Identification of Blueberry miRNAs and Their Targets Based on High-Throughput Sequencing and Degradome Analyses

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
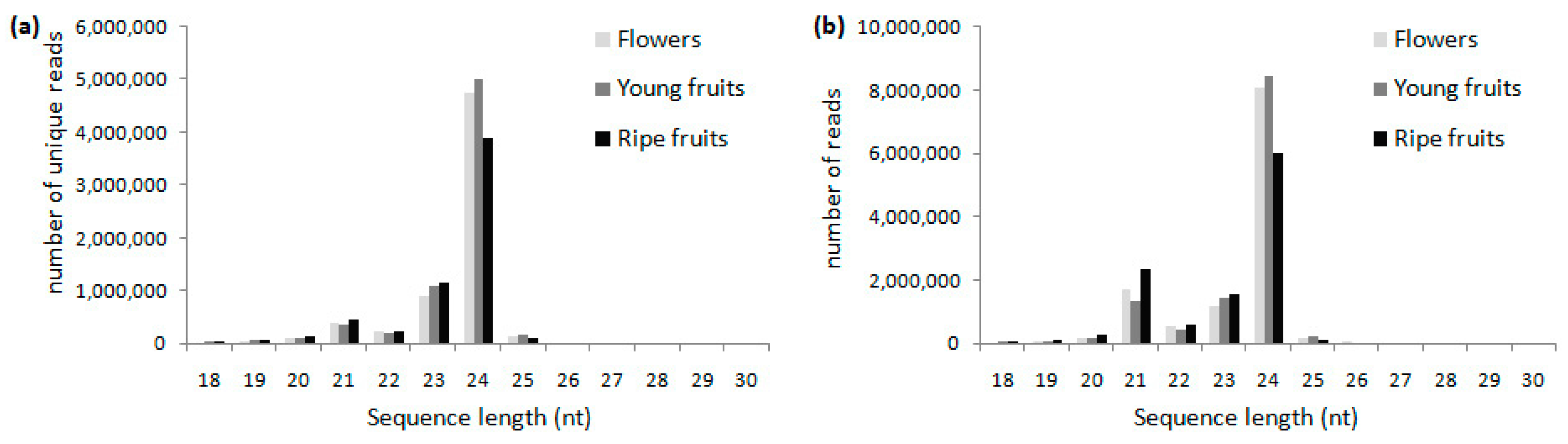
2.1. Small RNA Profiles in Blueberry
2.2. Identification of Known miRNAs
2.3. Identification of Novel miRNAs
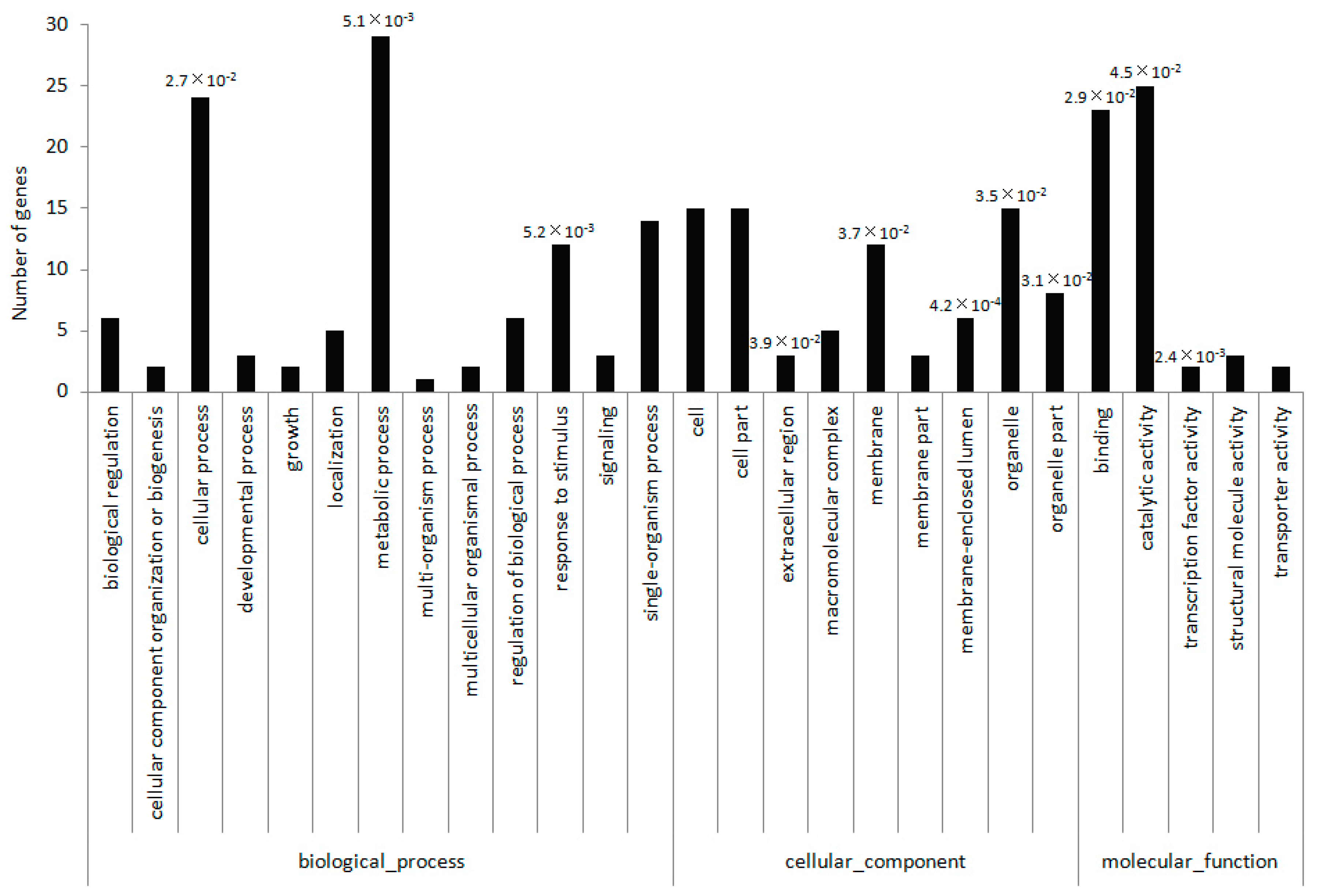
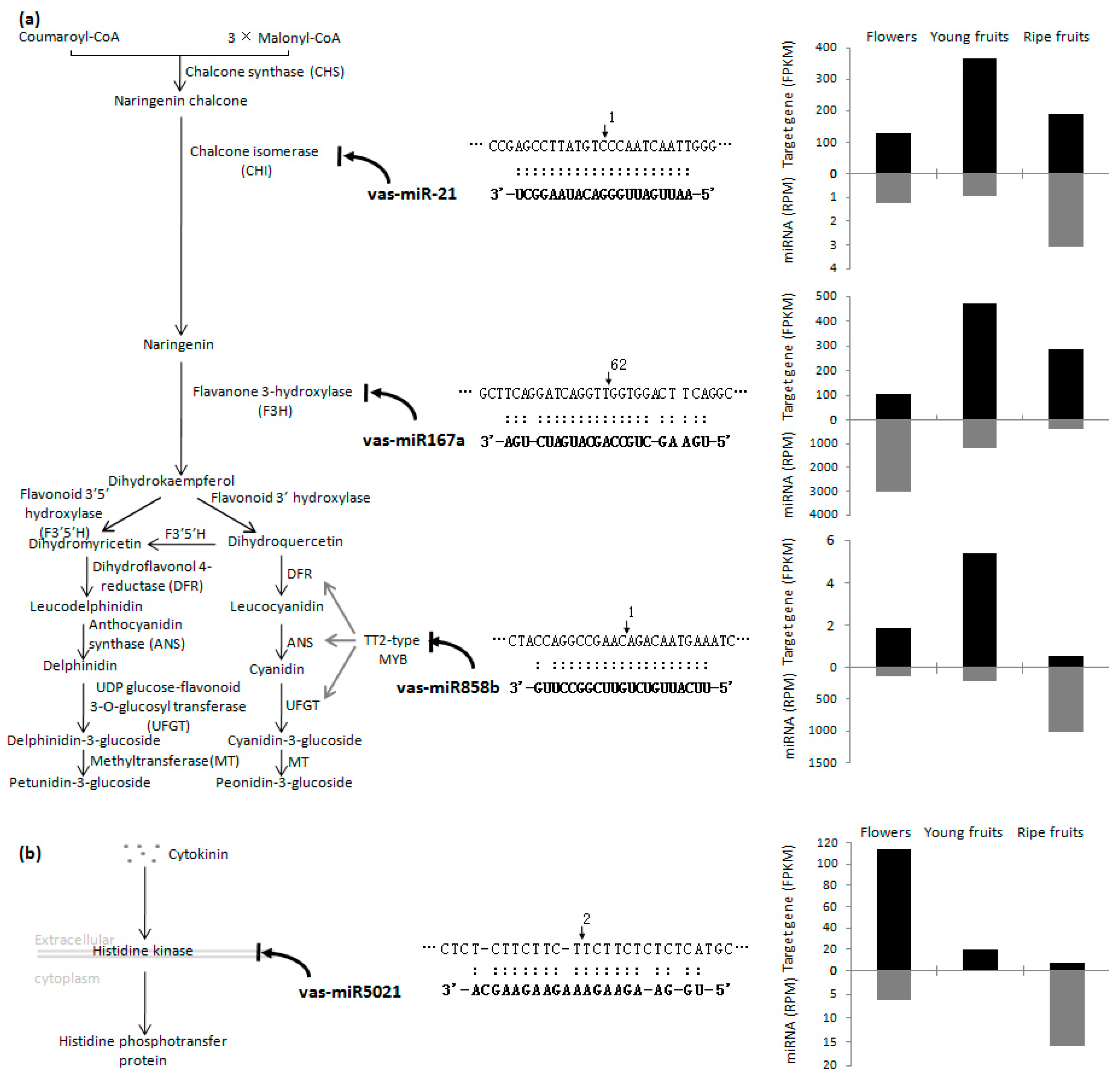
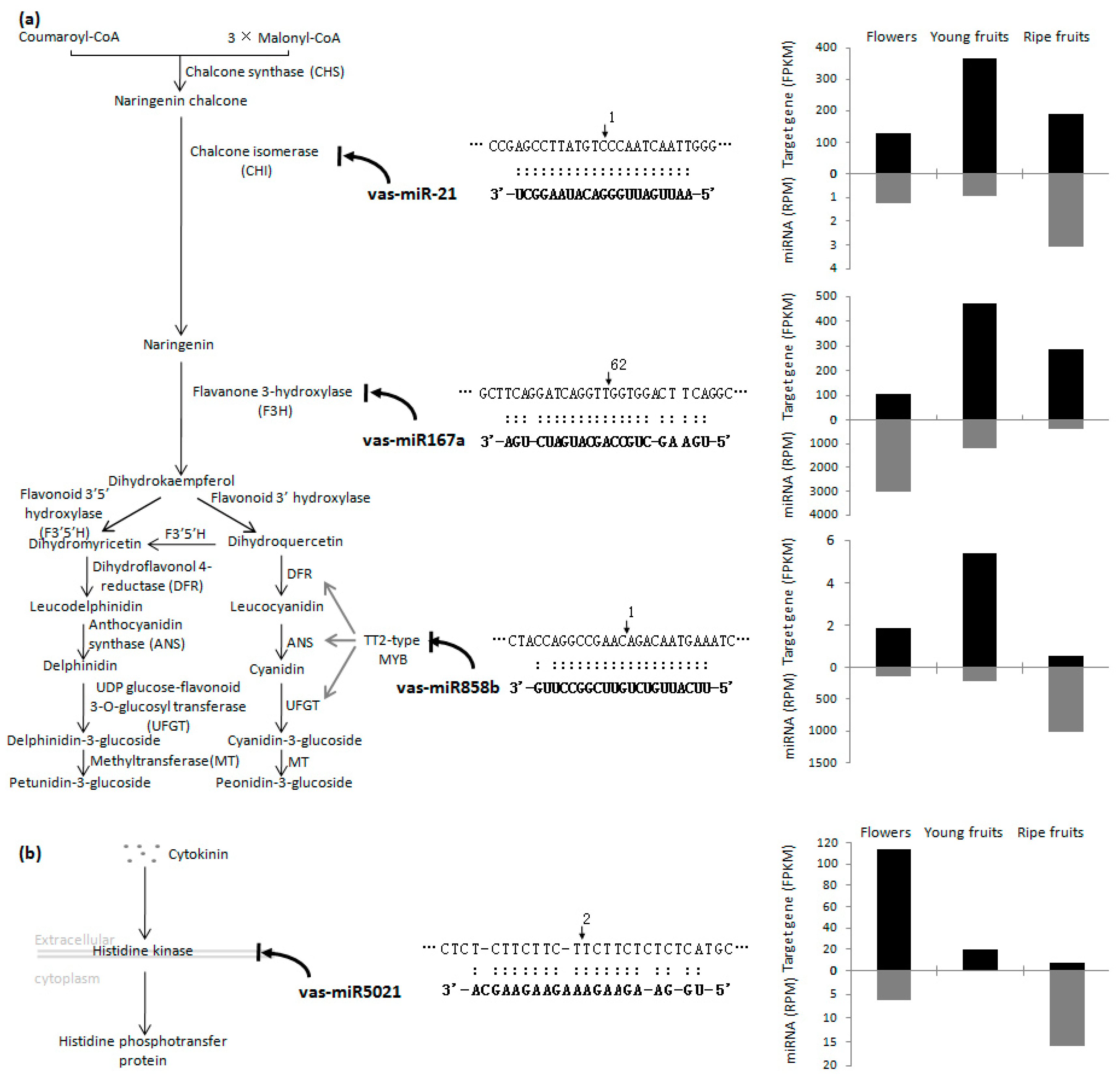
2.4. Identification of miRNA Targets Based on a Degradome Analysis
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Construction and Sequencing of Transcriptome, sRNA, and Degradome Libraries
4.3. Analysis of Transcriptome Sequencing Data
4.4. Analysis of sRNA Sequencing Data
4.5. Analysis of Degradome Sequencing Data
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Axtell, M.J. Classification and comparison of small RNAs from plants. Annu. Rev. Plant Biol. 2013, 64, 137–159. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Rhone, M.; Lyons, T.J. Berries: Emerging impact on cardiovascular health. Nutr. Rev. 2010, 68, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Lau, F.C.; Shukitt-Hale, B.; Joseph, J.A. The beneficial effects of fruit polyphenols on brain aging. Neurobiol. Aging 2005, 26, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Neto, C.C. Cranberry and blueberry: Evidence for protective effects against cancer and vascular diseases. Mol. Nutr. Food Res. 2007, 51, 652–664. [Google Scholar] [CrossRef] [PubMed]
- Gordillo, G.; Fang, H.; Khanna, S.; Harper, J.; Phillips, G.; Sen, C.K. Oral administration of blueberry inhibits angiogenic tumor growth and enhances survival of mice with endothelial cell neoplasm. Antioxid. Redox Signal. 2009, 11, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Yue, J.; Lu, X.; Zhang, H.; Ge, J.; Gao, X.; Liu, Y. Identification of conserved and novel microRNAs in blueberry. Front. Plant Sci. 2017, 8, 1155. