Brca1 Is Upregulated by 5-Aza-CdR and Promotes DNA Repair and Cell Survival, and Inhibits Neurite Outgrowth in Rat Retinal Neurons

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
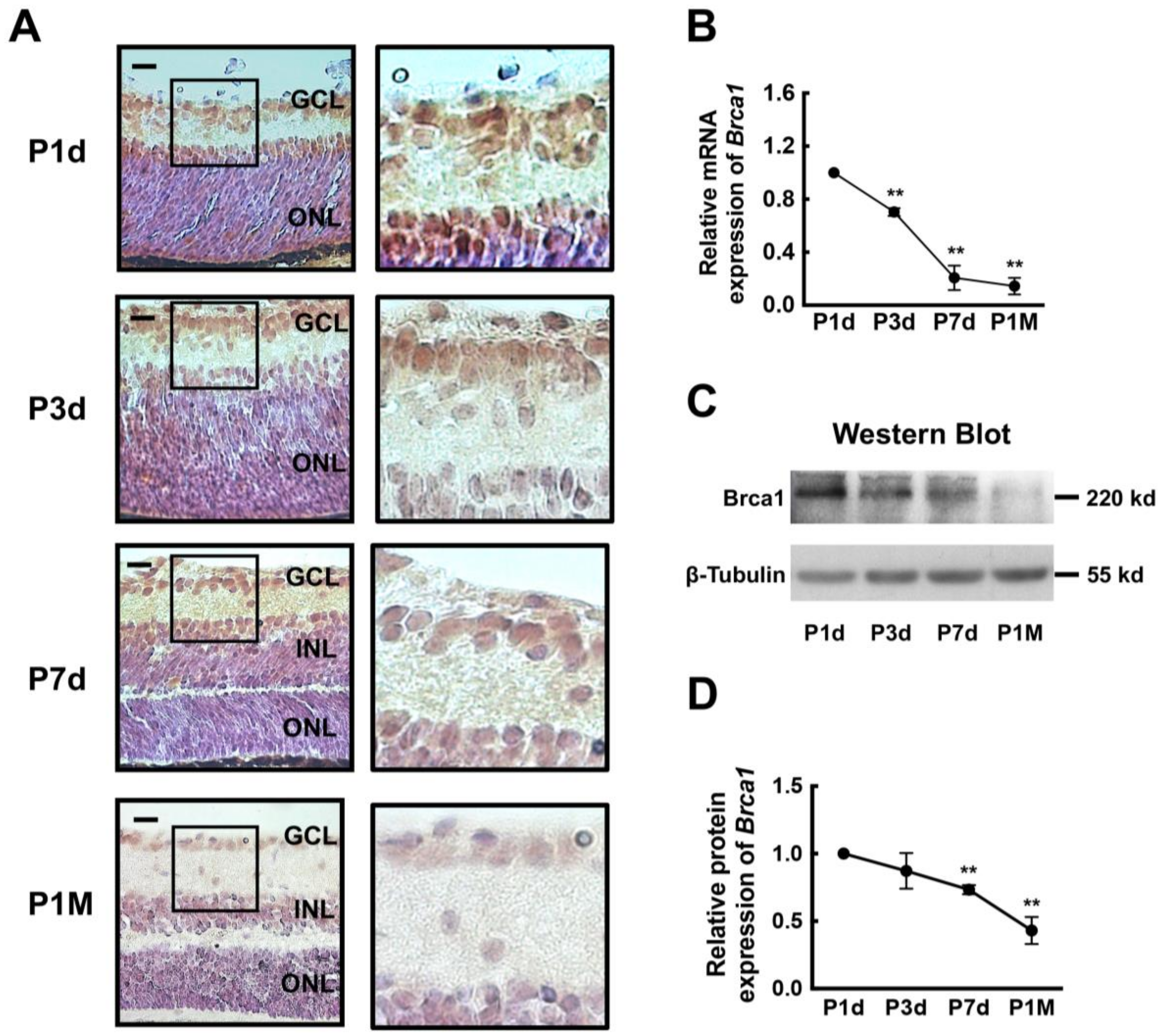
2.1. Breast Cancer Type 1 Susceptibility Protein (Brca1) Is Developmentally Downregulated in Rat Retina
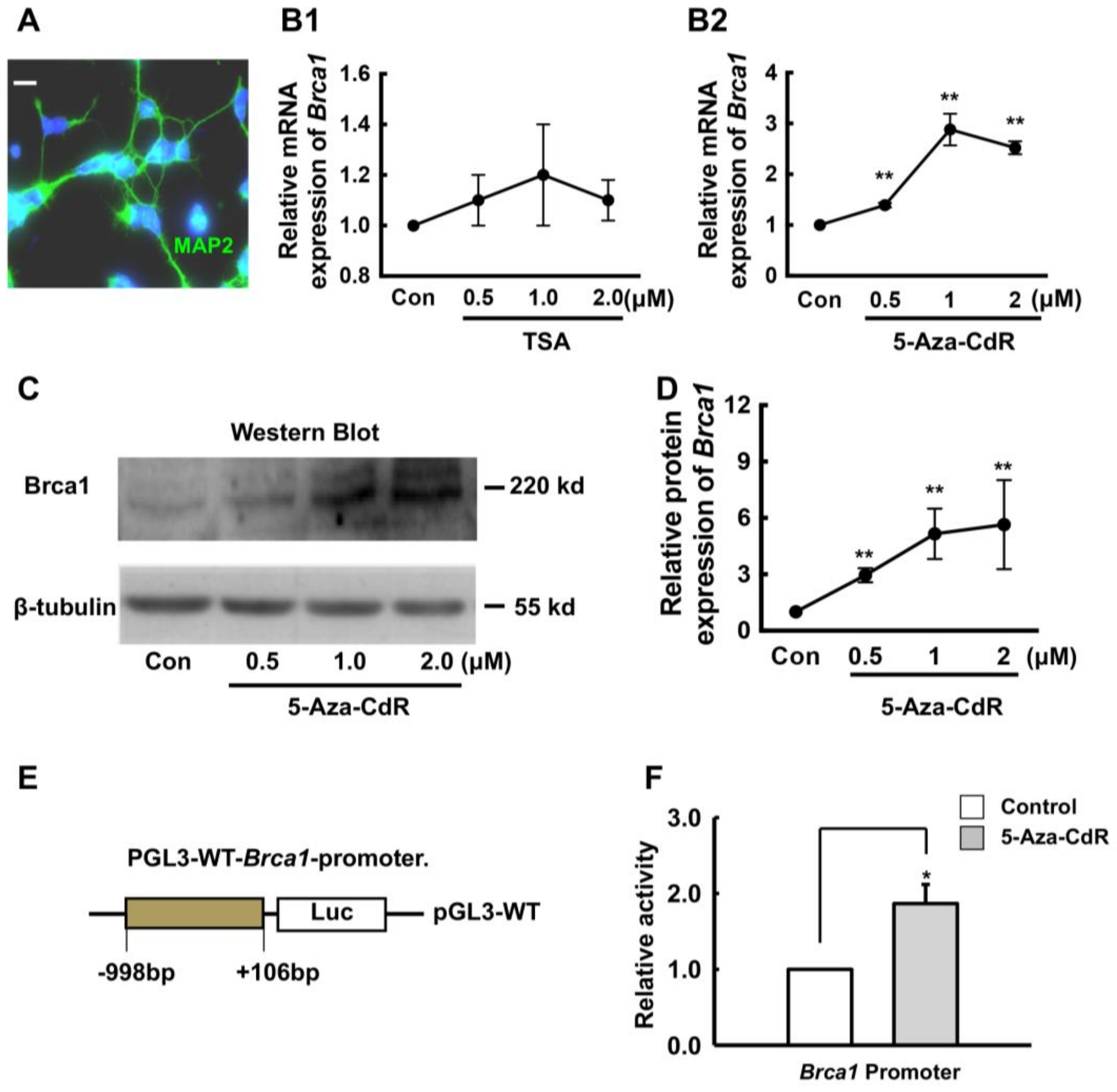
2.2. 5-Aza-CdR Upregulates Brca1 Expression in Retinal Neurons
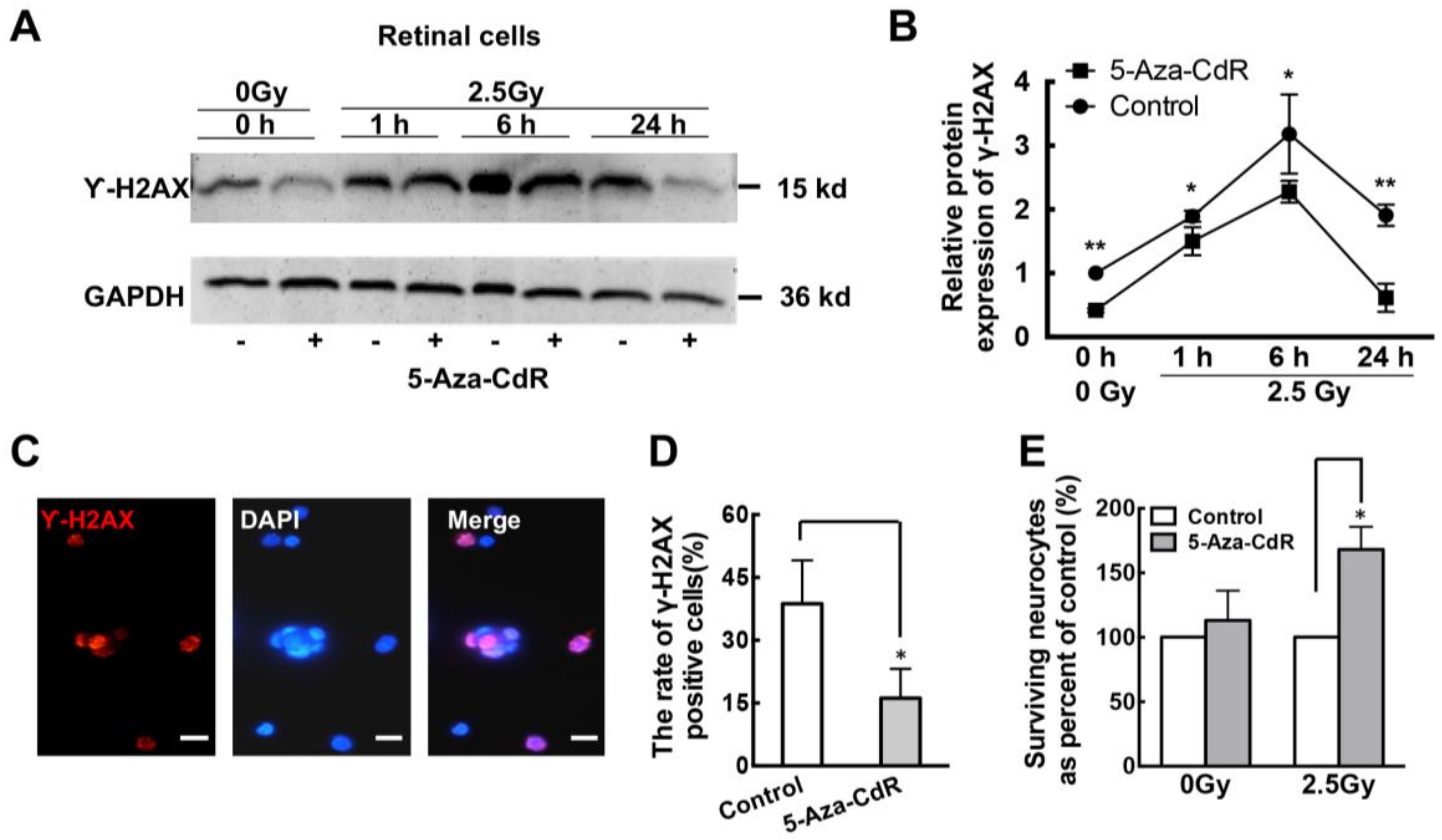
2.3. 5-Aza-CdR Promotes DNA Repair in Retinal Neurons after Ionizing Radiation (IR) Treatment
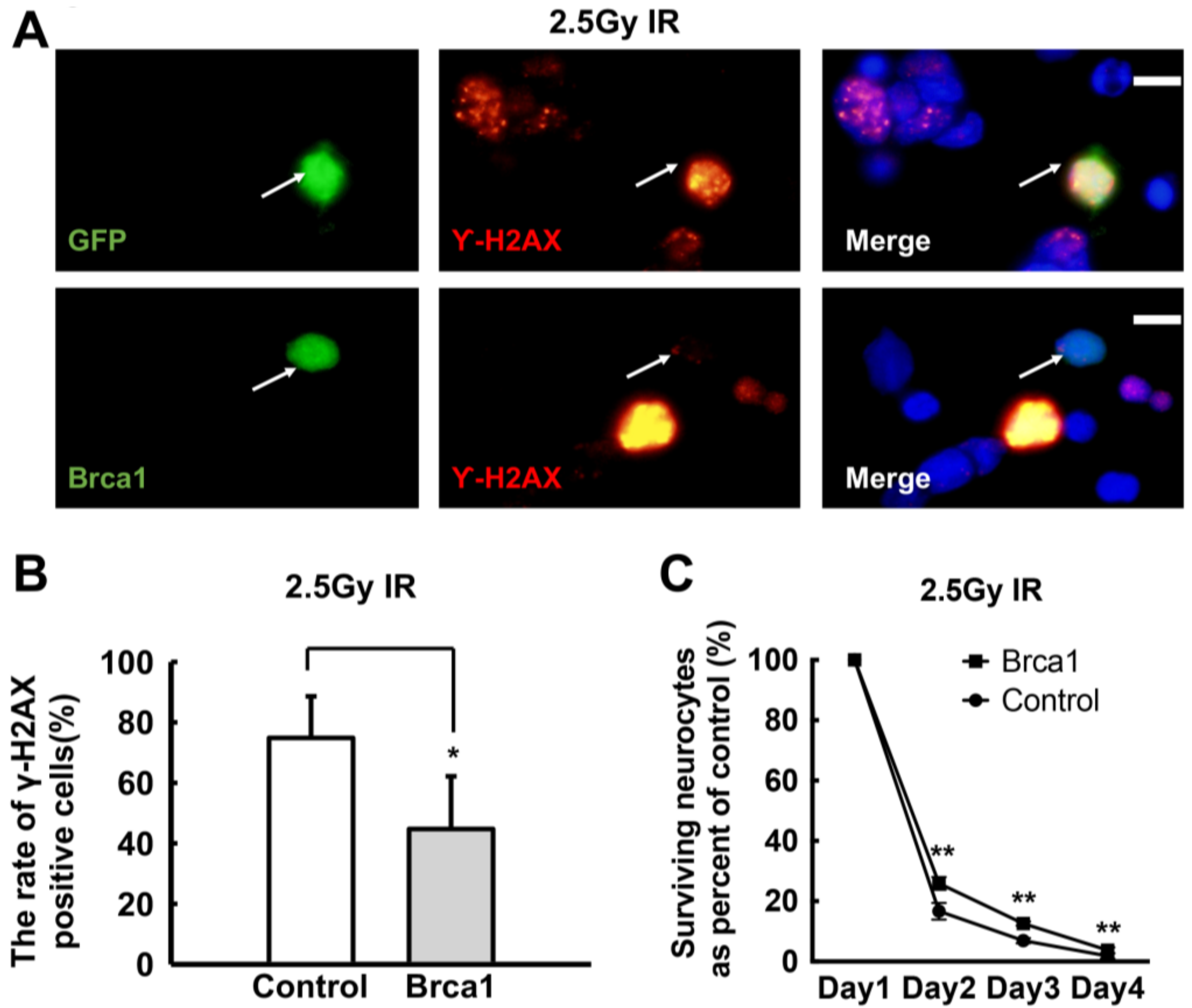
2.4. Transgenic Brca1 Promotes DNA Repair and Cell Survival of Retinal Neurons after IR Treatment
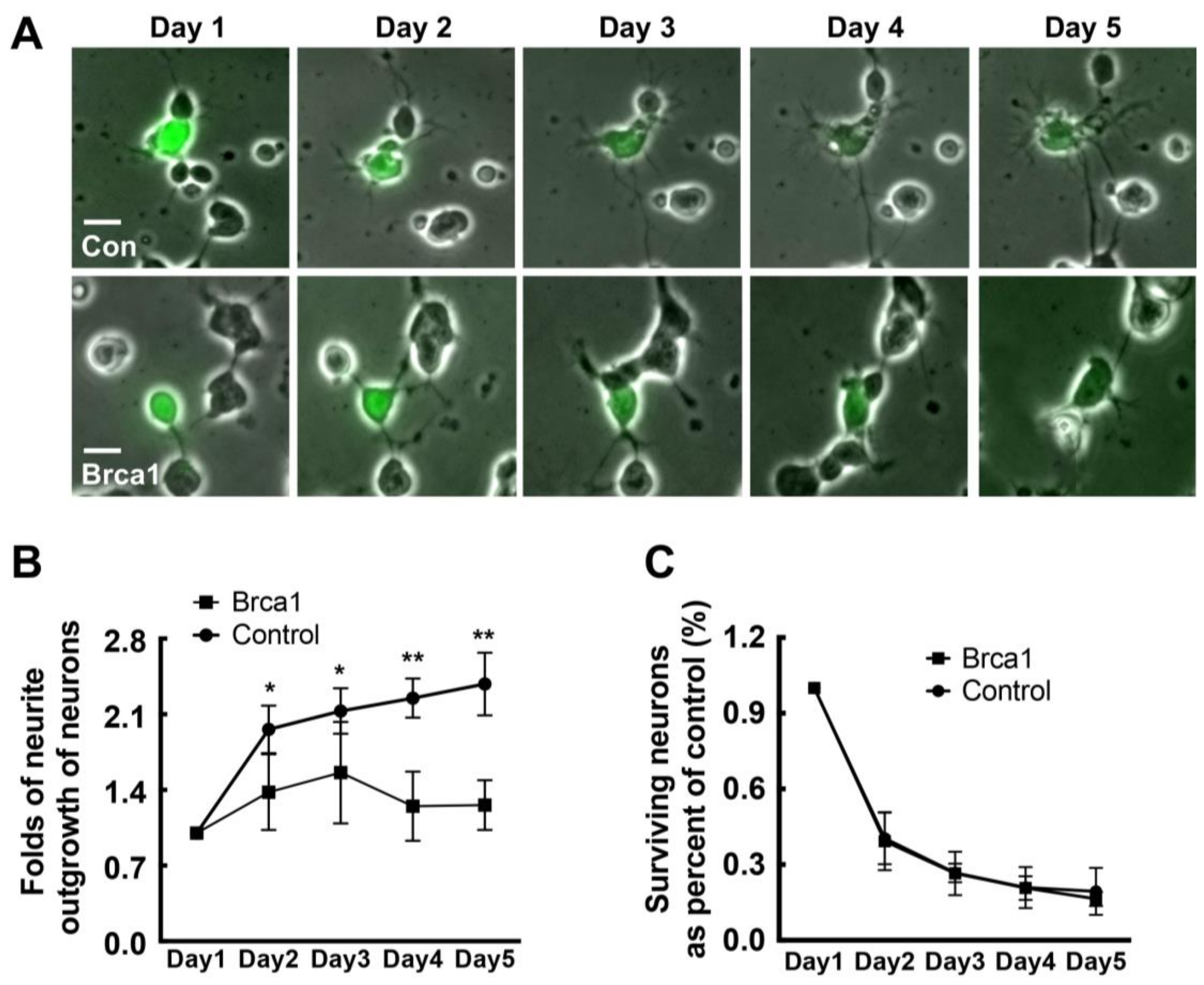
2.5. Transgenic Brca1 Inhibits Neurite Outgrowth in Retinal Neurons
3. Discussion
3.1. Expression Pattern of Brca1 in Retinal Development
3.2. Brca1 Promotes DNA Repair and Cell Viability of Retinal Neurons
3.3. Brca1 Silencing Might Promote Cell Maturation of Retinal Neurons
4. Materials and Methods
4.1. Ethics Statement
4.2. Immunohistochemistry
4.3. Primary Sprague-Dawley (SD) Rat Retinal Neurons Culture and Treatment
4.4. Real-Time PCR
4.5. Western Blot
4.6. Luciferase Activity Assays
4.7. Plasmid Construction and Transfection
