How to Process Sputum Samples and Extract Bacterial DNA for Microbiota Analysis

 , , , , and
, , , , and 
Abstract
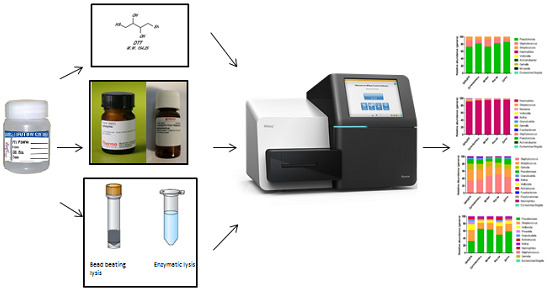
1. Introduction
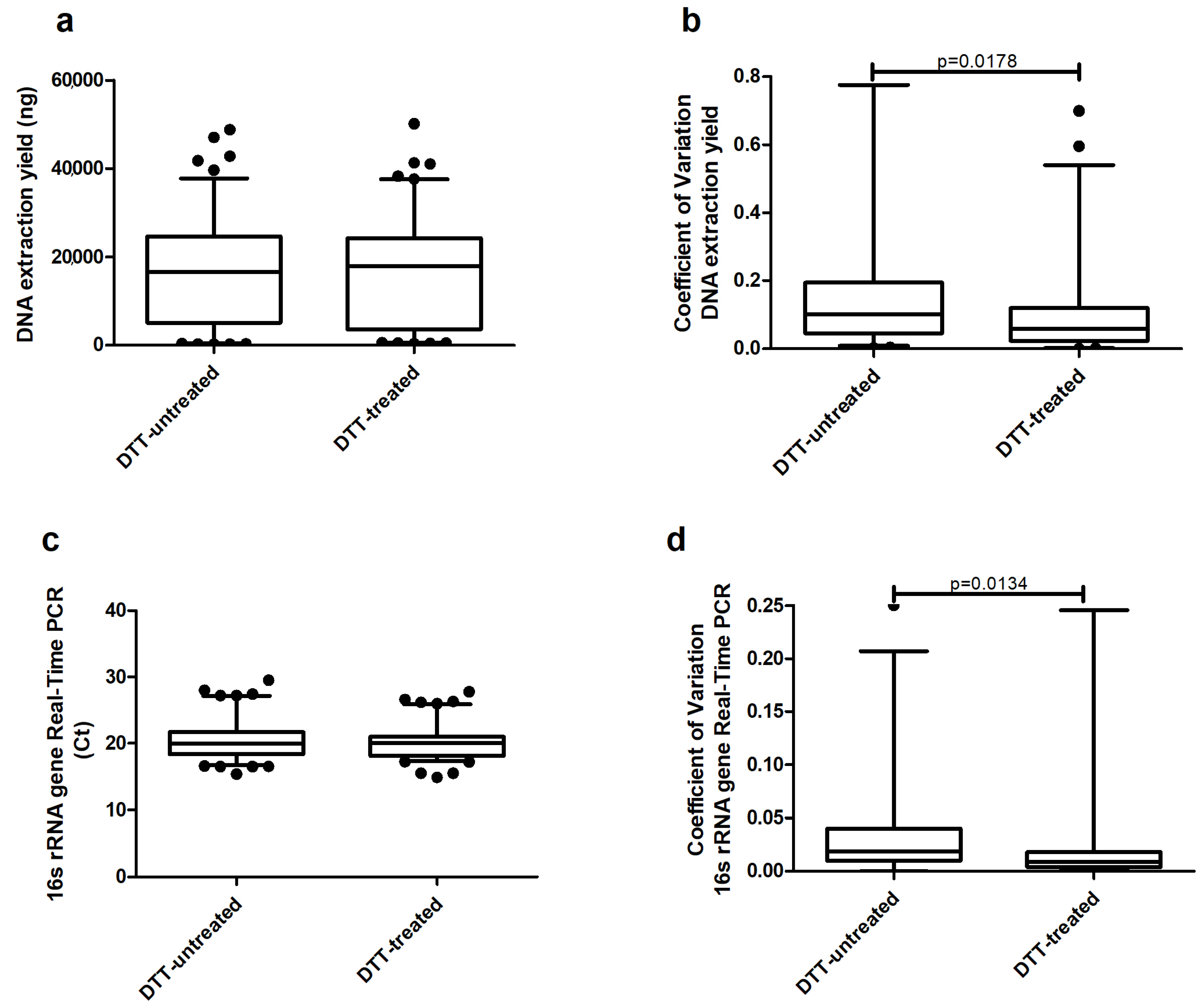
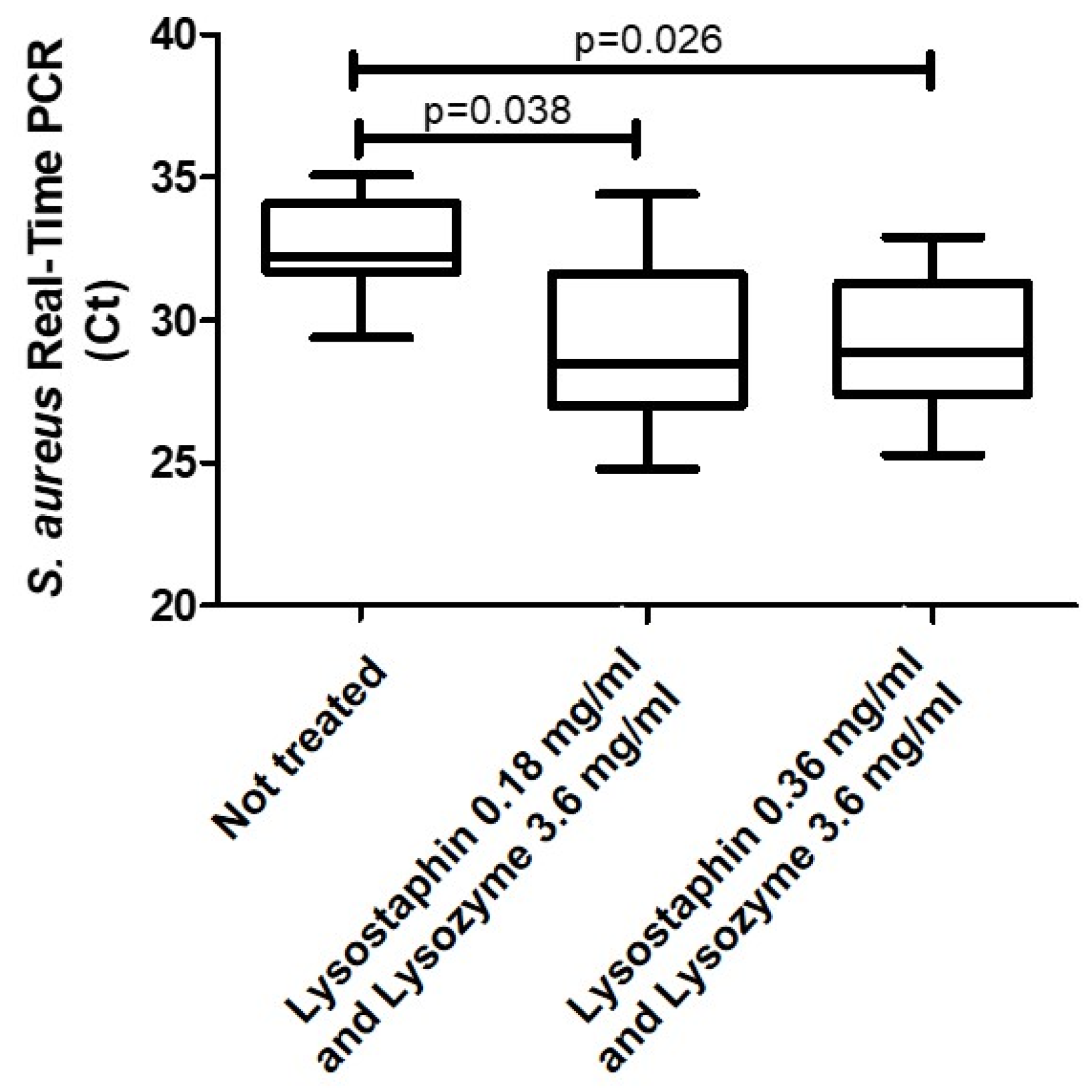
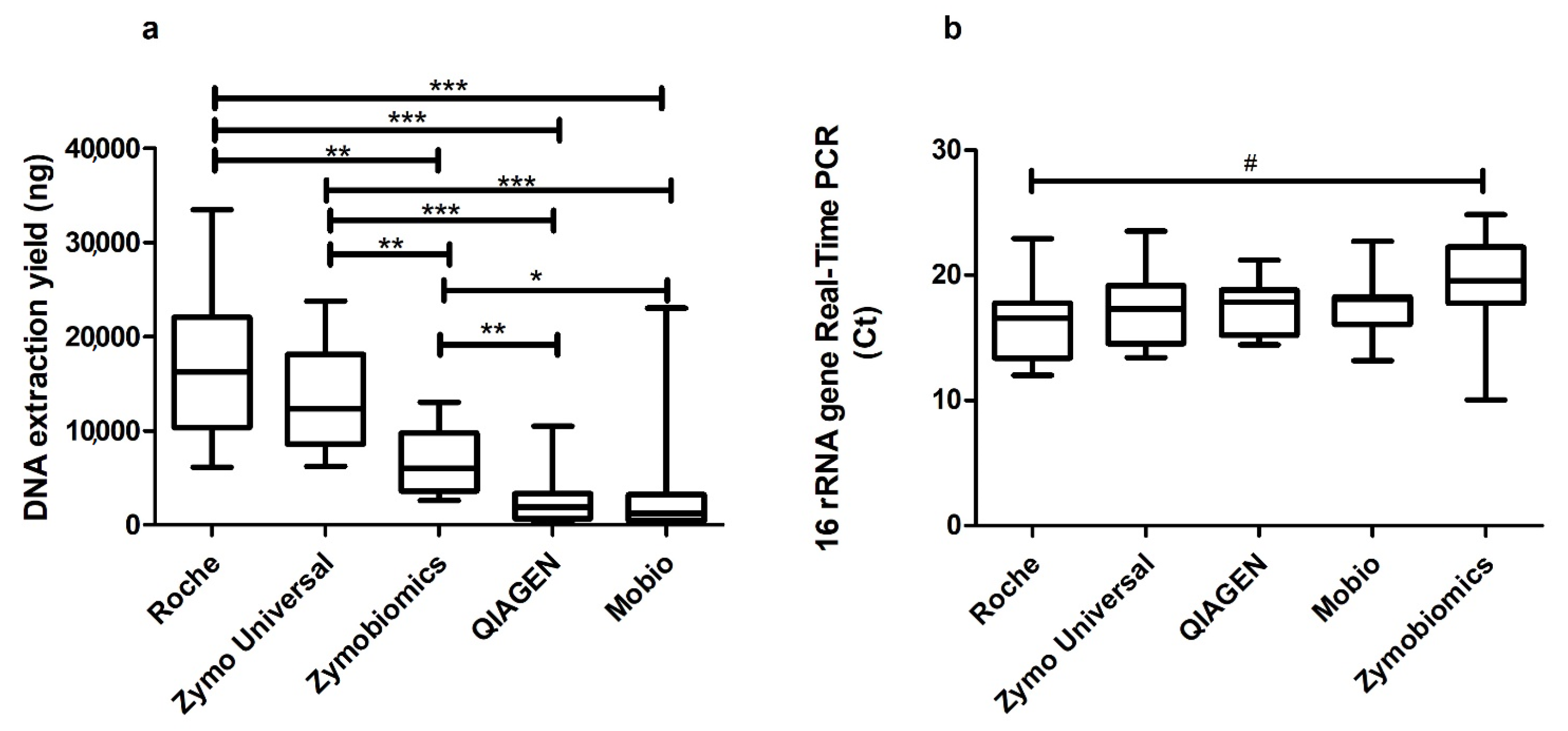
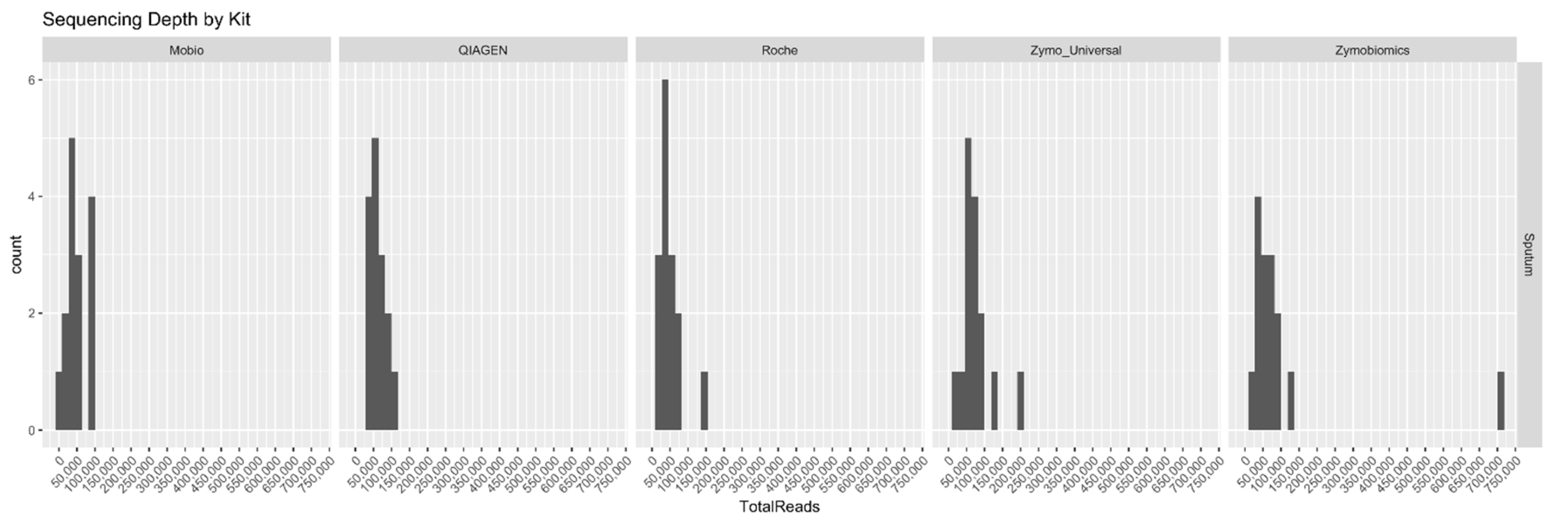
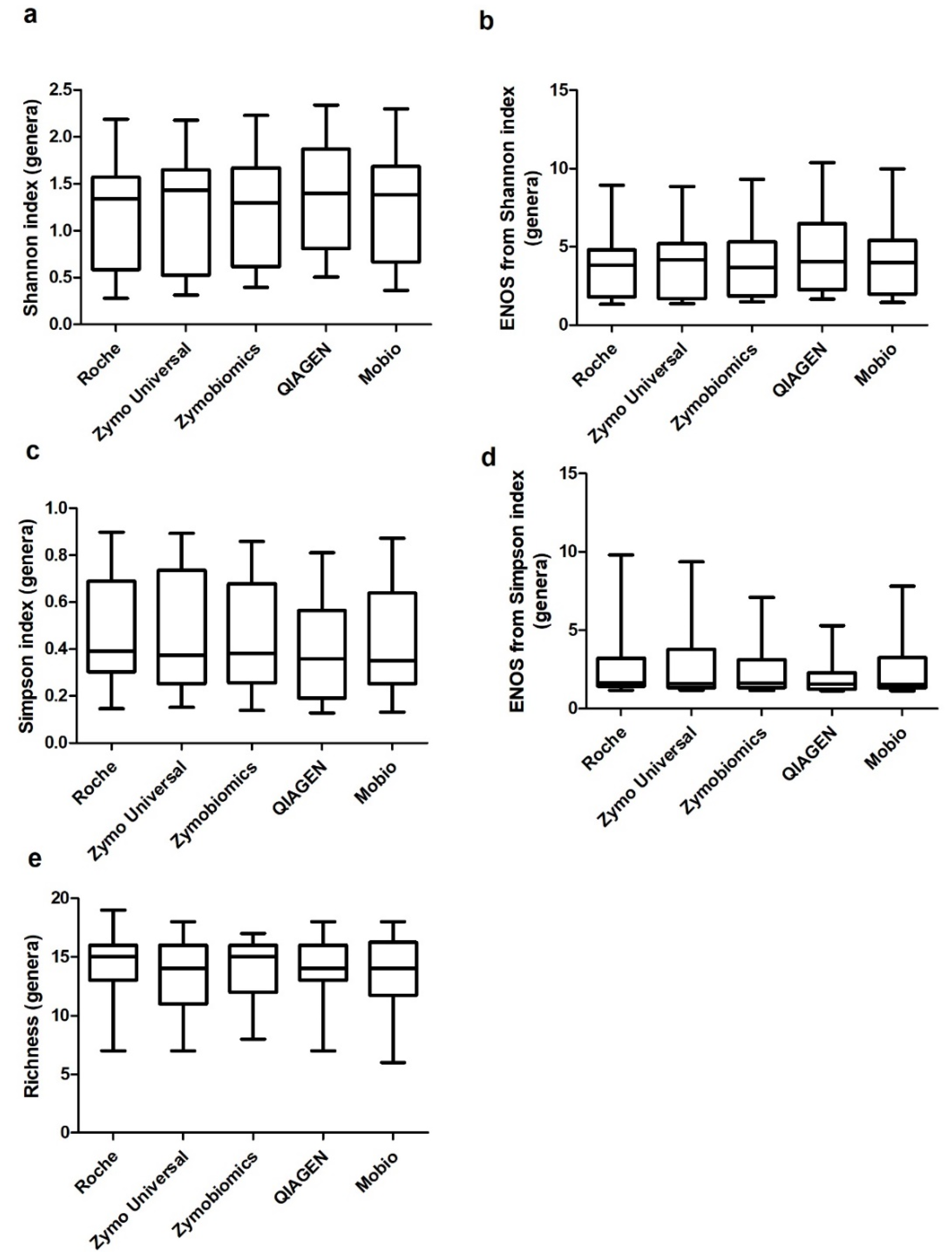
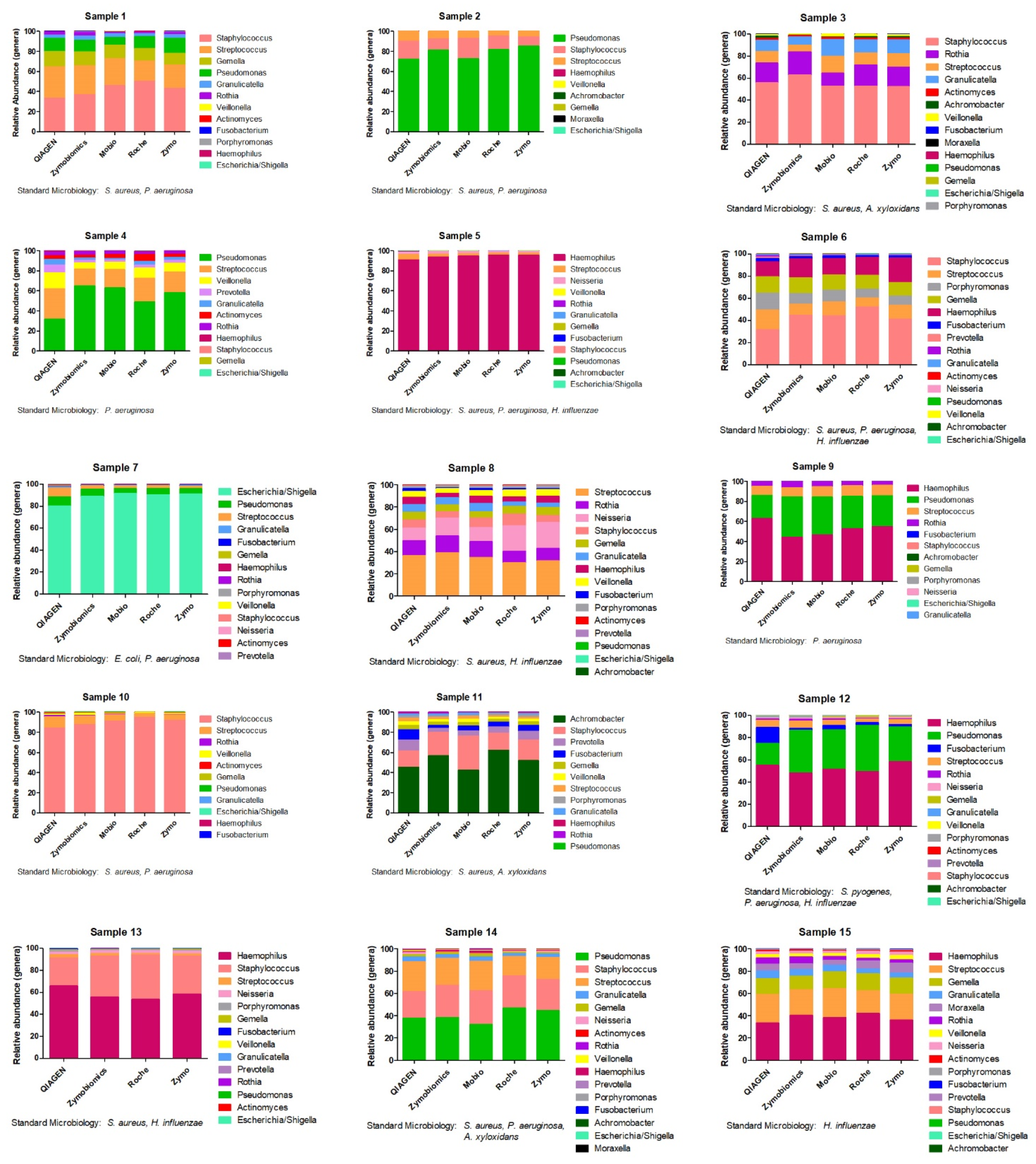
2. Results
3. Discussion
4. Materials and Methods
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Rogers, G.B.; Shaw, D.; Marsh, R.L.; Carroll, M.P.; Serisier, D.J.; Bruce, K.D. Respiratory microbiota: Addressing clinical questions, informing clinical practice. Thorax 2015, 70, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Muhlebach, M.S.; Zorn, B.T.; Esther, C.R.; Hatch, J.E.; Murray, C.P.; Turkovic, L.; Ranganathan, S.C.; Boucher, R.C.; Stick, S.M.; Wolfgang, M.C. Initial acquisition and succession of the cystic fibrosis lung microbiome is associated with disease progression in infants and preschool children. PLoS Pathog. 2018, 14, e1006798. [Google Scholar] [CrossRef] [PubMed]