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Zhai, L.; Li, X.; Xue, Y.; Wang, J.; Yang, P.; Cao, C.; Li, H.; Cui, Y.; Bian, S. Comparative analysis of fruit ripening-related mirnas and their targets in blueberry using small RNA and degradome sequencing. Int. J. Mol. Sci. 2017, 18, 2767. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Estrada, A.D.; Blakley, I.; Reid, R.; Patel, K.; Meyer, M.D.; Andersen, S.U.; Brown, A.F.; Lila, M.A.; Loraine, A.E. RNA-Seq analysis and annotation of a draft blueberry genome assembly identifies candidate genes involved in fruit ripening, biosynthesis of bioactive compounds, and stage-specific alternative splicing. Gigascience 2015, 4, 5. [Google Scholar] [CrossRef] [PubMed]
- Rowland, L.J.; Alkharouf, N.; Darwish, O.; Ogden, E.L.; Polashock, J.J.; Bassil, N.V.; Main, D. Generation and analysis of blueberry transcriptome sequences from leaves, developing fruit, and flower buds from cold acclimation through deacclimation. BMC Plant Biol. 2012, 12, 46. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Sun, H.; Pei, J.; Dong, Y.; Wang, F.; Chen, H.; Sun, Y.; Wang, N.; Li, H.; Li, Y. De novo sequencing and comparative analysis of the blueberry transcriptome to discover putative genes related to antioxidants. Gene 2012, 511, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Pantaleo, V.; Szittya, G.; Moxon, S.; Miozzi, L.; Moulton, V.; Dalmay, T.; Burgyan, J. Identification of grapevine microRNAs and their targets using high-throughput sequencing and degradome analysis. Plant J. 2010, 62, 960–976. [Google Scholar] [PubMed]
