Human Fibrinogen: Molecular and Genetic Aspects of Congenital Disorders



Abstract
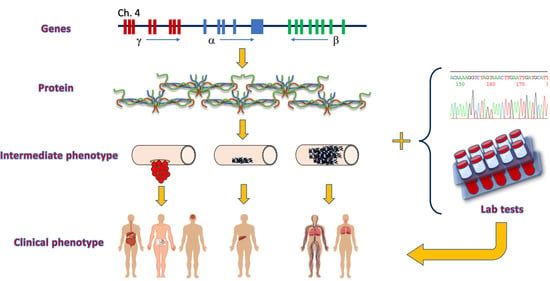
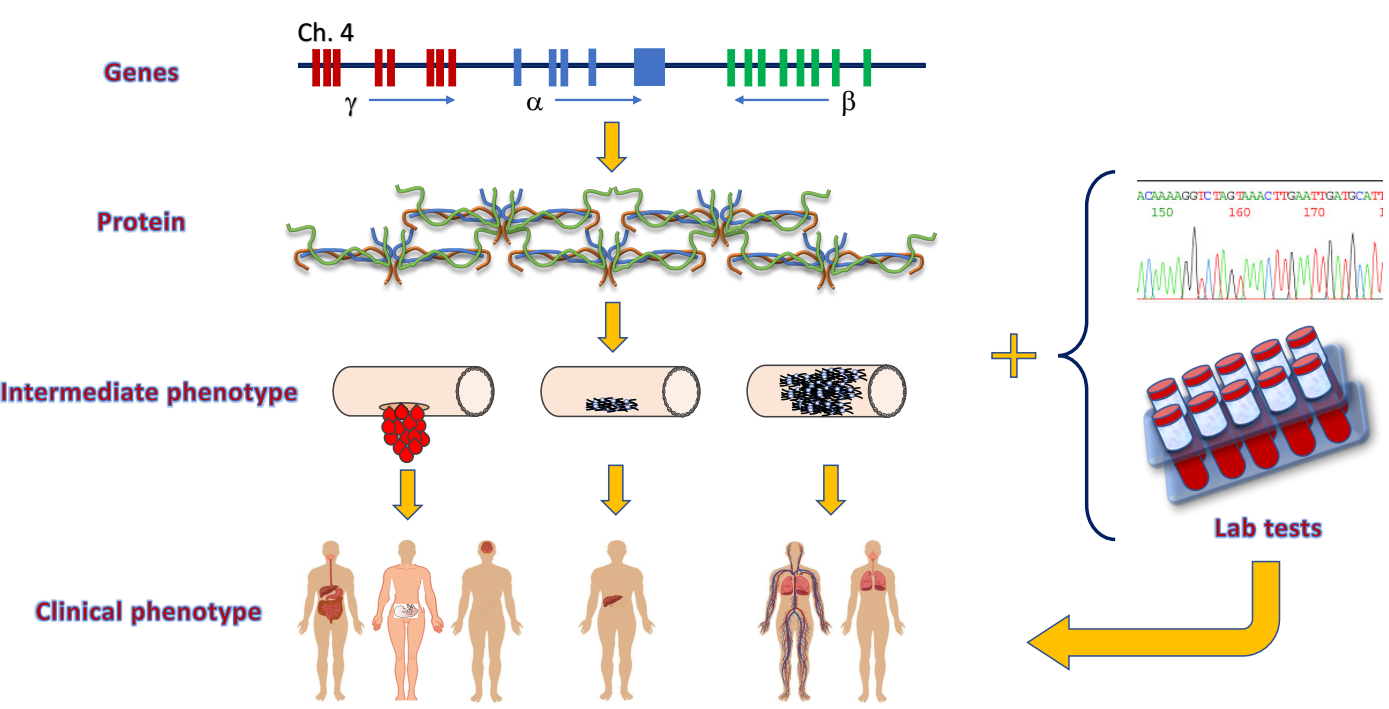
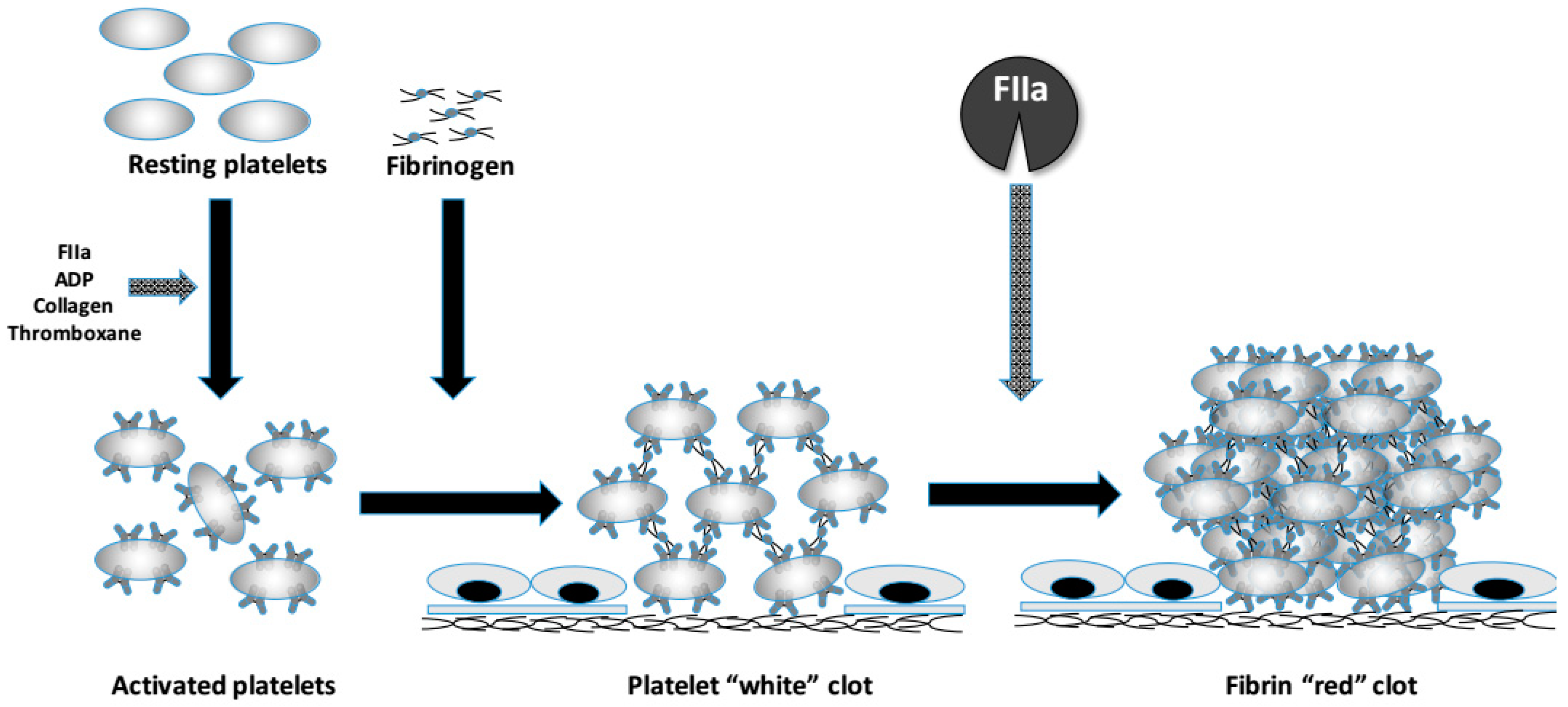
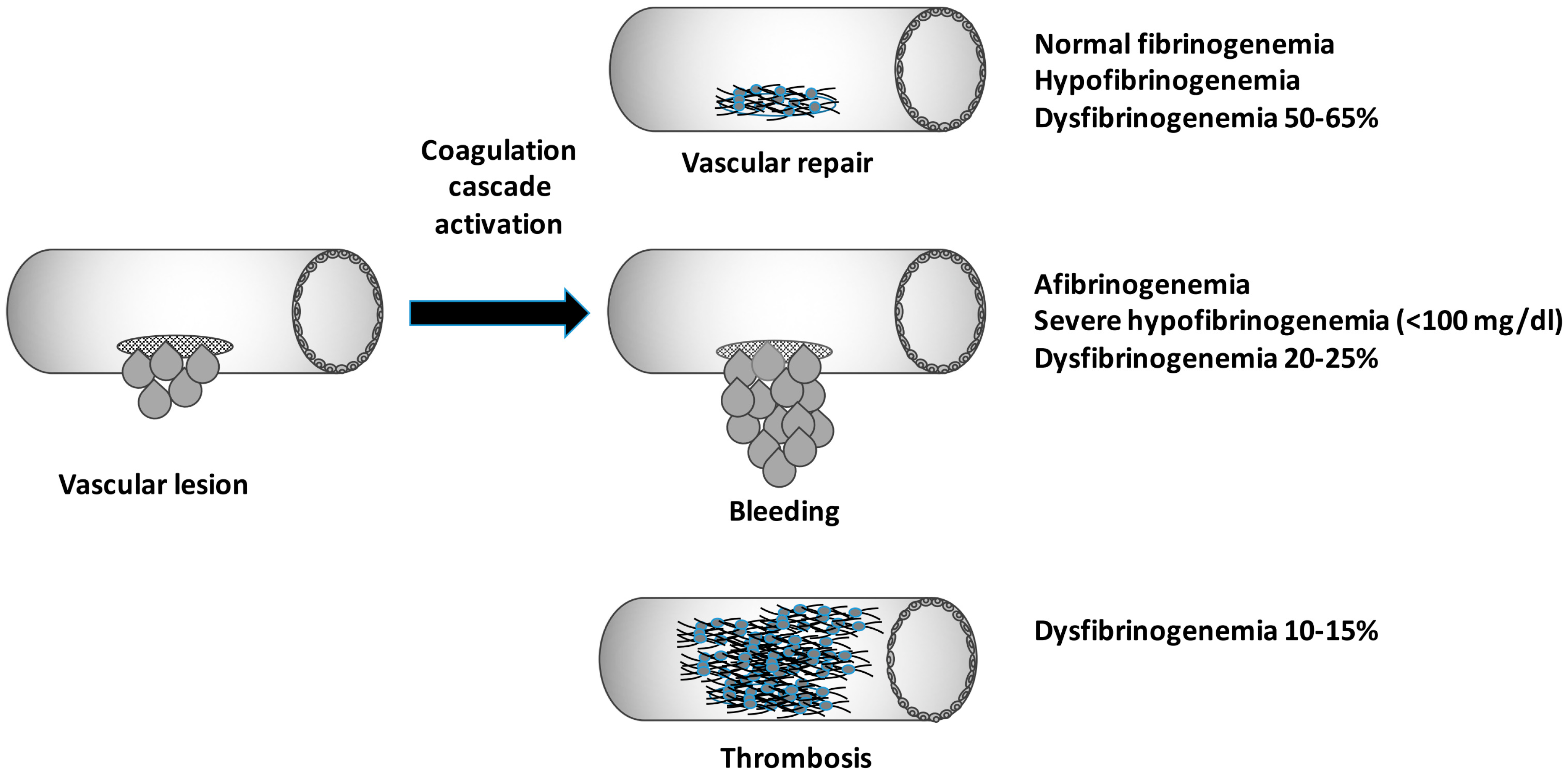
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Afibrinogenemia
3. Hypofibrinogenemia
4. Dysfibrinogenemia
5. Genotype-Phenotype Correlation in Fibrinogen Disorders
6. Conclusions
Author Contributions
Conflicts of Interest
References
- Tennent, G.A.; Brennan, S.O.; Stangou, A.J.; O’Grady, J.; Hawkins, P.N.; Pepys, M.B. Human plasma fibrinogen is synthesized in the liver. Blood 2007, 109, 1971–1974. [Google Scholar] [CrossRef] [PubMed]
- Blombäck, B. Fibrinogen and fibrin–proteins with complex roles in hemostasis and thrombosis. Thromb. Res. 1996, 83, 1–75. [Google Scholar] [CrossRef]
- Henschen, A.; Lottspeich, F.; Kehl, M.; Southan, C. Covalent structure of fibrinogen. Ann. N. Y. Acad. Sci. 1983, 408, 28–43. [Google Scholar] [CrossRef] [PubMed]
- Doolittle, R.F. Fibrinogen and fibrin. Annu. Rev. Biochem. 1984, 53, 195–229. [Google Scholar] [CrossRef] [PubMed]
- Blombäck, B.; Hessel, B.; Hogg, D. Disulfide bridges in NH2-terminal part of human fibrinogen. Thromb. Res. 1976, 8, 639–658. [Google Scholar] [CrossRef]
- Hoeprich, P.D., Jr.; Doolittle, R.F. Dimeric half-molecules of human fibrinogen are joined through disulfide bonds in an antiparallel orientation. Biochemistry 1983, 22, 2049–2055. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Z.; Redman, C. Fibrinogen assembly and secretion. Role of intrachain disulfide loops. J. Biol. Chem. 1996, 271, 30083–30088. [Google Scholar] [CrossRef] [PubMed]
- Doolittle, R.F.; Goldbaum, D.M.; Doolittle, L.R. Designation of sequences involved in the “coiled-coil” interdomainal connections in fibrinogen: Constructions of an atomic scale model. J. Mol. Biol. 1978, 120, 311–325. [Google Scholar] [CrossRef]
