)-3
7. GSK3 and Atherosclerosis
8. ER Stress Signaling through GSK3α/β
9. Targeting the ER Stress-GSK3α/β Pathway
10. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| ApoE | Apolipoprotein E |
| ATF6 | Activating transcription factor 6 |
| CHOP | C/EBP homologous protein |
| CVD | Cardiovascular disease |
| EC | Endothelial cell |
| ER | Endoplasmic reticulum |
| ERAD | ER stress associated protein degradation |
| GSK3α/β | Glycogen synthase kinase 3α/β |
| HDL | High-density lipoprotein |
| HFD | High fat diet |
| IFN γ | Interferon γ |
| IL | Interleukin |
| IRE1 | Inositol-requiring enzyme-1 |
| JAK | Janus kinase |
| JNK | c-Jun N-terminal kinase |
| LDL | Low-density lipoprotein |
| LDLR | Low-density lipoprotein receptor |
| MAPK | Mitogen-activated protein kinase |
| MMP | Matrix metalloprotease |
| NF κB | Nuclear factor κB |
| 4PBA | 4-phenylbutyrate |
| PERK | Protein kinase RNA-like ER kinase |
| SREBP | Sterol element binding protein |
| STAT | Signal transducer and activator of transcription protein |
| Th | T helper cell |
| TNFα | Tumor necrosis factor α |
| TUDCA | Tauroursodeoxycholic acid |
| UPR | Unfolded protein response |
| VCAM1 | Viral cell adhesion protein 1 |
| VLDL | Very low-density lipoprotein |
| VSMC | Vascular smooth muscle cell |
References
- Joseph, P.; Leong, D.; McKee, M.; Anand, S.S.; Schwalm, J.D.; Teo, K.; Mente, A.; Yusuf, S. Reducing the Global Burden of Cardiovascular Disease, Part 1: The Epidemiology and Risk Factors. Circ. Res. 2017, 121, 677–694. [Google Scholar] [CrossRef] [PubMed]
- Grundy, S.M.; Pasternak, R.; Greenland, P.; Smith, S.; Fuster, V. Assessment of Cardiovascular Risk by Use of Multiple-Risk-Factor Assessment Equations. Circulation 1999, 100, 1481–1492. [Google Scholar] [CrossRef] [PubMed]
- Lusis, A.J. Atherosclerosis. Nature 2000, 407, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Zarins, C.K.; Giddens, D.P.; Bharadvaj, B.K.; Sottiurai, V.S.; Mabon, R.F.; Glagov, S. Carotid bifurcation atherosclerosis. Quantitative correlation of plaque localization with flow velocity profiles and wall shear stress. Circ. Res. 1983, 53, 502–514. [Google Scholar] [CrossRef] [PubMed]
- Cybulsky, M.I.; Iiyama, K.; Li, H.; Zhu, S.; Chen, M.; Iiyama, M.; Davis, V.; Gutierrez-Ramos, J.C.; Connelly, P.W.; Milstone, D.S. A major role for VCAM-1, but not ICAM-1, in early atherosclerosis. J. Clin. Investig. 2001, 107, 1255–1262. [Google Scholar] [CrossRef] [PubMed]
- Johnson-Tidey, R.R.; McGregor, J.L.; Taylor, P.R.; Poston, R.N. Increase in the adhesion molecule P-selectin in endothelium overlying atherosclerotic plaques. Coexpression with intercellular adhesion molecule-1. Am. J. Pathol. 1994, 144, 952–961. [Google Scholar] [PubMed]
- Stary, H.C.; Chandler, A.B.; Glagov, S.; Guyton, J.R.; Insull, W., Jr.; Rosenfeld, M.E.; Schaffer, S.A.; Schwartz, C.J.; Wagner, W.D.; Wissler, R.W. A definition of initial, fatty streak, and intermediate lesions of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation 1994, 89, 2462–2478. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I.; Bornfeldt, K.E. Macrophage Phenotype and Function in Different Stages of Atherosclerosis. Circ. Res. 2016, 118, 653–667. [Google Scholar] [CrossRef] [PubMed]
