Pomalidomide Ameliorates H2O2-Induced Oxidative Stress Injury and Cell Death in Rat Primary Cortical Neuronal Cultures by Inducing Anti-Oxidative and Anti-Apoptosis Effects

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
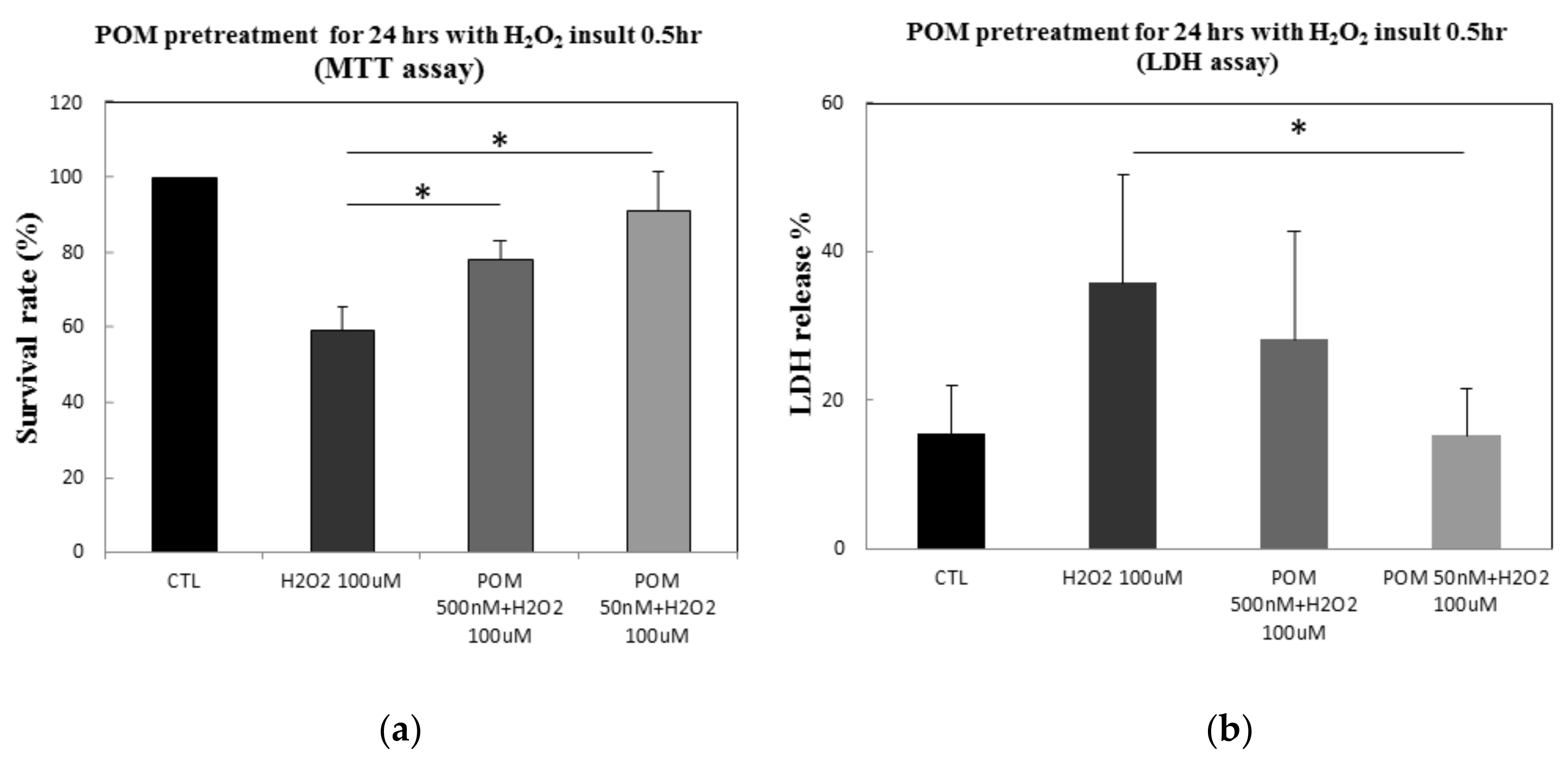
2.1. Protective Effects of POM Against H2O2-Induced Cell Damage in Primary Cultures of Rat Cortical Neurons
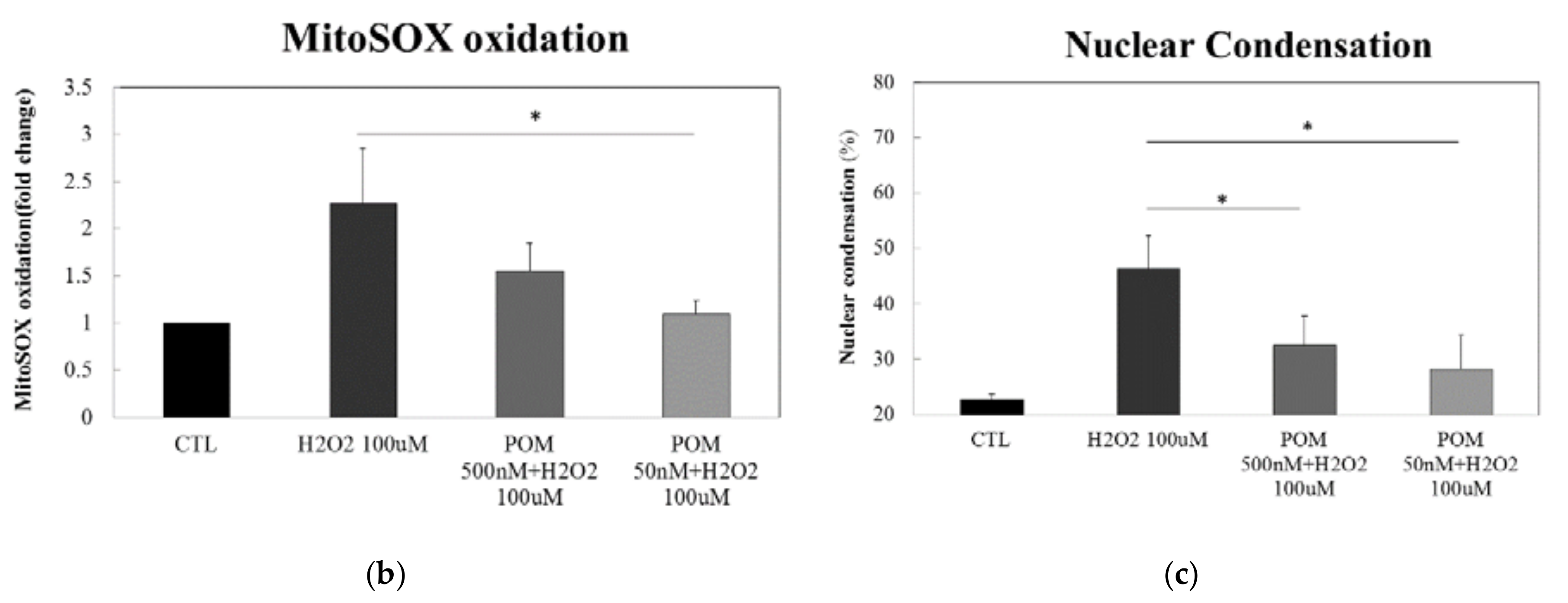
2.2. Pomalidomide Protects against H2O2-Induced Mitochondrial Superoxide Production and H2O2-Induced Cellular Apoptosis