4.8. Assay of Retinal Neurite Outgrowth, Cell Mortality, Cell Differentiation, Cell Proliferation, and DNA Stability
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| Brca1 | Breast cancer type 1 susceptibility protein |
| CNS | Central nervous system |
| NSLC | Neural stem-like cell |
| NHEJ | Non-homologues end joining |
| TSA | Trichostatin A |
| IR | Ionizing Radiation |
| PI | Propidium iodide |
| EdU | 5-ethynyl-2′-deoxyuridine |
| GCL | Ganglion cell layer |
References
- Miki, Y.; Swensen, J.; Shattuck-Eidens, D.; Futreal, P.A.; Harshman, K.; Tavtigian, S.; Liu, Q.; Cochran, C.; Bennett, L.M.; Ding, W.; et al. A strong candidate for the breast and ovarian cancer susceptibility gene brca1. Science 1994, 266, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Deng, C. Brca1: Cell cycle checkpoint, genetic instability, DNA damage response and cancer evolution. Nucleic Acids Res. 2006, 34, 1416–1426. [Google Scholar] [CrossRef] [PubMed]
- Yang, E.S.; Xia, F. Brca1 16 years later: DNA damage-induced brca1 shuttling. FEBS J. 2010, 277, 3079–3085. [Google Scholar] [CrossRef] [PubMed]