- Weisel, J.W.; Phillips, G.N., Jr.; Cohen, C. A model from electron microscopy for the molecular structure of fibrinogen and fibrin. Nature 1981, 289, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Mosesson, M.W.; Hainfeld, J.; Wall, J.; Haschemeyer, R.H. Identification and mass analysis of human fibrinogen molecules and their domains by scanning transmission electron microscopy. J. Mol. Biol. 1981, 153, 695–718. [Google Scholar] [CrossRef]
- Weisel, J.W.; Litvinov, R.I. Mechanisms of fibrin polymerization and clinical implications. Blood 2013, 121, 1712–1719. [Google Scholar] [CrossRef] [PubMed]
- Verhovsek, M.; Moffat, K.A.; Hayward, C.P. Laboratory testing for fibrinogen abnormalities. Am. J. Hematol. 2008, 83, 928–931. [Google Scholar] [CrossRef] [PubMed]
- Besser, M.W.; MacDonald, S.G. Acquired hypofibrinogenemia: Current perspectives. J. Blood Med. 2016, 7, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Kant, J.A.; Lord, S.T.; Crabtree, G.R. Partial mRNA sequences for human A alpha, B beta, and gamma fibrinogen chains: Evolutionary and functional implications. Proc. Natl. Acad. Sci. USA 1983, 80, 3953–3957. [Google Scholar] [CrossRef] [PubMed]
- Kant, J.A.; Fornace, A.J., Jr.; Saxe, D.; Simon, M.I.; McBride, O.W.; Crabtree, G.R. Evolution and organization of the fibrinogen locus on chromosome 4: Gene duplication accompanied by transposition and inversion. Proc. Natl. Acad. Sci. USA 1985, 82, 2344–2348. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Weissbach, L.; Plant, P.W.; Oddoux, C.; Cao, Y.; Liang, T.J.; Roy, S.N.; Redman, C.M.; Grieninger, G. Carboxy-terminal-extended variant of the human fibrinogen alpha subunit: A novel exon conferring marked homology to beta and gamma subunits. Biochemistry 1992, 31, 11968–11972. [Google Scholar] [CrossRef] [PubMed]
- Neerman-Arbez, M.; de Moerloose, P.; Casini, A. Laboratory and Genetic Investigation of Mutations Accounting for Congenital Fibrinogen Disorders. Semin. Thromb. Hemost. 2016, 42, 356–365. [Google Scholar] [PubMed]
- Rabe, F.; Solomon, E. Ueber Faserstoffmangel im Blute bei einen Falle von Hämophile. Arch. Klin. Med. 1920, 132, 240–244. [Google Scholar]
