Disease Modeling Using 3D Organoids Derived from Human Induced Pluripotent Stem Cells


Abstract
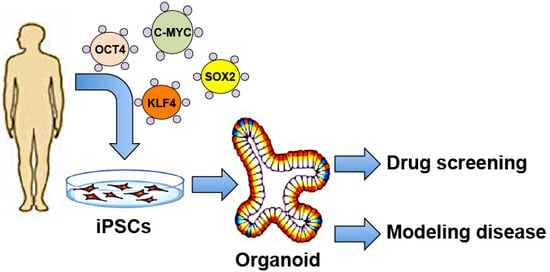
:
1. Introduction
2. Recognizing Limitations of both 2D Monolayer and 3D Organoid Models
2.1. Limitations of Conventional 2D Monolayer Models
2.1.1. Non-Natural and Static
2.1.2. Microtopography and Types of Cell Culture Surfaces
2.2. Limitations of Organoid Models
2.2.1. Reproducibility
2.2.2. Vascularization
2.2.3. Blood Perfusion and Inflammation
2.2.4. Humanized Mice Models Provide a Systemic Environment for Disease Modeling
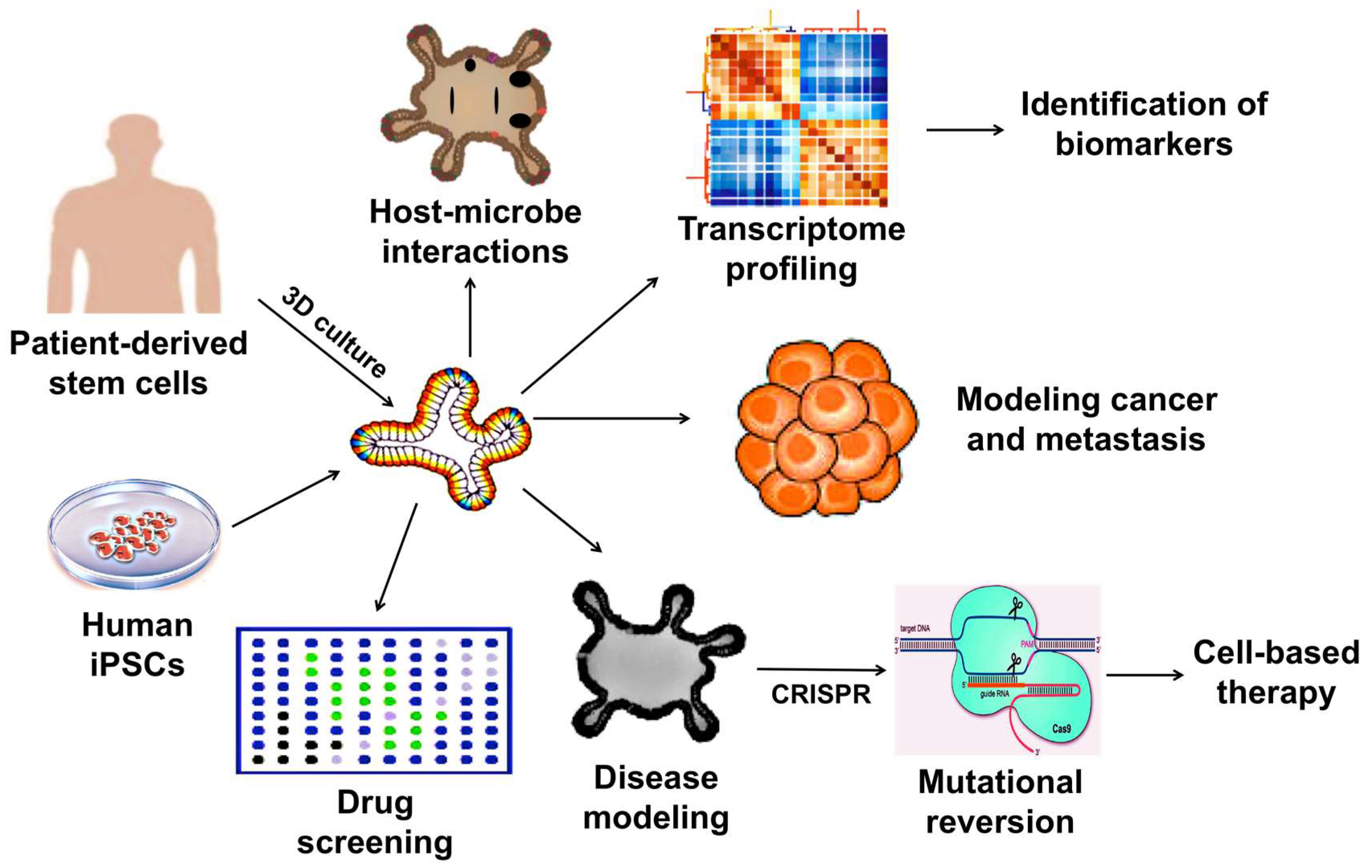
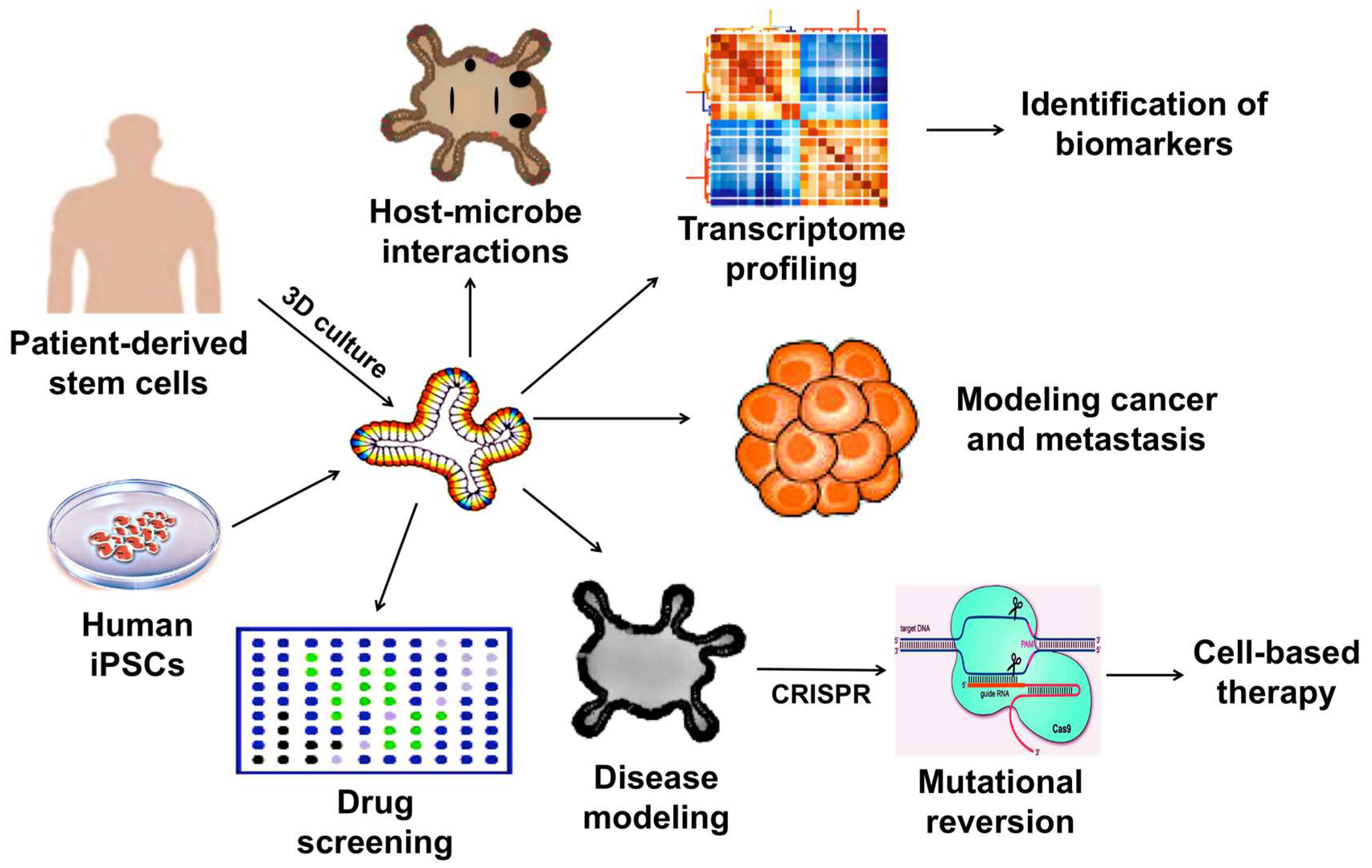
3. Therapeutic Applications of 3D Organoids
3.1. Development of Drug Screening Platforms
3.1.1. Cerebral Organoid Platforms for Drug Screening
3.1.2. Modeling Hepatic and Biliary Development for Drug Screening
3.2. Modeling Infectious Diseases
3.3. Modeling Cancer
3.3.1. Prostate Cancer
3.3.2. Colorectal Cancer
3.3.3. Ovarian Cancer
3.4. Modeling Hereditary Diseases
3.4.1. Cortical Organoids
3.4.2. Intestinal Organoids
3.4.3. Hepatic Organoids
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| 2D | two-dimensional |
| 3D | three-dimensional |
| hESCs | human embryonic stem cells |
| hiPSCs | human induced pluripotent stem cells |
| hPSCs | human pluripotent stem cells |
| CRISPR | clustered regularly interspaced short palindromic repeat |
| MSCs | mesenchymal stem cells |
| ECM | extracellular matrix |
| VSMCs | vascular smooth muscle cells |
| NSCs | neural stem cells |
| ZIKV | Zika virus |
| TLRs | Toll-like receptors |
| ASPM | abnormal spindle-like primary microcephaly |
| SVZ | sub-ventricular zone |
| IFL | inner fiber layer |
| TTX | tetrodotoxin |
| HP | Hippeastrine hydrobromide |
| AQ | Amodiaquine dihydrochloride dihydrate |
| hNPCs | human pluripotent stem cell-derived cortical neural progenitors cells |
| iHIOs | induced human intestinal organoids |
| DE | definitive endoderm |
| PCOs | prostate cancer organoids |
| CRPC | castrate-resistant prostate cancer |
| FTE | fallopian tube epithelium |
| FTO | fallopian tube organoid |
| HGSOC | High-grade serious ovarian cancer |
| FTSECs | fallopian tube secretory epithelial cells |
| ASD | autism spectrum disorder |
| CF | cystic fibrosis |
| CFTR | cystic fibrosis Transmembrane-conductor regulator |
| HO | hepatic organoids |
| ALG | Alagille syndrome |
| SCID | severe combined immunodeficiency |
References
- Yamanaka, S. Strategies and new developments in the generation of patient-specific pluripotent stem cells. Cell Stem Cell 2007, 1, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Brock, A.; Goh, H.T.; Yang, B.; Lu, Y.; Li, H.; Loh, Y.H. Cellular reprogramming: A new technology frontier in pharmaceutical research. Pharm. Res. 2012, 29, 35–52. [Google Scholar] [CrossRef] [PubMed]
- Kelava, I.; Lancaster, M.A. Dishing out mini-brains: Current progress and future prospects in brain organoid research. Dev. Biol. 2016, 420, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Ding, Q. Genome engineering of stem cell organoids for disease modeling. Protein Cell 2017, 8, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Firth, A.L.; Menon, T.; Parker, G.S.; Qualls, S.J.; Lewis, B.M.; Ke, E.; Dargitz, C.T.; Wright, R.; Khanna, A.; Gage, F.H.; et al. Functional gene correction for cystic fibrosis in lung epithelial cells generated from patient ipscs. Cell Rep. 2015, 12, 1385–1390. [Google Scholar] [CrossRef] [PubMed]