- Yun, M.H.; Hiom, K. Ctip-brca1 modulates the choice of DNA double-strand-break repair pathway throughout the cell cycle. Nature 2009, 459, 460–463. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, J.; Zhang, J.; Willers, H.; Wang, H.; Chung, J.H.; van Gent, D.C.; Hallahan, D.E.; Powell, S.N.; Xia, F. Checkpoint kinase 2-mediated phosphorylation of brca1 regulates the fidelity of nonhomologous end-joining. Cancer Res. 2006, 66, 1401–1408. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cortez, D.; Wang, Y.; Qin, J.; Elledge, S.J. Requirement of atm-dependent phosphorylation of Brca1 in the DNA damage response to double-strand breaks. Science 1999, 286, 1162–1166. [Google Scholar] [CrossRef] [PubMed]
- Penha, R.C.C.; Lima, S.C.S.; Boroni, M.; Ramalho-Oliveira, R.; Viola, J.P.; de Carvalho, D.P.; Fusco, A.; Pinto, L.F.R. Intrinsic line-1 hypomethylation and decreased brca1 expression are associated with DNA repair delay in irradiated thyroid cells. Radiat. Res. 2017, 188, 144–155. [Google Scholar] [CrossRef] [PubMed]
- Mullan, P.B.; Quinn, J.E.; Harkin, D.P. The role of brca1 in transcriptional regulation and cell cycle control. Oncogene 2006, 25, 5854–5863. [Google Scholar] [CrossRef] [PubMed]