- Martinez, J.; Palascak, J.; Peters, C. Functional and metabolic properties of human asialofibrinogen. J. Lab. Clin. Med. 1977, 89, 367–377. [Google Scholar] [PubMed]
- Bittles, A. Consanguinity and its relevance to clinical genetics. Clin. Genet. 2001, 60, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, B.P.; Nair, S.B.; Vijapurkar, M.; Mota, L.; Shanbhag, S.; Ali, S.; Shetty, S.D.; Ghosh, K. Molecular pathology of rare bleeding disorders (RBDs) in India: A systematic review. PLoS ONE 2014, 9, e108683. [Google Scholar] [CrossRef] [PubMed]
- Neerman-Arbez, M.; Honsberger, A.; Antonarakis, S.E.; Morris, M.A. Deletion of the fibrinogen [correction of fibrogen] alpha-chain gene (FGA) causes congenital afibrogenemia. J. Clin. Investig. 1999, 103, 215–218. [Google Scholar] [CrossRef] [PubMed]
- Werder, E. Congenital afibrinogenemia. Helv. Paediatr. Acta 1963, 18, 208–229. [Google Scholar] [PubMed]
- Neerman-Arbez, M.; de Moerloose, P. Mutations in the fibrinogen gene cluster accounting for congenital afibrinogenemia: An update and report of ten novel mutations. Hum. Mutat. 2007, 28, 540–553. [Google Scholar] [CrossRef] [PubMed]
- Galanakis, D.K.; Neerman-Arbez, M.; Scheiner, T.; Henschen, A.; Hubbs, D.; Nagaswami, C.; Weisel, J.W. Homophenotypic Aalpha R16H fibrinogen (Kingsport): Uniquely altered polymerization associated with slower fibrinopeptide A than fibrinopeptide B release. Blood Coagul. Fibrinolysis 2007, 18, 731–737. [Google Scholar] [CrossRef] [PubMed]
- Neerman-Arbez, M.; de Moerloose, P.; Bridel, C.; Honsberger, A.; Schönbörner, A.; Rossier, C.; Peerlinck, K.; Claeyssens, S.; Di Michele, D.; d’Oiron, R.; et al. Mutations in the fibrinogen aalpha gene account for the majority of cases of congenital afibrinogenemia. Blood 2000, 96, 149–152. [Google Scholar] [PubMed]
- Neerman-Arbez, M.; de Moerloose, P.; Honsberger, A.; Parlier, G.; Arnuti, B.; Biron, C.; Borg, J.Y.; Eber, S.; Meili, E.; Peter-Salonen, K.; et al. Molecular analysis of the fibrinogen gene cluster in 16 patients with congenital afibrinogenemia: Novel truncating mutations in the FGA and FGG genes. Hum. Genet. 2001, 108, 237–240. [Google Scholar] [CrossRef] [PubMed]