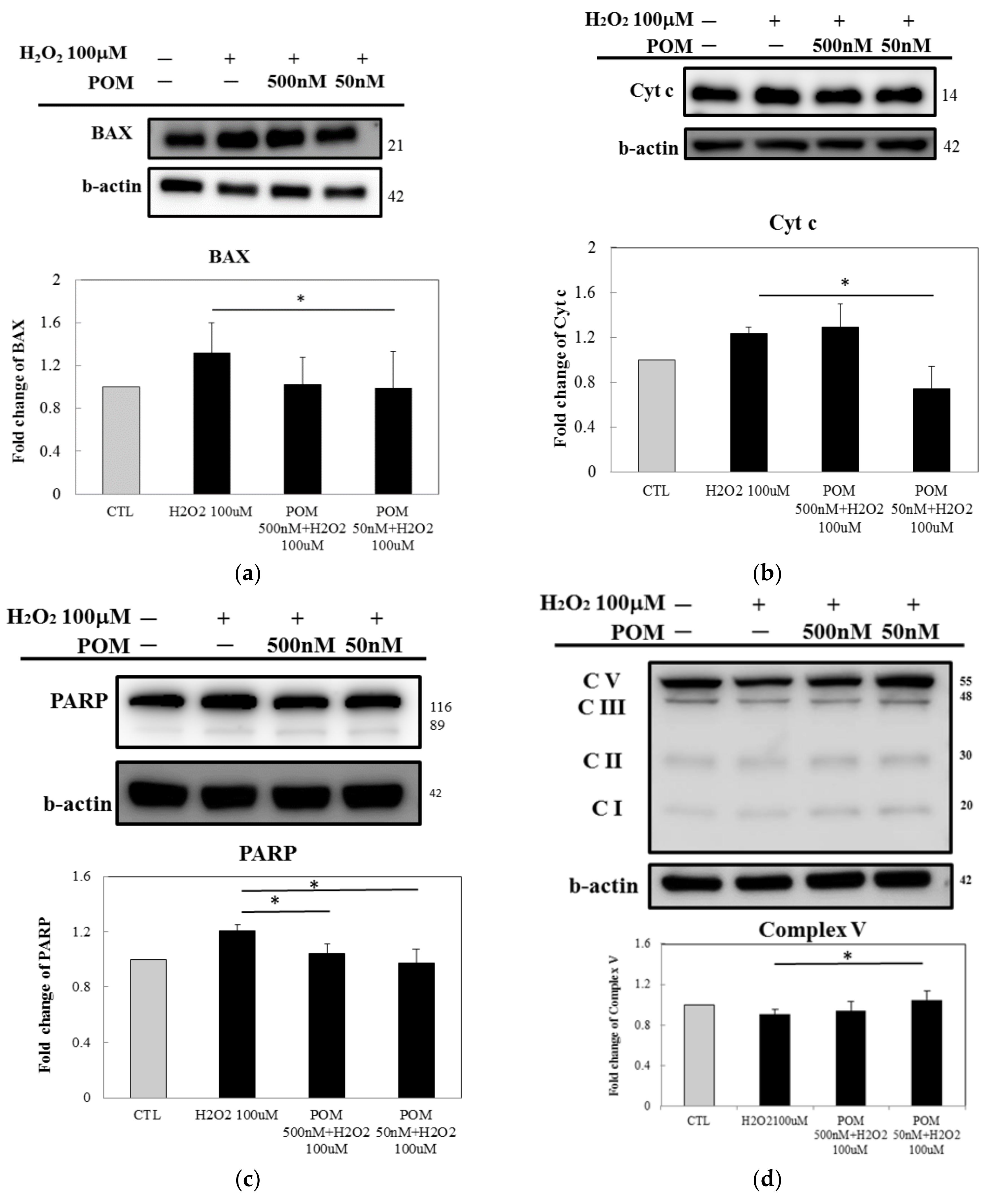
2.3. Protein Expression of ROS Defense System Enzymes and Anti-Inflammation Responses
2.4. Pomalidomide Antagonized the Cytochrome c-Mediated Apoptotic Signaling Pathway After H2O2-Induced Cellular Death and Mitochondrial Function
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Cell Cultures
4.2.1. Hydrogen Peroxide-Induced Injury and Pomalidomide Pretreatment
4.2.2. Cell Viability
4.3. The H2O2 Production Assay
4.4. Quantification of ATP
4.5. Western Blot Analysis
4.6. Determination of Cellular Reactive Oxygen Species (ROS) Production
4.7. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Collins, T.R. Neurologic Diseases Found to Be the Largest Cause of Disability Worldwide. Neurol. Today 2017, 17, 132–135. [Google Scholar] [CrossRef]
- World Health Organization. The Top 10 Causes of Death. Available online: http://www.who.int/mediacentre/factsheets/fs310/en/ (accessed on 12 September 2018).