- Korhonen, L.; Brännvall, K.; Skoglösa, Y.; Lindholm, D. Tumor suppressor gene Brca-1 is expressed by embryonic and adult neural stem cells and involved in cell proliferation. J. Neurosci. Res. 2003, 71, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Pulvers, J.N.; Huttner, W.B. Brca1 is required for embryonic development of the mouse cerebral cortex to normal size by preventing apoptosis of early neural progenitors. Development 2009, 136, 1859–1868. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Raff, M. Chromatin remodeling and histone modification in the conversion of oligodendrocyte precursors to neural stem cells. Genes Dev. 2004, 18, 2963–2972. [Google Scholar] [CrossRef] [PubMed]
- Keimpema, E.; Tortoriello, G.; Alpár, A.; Capsoni, S.; Arisi, I.; Calvigioni, D.; Hu, S.S.-J.; Cattaneo, A.; Doherty, P.; Mackie, K. Nerve growth factor scales endocannabinoid signaling by regulating monoacylglycerol lipase turnover in developing cholinergic neurons. Proc. Natl. Acad. Sci. USA 2013, 110, 1935–1940. [Google Scholar] [CrossRef] [PubMed]
- Mckinnon, P.J. Maintaining genome stability in the nervous system. Nat. Neurosci. 2013, 16, 1523–1529. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Pan, Y.; Kao, S.Y.; Li, C.; Kohane, I.S.; Chan, J.A.; Yankner, B.A. Gene regulation and DNA damage in the ageing human brain. Nature 2004, 429, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Rulten, S.L.; Caldecott, K.W. DNA strand break repair and neurodegeneration. DNA Repair 2013, 12, 558–567. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Hu, H.; Chen, Z.; Cai, X.; Zhang, Z.; Yang, Y.; Yu, N.; Zhang, J.; Xia, L.; Ge, J. Brca1 silencing is associated with failure of DNA repairing in retinal neurocytes. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Evans, T.A.; Raina, A.K.; Delacourte, A.; Aprelikova, O.; Lee, H.G.; Zhu, X.; Perry, G.; Smith, M.A. Brca1 may modulate neuronal cell cycle re-entry in alzheimer disease. Int. J. Med. Sci. 2007, 4, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Bromberg, K.D.; Maayan, A.; Neves, S.R.; Iyengar, R. Design logic of a cannabinoid receptor signaling network that triggers neurite outgrowth. Science 2008, 320, 903–909. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.A.; Sweatt, J.D. Covalent modification of DNA regulates memory formation. Neuron 2007, 53, 857–869. [Google Scholar] [CrossRef] [PubMed]
- Aldiri, I.; Xu, B.; Wang, L.; Chen, X.; Hiler, D.; Griffiths, L.; Valentine, M.; Shirinifard, A.; Thiagarajan, S.; Sablauer, A. The dynamic epigenetic landscape of the retina during development, reprogramming, and tumorigenesis. Neuron 2017, 94, 550–568. [Google Scholar] [CrossRef] [PubMed]