- Angles-Cano, E.; Mathonnet, F.; Dreyfus, M.; Claeyssens, S.; de Mazancourt, P. A case of fibrinogenemia associated with A-alpha chain gene compound heterozygosity (HUMFIBRA c.[4110delA]+[3200+1G>T]). Blood Coagul. Fibrinolysis 2007, 18, 73–75. [Google Scholar] [CrossRef] [PubMed]
- Berens, C.; Rühl, H.; Ivaškevičius, V.; Oldenburg, J.; Hertfelder, H.J.; Pötzsch, B. Recurrent VTE in a heterozygote of the fibrinogen Aα IVS4+1G>T and Aα p.Arg168Ter mutation. Thromb. Haemost. 2016, 115, 1073–1075. [Google Scholar] [CrossRef] [PubMed]
- Polack, B.; Pouzol, P.; de Mazancourt, P.; Gay, V.; Hanss, M. Is primary prophylaxisrequired in afibrinogenemia? Transfusion 2010, 50, 1401–1403. [Google Scholar] [CrossRef] [PubMed]
- Santacroce, R.; Cappucci, F.; Pisanelli, D.; Perricone, F.; Papa, M.L.; Santoro, R.; Grandone, E.; Margaglione, M. Inherited abnormalities of fibrinogen: 10-year clinical experience of an Italian group. Blood Coagul. Fibrinolysis 2006, 17, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Attanasio, C.; de Moerloose, P.; Antonarakis, S.E.; Morris, M.A.; Neerman-Arbez, M. Activation of multiple cryptic donor splice sites by the common congenital afibrinogenemia mutation, FGA IVS4 1 G3T. Blood 2001, 97, 1879–1881. [Google Scholar] [CrossRef] [PubMed]
- Monaldini, L.; Asselta, R.; Duga, S.; Peyvandi, F.; Karimi, M.; Malcovati, M.; Tenchini, M.L. Mutational screening of six afibrinogenemic patients: Identification and characterization of four novel molecular defects. Thromb. Haemost. 2007, 97, 546–551. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Shibuya, A.; Ishii, E.; Kurihara, M.; Inoue, S.; Ono, M.; Wada, Y.; Wakiyama, M.; Zaitsu, M.; Iida, H.; et al. Identification of simultaneous mutation of fibrinogen alpha chain and protein C genes in a Japanese kindred. Br. J. Haematol. 2003, 120, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Spena, S.; Duga, S.; Asselta, R.; Peyvandi, F.; Mahasandana, C.; Malcovati, M.; Tenchini, M.L. Congenital afibrinogenaemia caused by uniparental isodisomy of chromosome 4 containing a novel 15-kb deletion involving fibrinogen Aalpha-chain gene. Eur. J. Hum. Genet. 2004, 12, 891–898. [Google Scholar] [CrossRef] [PubMed]