- Robbins, C.S.; Hilgendorf, I.; Weber, G.F.; Theurl, I.; Iwamoto, Y.; Figueiredo, J.L.; Gorbatov, R.; Sukhova, G.K.; Gerhardt, L.M.; Smyth, D.; et al. Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat. Med. 2013, 19, 1166–1172. [Google Scholar] [CrossRef] [PubMed]
- Allahverdian, S.; Chehroudi, A.C.; McManus, B.M.; Abraham, T.; Francis, G.A. Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis. Circulation 2014, 129, 1551–1559. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef] [PubMed]
- De Paoli, F.; Staels, B.; Chinetti-Gbaguidi, G. Macrophage Phenotypes and Their Modulation in Atherosclerosis. Circ. J. 2014, 78, 1775–1781. [Google Scholar] [CrossRef] [PubMed]
- Adorni, M.P.; Zimetti, F.; Billheimer, J.T.; Wang, N.; Rader, D.J.; Phillips, M.C.; Rothblat, G.H. The roles of different pathways in the release of cholesterol from macrophages. J. Lipid Res. 2007, 48, 2453–2462. [Google Scholar] [CrossRef] [PubMed]
- Van Gils, J.M.; Derby, M.C.; Fernandes, L.R.; Ramkhelawon, B.; Ray, T.D.; Rayner, K.J.; Parathath, S.; Distel, E.; Feig, J.L.; Alvarez-Leite, J.I.; et al. The neuroimmune guidance cue netrin-1 promotes atherosclerosis by inhibiting the emigration of macrophages from plaques. Nat. Immunol. 2012, 13, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Yao, P.M.; Tabas, I. Free cholesterol loading of macrophages induces apoptosis involving the fas pathway. J. Biol. Chem. 2000, 275, 23807–23813. [Google Scholar] [CrossRef] [PubMed]
- To, K.; Agrotis, A.; Besra, G.; Bobik, A.; Toh, B. NKT Cell Subsets Mediate Differential Proatherogenic Effects in ApoE(−/−) Mice. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Amorino, G.; Hoover, R. Interactions of monocytic cells with human endothelial cells stimulate monocytic metalloproteinase production. Am. J. Pathol. 1998, 152, 199–207. [Google Scholar] [PubMed]
- Thim, T.; Hagensen, M.K.; Bentzon, J.F.; Falk, E. From vulnerable plaque to atherothrombosis. J. Intern. Med. 2008, 263, 506–516. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I. The role of endoplasmic reticulum stress in the progression of atherosclerosis. Circ. Res. 2010, 107, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Zinszner, H.; Kuroda, M.; Wang, X.; Batchvarova, N.; Lightfoot, R.T.; Remotti, H.; Stevens, J.L.; Ron, D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998, 12, 982–995. [Google Scholar] [CrossRef] [PubMed]
- Lindholm, D.; Korhonen, L.; Eriksson, O.; Kõks, S. Recent Insights into the Role of Unfolded Protein Response in ER Stress in Health and Disease. Front. Cell Dev. Biol. 2017, 5, 48. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, M.A.; Lalla, E.; Lu, Y.; Gleason, M.R.; Wolf, B.M.; Tanji, N.; Ferran, L.J., Jr.; Kohl, B.; Rao, V.; Kisiel, W.; et al. Hyperhomocysteinemia enhances vascular inflammation and accelerates atherosclerosis in a murine model. J. Clin. Investig. 2001, 107, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Lhotak, S.; Hilditch, B.A.; Austin, R.C. Activation of the unfolded protein response occurs at all stages of atherosclerotic lesion development in apolipoprotein E-deficient mice. Circulation 2005, 111, 1814–1821. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.I.; Pichna, B.A.; Shi, Y.; Bowes, A.J.; Werstuck, G.H. Evidence supporting a role for endoplasmic reticulum stress in the development of atherosclerosis in a hyperglycaemic mouse model. Antioxid. Redox Signal. 2009, 11, 2289–2298. [Google Scholar] [CrossRef] [PubMed]