- Karlova, R.; van Haarst, J.C.; Maliepaard, C.; van de Geest, H.; Bovy, A.G.; Lammers, M.; Angenent, G.C.; de Maagd, R.A. Identification of microRNA targets in tomato fruit development using high-throughput sequencing and degradome analysis. J. Exp. Bot. 2013, 64, 1863–1878. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Able, A.J.; Able, J.A. SMARTER De-Stressed Cereal Breeding. Trends Plant Sci. 2016, 21, 909–925. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Wu, G.; Yuan, H.; Cheng, L.; Zhao, D.; Huang, W.; Zhang, S.; Zhang, L.; Chen, H.; Zhang, J.; et al. Identification and characterization of miRNAs and targets in flax (Linum usitatissimum) under saline, alkaline, and saline-alkaline stresses. BMC Plant Biol. 2016, 16, 124. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Pan, X.; Stellwag, E.J. Identification of soybean microRNAs and their targets. Planta 2008, 229, 161–182. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Frazier, T.P.; Zhang, B. Identification and characterization of microRNAs and their targets in the bioenergy plant switchgrass (Panicum virgatum). Planta 2010, 232, 417–434. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Wang, M.; Ma, Y.; Yuan, L.; Lu, S. High-throughput sequencing and characterization of the small RNA transcriptome reveal features of novel and conserved microRNAs in Panax ginseng. PLoS ONE 2012, 7, e44385. [Google Scholar] [CrossRef] [PubMed]
- Umbrasaite, J.; Schweighofer, A.; Meskiene, I. Substrate analysis of Arabidopsis PP2C-type protein phosphatases. Methods Mol. Biol. 2011, 779, 149–161. [Google Scholar] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Lakhotia, N.; Joshi, G.; Bhardwaj, A.R.; Katiyar-Agarwal, S.; Agarwal, M.; Jagannath, A.; Goel, S.; Kumar, A. Identification and characterization of miRNAome in root, stem, leaf and tuber developmental stages of potato (Solanum tuberosum L.) by high-throughput sequencing. BMC Plant Biol. 2014, 14, 6. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Wang, C.; Zhang, C.; Korir, N.K.; Yu, H.; Ma, Z.; Fang, J. Deep sequencing discovery of novel and conserved microRNAs in trifoliate orange (Citrus trifoliata). BMC Genom. 