- Attanasio, C.; David, A.; Neerman-Arbez, M. Outcome of donor splice site mutations accounting for congenital afibrinogenemia reflects order of intron removal in the fibrinogen alpha gene (FGA). Blood 2003, 101, 1851–1856. [Google Scholar] [CrossRef] [PubMed]
- Margaglione, M.; Santacroce, R.; Colaizzo, D.; Seripa, D.; Vecchione, G.; Lupone, M.R.; De Lucia, D.; Fortina, P.; Grandone, E.; Perricone, C.; et al. A G-to-A mutation in IVS-3 of the human gamma fibrinogen gene causing afibrinogenemia due to abnormal RNA splicing. Blood 2000, 96, 2501–2505. [Google Scholar] [PubMed]
- Asselta, R.; Duga, S.; Simonic, T.; Malcovati, M.; Santagostino, E.; Giangrande, P.L.; Mannucci, P.M.; Tenchini, M.L. Afibrinogenemia: First identification of a splicing mutation in the fibrinogen gamma chain gene leading to a major gamma chain truncation. Blood 2000, 96, 2496–2500. [Google Scholar] [PubMed]
- Spena, S.; Duga, S.; Asselta, R.; Malcovati, M.; Peyvandi, F.; Tenchini, M.L. Congenitalafibrinogenemia: First identification of splicing mutations in the fibrinogenBbeta-chain gene causing activation of cryptic splice sites. Blood 2002, 100, 4478–4484. [Google Scholar] [CrossRef] [PubMed]
- Spena, S.; Asselta, R.; Platé, M.; Castaman, G.; Duga, S.; Tenchini, M.L. Pseudo-exon activation caused by a deep-intronic mutation in the fibrinogen gamma-chain gene as a novel mechanism for congenital afibrinogenaemia. Br. J. Haematol. 2007, 139, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Evans, K.; Hayden, M.; Heywood, S.; Hussain, M.; Phillips, A.D.; Cooper, D.N. The Human Gene Mutation Database: Towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum. Genet. 2017, 136, 665–677. [Google Scholar] [CrossRef] [PubMed]
- The Human Fibrinogen Database. Available online: http://site.geht.org/base-de-donnees-fibrinogene/ (accessed on 28 September 2017).