- Ubags, N.D.J.; Marsland, B.J. Mechanistic insight into the function of the microbiome in lung diseases. Eur. Respir. J. 2017, 50, 1602467. [Google Scholar] [CrossRef] [PubMed]
- Tunney, M.M.; Einarsson, G.G.; Wei, L.; Drain, M.; Klem, E.R.; Cardwell, C.; Ennis, M.; Boucher, R.C.; Wolfgang, M.C.; Elborn, S.E. Lung microbiota and bacterial abundance in patients with bronchiectasis when clinically stable and during exacerbation. Am. J. Respir. Crit. Care Med. 2013, 187, 1118–1126. [Google Scholar] [CrossRef] [PubMed]
- Williamson, K.M.; Wagner, B.D.; Robertson, C.E.; Johnson, E.J.; Zemanick, E.T.; Kirk Harris, J. Impact of enzymatic digestion on bacterial community composition in CF airway samples. PeerJ 2017, 5, e3362. [Google Scholar] [CrossRef] [PubMed]
- Pragman, A.A.; Lyu, T.; Baller, J.A.; Gould, T.J.; Kelly, R.F.; Reilly, C.S.; Isaacson, R.E.; Wendt, C.H. The lung tissue microbiota of mild and moderate chronic obstructive pulmonary disease. Microbiome 2018, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- Bacci, G.; Paganin, P.; Lopez, L.; Vanni, C.; Dalmastri, C.; Daddiego, L.; Perrotta, G.; Dolce, D.; Morelli, P.; Tuccio, V.; et al. Pyrosequencing Unveils Cystic Fibrosis Lung Microbiome Differences Associated with a Severe Lung Function Decline. PLoS ONE 2016, 11, e0156807. [Google Scholar] [CrossRef]