- Beriault, D.R.; Sharma, S.; Shi, Y.; Khan, M.I.; Werstuck, G.H. Glucosamine-supplementation promotes endoplasmic reticulum stress, hepatic steatosis and accelerated atherogenesis in apoE−/− mice. Atherosclerosis 2011, 219, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Werstuck, G.H.; Lentz, S.R.; Dayal, S.; Shi, Y.; Hossain, G.S.; Sood, S.K.; Krisans, S.K.; Austin, R.C. Homocysteine-induced endoplasmic reticulum stress causes dysregulation of the cholesterol and triglyceride biosynthetic pathways. J. Clin. Investig. 2001, 107, 1263–1273. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, U.; Cao, Q.; Yilmaz, E.; Lee, A.H.; Iwakoshi, N.N.; Ozdelen, E.; Tuncman, G.; Görgün, C.; Glimcher, L.H.; Hotamisligil, G.S. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004, 306, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Schwabe, R.F.; Devries-Seimon, T.; Yao, P.M.; Gerbod-Giannone, M.C.; Tall, A.R.; Davis, R.J.; Flavell, R.; Brenner, D.A.; Tabas, I. Free cholesterol-loaded macrophages are an abundant source of TNF-alpha and IL-6. Model of NF-kappa B- and MAP kinase-dependent inflammation in advanced atherosclerosis. J. Biol. Chem. 2005, 280, 21763–21772. [Google Scholar] [CrossRef] [PubMed]
- Karaskov, E.; Scott, C.; Zhang, L.; Teodoro, T.; Ravazzola, M.; Volchuk, A. Chronic palmitate but not oleate exposure induces endoplasmic reticulum stress, which may contribute to INS-1 pancreatic beta-cell apoptosis. Endocrinology 2006, 147, 3398–3407. [Google Scholar] [CrossRef] [PubMed]
- Young, C.N.; Cao, X.; Guruju, M.R.; Pierce, J.P.; Morgan, D.A.; Wang, G.; Iadecola, C.; Mark, A.L.; Davisson, R.L. ER stress in the brain subfornical organ mediates angiotensin-dependent hypertension. J. Clin. Investig. 2012, 122, 3960–3964. [Google Scholar] [CrossRef] [PubMed]
- Somborac-Bacura, A.; van der Toorn, M.; Franciosi, L.; Slebos, D.J.; Zanic-Grubisic, T.; Bischoff, R.; van Oosterhout, A.J. Cigarette smoke induces endoplasmic reticulum stress response and proteasomal dysfunction in human alveolar epithelial cells. Exp. Physiol. 2013, 98, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Myoishi, M.; Hao, H.; Minamino, T.; Watanabe, K.; Nishihira, K.; Hatakeyama, K.; Asada, Y.; Okada, K.; Ishibashi-Ueda, H.; Gabbiani, G.; et al. Increased endoplasmic reticulum stress in atherosclerotic plaques associated with acute coronary syndrome. Circulation 2007, 116, 1226–1233. [Google Scholar] [CrossRef] [PubMed]
- Thorp, E.; Li, G.; Seimon, T.A.; Kuriakose, G.; Ron, D.; Tabas, I. Reduced Apoptosis and Plaque Necrosis in Advanced Atherosclerotic Lesions of Apoe−/− and Ldlr−/− Mice Lacking CHOP. Cell Metab. 2009, 9, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Ishimura, S.; Furuhashi, M.; Mita, T.; Fuseya, T.; Watanabe, Y.; Hoshina, K.; Kokubu, N.; Inoue, K.; Yoshida, H.; Miura, T. Reduction of endoplasmic reticulum stress inhibits neointima formation after vascular injury. Sci. Rep. 2014, 4, 6943. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Zhang, M.; Liang, B.; Xie, Z.; Zhao, Z.; Asfa, S.; Choi, H.C.; Zou, M.H. Reduction of AMP-activated protein kinase alpha2 increases endoplasmic reticulum stress and atherosclerosis in vivo. Circulation 2010, 121, 792–803. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Young, T.L.; Dang, V.T.; Shi, Y.; McAlpine, C.S.; Werstuck, G.H. 4-phenylbutyrate and valproate treatment attenuates the progression of atherosclerosis and stabilizes existing plaques. Atherosclerosis 2017, 266, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Dang, V.T.; Beriault, D.R.; Deng, A.; Shi, Y.; Werstuck, G.H. Glucosamine-induced ER stress accelerates atherogenesis: A potential link between diabetes and cardiovascular disease. J. Mol. Genet. Med. 2016, 9, 197. [Google Scholar] [CrossRef]
- Zeng, L.; Lu, M.; Mori, K.; Luo, S.; Lee, A.S.; Zhu, Y.; Shyy, J.Y. ATF6 modulates SREBP2-mediated lipogenesis. EMBO J. 2004, 23, 950–958. [Google Scholar] [CrossRef] [PubMed]
- Colgan, S.M.; Tang, D.; Werstuck, G.H.; Austin, R.C. Endoplasmic reticulum stress activation of sterol regulatory element binding protein-2. Int. J. Biochem. Cell Biol. 2007, 39, 1843–1851. [Google Scholar] [CrossRef] [PubMed]
- Pahl, H.L.; Baeuerle, P.A. A novel signal transduction pathway from the endoplasmic reticulum to the nucleus is mediated by the transcription factor NF-kappaB. EMBO J. 1995, 14, 2580–2588. [Google Scholar] [PubMed]
- Jiang, H.Y.; Wek, S.A.; McGrath, B.C.; Scheumer, D.; Kaufman, R.J.; Cavener, D.R.; Wek, R.C. Phosphorylation of the alpha subunit of eukaryotic initiation factor 2 is required for activation of NF-kappaB in response to diverse cellular stresses. Mol. Cell Biol. 2003, 23, 5651–5663. [Google Scholar] [CrossRef] [PubMed]
- DeVries-Seimon, T.; Li, Y.; Yao, P.M.; Stone, E.; Wang, Y.; Davis, R.S.; Flavell, R.; Tabas, I. Cholesterol-induced macrophage apoptosis requires ER stress pathways and engagement of the type A scavenger receptor. J. Cell Biol. 2005, 171, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Hossain, G.S.; van Thienen, J.V.; Werstuck, G.H.; Zhou, J.; Sood, S.K.; Dickhout, J.G.; de Koning, A.B.; Tang, D.; Wu, D.; Falk, E.; et al. TDAG51 is induced by homocysteine, promotes detachment-mediated programmed cell death and contributes to the development of atherosclerosis in hyperhomocysteinemia. J. Biol. Chem. 2003, 278, 30317–30327. [Google Scholar] [CrossRef] [PubMed]
- Seimon, T.A.; Obstfeld, A.; Moore, K.J.; Golenbock, D.T.; Tabas, I. Combinatorial pattern recognition receptor signaling alters the balance of life and death in macrophages. Proc. Natl. Acad. Sci. USA 2006, 103, 19794–19799. [Google Scholar] [CrossRef] [PubMed]
- Bowes, A.J.; Khan, M.I.; Shi, Y.Y.; Robertson, L.; Werstuck, G.H. A role for glycogen synthase kinase–3 in endoplasmic reticulum stress-associated lipid accumulation and accelerated atherogenesis in hyperglycemic ApoE-deficient mice. Am. J. Pathol. 2009, 174, 330–342. [Google Scholar] [CrossRef] [PubMed]
- McAlpine, C.S.; Bowes, A.J.; Khan, M.I.; Shi, Y.Y.; Werstuck, G.H. Endoplasmic reticulum stress and glycogen synthase kinase-3β activation in apolipoprotein E-deficient mouse models of accelerated atherosclerosis. Arterioscler. Throm. Vasc. Biol. 2012, 32, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Banko, N.; McAlpine, C.S.; Raja, P.; Shi, Y.; Khan, M.I.; Venegas-Pino, D.E.; Werstuck, G.H. Glycogen Synthase Kinase 3 alpha deficiency protects LDLR knockout mice from high fat diet induced accelerated atherosclerosis. Am. J. Pathol. 2014, 184, 3394–3404. [Google Scholar] [CrossRef] [PubMed]