- Simurda, T.; Zolkova, J.; Snahnicanova, Z.; Loderer, D.; Skornova, I.; Sokol, J.; Hudecek, J.; Stasko, J.; Lasabova, Z.; Kubisz, P. Identification of Two Novel Fibrinogen Bβ Chain Mutations in Two Slovak Families with Quantitative Fibrinogen Disorders. Int. J. Mol. Sci. 2018, 19, 100. [Google Scholar] [CrossRef] [PubMed]
- Simurda, T.; Snahnicanova, Z.; Loderer, D.; Sokol, J.; Stasko, J.; Lasabova, Z.; Kubisz, P. Fibrinogen Martin: A Novel Mutation in FGB (Gln180Stop) Causing Congenital Afibrinogenemia. Semin. Thromb. Hemost. 2016, 42, 455–458. [Google Scholar] [PubMed]
- Naz, A.; Biswas, A.; Khan, T.N.; Goodeve, A.; Ahmed, N.; Saqlain, N.; Ahmed, S.; Ujjan, I.D.; Shamsi, T.S.; Oldenburg, J. Identification of novel mutations in congenital afibrinogenemia patients and molecular modeling of missense mutations in Pakistani population. Thromb. J. 2017, 15, 24. [Google Scholar] [CrossRef] [PubMed]
- Neerman-Arbez, M.; Vu, D.; Abu-Libdeh, B.; Bouchardy, I.; Morris, M.A. Prenatal diagnosis for congenital afibrinogenemia caused by a novel nonsense mutation in the FGB gene in a Palestinian family. Blood 2003, 101, 3492–3494. [Google Scholar] [CrossRef] [PubMed]
- Vu, D.; Di Sanza, C.; Caille, D.; de Moerloose, P.; Scheib, H.; Meda, P.; Neerman-Arbez, M. Quality control of fibrinogen secretion in the molecular pathogenesis of congenital afibrinogenemia. Hum. Mol. Genet. 2005, 14, 3271–3280. [Google Scholar] [CrossRef] [PubMed]
- Platé, M.; Asselta, R.; Peyvandi, F.; Tenchini, M.L.; Duga, S. Molecular characterization of the first missense mutation in the fibrinogen Aalpha-chain gene identified in a compound heterozygous afibrinogenemic patient. Biochim. Biophys. Acta 2007, 1772, 781–787. [Google Scholar] [CrossRef] [PubMed]
- Tirefort, Y.; Alson, O.R.; de Moerloose, P.; Neerman-Arbez, M. Mutation of the translation initiation codon in FGA causes congenital afibrinogenemia. Blood Coagul. Fibrinolysis 2012, 23, 556–558. [Google Scholar] [CrossRef] [PubMed]
- Okumura, N.; Terasawa, F.; Tanaka, H.; Hirota, M.; Ota, H.; Kitano, K.; Kiyosawa, K.; Lord, S.T. Analysis of fibrinogen gamma-chain truncations shows the C-terminus, particularly gammaIle387, is essential for assembly and secretion of this multichain protein. Blood 2002, 99, 3654–3660. [Google Scholar] [CrossRef] [PubMed]
- Casini, A.; Lukowski, S.; Quintard, V.L.; Crutu, A.; Zak, M.; Regazzoni, S.; de Moerloose, P.; Neerman-Arbez, M. FGB mutations leading to congenital quantitative fibrinogen deficiencies: An update and report of four novel mutations. Thromb. Res. 2014, 133, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Casini, A.; Vilar, R.; Beauverd, Y.; Aslan, D.; Devreese, K.; Mondelaers, V.; Alberio, L.; Gubert, C.; de Moerloose, P.; Neerman-Arbez, M. Protein modelling to understand FGB mutations leading to congenital hypofibrinogenaemia. Haemophilia 2017, 23, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Casini, A.; Brungs, T.; Lavenu-Bombled, C.; Vilar, R.; Neerman-Arbez, M.; de Moerloose, P. Genetics, diagnosis and clinical features of congenital hypodysfibrinogenemia: A systematic literature review and report of a novel mutation. J. Thromb. Haemost. 2017, 15, 876–888. [Google Scholar] [CrossRef] [PubMed]
- Imperato, C.; Dettori, A.G. Congenital hypofibrinogenemia with fibrinoasthenia. Helv. Paediatr. Acta 1958, 13, 380–399. [Google Scholar] [PubMed]
- Blombäck, M.; Blombäck, B.; Mammen, E.F.; Prasad, A.S. Fibrinogen Detroit--a molecular defect in the N-terminal disulphide knot of human fibrinogen? Nature 1968, 218, 134–137. [Google Scholar] [CrossRef] [PubMed]
- Ridgway, H.J.; Brennan, S.O.; Gibbons, S.; George, P.M. Fibrinogen Lincoln: A newtruncated alpha chain variant with delayed clotting. Br. J. Haematol. 1996, 93, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Dempfle, C.E.; George, P.M.; Borggrefe, M.; Neumaier, M.; Brennan, S.O. Demonstration ofheterodimeric fibrinogen molecules partially conjugated with albumin in a novel dysfibrinogen: Fibrinogen Mannheim V. Thromb. Haemost. 2009, 102, 29–34. [Google Scholar] [PubMed]
- Margaglione, M.; Vecchione, G.; Santacroce, R.; D’Angelo, F.; Casetta, B.; Papa, M.L.; Grandone, E.; Di Minno, G. A frameshift mutation in the human fibrinogen Aalpha-chain gene (Aalpha(499)Ala frameshift stop) leading to dysfibrinogen San Giovanni Rotondo. Thromb. Haemost. 2001, 86, 1483–1488. [Google Scholar] [PubMed]