- Dapp, M.J.; Clouser, C.L.; Patterson, S.; Mansky, L.M. 5-azacytidine can induce lethal mutagenesis in human immunodeficiency virus type 1. J. Virol. 2009, 83, 11950–11958. [Google Scholar] [CrossRef] [PubMed]
- Diamantopoulos, P.T.; Michael, M.; Benopoulou, O.; Bazanis, E.; Tzeletas, G.; Meletis, J.; Vayopoulos, G.; Viniou, N. Antiretroviral activity of 5-azacytidine during treatment of a htlv-1 positive myelodysplastic syndrome with autoimmune manifestations. Virol. J. 2012, 9, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Komashko, V.M.; Farnham, P.J. 5-azacytidine treatment reorganizes genomic histone modification patterns. Epigenetics 2010, 5, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Pao, G.M.; Zhu, Q.; Perez-Garcia, C.G.; Chou, S.-J.; Suh, H.; Gage, F.H.; O’Leary, D.D.; Verma, I.M. Role of Brca1 in brain development. Proc. Natl. Acad. Sci. USA 2014, 111, E1240–E1248. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, A.M.; Miller-Pinsler, L.; Wells, P.G. Breast cancer 1 (Brca1)-deficient embryos develop normally but are more susceptible to ethanol-initiated DNA damage and embryopathies. Redox Biol. 2016, 7, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Brooks, P.; Marietta, C.; Goldman, D. DNA mismatch repair and DNA methylation in adult brain neurons. J. Neurosci. 