- Parkins, M.D.; Floto, R.A. Emerging bacterial pathogens and changing concepts of bacterial pathogenesis in cystic fibrosis. J. Cyst. Fibros. 2015, 14, 293–304. [Google Scholar] [CrossRef] [PubMed]
- King, P.T.; Holdsworth, S.R.; Freezer, N.J.; Villanueva, E.; Holmes, P.W. Microbiologic follow-up study in adult bronchiectasis. Respir. Med. 2007, 101, 1633–1638. [Google Scholar] [CrossRef] [PubMed]
- Hogan, D.A.; Willger, S.D.; Dolben, E.L.; Hampton, T.H.; Stanton, B.A.; Morrison, H.G.; Sogin, M.L.; Czum, J.; Ashare, A. Analysis of lung microbiota in bronchoalveolar lavage, protected brush and sputum samples from subjects with mild-to- moderate cystic fibrosis lung disease. PLoS ONE 2016, 11, e0149998. [Google Scholar] [CrossRef] [PubMed]
- Lim, M.Y.; Song, E.J.; Kim, S.H.; Lee, J.; Nam, Y.D. Comparison of DNA extraction methods for human gut microbial community profiling. Syst. Appl. Microbiol. 2018, 41, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Xiao, F.; Wang, C.; Wang, Z. The impact of different methods of DNA extraction on microbial community measures of BALF samples based on metagenomic data. Am. J. Transl. Res. 2016, 8, 1412–1425. [Google Scholar] [PubMed]