- Woodgett, J.R. Molecular cloning and expression of glycogen synthase kinase-3/factor A. EMBO J. 1990, 9, 2431–2438. [Google Scholar] [PubMed]
- Phiel, C.J.; Wilson, C.A.; Lee, V.M.-Y.; Klein, P.S. GSK-3α regulates production of Alzheimer’s disease amyloid-β peptides. Nature 2003, 423, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Nikoulina, S.E.; Ciaraldi, T.P.; Mudaliar, S.; Mohideen, P.; Carter, L.; Henry, R.R. Potential role of glycogen synthase kinase-3 in skeletal muscle insulin resistance of type 2 diabetes. Diabetes 2000, 49, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Klein, P.S.; Melton, D.A. A molecular mechanism for the effect of lithium on development. Proc. Natl. Acad. Sci. USA 1996, 93, 8455–8459. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, C. What are the bona fide GSK3 Substrates? Int. J. Alzheimers Dis. 2011, 2011, 505607. [Google Scholar] [PubMed]
- MacAulay, K.; Doble, B.W.; Patel, S.; Hansotia, T.; Sinclair, E.M.; Drucker, D.J.; Nagy, A.; Woodgett, J.R. Glycogen synthase kinase 3alpha-specific regulation of murine hepatic glycogen metabolism. Cell Metab. 2007, 6, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Macaulay, K.; Woodgett, J.R. Tissue-specific analysis of glycogen synthase kinase-3α (GSK-3α) in glucose metabolism: Effect of strain variation. PLoS ONE 2011, 6, e15845. [Google Scholar] [CrossRef] [PubMed]
- Hoeflich, K.P.; Luo, J.; Rubie, E.A.; Tsao, M.S.; Jin, O.; Woodgett, J.R. Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature 2000, 406, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Demarchi, F.; Bertoli, C.; Sandy, P.; Schneider, C. Glycogen synthase kinase-3 beta regulates NF-kappa B1/p105 stability. J. Biol. Chem. 2003, 278, 39583–39590. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; De Sarno, P.; Jope, R.S. Central role of glycogen synthase kinase-3beta in endoplasmic reticulum stress-induced caspase-3 activation. J. Biol. Chem. 2002, 277, 44701–44708. [Google Scholar] [CrossRef] [PubMed]
- Baltzis, D.; Pluquet, O.; Papadakis, A.I.; Kazemi, S.; Qu, L.K.; Koromilas, A.E. The eIF2alpha kinases PERK and PKR activate glycogen synthase kinase 3 to promote the proteasomal degradation of p53. J. Biol. Chem. 2007, 282, 31675–31687. [Google Scholar] [CrossRef] [PubMed]
- Qu, L.; Huang, S.; Baltzis, D.; Rivas-Estilla, A.M.; Pluquet, O.; Hatzoglou, M.; Koumenis, C.; Taya, Y.; Yoshimura, A.; Koromilas, A.E. Endoplasmic reticulum stress induces p53 cytoplasmic localization and prevents p53-dependent apoptosis by a pathway involving glycogen synthase kinase-3beta. Genes Dev. 2004, 18, 261–277. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Hoeflich, K.P.; Woodgett, J.R. Glycogen Synthase Kinase-3: Properties, Functions, and Regulation. Chem. Rev. 2001, 101, 2527–2540. [Google Scholar] [CrossRef] [PubMed]
- McAlpine, C.S.; Huang, A.; Emdin, A.; Banko, N.S.; Beriault, D.R.; Shi, Y.; Werstuck, G.H. Deletion of myeloid GSK3α attenuates atherosclerosis and promotes an M2 macrophage phenotype. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1113–1122. [Google Scholar] [CrossRef] [PubMed]