- Santos, C.Y.; Snyder, P.J.; Wu, W.C.; Zhang, M.; Echeverria, A.; Alber, J. Pathophysiologic relationship between Alzheimer’s disease, cerebrovascular disease and cardiovascular risk: A review and synthesis. Alzheimers Dement. 2017, 7, 69–87. [Google Scholar] [CrossRef] [PubMed]
- Kalaria, R.N.; Bhatti, S.U.; Lust, W.D.; Perry, G. The amyloid precursor protein in ischemic brain injury and chronic hypoperfusion. Ann. N. Y. Acad. Sci. 1993, 695, 190–193. [Google Scholar] [CrossRef] [PubMed]
- Kalaria, R.N. The role of cerebral ischemia in Alzheimer’s disease. Neurobiol. Aging 2000, 21, 321–330. [Google Scholar] [CrossRef]
- Kalaria, R.N. The pathology and pathophysiology of vascular dementia. Neuropharmacology 2018, 134, 226–239. [Google Scholar] [CrossRef] [PubMed]
- Doyle, K.P.; Simon, R.P.; Stenzel-Poore, M.P. Mechanisms of ischemic brain damage. Neuropharmacology 2008, 55, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Rosamond, W.; Flegal, K.; Friday, G.; Furie, K.; Go, A.; Greenlund, K.; Haase, N.; Ho, M.; Howard, V.; Kissela, B.; et al. Heart Disease and Stroke Statistics—2007 Update. Circulation 2007. [Google Scholar] [CrossRef] [PubMed]
- Neuhaus, A.A.; Couch, Y.; Hadley, G.; Buchan, A.M. Neuroprotection in stroke: The importance of collaboration and reproducibility. Brain 2017, 140, 2079–2092. [Google Scholar] [CrossRef] [PubMed]
- Chamorro, Á.; Dirnagl, U.; Urra, X.; Planas, A.M. Neuroprotection in acute stroke: Targeting excitotoxicity, oxidative and nitrosative stress and inflammation. Lancet Neurol. 2016, 15, 869–881. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; Khoury, J.E.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Clark, I.A.; Vissel, B. Therapeutic implications of how TNF links apolipoprotein E, phosphorylated tau, α-synuclein, amyloid-β and insulin resistance in neurodegenerative diseases. Br. J. Pharmacol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Kalogeris, T.; Bao, Y.; Korthuis, R.J. Mitochondrial reactive oxygen species: A double edged sword in ischemia/reperfusion vs. preconditioning. Redox Biol. 2014, 2, 702–714. [Google Scholar] [CrossRef] [PubMed]
- Brouns, R.; De Deyn, P.P. The complexity of neurobiological processes in acute ischemic stroke. Clin. Neurol. Neurosurg. 2009, 111, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Beckman, J.S.; Koppenol, W.H. Nitric oxide, superoxide and peroxynitrite: The good, the bad and ugly. Am. J. Physiol. 1996, 271, 1424–1437. [Google Scholar] [CrossRef] [PubMed]
- Lennon, S.V.; Martin, S.J.; Cotter, T.G. Dose-dependent induction of apoptosis in human tumour cell lines by widely diverging stimuli. Cell Prolif. 1991, 24, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.D.; Cooke, M.S. Factors contributing to the outcome of oxidative damage to nucleic acids. Bioessays 2004, 26, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.K.; Hsu, C.Y.; Dizdaroglu, M.; Floyd, R.A.; Kow, Y.W.; Karakaya, A.; Rabow, L.E.; Cui, J.K. Damage, repair and mutagenesis in nuclear genes after mouse forebrain ischemia-reperfusion. J. Neurosci. 1996, 16, 6795–6806. [Google Scholar] [CrossRef] [PubMed]
- Endres, M.; Wang, Z.Q.; Namura, S.; Waeber, C.; Moskowitz, M.A. Ischemic brain injury is mediated by the activation of poly(ADP-ribose)polymerase. J. Cereb. Blood Flow Metab. 1997, 17, 1143–1151. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. Committee for Medicinal Products for Human Use, Assessment Report: Pomalidomide Celgene. Available online: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002682/WC500147721.pdf (accessed on 12 September 2018).