- Oriano, M.; Terranova, L.; Ruggiero, L.; Tafuro, C.; Teri, A.; Franceschi, E.; Amati, F.; Gramegna, A.; Contarini, M.; Cariani, L.; et al. What Is the Best Technique to Extract Bacterial DNA from Sputum? ERS International Congress: Paris, France, 2018. [Google Scholar]
- Dicker, A.J.; Crichton, M.L.; Pumphrey, E.G.; Cassidy, A.J.; Suarez-Cuartin, G.; Sibila, O.; Furrie, E.; Fong, C.J.; Ibrahim, W.; Brady, G.; et al. Neutrophil extracellular traps are associated with disease severity and microbiota diversity in patients with chronic obstructive pulmonary disease. J. Allergy. Clin. Immunol. 2018, 141, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Denton, M.; Doherty, C.; Foweraker, J.; Govan, J.; Hall, M.; Isalska, B.; Jones, A.; Kenna, D.; Larsen, A.; Pitt, T.; et al. Laboratory standards for processing microbiological samples from people with cystic fibrosis. In Cystic Fibrosis Trust; Report of the UK Cystic Fibrosis Trust Microbiology Laboratory Standards Working Group: Kent, UK, 2010. [Google Scholar]
- Zhao, J.; Carmody, L.A.; Kalikin, L.M.; Li, J.; Petrosino, J.F.; Schloss, P.D.; Young, V.B.; LiPuma, J.J. Impact of Enhanced Staphylococcus DNA extraction on microbial community measures in cystic fibrosis sputum. PLoS ONE 2012, 7, e33127. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Lin, P.; Li, Q.; Han, L.; Zheng, H.; Wei, Y.; Cui, Z.; Ni, Y.; Guo, X. Analysis of the microbiota of sputum samples from patients with lower respiratory tract infections. Acta Biochim. Biophys. Sin. 2010, 42, 754–761. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Shen, T.; Tian, F.; Lin, P.; Li, Q.; Cui, Z.; Zhang, Y.; Xue, M.; Ye, J.; Guo, X.; et al. New microbiota found in sputum from patients with community-acquired pneumonia. Acta Biochim. Biophys. Sin. 2013, 45, 1039–1048. [Google Scholar] [CrossRef] [PubMed]
- Dickson, R.P.; Erb-Downward, J.R.; Prescott, H.C.; Martinez, F.J.; Curtis, J.L.; Lama, V.N.; Huffnagle, G.B. Cell-associated bacteria in the human lung microbiome. Microbiome 2014, 2, 28. [Google Scholar] [CrossRef] [PubMed]
- Rogers, G.B.; van der Gast, C.J.; Cuthbertson, L.; Thomson, S.K.; Bruce, K.D.; Martin, M.L.; Serisier, D.J. Clinical measures of disease in adult non-CF bronchiectasis correlate with airway microbiota composition. Thorax 2013, 68, 731–737. [Google Scholar] [CrossRef] [PubMed]