2010, 11, 431. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Searle, I.R.; Watson-Haigh, N.S.; Baumann, U.; Mather, D.E.; Able, A.J.; Able, J.A. Genome-Wide Identification of MicroRNAs in Leaves and the Developing Head of Four Durum Genotypes during Water Deficit Stress. PLoS ONE 2015, 10, e0142799. [Google Scholar] [CrossRef] [PubMed]
- Peng, T.; Lv, Q.; Zhang, J.; Li, J.; Du, Y.; Zhao, Q. Differential expression of the microRNAs in superior and inferior spikelets in rice (Oryza sativa). J. Exp. Bot. 2011, 62, 4943–4954. [Google Scholar] [CrossRef] [PubMed]
- Moxon, S.; Jing, R.; Szittya, G.; Schwach, F.; Rusholme Pilcher, R.L.; Moulton, V.; Dalmay, T. Deep sequencing of tomato short RNAs identifies microRNAs targeting genes involved in fruit ripening. Genome Res. 2008, 18, 1602–1609. [Google Scholar] [CrossRef] [PubMed]
- Solofoharivelo, M.C.; van der Walt, A.P.; Stephan, D.; Burger, J.T.; Murray, S.L. MicroRNAs in fruit trees: Discovery, diversity and future research directions. Plant Biol. 2014, 16, 856–865. [Google Scholar] [CrossRef] [PubMed]
- Zifkin, M.; Jin, A.; Ozga, J.A.; Zaharia, L.I.; Schernthaner, J.P.; Gesell, A.; Abrams, S.R.; Kennedy, J.A.; Constabel, C.P. Gene expression and metabolite profiling of developing highbush blueberry fruit indicates transcriptional regulation of flavonoid metabolism and activation of abscisic acid metabolism. Plant Physiol. 2012, 158, 200–224. [Google Scholar] [CrossRef] [PubMed]
- Terrier, N.; Torregrosa, L.; Ageorges, A.; Vialet, S.; Verries, C.; Cheynier, V.; Romieu, C. Ectopic expression of VvMybPA2 promotes proanthocyanidin biosynthesis in grapevine and suggests additional targets in the pathway. Plant Physiol. 2009, 149, 1028–1041. [Google Scholar] [CrossRef] [PubMed]