1996, 16, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Holmes, E.H.; Liu, P.K. Oxidative damage to the c-fos gene and reduction of its transcription after focal cerebral ischemia. J. Neurochem. 1999, 73, 1164–1174. [Google Scholar] [CrossRef] [PubMed]
- Brooks, P. Detection of excision nuclease in cell-free extracts from the adult mammalian brain. Mutat. Res./DNA Repair 1998, 408, 37–46. [Google Scholar] [CrossRef]
- Liu, P.K.; Hsu, C.Y.; Dizdaroglu, M.; Floyd, R.A.; Kow, Y.W.; Karakaya, A.; Rabow, L.E.; Cui, J.-K. Damage, repair, and mutagenesis in nuclear genes after mouse forebrain ischemia-reperfusion. J. Neurosci. 1996, 16, 6795–6806. [Google Scholar] [CrossRef] [PubMed]
- Di, L.-J.; Fernandez, A.G.; De Siervi, A.; Longo, D.L.; Gardner, K. Transcriptional regulation of Brca1 expression by a metabolic switch. Nat. Struct. Mol. Biol. 2010, 17, 1406. [Google Scholar] [CrossRef] [PubMed]
- Jenke, A.C.; Stehle, I.M.; Herrmann, F.; Eisenberger, T.; Baiker, A.; Bode, J.; Fackelmayer, F.O.; Lipps, H.J. Nuclear scaffold/matrix attached region modules linked to a transcription unit are sufficient for replication and maintenance of a mammalian episome. Proc. Natl. Acad. Sci. USA 2004, 101, 11322–11327. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Q.; Xu, L.; Chen, P.; Xu, Z.; Qiu, J.; Ge, J.; Yu, K.; Zhuang, J. Brca1 Is Upregulated by 5-Aza-CdR and Promotes DNA Repair and Cell Survival, and Inhibits Neurite Outgrowth in Rat Retinal Neurons. Int. J. Mol. Sci. 2018, 19, 1214. https://doi.org/10.3390/ijms19041214
Wang Q, Xu L, Chen P, Xu Z, Qiu J, Ge J, Yu K, Zhuang J. Brca1 Is Upregulated by 5-Aza-CdR and Promotes DNA Repair and Cell Survival, and Inhibits Neurite Outgrowth in Rat Retinal Neurons. International Journal of Molecular Sciences. 2018; 19(4):1214. https://doi.org/10.3390/ijms19041214
Chicago/Turabian StyleWang, Qiyun, Lijun Xu, Pei Chen, Zhuojun Xu, Jin Qiu, Jian Ge, Keming Yu, and Jing Zhuang. 2018. "Brca1 Is Upregulated by 5-Aza-CdR and Promotes DNA Repair and Cell Survival, and Inhibits Neurite Outgrowth in Rat Retinal Neurons" International Journal of Molecular Sciences 19, no. 4: 1214. https://doi.org/10.3390/ijms19041214
APA StyleWang, Q., Xu, L., Chen, P., Xu, Z., Qiu, J., Ge, J., Yu, K., & Zhuang, J. (2018). Brca1 Is Upregulated by 5-Aza-CdR and Promotes DNA Repair and Cell Survival, and Inhibits Neurite Outgrowth in Rat Retinal Neurons. International Journal of Molecular Sciences, 19(4), 1214. https://doi.org/10.3390/ijms19041214