- Bassett, A.R. Editing the genome of hiPSC with CRISPR/Cas9: Disease models. Mamm. Genome 2017, 28, 348–364. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Kang, K.H.; Ju, J.H. CRISPR-Cas9: A promising tool for gene editing on induced pluripotent stem cells. Korean J. Intern. Med. 2017, 32, 42–61. [Google Scholar] [CrossRef] [PubMed]
- Musunuru, K. Genome editing of human pluripotent stem cells to generate human cellular disease models. Dis. Model. Mech. 2013, 6, 896–904. [Google Scholar] [CrossRef] [PubMed]
- Randolph, L.N.; Jiang, Y.; Lian, X. Stem cell engineering and differentiation for disease modeling and cell-based therapies. AIMS Cell Tissue Eng. 2017, 1, 140–157. [Google Scholar] [CrossRef]
- Yu, H.; Cowan, C.A. Minireview: Genome editing of human pluripotent stem cells for modeling metabolic disease. Mol. Endocrinol. 2016, 30, 575–586. [Google Scholar] [CrossRef] [PubMed]
- Dang, J.; Tiwari, S.K.; Lichinchi, G.; Qin, Y.; Patil, V.S.; Eroshkin, A.M.; Rana, T.M. Zika virus depletes neural progenitors in human cerebral organoids through activation of the innate immune receptor TLR3. Cell Stem Cell 2016, 19, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Garcez, P.P.; Loiola, E.C.; Madeiro da Costa, R.; Higa, L.M.; Trindade, P.; Delvecchio, R.; Nascimento, J.M.; Brindeiro, R.; Tanuri, A.; Rehen, S.K. Zika virus impairs growth in human neurospheres and brain organoids. Science 2016, 352, 3. [Google Scholar] [CrossRef] [PubMed]
- Lou, Y.R.; Leung, A.W. Next generation organoids for biomedical research and applications. Biotechnol. Adv. 2018, 36, 132–149. [Google Scholar] [CrossRef] [PubMed]
- Cugola, F.R.; Fernandes, I.R.; Russo, F.B.; Freitas, B.C.; Dias, J.L.; Guimaraes, K.P.; Benazzato, C.; Almeida, N.; Pignatari, G.C.; Romero, S.; et al. The brazilian zika virus strain causes birth defects in experimental models. Nature 2016, 534, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.T.; Bendriem, R.M.; Wu, W.W.; Shen, R.F. 3D brain organoids derived from pluripotent stem cells: Promising experimental models for brain development and neurodegenerative disorders. J. Biomed. Sci. 2017, 24, 59. [Google Scholar] [CrossRef] [PubMed]
- Dutta, D.; Heo, I.; Clevers, H. Disease modeling in stem cell-derived 3D organoid systems. Trends Mol. Med. 2017, 23, 393–410. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Renner, M.; Martin, C.A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Knoblich, J.A. Organogenesis in a dish: Modeling development and disease using organoid technologies. Science 2014, 345, 1247125. [Google Scholar] [CrossRef] [PubMed]
- Masumoto, H.; Yamashita, J.K. Human ips cell-derived cardiac tissue sheets: A platform for cardiac regeneration. Curr. Treat. Opt. Cardiovasc. Med. 2016, 18, 65. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Sun, L.; Fang, A.; Li, P.; Wu, Q.; Wang, X. Recapitulating cortical development with organoid culture in vitro and modeling abnormal spindle-like (aspm related primary) microcephaly disease. Protein Cell 2017, 8, 823–833. [Google Scholar] [CrossRef] [PubMed]
- Mariani, J.; Coppola, G.; Zhang, P.; Abyzov, A.; Provini, L.; Tomasini, L.; Amenduni, M.; Szekely, A.; Palejev, D.; Wilson, M.; et al. FOXG1-dependent dysregulation of GABA/glutamate neuron differentiation in autism spectrum disorders. Cell 2015, 162, 375–390. [Google Scholar] [CrossRef] [PubMed]