- Chanan-Khan, A.A.; Swaika, A.; Paulus, A.; Kumar, S.K.; Mikhael, J.R.; Rajkumar, S.V.; Dispenzieri, A.; Lacy, M.Q. Pomalidomide: The new immunomodulatory agent for the treatment of multiple myeloma. Blood Cancer J. 2013. [Google Scholar] [CrossRef] [PubMed]
- Terpos, E.; Kanellias, N.; Christoulas, D.; Kastritis, E.; Dimopoulos, M.A. Pomalidomide: A novel drug to treat relapsed and refractory multiple myeloma. Oncol. Targets Ther. 2013, 6, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Mahony, C.; Erskine, L.; Niven, J.; Greig, N.H.; Figg, W.D.; Vargesson, N. Pomalidomide is nonteratogenic in chicken and zebrafish embryos and nonneurotoxic in vitro. Proc. Natl. Acad. Sci. USA 2013, 110, 12703–12708. [Google Scholar] [CrossRef] [PubMed]
- Vargesson, N.; Mahony, C.; Erskine, L.; Niven, J.; Greig, N.H.; Figg, W.D. Screening of thalidomide derivatives in chicken and zebrafish embryos. Proc. Natl. Acad. Sci. USA 2013, 110, 4820. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kasserra, C.; Reyes, J.; Schafer, P.; Kosek, J.; Capone, L.; Parton, A.; Kim-Kang, H.; Surapaneni, S.; Kumar, G. Absorption, metabolism and excretion of pomalidomide in humans following oral administration. Cancer Chemother. Pharmacol. 2013, 71, 489–501. [Google Scholar] [PubMed]
- Han, Z.; Gao, L.Y.; Lin, Y.H.; Chang, L.; Wu, H.Y.; Luo, C.X.; Zhu, D.Y. Neuroprotection of taurine against reactive oxygen species is associated with inhibiting NADPH oxidases. Eur. J. Pharmacol. 2016, 777, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Tweedie, D.; Fukui, K.; Li, Y.; Yu, Q.-S.; Barak, S.; Tamargo, I.A.; Rubovitch, V.; Holloway, H.W.; Lehrmann, E.; Wood, W.H.; et al. Cognitive Impairments Induced by Concussive Mild Traumatic Brain Injury in Mouse Are Ameliorated by Treatment with Phenserine via Multiple Non-Cholinergic and Cholinergic Mechanisms. PLoS ONE 2016. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wu, K.J.; Yu, S.J.; Tamargo, I.A.; Wang, Y.; Greig, N.H. Neurotrophic and neuroprotective effects of oxyntomodulin in neuronal cells and a rat model of stroke. Exp. Neurol. 2017, 288, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Perry, T.; Kindy, M.S.; Harvey, B.K.; Tweedie, D.; Holloway, H.W.; Powers, K.; Shen, H.; Egan, J.M.; Sambamurti, K.; et al. GLP-1 receptor stimulation preserves primary cortical and dopaminergic neurons in cellular and rodent models of stroke and Parkinsonism. Proc. Natl. Acad. Sci. USA 2009, 106, 1285–1290. [Google Scholar] [CrossRef] [PubMed]
- Lobner, D. Comparison of the LDH and MTT assays for quantifying cell death: Validity for neuronal apoptosis? J. Neurosci. Methods 2000, 96, 147–152. [Google Scholar] [CrossRef]