- Beurel, E.; Kaidanovich-Beilin, O.; Yeh, W.I.; Song, L.; Palomo, V.; Michalek, S.M.; Woodgett, J.R.; Harrington, L.E.; Eldar-Finkelman, H.; Martinez, A.; et al. Regulation of Th1 Cells and Experimental Autoimmune Encephalomyelitis by Glycogen Synthase Kinase-3. J. Immunol. 2013, 190, 5000–5011. [Google Scholar] [CrossRef] [PubMed]
- Babaev, V.R.; Yancey, P.G.; Ryzhov, S.V.; Kon, V.; Breyer, M.D.; Magnuson, M.A.; Fazio, S.; Linton, M.F. Conditional Knockout of Macrophage PPAR Increases Atherosclerosis in C57BL/6 and Low-Density Lipoprotein Receptor-Deficient Mice. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1647–1653. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, T.; Zhai, P.; Maejima, Y.; Hong, C.; Gao, S.; Tian, B.; Goto, K.; Takagi, H.; Tamamori-Adachi, M.; Kitajima, S.; et al. Distinct roles of GSK-3alpha and GSK-3beta phosphorylation in the heart under pressure overload. Proc. Natl. Acad. Sci. USA 2008, 105, 20900–20905. [Google Scholar] [CrossRef] [PubMed]
- Hardt, S.E.; Tomita, H.; Katus, H.A.; Sadoshima, J. Phosphorylation of eukaryotic initiation factor 2Bepsilon by glycogen synthase kinase-3beta regulates beta-adrenergic cardiac myocyte hypertrophy. Circ. Res. 2004, 94, 926–935. [Google Scholar] [CrossRef] [PubMed]
- Badorff, C.; Seeger, F.H.; Zeiher, A.M.; Dimmeler, S. Glycogen synthase kinase 3beta inhibits myocardin-dependent transcription and hypertrophy induction through site-specific phosphorylation. Circ. Res. 2005, 97, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Kerkela, R.; Kockeritz, L.; Macaulay, K.; Zhou, J.; Doble, B.W.; Beahm, C.; Greytak, S.; Woulfe, K.; Trivedi, C.M.; Woodgett, J.R.; et al. Deletion of GSK-3beta in mice leads to hypertrophic cardiomyopathy secondary to cardiomyoblast hyperproliferation. J. Clin. Investig. 2008, 118, 3609–3618. [Google Scholar] [CrossRef] [PubMed]
- Juhaszova, M.; Zorov, D.B.; Kim, S.H.; Pepe, S.; Fu, Q.; Fishbein, K.W.; Ziman, B.D.; Wang, S.; Ytrehus, K.; Antos, C.L.; et al. Glycogen synthase kinase-3beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J. Clin. Investig. 2004, 113, 1535–1549. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.; Rehani, K.; Jope, R.S.; Michalek, S.M. Toll-like receptor–mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat. Immunol. 2005, 6, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Beurel, E.; Jope, R.S. Glycogen synthase kinase-3 promotes the synergistic action of interferon-gamma on lipopolysaccharide-induced IL-6 production in RAW264.7 cells. Cell Signal. 2009, 21, 978–985. [Google Scholar] [CrossRef] [PubMed]
- Beurel, E.; Jope, R.S. Differential regulation of STAT family members by glycogen synthase kinase-3. J. Biol. Chem. 2008, 283, 21934–21944. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Devgan, G.; Darnell, J.; Bromberg, J. Constitutively activated Stat3 protects fibroblasts from serum withdrawal and UV-induced apoptosis and antagonizes the proapoptotic effects of activated Stat1. Proc. Natl. Acad. Sci. USA 2001, 98, 1543–1548. [Google Scholar] [CrossRef] [PubMed]