- Raja, W.K.; Mungenast, A.E.; Lin, Y.T.; Ko, T.; Abdurrob, F.; Seo, J.; Tsai, L.H. Self-organizing 3D human neural tissue derived from induced pluripotent stem cells recapitulate alzheimer’s disease phenotypes. PLoS ONE 2016, 11, e0161969. [Google Scholar] [CrossRef] [PubMed]
- Monzel, A.S.; Smits, L.M.; Hemmer, K.; Hachi, S.; Moreno, E.L.; van Wuellen, T.; Jarazo, J.; Walter, J.; Bruggemann, I.; Boussaad, I.; et al. Derivation of human midbrain-specific organoids from neuroepithelial stem cells. Stem Cell Rep. 2017, 8, 1144–1154. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Xu, D.; Garfin, P.M.; Ehmer, U.; Hurwitz, M.; Enns, G.; Michie, S.; Wu, M.; Zheng, M.; Nishimura, T.; et al. Human hepatic organoids for the analysis of human genetic diseases. JCI Insight 2017, 2, 94954. [Google Scholar] [CrossRef] [PubMed]
- Gomez, N.C.; Hepperla, A.J.; Dumitru, R.; Simon, J.M.; Fang, F.; Davis, I.J. Widespread chromatin accessibility at repetitive elements links stem cells with human cancer. Cell Rep. 2016, 17, 1607–1620. [Google Scholar] [CrossRef] [PubMed]
- Hohwieler, M.; Illing, A.; Hermann, P.C.; Mayer, T.; Stockmann, M.; Perkhofer, L.; Eiseler, T.; Antony, J.S.; Muller, M.; Renz, S.; et al. Human pluripotent stem cell-derived acinar/ductal organoids generate human pancreas upon orthotopic transplantation and allow disease modelling. Gut 2017, 66, 473–486. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.Y.; Sweeney, E.G.; Sigal, M.; Zhang, H.C.; Remington, S.J.; Cantrell, M.A.; Kuo, C.J.; Guillemin, K.; Amieva, M.R. Chemodetection and destruction of host urea allows helicobacter pylori to locate the epithelium. Cell Host Microbe 2015, 18, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Baker, L.A.; Tiriac, H.; Clevers, H.; Tuveson, D.A. Modeling pancreatic cancer with organoids. Trends Cancer 2016, 2, 176–190. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kim, H.; Ko, U.H.; Oh, Y.; Lim, A.; Sohn, J.W.; Shin, J.H.; Kim, H.; Han, Y.M. Islet-like organoids derived from human pluripotent stem cells efficiently function in the glucose responsiveness in vitro and in vivo. Sci. Rep. 2016, 6, 35145. [Google Scholar] [CrossRef] [PubMed]
- Finkbeiner, S.R.; Zeng, X.L.; Utama, B.; Atmar, R.L.; Shroyer, N.F.; Estes, M.K. Stem cell-derived human intestinal organoids as an infection model for rotaviruses. MBio 2012, 3, e00159-00112. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Bijvelds, M.; Dang, W.; Xu, L.; van der Eijk, A.A.; Knipping, K.; Tuysuz, N.; Dekkers, J.F.; Wang, Y.; de Jonge, J.; et al. Modeling rotavirus infection and antiviral therapy using primary intestinal organoids. Antiviral Res. 2015, 123, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Ettayebi, K.; Crawford, S.E.; Murakami, K.; Broughman, J.R.; Karandikar, U.; Tenge, V.R.; Neill, F.H.; Blutt, S.E.; Zeng, X.L.; Qu, L.; et al. Replication of human noroviruses in stem cell-derived human enteroids. Science 2016, 353, 1387–1393. [Google Scholar] [CrossRef] [PubMed]
- Dekkers, J.F.; Wiegerinck, C.L.; de Jonge, H.R.; Bronsveld, I.; Janssens, H.M.; de Winter-de Groot, K.M.; Brandsma, A.M.; de Jong, N.W.; Bijvelds, M.J.; Scholte, B.J.; et al. A functional cftr assay using primary cystic fibrosis intestinal organoids. Nat. Med. 2013, 19, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Schwank, G.; Koo, B.K.; Sasselli, V.; Dekkers, J.F.; Heo, I.; Demircan, T.; Sasaki, N.; Boymans, S.; Cuppen, E.; van der Ent, C.K.; et al. Functional repair of cftr by crispr/cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell 2013, 13, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Drost, J.; van Jaarsveld, R.H.; Ponsioen, B.; Zimberlin, C.; van Boxtel, R.; Buijs, A.; Sachs, N.; Overmeer, R.M.; Offerhaus, G.J.; Begthel, H.; et al. Sequential cancer mutations in cultured human intestinal stem cells. Nature 2015, 521, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Van de Wetering, M.; Francies, H.E.; Francis, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; van Houdt, W.; van Gorp, J.; Taylor-Weiner, A.; Kester, L.; et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015, 161, 933–945. [Google Scholar] [CrossRef] [PubMed]
- Amieva, M.; Peek, R.M., Jr. Pathobiology of helicobacter pylori-induced gastric cancer. Gastroenterology 2016, 150, 64–78. [Google Scholar] [CrossRef] [PubMed]
- Boj, S.F.; Vonk, A.M.; Statia, M.; Su, J.; Vries, R.R.; Beekman, J.M.; Clevers, H. Forskolin-induced swelling in intestinal organoids: An in vitro assay for assessing drug response in cystic fibrosis patients. J. Vis. Exp. 2017. [Google Scholar] [CrossRef] [PubMed]
- Takasato, M.; Er, P.X.; Chiu, H.S.; Maier, B.; Baillie, G.J.; Ferguson, C.; Parton, R.G.; Wolvetang, E.J.; Roost, M.S.; Chuva de Sousa Lopes, S.M.; et al. Kidney organoids from human ips cells contain multiple lineages and model human nephrogenesis. Nature 2015, 526, 564–568. [Google Scholar] [CrossRef] [PubMed]
- Freedman, B.S.; Brooks, C.R.; Lam, A.Q.; Fu, H.; Morizane, R.; Agrawal, V.; Saad, A.F.; Li, M.K.; Hughes, M.R.; Werff, R.V.; et al. Modelling kidney disease with crispr-mutant kidney organoids derived from human pluripotent epiblast spheroids. Nat. Commun. 2015, 6, 8715. [Google Scholar] [CrossRef] [PubMed]
- Yucer, N.; Holzapfel, M.; Jenkins Vogel, T.; Lenaeus, L.; Ornelas, L.; Laury, A.; Sareen, D.; Barrett, R.; Karlan, B.Y.; Svendsen, C.N. Directed differentiation of human induced pluripotent stem cells into fallopian tube epithelium. Sci. Rep. 2017, 7, 10741. [Google Scholar] [CrossRef] [PubMed]
- Lawrenson, K.; Notaridou, M.; Lee, N.; Benjamin, E.; Jacobs, I.J.; Jones, C.A.; Gayther, S.A. In vitro three-dimensional modeling of fallopian tube secretory epithelial cells. BMC Cell Biol. 2013, 14, 15. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Vela, I.; Sboner, A.; Iaquinta, P.J.; Karthaus, W.R.; Gopalan, A.; Dowling, C.; Wanjala, J.N.; Undvall, E.A.; Arora, V.K.; et al. Organoid cultures derived from patients with advanced prostate cancer. Cell 2014, 159, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Chen, Y. Organoid development in cancer genome discovery. Curr. Opin. Genet. Dev. 2015, 30, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Dye, B.R.; Hill, D.R.; Ferguson, M.A.; Tsai, Y.H.; Nagy, M.S.; Dyal, R.; Wells, J.M.; Mayhew, C.N.; Nattiv, R.; Klein, O.D.; et al. In Vitro generation of human pluripotent stem cell derived lung organoids. Elife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Barkauskas, C.E.; Chung, M.I.; Fioret, B.; Gao, X.; Katsura, H.; Hogan, B.L. Lung organoids: Current uses and future promise. Development 2017, 144, 986–997. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.W.; Huang, S.X.; de Carvalho, A.; Ho, S.H.; Islam, M.N.; Volpi, S.; Notarangelo, L.D.; Ciancanelli, M.; Casanova, J.L.; Bhattacharya, J.; et al. A three-dimensional model of human lung development and disease from pluripotent stem cells. Nat. Cell Biol. 2017, 19, 542–549. [Google Scholar] [CrossRef] [PubMed]