- Cox, M.J.; Turek, E.M.; Hennessy, C.; Mirza, G.; James, P.L.; Coleman, M.; Jones, A.; Wilson, R.; Bilton, D.; Cookson, W.O.C.; et al. Longitudinal assessment of sputum microbiome by sequencing of the 16S rRNA gene in non-cystic fibrosis bronchiectasis patients. PLoS ONE 2017, 12, e0170622. [Google Scholar] [CrossRef] [PubMed]
- Rogers, G.B.; Zain, N.M.M.; Bruce, K.D.; Burr, L.D.; Chen, A.C.; Rivett, D.W.; McGuckin, M.A.; Serisier, D.J. A novel microbiota stratification system predicts future exacerbations in bronchiectasis. Ann. Am. Thorac. Soc. 2014, 11, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Purcell, P.; Jary, H.; Perry, A.; Perry, J.D.; Stewart, C.J.; Nelson, A.; Lanyon, C.; Smith, D.L.; Cumming, S.P.; De Soyza, A. Polymicrobial airway bacterial communities in adult bronchiectasis patients. BMC Microbiol. 2014, 14, 130. [Google Scholar] [CrossRef] [PubMed]
- Farrell, P.M.; White, T.B.; Ren, C.; Hempstead, S.E.; Accurso, F.; Derichs, N.; Howenstine, M.; McColley, S.A.; Rock, M.; Rosenfeld, M.; et al. Diagnosis of Cystic Fibrosis: Consensus Guidelines from the Cystic Fibrosis Foundation. J. Pediatr. 2018, 181, S4–S15.e1. [Google Scholar] [CrossRef] [PubMed]