- Lee, S.; Tak, E.; Lee, J.; Rashid, M.A.; Murphy, M.P.; Ha, J.; Kim, S.S. Mitochondrial H2O2 generated from electron transport chain complex I stimulates muscle differentiation. Cell Res. 2011, 21, 817–834. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.X.; Wang, R.; Xi, J.; Shen, L.; Zhu, A.Y.; Qi, Q.; Wang, Q.Y.; Zhang, L.J.; Wang, F.C.; Lü, H.Z.; et al. Morroniside protects SK-N-SH human neuroblastoma cells against H2O2-induced damage. Int. J. Mol. Med. 2017, 39, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Nioi, P.; Pickett, C.B. The Nrf2-Antioxidant Response Element Signaling Pathway and Its Activation by Oxidative Stress. J. Biol. Chem. 2009, 284, 13291–13295. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, T.; Chan, P.H. Reactive oxygen radicals and pathogenesis of neuronal death after cerebral ischemia. Antioxid. Redox Signal. 2003, 5, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Kowaltowski, A.J.; de Souza-Pinto, N.C.; Castilho, R.F.; Vercesi, A.E. Mitochondria and reactive oxygen species. Free Radic. Biol. Med. 2009, 47, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Deng, H.; Xu, S.; Zhang, J. MicroRNAs Regulate Mitochondrial Function in Cerebral Ischemia-Reperfusion Injury. Int. J. Mol. Sci. 2015, 16, 24895–24917. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, T.; Fujimura, M.; Noshita, N.; Kim, G.W.; Saito, A.; Hayashi, T.; Narasimhan, P.; Maier, C.M.; Chan, P.H. Neuronal death/survival signaling pathways in cerebral ischemia. NeuroRx 2004, 1, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.; Ambrosone, C.B.; Kanetsky, P.A.; Tian, C.; Lehman, T.A.; Kropp, S.; Helmbold, I.; von Fournier, D.; Haase, W.; Sautter-Bihl, M.L.; et al. Polymorphisms in Genes Related to Oxidative Stress (CAT, MnSOD, MPO and eNOS) and Acute Toxicities from Radiation Therapy following Lumpectomy for Breast Cancer. Clin. Cancer Res. 2006, 12, 7063–7070. [Google Scholar] [CrossRef] [PubMed]
- Jurgensmeier, J.M.; Xie, Z.; Deveraux, Q.; Ellerby, L.; Bredesen, D.; Reed, J.C. Bax directly induces release of cytochrome c from isolated mitochondria. Proc. Natl. Acad. Sci. USA 1998, 95, 4997–5002. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.; Li, Y.; Liu, X.; Wang, X. An APAF-1 cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J. Biol. Chem. 1999, 274, 11549–11556. [Google Scholar] [CrossRef] [PubMed]
- Deb, P.; Sharma, S.; Hassan, K.M. Pathophysiologic mechanisms of acute ischemic stroke: An overview with emphasis on therapeutic significance beyond thrombolysis. Pathophysiology 2010, 17, 197–218. [Google Scholar] [CrossRef] [PubMed]
- Popa-Wagner, A.; Glavan, D.G.; Olaru, A.; Olaru, D.G.; Margaritescu, O.; Tica, O.; Surugiu, R.; Sandu, R. Present Status and Future Challenges of New Therapeutic Targets in Preclinical Models of Stroke in Aged Animals with/without Comorbidities. Int. J. Mol. Sci. 2018. [Google Scholar] [CrossRef] [PubMed]
- Weinberger, J.M. Evolving therapeutic approaches to treating acute ischemic stroke. J. Neurol. Sci. 2006, 249, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Perry, G.; Nunomura, A.; Hirai, K.; Zhu, X.; Perez, M.; Avila, J.; Castellani, R.J.; Atwood, C.S.; Aliev, G.; Sayre, L.M.; et al. Is oxidative damage the fundamental pathogenic mechanism of Alzheimer’s and other neurodegenerative diseases? Free Radic. Biol. Med. 2002, 33, 1475–1479. [Google Scholar] [CrossRef]