- Wahlin, K.J.; Maruotti, J.A.; Sripathi, S.R.; Ball, J.; Angueyra, J.M.; Kim, C.; Grebe, R.; Li, W.; Jones, B.W.; Zack, D.J. Photoreceptor outer segment-like structures in long-term 3d retinas from human pluripotent stem cells. Sci. Rep. 2017, 7, 766. [Google Scholar] [CrossRef] [PubMed]
- Llonch, S.; Carido, M.; Ader, M. Organoid technology for retinal repair. Dev. Biol. 2018, 433, 132–143. [Google Scholar] [CrossRef] [PubMed]
- DiStefano, T.; Chen, H.Y.; Panebianco, C.; Kaya, K.D.; Brooks, M.J.; Gieser, L.; Morgan, N.Y.; Pohida, T.; Swaroop, A. Accelerated and improved differentiation of retinal organoids from pluripotent stem cells in rotating-wall vessel bioreactors. Stem Cell Rep. 2018, 10, 300–313. [Google Scholar] [CrossRef] [PubMed]
- Gasiorowski, J.Z.; Murphy, C.J.; Nealey, P.F. Biophysical cues and cell behavior: The big impact of little things. Annu. Rev. Biomed. Eng. 2013, 15, 155–176. [Google Scholar] [CrossRef] [PubMed]
- Duval, K.; Grover, H.; Han, L.H.; Mou, Y.; Pegoraro, A.F.; Fredberg, J.; Chen, Z. Modeling physiological events in 2D vs. 3D cell culture. Physiology (Bethesda) 2017, 32, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Saha, K.; Mei, Y.; Reisterer, C.M.; Pyzocha, N.K.; Yang, J.; Muffat, J.; Davies, M.C.; Alexander, M.R.; Langer, R.; Anderson, D.G.; et al. Surface-engineered substrates for improved human pluripotent stem cell culture under fully defined conditions. Proc. Natl. Acad. Sci. USA 2011, 108, 18714–18719. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, Y.; Furue, M.K. Biological effects of culture substrates on human pluripotent stem cells. Stem Cells Int. 2016, 2016, 5380560. [Google Scholar] [CrossRef] [PubMed]
- Boyan, B.D.; Bonewald, L.F.; Paschalis, E.P.; Lohmann, C.H.; Rosser, J.; Cochran, D.L.; Dean, D.D.; Schwartz, Z.; Boskey, A.L. Osteoblast-mediated mineral deposition in culture is dependent on surface microtopography. Calcif. Tissue Int. 2002, 71, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Gomez, N.; Lu, Y.; Chen, S.; Schmidt, C.E. Immobilized nerve growth factor and microtopography have distinct effects on polarization versus axon elongation in hippocampal cells in culture. Biomaterials 2007, 28, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.A.; Desai, T.A.; Kumar, S. Microtopographical assembly of cardiomyocytes. Integr. Biol. (Camb.) 2011, 3, 1011–1019. [Google Scholar] [CrossRef] [PubMed]
- Liberio, M.S.; Sadowski, M.C.; Soekmadji, C.; Davis, R.A.; Nelson, C.C. Differential effects of tissue culture coating substrates on prostate cancer cell adherence, morphology and behavior. PLoS ONE 2014, 9, e112122. [Google Scholar] [CrossRef] [PubMed]
- Edmondson, R.; Broglie, J.J.; Adcock, A.F.; Yang, L. Three-dimensional cell culture systems and their applications in drug discovery and cell-based biosensors. Assay Drug Dev. Technol. 2014, 12, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Huch, M.; Knoblich, J.A.; Lutolf, M.P.; Martinez-Arias, A. The hope and the hype of organoid research. Development 2017, 144, 938–941. [Google Scholar] [CrossRef] [PubMed]
- Krefft, O.; Jabali, A.; Iefremova, V.; Koch, P.; Ladewig, J. Generation of standardized and reproducible forebrain-type cerebral organoids from human induced pluripotent stem cells. J. Vis. Exp. 2018. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Mead, B.E.; Safaee, H.; Langer, R.; Karp, J.M.; Levy, O. Engineering stem cell organoids. Cell Stem Cell 2016, 18, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Magdeldin, T.; Lopez-Davila, V.; Pape, J.; Cameron, G.W.; Emberton, M.; Loizidou, M.; Cheema, U. Engineering a vascularised 3D in vitro model of cancer progression. Sci. Rep. 2017, 7, 44045. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.H.; Gimble, J.M.; Kaplan, D.L. In Vitro 3d model for human vascularized adipose tissue. Tissue Eng. Part A 2009, 15, 2227–2236. [Google Scholar] [CrossRef] [PubMed]