- Vega, M.F.; Dieguez, S.N.; Riccio, B.; Aranguren, S.; Giordano, A.; Denzoin, L.; Soraci, A.L.; Tapia, M.O.; Ross, R.; Apás, A.; et al. Zearalenone adsorption capacity of lactic acid bacteria isolated from pigs. Braz. J. Microbiol. 2017, 48, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Gadsby, N.J.; McHugh, M.P.; Russell, C.D.; Mark, H.; Morris, A.C.; Laurenson, I.F.; Hill, A.T.; Templeton, K.E. Development of two real-time multiplex PCR assays for the detection and quantification of eight key bacterial pathogens in lower respiratory tract infections. Clin. Microbiol. Infect. 2015, 21, 788.e1–788.e13. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 5 February 2018).
- Ewels, P.; Ankenbrand, M.J.; Hammarén, R.; Saveliev, V.; Booth, T.; Jones, A.; Neumann, T.; Magnusson, M.; Alexanderscholz; Andeer, R.; et al. MultiQC Version 1.4. 2016. Available online: https://zenodo.org/record/1145834#.W2FsDi1aZBw (accessed on 5 February 2018).
- Edgar, R.C. SINTAX: A Simple Non-BAYESIAN Taxonomy Classifier for 16S and ITS Sequences. bioRxiv. 2016. Available online: https://www.biorxiv.org/content/early/2016/09/09/074161 (accessed on 4 July 2017).