- Okumura, N.; Terasawa, F.; Hirota-Kawadobora, M.; Yamauchi, K.; Nakanishi, K.; Shiga, S.; Ichiyama, S.; Saito, M.; Kawai, M.; Nakahata, T. A novel variant fibrinogen, deletion ofBbeta111Ser in coiled-coil region, affecting fibrin lateral aggregation. Clin. Chim. Acta 2006, 365, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Hogan, K.A.; Gorkun, O.V.; Lounes, K.C.; Coates, A.I.; Weisel, J.W.; Hantgan, R.R.; Lord, S.T. Recombinant fibrinogen Vlissingen/Frankfurt IV. The deletion of residues 319 and 320 from thegamma chain of fibrinogen alters calcium binding, fibrin polymerization, cross-linking, and platelet aggregation. J. Biol. Chem. 2000, 275, 17778–17785. [Google Scholar] [CrossRef] [PubMed]
- Koopman, J.; Haverkate, F.; Briët, E.; Lord, S.T. A congenitally abnormal fibrinogen(Vlissingen) with a 6-base deletion in the gamma-chain gene, causing defective calcium binding and impaired fibrin polymerization. J. Biol. Chem. 1991, 266, 13456–13461. [Google Scholar] [PubMed]
- Furlan, M.; Steinmann, C.; Jungo, M.; Bögli, C.; Baudo, F.; Redaelli, R.; Fedeli, F.; Lämmle, B. A frameshift mutation in Exon V of the A alpha-chain gene leading to truncated A alpha-chains in the homozygous dysfibrinogen Milano III. J. Biol. Chem. 1994, 269, 33129–33134. [Google Scholar] [PubMed]
- Collen, A.; Maas, A.; Kooistra, T.; Lupu, F.; Grimbergen, J.; Haas, F.J.; Biesma, D.H.; Koolwijk, P.; Koopman, J.; van Hinsbergh, V.W. Aberrant fibrin formation and cross-linking of fibrinogen Nieuwegein, a variant with a shortened Aalpha-chain, alters endothelial capillary tube formation. Blood 2001, 97, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, J.B.; Newman, P.J.; Mosesson, M.W.; Guillin, M.C.; Amrani, D.L. Paris I dysfibrinogenemia: A point mutation in intron 8 results in insertion of a 15 amino acid sequence in the fibrinogen gamma-chain. Thromb. Haemost. 1993, 69, 217–220. [Google Scholar] [PubMed]
- Amri, Y.; Jouini, H.; Becheur, M.; Dabboubi, R.; Mahjoub, B.; Messaoud, T.; Sfar, M.T.; Casini, A.; de Moerloose, P.; Toumi, N.E.H. Fibrinogen Mahdia: A congenitally abnormal fibrinogen characterized by defective fibrin polymerization. Haemophilia 2017, 23, e340–e347. [Google Scholar] [CrossRef] [PubMed]
- Casini, A.; Blondon, M.; Lebreton, A.; Koegel, J.; Tintillier, V.; de Maistre, E.; Gautier, P.; Biron, C.; Neerman-Arbez, M.; de Moerloose, P. Natural history of patients with congenital dysfibrinogenemia. Blood 2015, 125, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.; Dolan, G. Diagnosis, clinical features and molecular assessment of the dysfibrinogenaemias. Haemophilia 2008, 14, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.A.; Southan, C.; Ireland, H.; Thompson, E.; Kehl, M.; Henschen, A. Delayed release of an abnormal fibrinopeptide A from fibrinogen Manchester: Effect of the A alpha 16 Arg leads toHis substitution upon fibrin monomer polymerization and the immunological crossreactivity of the peptide. Br. J. Haematol. 1983, 53, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Galanakis, D.K.; Henschen, A.; Peerschke, E.I.; Kehl, M. Fibrinogen Stony Brook, a heterozygous A alpha 16Arg—Cys dysfibrinogenemia. Evaluation of diminished platelet aggregation support and of enhanced inhibition of fibrin assembly. J. Clin. Investig. 1989, 84, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Flood, V.H.; Al-Mondhiry, H.A.; Farrell, D.H. The fibrinogen Aalpha R16C mutation results in fibrinolytic resistance. Br. J. Haematol. 2006, 134, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Cote, H.C.; Lord, S.T.; Pratt, K.P. Gamma-Chain dysfibrinogenemias: Molecular structure–function relationships of naturally occurring mutations in the gamma chain of human fibrinogen. Blood 1998, 92, 2195–2212. [Google Scholar] [PubMed]
- Brennan, S.O.; Wyatt, J.; Medicina, D.; Callea, F.; George, P.M. Fibrinogen Brescia: Hepatic endoplasmic reticulum storage and hypofibrinogenemia because of a gamma 284 Gly→Arg mutation. Am. J. Pathol. 2000, 157, 189–196. [Google Scholar] [CrossRef]