- Massa, S.; Sakr, M.A.; Seo, J.; Bandaru, P.; Arneri, A.; Bersini, S.; Zare-Eelanjegh, E.; Jalilian, E.; Cha, B.H.; Antona, S.; et al. Bioprinted 3D vascularized tissue model for drug toxicity analysis. Biomicrofluidics 2017, 11, 044109. [Google Scholar] [CrossRef] [PubMed]
- Ley, K.; Laudanna, C.; Cybulsky, M.I.; Nourshargh, S. Getting to the site of inflammation: The leukocyte adhesion cascade updated. Nat. Rev. Immunol. 2007, 7, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Anacker, D.; Moody, C. Generation of organotypic raft cultures from primary human keratinocytes. J. Vis. Exp. 2012. [Google Scholar] [CrossRef] [PubMed]
- Linde, N.; Gutschalk, C.M.; Hoffmann, C.; Yilmaz, D.; Mueller, M.M. Integrating macrophages into organotypic co-cultures: A 3D in vitro model to study tumor-associated macrophages. PLoS ONE 2012, 7, e40058. [Google Scholar] [CrossRef] [PubMed]
- McGuigan, A.P.; Sefton, M.V. Vascularized organoid engineered by modular assembly enables blood perfusion. Proc. Natl. Acad. Sci. USA 2006, 103, 11461–11466. [Google Scholar] [CrossRef] [PubMed]
- Wufuer, M.; Lee, G.; Hur, W.; Jeon, B.; Kim, B.J.; Choi, T.H.; Lee, S. Skin-on-a-chip model simulating inflammation, edema and drug-based treatment. Sci. Rep. 2016, 6, 37471. [Google Scholar] [CrossRef] [PubMed]
- Shultz, L.D.; Brehm, M.A.; Garcia-Martinez, J.V.; Greiner, D.L. Humanized mice for immune system investigation: Progress, promise and challenges. Nat. Rev. Immunol. 2012, 12, 786–798. [Google Scholar] [CrossRef] [PubMed]
- Ito, R.; Takahashi, T.; Katano, I.; Ito, M. Current advances in humanized mouse models. Cell. Mol. Immunol. 2012, 9, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Mestas, J.; Hughes, C.C. Of mice and not men: Differences between mouse and human immunology. J. Immunol. 2004, 172, 2731–2738. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Nguyen, H.N.; Song, M.M.; Hadiono, C.; Ogden, S.C.; Hammack, C.; Yao, B.; Hamersky, G.R.; Jacob, F.; Zhong, C.; et al. Brain-region-specific organoids using mini-bioreactors for modeling zikv exposure. Cell 2016, 165, 1238–1254. [Google Scholar] [CrossRef] [PubMed]
- Camp, J.G.; Badsha, F.; Florio, M.; Kanton, S.; Gerber, T.; Wilsch-Brauninger, M.; Lewitus, E.; Sykes, A.; Hevers, W.; Lancaster, M.; et al. Human cerebral organoids recapitulate gene expression programs of fetal neocortex development. Proc. Natl. Acad. Sci. USA 2015, 112, 15672–15677. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Tan, L.; Cederquist, G.Y.; Fan, Y.; Hartley, B.J.; Mukherjee, S.; Tomishima, M.; Brennand, K.J.; Zhang, Q.; Schwartz, R.E.; et al. High-content screening in hpsc-neural progenitors identifies drug candidates that inhibit zika virus infection in fetal-like organoids and adult brain. Cell Stem Cell 2017, 21, 274–283.e5. [Google Scholar] [CrossRef] [PubMed]