- Frankola, K.A.; Greig, N.H.; Luo, W.; Tweedie, D. Targeting TNF-alpha to elucidate and ameliorate neuroinflammation in neurodegenerative diseases. CNS Neurol. Disord. Drug Targets 2011, 10, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Vila, M.; Przedborski, S. Targeting programmed cell death in neurodegenerative diseases. Nat. Rev. Neurosci. 2003, 4, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Liska, G.M.; Lippert, T.; Russo, E.; Nieves, N.; Borlongan, C.V. A Dual Role for Hyperbaric Oxygen in Stroke Neuroprotection: Preconditioning of the Brain and Stem Cells. Cond. Med. 2018, 1, 151–166. [Google Scholar] [PubMed]
- Giunta, B.; Obregon, D.; Velisetty, R.; Sanberg, P.R.; Borlongan, C.V.; Tan, J. The immunology of traumatic brain injury: A prime target for Alzheimer’s disease prevention. J. Neuroinflamm. 2012, 9, 185. [Google Scholar] [CrossRef] [PubMed]
- Knecht, T.; Story, J.; Liu, J.; Davis, W.; Borlongan, C.; dela Peña, I. Adjunctive Therapy Approaches for Ischemic Stroke: Innovations to Expand Time Window of Treatment. Int. J. Mol. Sci. 2017. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C.; Anrather, J. Stroke research at a crossroad: Asking the brain for directions. Nat. Neurosci. 2013, 14, 1363–1368. [Google Scholar] [CrossRef] [PubMed]
- Peña, I.D.; Borlongan, C.; Shen, G.; Davis, W. Strategies to Extend Thrombolytic Time Window for Ischemic Stroke Treatment: An Unmet Clinical Need. J. Stroke 2017, 19, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y.; Huang, Y.N.; Chiu, C.C.; Tweedie, D.; Luo, W.; Pick, C.G.; Chou, S.Y.; Luo, Y.; Hoffer, B.J.; Greig, N.H.; et al. Pomalidomide mitigates neuronal loss, neuroinflammation and behavioral impairments induced by traumatic brain injury in rat. J. Neuroinflamm. 2016. [Google Scholar] [CrossRef]
- Chiu, C.C.; Liao, Y.E.; Yang, L.Y.; Wang, J.Y.; Tweedie, D.; Karnati, H.K.; Greig, N.H.; Wang, J.Y. Neuroinflammation in animal models of traumatic brain injury. J. Neurosci. Methods 2016, 272, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 2013, 8, 2003–2014. [Google Scholar] [PubMed]
- Kunz, A.; Abe, T.; Hochrainer, K.; Shimamura, M.; Anrather, J.; Racchumi, G.; Zhou, P.; Iadecola, C. Nuclear factor-kappaB activation and postischemic inflammation are suppressed in CD36-null mice after middle cerebral artery occlusion. J. Neurosci. 2008, 28, 1649–1658. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chang, C.F.; Morales, M.; Chou, J.; Chen, H.L.; Chiang, Y.H.; Lin, S.Z.; Cadet, J.L.; Deng, X.; Wang, J.Y.; et al. Bone morphogenetic protein-6 reduces ischemia-induced brain damage in rats. Stroke 2001, 32, 2170–2178. [Google Scholar] [CrossRef] [PubMed]