- Akagi, T.; Ikegami, A.; Yonemori, K. DkMyb2 wound-induced transcription factor of persimmon (Diospyros kaki Thunb.), contributes to proanthocyanidin regulation. Planta 2010, 232, 1045–1059. [Google Scholar] [CrossRef] [PubMed]
- Qu, D.; Yan, F.; Meng, R.; Jiang, X.; Yang, H.; Gao, Z.; Dong, Y.; Yang, Y.; Zhao, Z. Identification of microRNAs and their targets associated with fruit-bagging and subsequent sunlight re-exposure in the ”Granny Smith” apple exocarp using high-throughput sequencing. Front. Plant Sci. 2016, 7, 27. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, N.; Wang, H.; Kasahara, H.; Liu, J.; Macpherson, C.; Machida, Y.; Kamiya, Y.; Hannah, M.A.; Chua, N.H. IAA-Ala Resistant3, an evolutionarily conserved target of miR167, mediates Arabidopsis root architecture changes during high osmotic stress. Plant Cell 2012, 24, 3590–3602. [Google Scholar] [CrossRef] [PubMed]
- Rolle, R.S.; Chism, G.W. Kinetic comparison of cytokinin nucleosidase activity isolated from normally ripening and mutant tomato varieties. Plant Physiol. 1989, 91, 148–150. [Google Scholar] [CrossRef] [PubMed]
- Saavedra, X.; Modrego, A.; Rodriguez, D.; Gonzalez-Garcia, M.P.; Sanz, L.; Nicolas, G.; Lorenzo, O. The nuclear interactor PYL8/RCAR3 of Fagus sylvatica FsPP2C1 is a positive regulator of abscisic acid signaling in seeds and stress. Plant Physiol. 2010, 152, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Cheah, B.H.; Nadarajah, K.; Divate, M.D.; Wickneswari, R. Identification of four functionally important microRNA families with contrasting differential expression profiles between drought-tolerant and susceptible rice leaf at vegetative stage. BMC Genom. 2015, 16, 692. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Able, A.J.; Able, J.A. Water-deficit stress-responsive microRNAs and their targets in four durum wheat genotypes. Funct. Integr. Genom. 