- Hindley, C.J.; Cordero-Espinoza, L.; Huch, M. Organoids from adult liver and pancreas: Stem cell biology and biomedical utility. Dev. Biol. 2016, 420, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Takebe, T.; Sekine, K.; Enomura, M.; Koike, H.; Kimura, M.; Ogaeri, T.; Zhang, R.R.; Ueno, Y.; Zheng, Y.W.; Koike, N.; et al. Vascularized and functional human liver from an ipsc-derived organ bud transplant. Nature 2013, 499, 481–484. [Google Scholar] [CrossRef] [PubMed]
- Takebe, T.; Enomura, M.; Yoshizawa, E.; Kimura, M.; Koike, H.; Ueno, Y.; Matsuzaki, T.; Yamazaki, T.; Toyohara, T.; Osafune, K.; et al. Vascularized and complex organ buds from diverse tissues via mesenchymal cell-driven condensation. Cell Stem Cell 2015, 16, 556–565. [Google Scholar] [CrossRef] [PubMed]
- Nantasanti, S.; de Bruin, A.; Rothuizen, J.; Penning, L.C.; Schotanus, B.A. Concise review: Organoids are a powerful tool for the study of liver disease and personalized treatment design in humans and animals. Stem Cells Transl. Med. 2016, 5, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Spence, J.R.; Mayhew, C.N.; Rankin, S.A.; Kuhar, M.F.; Vallance, J.E.; Tolle, K.; Hoskins, E.E.; Kalinichenko, V.V.; Wells, S.I.; Zorn, A.M.; et al. Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature 2011, 470, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Bartfeld, S. Modeling infectious diseases and host-microbe interactions in gastrointestinal organoids. Dev. Biol. 2016, 420, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Weeber, F.; Ooft, S.N.; Dijkstra, K.K.; Voest, E.E. Tumor organoids as a pre-clinical cancer model for drug discovery. Cell Chem. Biol. 2017, 24, 1092–1100. [Google Scholar] [CrossRef] [PubMed]
- Praharaj, P.P.; Bhutia, S.K.; Nagrath, S.; Bitting, R.L.; Deep, G. Circulating tumor cell-derived organoids: Current challenges and promises in medical research and precision medicine. Biochim. Biophys. Acta 2018, 1869, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Saeed, K.; Rahkama, V.; Eldfors, S.; Bychkov, D.; Mpindi, J.P.; Yadav, B.; Paavolainen, L.; Aittokallio, T.; Heckman, C.; Wennerberg, K.; et al. Comprehensive drug testing of patient-derived conditionally reprogrammed cells from castration-resistant prostate cancer. Eur. Urol. 2017, 71, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Kessler, M.; Hoffmann, K.; Brinkmann, V.; Thieck, O.; Jackisch, S.; Toelle, B.; Berger, H.; Mollenkopf, H.J.; Mangler, M.; Sehouli, J.; et al. The notch and wnt pathways regulate stemness and differentiation in human fallopian tube organoids. Nat. Commun. 2015, 6, 8989. [Google Scholar] [CrossRef] [PubMed]
- Courchesne, E.; Mouton, P.R.; Calhoun, M.E.; Semendeferi, K.; Ahrens-Barbeau, C.; Hallet, M.J.; Barnes, C.C.; Pierce, K. Pierce Neuron number and size in prefrontal cortex of children with autism. JAMA 2011, 306, 2001–2010. [Google Scholar] [CrossRef] [PubMed]
- Casanova, M.F.; Buxhoeveden, D.P.; Switala, A.E.; Roy, E. Minicolumnar pathology in autism. Neurology 2002, 58, 428–432. [Google Scholar] [CrossRef] [PubMed]
- Hutsler, J.J.; Zhang, H. Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brain Res. 2010, 1309, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Hannan, N.R.; Segeritz, C.P.; Touboul, T.; Vallier, L. Production of hepatocyte like cells from human pluripotent stem cells. Nat. Protocols 2013, 8, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Kruitwagen, H.S.; Oosterhoff, L.A.; Vernooij, I.; Schrall, I.M.; van Wolferen, M.E.; Bannink, F.; Roesch, C.; van Uden, L.; Molenaar, M.R.; Helms, J.B.; et al. Long-term adult feline liver organoid cultures for disease modeling of hepatic steatosis. Stem Cell Rep. 2017, 8, 822–830. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Technical and Experimental Considerations | 2D Model | 3D Organoid Model |