- Adibhatla, R.M.; Hatcher, J.F. Phospholipase A2, reactive oxygen species and lipid peroxidation in cerebral ischemia. Free Radic. Biol. Med. 2006, 40, 376–387. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Reactive oxygen species and the central nervous system. J. Neurochem. 1992, 59, 1609–1623. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liu, Y.; Cui, Y. Pathways to caspase activation. Cell Biol. Int. 2005, 29, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.F.; Morales, M.; Chou, J.; Chen, H.L.; Hoffer, B.; Wang, Y. Bone morphogenetic proteins are involved in fetal kidney tissue transplantation-induced neuroprotection in stroke rats. Neuropharmacology 2002, 43, 418–426. [Google Scholar] [CrossRef]
- Becker, R.E.; Kapogiannis, D.; Greig, N.H. Does traumatic brain injury hold the key to the Alzheimer’s disease puzzle? Alzheimers Dement. 2018, 14, 431–443. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.Y.; Weng, J.Y.; Lai, H.L.; Liao, F.; Sun, S.H.; Tu, P.H.; Dickson, D.W.; Chern, Y. Expanded-Polyglutamine Huntingtin Protein Suppresses the Secretion and Production of a Chemokine (CCL5/RANTES) by Astrocytes. J. Neurosci. 2008, 28, 3277–3290. [Google Scholar] [CrossRef] [PubMed]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsai, Y.-R.; Chang, C.-F.; Lai, J.-H.; Wu, J.C.-C.; Chen, Y.-H.; Kang, S.-J.; Hoffer, B.J.; Tweedie, D.; Luo, W.; Greig, N.H.; et al. Pomalidomide Ameliorates H2O2-Induced Oxidative Stress Injury and Cell Death in Rat Primary Cortical Neuronal Cultures by Inducing Anti-Oxidative and Anti-Apoptosis Effects. Int. J. Mol. Sci. 2018, 19, 3252. https://doi.org/10.3390/ijms19103252
Tsai Y-R, Chang C-F, Lai J-H, Wu JC-C, Chen Y-H, Kang S-J, Hoffer BJ, Tweedie D, Luo W, Greig NH, et al. Pomalidomide Ameliorates H2O2-Induced Oxidative Stress Injury and Cell Death in Rat Primary Cortical Neuronal Cultures by Inducing Anti-Oxidative and Anti-Apoptosis Effects. International Journal of Molecular Sciences. 2018; 19(10):3252. https://doi.org/10.3390/ijms19103252
Chicago/Turabian StyleTsai, Yan-Rou, Cheng-Fu Chang, Jing-Huei Lai, John Chung-Che Wu, Yen-Hua Chen, Shuo-Jhen Kang, Barry J. Hoffer, David Tweedie, Weiming Luo, Nigel H. Greig, and et al. 2018. "Pomalidomide Ameliorates H2O2-Induced Oxidative Stress Injury and Cell Death in Rat Primary Cortical Neuronal Cultures by Inducing Anti-Oxidative and Anti-Apoptosis Effects" International Journal of Molecular Sciences 19, no. 10: 3252. https://doi.org/10.3390/ijms19103252
APA StyleTsai, Y.-R., Chang, C.-F., Lai, J.-H., Wu, J. C.-C., Chen, Y.-H., Kang, S.-J., Hoffer, B. J., Tweedie, D., Luo, W., Greig, N. H., Chiang, Y.-H., & Chen, K.-Y. (2018). Pomalidomide Ameliorates H2O2-Induced Oxidative Stress Injury and Cell Death in Rat Primary Cortical Neuronal Cultures by Inducing Anti-Oxidative and Anti-Apoptosis Effects. International Journal of Molecular Sciences, 19(10), 3252. https://doi.org/10.3390/ijms19103252