- Callea, F.; Giovannoni, I.; Sari, S.; Aksu, A.U.; Esendagly, G.; Dalgic, B.; Boldrini, R.; Akyol, G.; Francalanci, P.; Bellacchio, E. A novel fibrinogen gamma chain mutation (c.1096C>G; p.His340Asp), fibrinogen Ankara, causing hypofibrinogenaemia and hepatic storage. Pathology 2017, 49, 534–537. [Google Scholar] [CrossRef] [PubMed]
- Simurda, T.; Stanciakova, L.; Stasko, J.; Dobrotova, M.; Kubisz, P. Yes or no for secondary prophylaxis in afibrinogenemia? Blood Coagul. Fibrinolysis 2015, 26, 978–980. [Google Scholar] [CrossRef] [PubMed]
- Casini, A.; de Moerloose, P. Congenital Fibrinogen Disorders Group. Management of congenital quantitative fibrinogen disorders: A Delphi consensus. Haemophilia 2016, 22, 898–905. [Google Scholar] [CrossRef] [PubMed]
- James, A.H. More than menorrhagia: A review of the obstetric and gynaecological manifestations of bleeding disorders. Haemophilia 2005, 11, 295–307. [Google Scholar] [CrossRef] [PubMed]
- Patil, R.; Mukaddam, A.; Ghosh, K.; Shetty, S. Management of pregnancy in dysfibrinogenemia cases: A dilemma. Blood Coagul. Fibrinolysis 2017, 28, 91–93. [Google Scholar] [CrossRef] [PubMed]
- Munoz, J.; Schering, J.; Lambing, A.; Neal, S.; Goyert, G.; Green, P.M.; Hanbali, A.; Raman, S.; Kuriakose, P. The dilemma of inherited dysfibrinogenemia during pregnancy. Blood Coagul. Fibrinolysis 2012, 23, 775–777. [Google Scholar] [CrossRef] [PubMed]
- Marchi, R.; Lundberg, U.; Grimbergen, J.; Koopman, J.; Torres, A.; de Bosch, N.B.; Haverkate, F.; Arocha Piñango, C.L. Fibrinogen Caracas V, an abnormal fibrinogen with an Aalpha 532 Ser→Cys substitution associated with thrombosis. Thromb. Haemost. 2000, 84, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Koopman, J.; Haverkate, F.; Grimbergen, J.; Lord, S.T.; Mosesson, M.W.; DiOrio, J.P.; Siebenlist, K.S.; Legrand, C.; Soria, J.; Soria, C.; et al. Molecular basis for fibrinogenDusart (A alpha 554 Arg→Cys) and its association with abnormal fibrin polymerization and thrombophilia. J. Clin. Investig. 1993, 91, 1637–1643. [Google Scholar] [CrossRef] [PubMed]
- Koopman, J.; Haverkate, F.; Grimbergen, J.; Engesser, L.; Nováková, I.; Kerst, A.F.; Lord, S.T. Abnormal fibrinogens IJmuiden (B beta Arg14—Cys) and Nijmegen (B betaArg44—Cys) form disulfide-linked fibrinogen-albumin complexes. Proc. Natl. Acad. Sci. USA 1992, 89, 3478–3482. [Google Scholar] [CrossRef] [PubMed]
- Engesser, L.; Koopman, J.; de Munk, G.; Haverkate, F.; Nováková, I.; Verheijen, J.H.; Briët, E.; Brommer, E.J. Fibrinogen Nijmegen: Congenital dysfibrinogenemia associated with impaired t-PA mediated plasminogen activation and decreased binding of t-PA. Thromb. Haemost. 1988, 60, 113–120. [Google Scholar] [PubMed]
- De Bosch, N.B.; Mosesson, M.W.; Ruiz-Saez, A.; Echenagucia, M.; Rodriguez-Lemoin, A. Inhibition of thrombin generation in plasma by fibrin formation (antithrombin I). Thromb. Haemost. 2002, 88, 253–258. [Google Scholar] [CrossRef]
- Dupuy, E.; Soria, C.; Molho, P.; Zini, J.M.; Rosenstingl, S.; Laurian, C.; Bruneval, P.; Tobelem, G. Embolized ischemic lesions of toes in an afibrinogenemic patient: Possible relevance to in vivo circulating thrombin. Thromb. Res. 2001, 102, 211–219. [Google Scholar] [CrossRef]
- Nagler, M.; Kremer Hovinga, J.A.; Alberio, L.; Peter-Salonen, K.; von Tengg-Kobligk, H.; Lottaz, D.; Neerman-Arbez, M.; Lämmle, B. Thromboembolism in patients with congenital afibrinogenaemia. Long-term observational data and systematic review. Thromb. Haemost. 2016, 116, 722–732. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tiscia, G.L.; Margaglione, M. Human Fibrinogen: Molecular and Genetic Aspects of Congenital Disorders. Int. J. Mol. Sci. 2018, 19, 1597. https://doi.org/10.3390/ijms19061597
Tiscia GL, Margaglione M. Human Fibrinogen: Molecular and Genetic Aspects of Congenital Disorders. International Journal of Molecular Sciences. 2018; 19(6):1597. https://doi.org/10.3390/ijms19061597
Chicago/Turabian StyleTiscia, Giovanni Luca, and Maurizio Margaglione. 2018. "Human Fibrinogen: Molecular and Genetic Aspects of Congenital Disorders" International Journal of Molecular Sciences 19, no. 6: 1597. https://doi.org/10.3390/ijms19061597
APA StyleTiscia, G. L., & Margaglione, M. (2018). Human Fibrinogen: Molecular and Genetic Aspects of Congenital Disorders. International Journal of Molecular Sciences, 19(6), 1597. https://doi.org/10.3390/ijms19061597