2017, 17, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Candar-Cakir, B.; Arican, E.; Zhang, B. Small RNA and degradome deep sequencing reveals drought-and tissue-specific micrornas and their important roles in drought-sensitive and drought-tolerant tomato genotypes. Plant Biotechnol. J. 2016, 14, 1727–1746. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Liu, F.; Zhang, Y.; Wang, L.; Cheng, Y.F. Cold-responsive miRNAs and their target genes in the wild eggplant species Solanum aculeatissimum. BMC Genom. 2017, 18, 1000. [Google Scholar] [CrossRef] [PubMed]
- Deng, P.; Wang, L.; Cui, L.; Feng, K.; Liu, F.; Du, X.; Tong, W.; Nie, X.; Ji, W.; Weining, S. Global identification of microRNAs and their targets in barley under salinity stress. PLoS ONE 2015, 10, e0137990. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, G.; Wang, Y.; Lou, X.; Li, H.; Zhang, C. Identification of Blueberry miRNAs and Their Targets Based on High-Throughput Sequencing and Degradome Analyses. Int. J. Mol. Sci. 2018, 19, 983. https://doi.org/10.3390/ijms19040983
Li G, Wang Y, Lou X, Li H, Zhang C. Identification of Blueberry miRNAs and Their Targets Based on High-Throughput Sequencing and Degradome Analyses. International Journal of Molecular Sciences. 2018; 19(4):983. https://doi.org/10.3390/ijms19040983
Chicago/Turabian StyleLi, Guangping, Yun Wang, Xiaoming Lou, Hailing Li, and Changqing Zhang. 2018. "Identification of Blueberry miRNAs and Their Targets Based on High-Throughput Sequencing and Degradome Analyses" International Journal of Molecular Sciences 19, no. 4: 983. https://doi.org/10.3390/ijms19040983
APA StyleLi, G., Wang, Y., Lou, X., Li, H., & Zhang, C. (2018). Identification of Blueberry miRNAs and Their Targets Based on High-Throughput Sequencing and Degradome Analyses. International Journal of Molecular Sciences, 19(4), 983. https://doi.org/10.3390/ijms19040983