|---|---|---|
| Cost | Low | Moderate to high |
| Ease of setup | Easy to moderately challenging | Very challenging |
| Time required | Low | Moderate to high |
| Cell–ECM interactions | Artificial environment | Mimics natural environment |
| Reproducibility | High | Low |
| Vascularization | No | Yes |
| Blood perfusion | No | Highly possible |
| Inflammation | Co-culturing techniques allow for a simple way to model inflammation | More improved technologies allow for modeling the complexity of inflammation |
| Tissue/Organ | Disease Modeled | References |
|---|---|---|
| Brain | Zika virus and congenital brain malformations | Kelava et al., 2016 [3]; Dang et al., 2016 [11]; Garcez et al., 2016 [12]; Cugola et al., 2016 [14] |
| Primary microencephaly | Kelava et al., 2016 [3]; Dang et al., 2016 [11]; Lancaster et al., 2013 [17]; Li et al., 2017 [20] | |
| Autism/macrocephaly | Mariani et al., 2015 [21] | |
| Alzheimer’s disease | Raja et al., 2016 [22] | |
| Parkinson’s disease | Monzel et al., 2017 [23] | |
| Liver | Alagille syndrome A1AT deficiency Cystic fibrosis | Guan et al., 2017 [24]; Gomez et al., 2016 [25] |
| Pancreas | Cystic fibrosis | Hohwieler et al., 2017 [26] |
| Pancreatic ductal adenocarcinoma | Huang et al., 2015 [27]; Baker et al., 2016 [28] | |
| Diabetes mellitus | Kim et al., 2016 [29] | |
| Intestinal | Host–microbe interactions e.g., human norovirus | Finkbeiner et al., 2012 [30]; Yin et al., 2015 [31]; Ettayebi et al., 2016 [32] |
| Cystic fibrosis (CF) | Dekkers et al., 2013 [33]; Schwank et al., 2013 [34] | |
| Colorectal cancer | Drost et al., 2015 [35]; van de Wetering et al., 2015 [36] | |
| Host–microbial interactions (e.g., Helicobacter pylori) | Finkbeiner et al., 2012 [30]; Huang et al., 2015 [27]; Amieva et al., 2016 [37]; Boj et al., 2017 [38] | |
| Stomach | Cancer | Takasato et al., 2015 [39] |
| Kidney | Polycystic kidney disease | Freedman et al., 2015 [40] |
| Ovarian cancer | Yucer et al., 2017 [41]; Lawrenson et al., 2013 [42] | |
| Urological | Prostate cancer | Gao et al., 2014 [43]; Gao et al., 2015 [44] |
| Lung | Fibrotic lung disease | Dye et al., 2015 [45]; Barkauskas et al., 2017 [46]; Chen et al., 2017 [47] |
| Retinal | Leber congenital amaurosis (LCA), Retinitis pigmentosa, Age-related macular degeneration | Wahlin et al., 2017 [48]; Llonch et al., 2018 [49]; DiStefano et al., 2018 [50] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ho, B.X.; Pek, N.M.Q.; Soh, B.-S. Disease Modeling Using 3D Organoids Derived from Human Induced Pluripotent Stem Cells. Int. J. Mol. Sci. 2018, 19, 936. https://doi.org/10.3390/ijms19040936
Ho BX, Pek NMQ, Soh B-S. Disease Modeling Using 3D Organoids Derived from Human Induced Pluripotent Stem Cells. International Journal of Molecular Sciences. 2018; 19(4):936. https://doi.org/10.3390/ijms19040936
Chicago/Turabian StyleHo, Beatrice Xuan, Nicole Min Qian Pek, and Boon-Seng Soh. 2018. "Disease Modeling Using 3D Organoids Derived from Human Induced Pluripotent Stem Cells" International Journal of Molecular Sciences 19, no. 4: 936. https://doi.org/10.3390/ijms19040936
APA StyleHo, B. X., Pek, N. M. Q., & Soh, B.-S. (2018). Disease Modeling Using 3D Organoids Derived from Human Induced Pluripotent Stem Cells. International Journal of Molecular Sciences, 19(4), 936. https://doi.org/10.3390/ijms19040936