- Ho, H.; Ivashkiv, L. Role of STAT3 in type I interferon responses—Negative regulation of STAT1-dependent inflammatory gene activation. J. Biol. Chem. 2006, 281, 14111–14118. [Google Scholar] [CrossRef] [PubMed]
- McAlpine, C.S.; Werstuck, G.H. Protein kinase R-like endoplasmic reticulum kinase and glycogen synthase kinase-3α/β regulate foam cell formation. J. Lipid Res. 2014, 55, 2320–2333. [Google Scholar] [CrossRef] [PubMed]
- Petri, S.; Kiaei, M.; Kipiani, K.; Chen, J.; Calingasan, N.Y.; Crow, J.P.; Beal, M.F. Additive neuroprotective effects of a histone deacetylase inhibitor and a catalytic antioxidant in a transgenic mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 2006, 22, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Akerfeldt, M.C.; Howes, J.; Chan, J.Y.; Stevens, V.A.; Boubenna, N.; McGuire, H.M.; King, C.; Biden, T.J.; Laybutt, D.R. Cytokine-induced beta-cell death is independent of endoplasmic reticulum stress signaling. Diabetes 2008, 57, 3034–3044. [Google Scholar] [CrossRef] [PubMed]
- Özcan, L.; Ergin, A.S.; Lu, A.; Chung, J.; Sarkar, S.; Nie, D.; Myers, M.G.Jr.; Özcan, U. Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab. 2009, 9, 35–51. [Google Scholar] [CrossRef] [PubMed]
- Özcan, U.; Yilmaz, E.; Özcan, L.; Furuhashi, M.; Vaillancourt, E.; Smith, R.O.; Görgün, C.Z.; Hotamisligil, G.S. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 2006, 313, 1137–1140. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Bowes, R.C., 3rd; van de Water, B.; Sillence, C.; Nagelkerke, J.F.; Stevens, J.L. Endoplasmic reticulum chaperones GRP78 and calreticulin prevent oxidative stress, Ca2+ disturbances, and cell death in renal epithelial cells. J. Biol. Chem. 2007, 272, 21751–21759. [Google Scholar] [CrossRef]
- Kudo, T.; Kanemoto, S.; Hara, H.; Morimoto, N.; Morihara, T.; Kimura, R.; Tabira, T.; Imaizumi, K.; Takeda, M. A molecular chaperone inducer protects neurons from ER stress. Cell Death Differ. 2008, 15, 364–375. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.E.; Jang, H.J.; Kang, Y.; Jung, J.G.; Han, S.J.; Kim, H.J.; Kim, D.J.; Lee, K.W. Atherosclerosis induced by a high-fat diet is alleviated by lithium chloride via reduction of VCAM expression in ApoE-deficient mice. Vascul. Pharmacol. 2010, 53, 264–272. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, A.; Patel, S.; McAlpine, C.S.; Werstuck, G.H. The Role of Endoplasmic Reticulum Stress-Glycogen Synthase Kinase-3 Signaling in Atherogenesis. Int. J. Mol. Sci. 2018, 19, 1607. https://doi.org/10.3390/ijms19061607
Huang A, Patel S, McAlpine CS, Werstuck GH. The Role of Endoplasmic Reticulum Stress-Glycogen Synthase Kinase-3 Signaling in Atherogenesis. International Journal of Molecular Sciences. 2018; 19(6):1607. https://doi.org/10.3390/ijms19061607
Chicago/Turabian StyleHuang, Aric, Sarvatit Patel, Cameron S. McAlpine, and Geoff H. Werstuck. 2018. "The Role of Endoplasmic Reticulum Stress-Glycogen Synthase Kinase-3 Signaling in Atherogenesis" International Journal of Molecular Sciences 19, no. 6: 1607. https://doi.org/10.3390/ijms19061607
APA StyleHuang, A., Patel, S., McAlpine, C. S., & Werstuck, G. H. (2018). The Role of Endoplasmic Reticulum Stress-Glycogen Synthase Kinase-3 Signaling in Atherogenesis. International Journal of Molecular Sciences, 19(6), 1607. https://doi.org/10.3390/ijms19061607