- Chao, A.; Chiu, C.-H.; Jost, L. Phylogenetic diversity measures based on Hill numbers. Philos. Trans. R Soc. B Biol. Sci. 2010, 365, 3599–3609. [Google Scholar] [CrossRef] [PubMed]
- Feigelman, R.; Kahlert, C.R.; Baty, F.; Rassouli, F.; Kleiner, R.L.; Kohler, P.; Brutsche, M.H.; von Mering, C. Sputum DNA sequencing in cystic fibrosis: Non-invasive access to the lung microbiome and to pathogen details. Microbiome 2017, 5, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Blow, F.; Vontas, J.; Darby, A.C. Draft Genome Sequence of Chryseobacterium Strain CBo1 Isolated from Bactrocera oleae. Genome Announc. 2017, 5, e00177-17. [Google Scholar] [CrossRef] [PubMed]
- Willner, D.; Daly, J.; Whiley, D.; Grimwood, K.; Wainwright, C.E.; Hugenholtz, P. Comparison of DNA extraction methods for microbial community profiling with an application to pediatric bronchoalveolar lavage samples. PLoS ONE 2012, 7, e34605. [Google Scholar] [CrossRef] [PubMed]
- Hart, M.L.; Meyer, A.; Johnson, P.J.; Ericsson, A.C. Comparative Evaluation of DNA Extraction Methods from Feces of Multiple Host Species for Downstream Next-Generation Sequencing. PLoS ONE 2015, 10, e0143334. [Google Scholar] [CrossRef] [PubMed]
- Sohrabi, M.; Nair, R.G.; Samaranayake, L.P.; Zhang, L.; Zulfiker, A.H.M.; Ahmetagic, A.; Good, D.; Wei, M.Q. The yield and quality of cellular and bacterial DNA extracts from human oral rinse samples are variably affected by the cell lysis methodology. J. Microbiol. Methods 2016, 122, 64–72. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Basis of Function | Cost per Sample | Time per Sample (min) | Advantages | Disadvantages | Reference |
|---|---|---|---|---|---|---|
| Roche High Pure PCR Template Preparation Kit | Lysis Buffer and proteinase K digestion | 2.62 € | 16 | Easy to use and quick protocol. | Milder extraction method than mechanical | Feigelman et al. 2017 [34] |
| Zymo Quick-DNA Universal Kit | Lysis Buffer and proteinase K digestion | 2.49 € | 18 | Easy to use and quick protocol | Milder extraction method than mechanical | Blow et al. 2017 [35] |
| MoBio PowerLyzer PowerSoil DNA isolation kit | Bead beating | 6.16 € | 41 | Possibility to lyse hard-to-lyse bacteria | Time consuming, possibility of DNA loss because of high number of steps | Willner et al. 2012 [36] |
| QIAGEN QIAmp Cador Pathogen Mini kit | Bead beating and proteinase K digestion | 4.6 € | 20 | Possibility to lyse hard-to-lyse bacteria. Possibility to combine different methods. | Possibility of DNA loss | Hart et al. 2015 [37] |
| ZymoBIOMICS DNA Miniprep Kit | Bead beating and proteinase K digestion | 5.12 € | 63 | Combination of enzymatic and mechanical distruption. Option to use different protocols | Time consuming, possibility of DNA loss because of high number of steps | Sohrabi et al. 2016 [38] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Terranova, L.; Oriano, M.; Teri, A.; Ruggiero, L.; Tafuro, C.; Marchisio, P.; Gramegna, A.; Contarini, M.; Franceschi, E.; Sottotetti, S.; et al. How to Process Sputum Samples and Extract Bacterial DNA for Microbiota Analysis. Int. J. Mol. Sci. 2018, 19, 3256. https://doi.org/10.3390/ijms19103256
Terranova L, Oriano M, Teri A, Ruggiero L, Tafuro C, Marchisio P, Gramegna A, Contarini M, Franceschi E, Sottotetti S, et al. How to Process Sputum Samples and Extract Bacterial DNA for Microbiota Analysis. International Journal of Molecular Sciences. 2018; 19(10):3256. https://doi.org/10.3390/ijms19103256
Chicago/Turabian StyleTerranova, Leonardo, Martina Oriano, Antonio Teri, Luca Ruggiero, Camilla Tafuro, Paola Marchisio, Andrea Gramegna, Martina Contarini, Elisa Franceschi, Samantha Sottotetti, and et al. 2018. "How to Process Sputum Samples and Extract Bacterial DNA for Microbiota Analysis" International Journal of Molecular Sciences 19, no. 10: 3256. https://doi.org/10.3390/ijms19103256
APA StyleTerranova, L., Oriano, M., Teri, A., Ruggiero, L., Tafuro, C., Marchisio, P., Gramegna, A., Contarini, M., Franceschi, E., Sottotetti, S., Cariani, L., Bevivino, A., Chalmers, J. D., Aliberti, S., & Blasi, F. (2018). How to Process Sputum Samples and Extract Bacterial DNA for Microbiota Analysis. International Journal of Molecular Sciences, 19(10), 3256. https://doi.org/10.3390/ijms19103256