Fullerene Derivatives of Nucleoside HIV Reverse Transcriptase Inhibitors—In Silico Activity Prediction



Abstract
:
1. Introduction
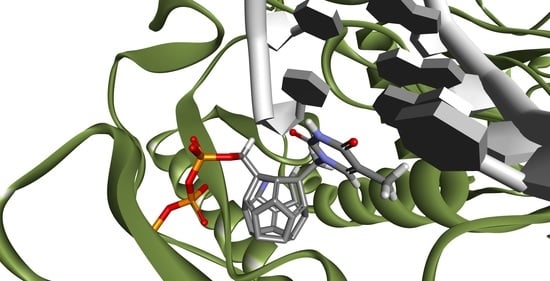
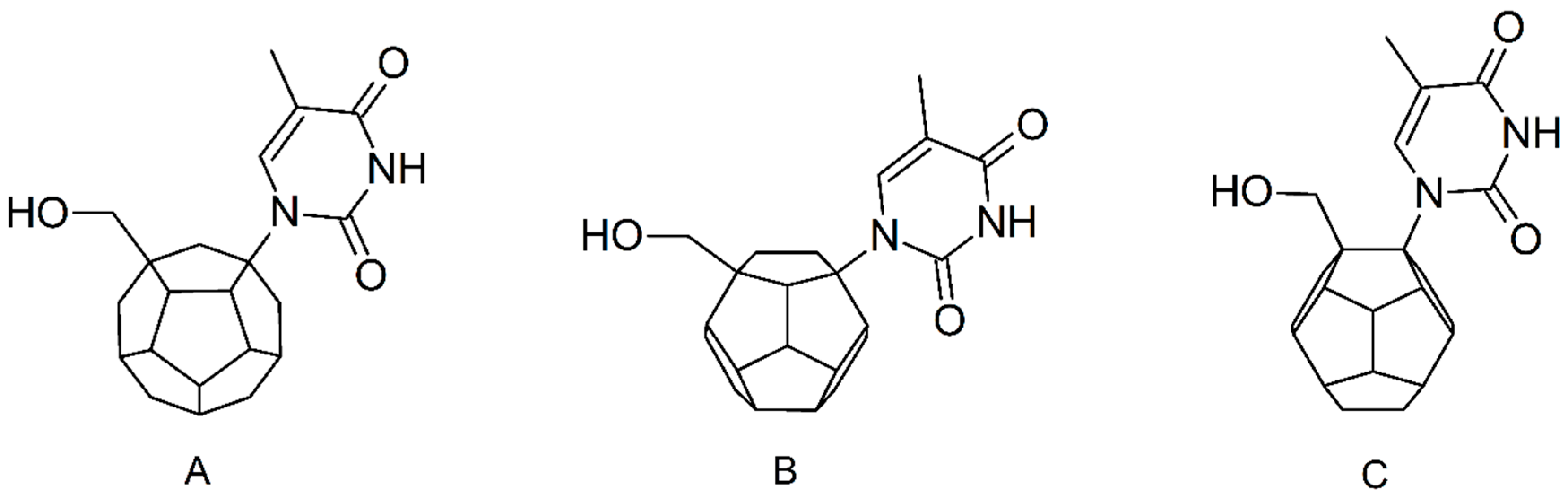
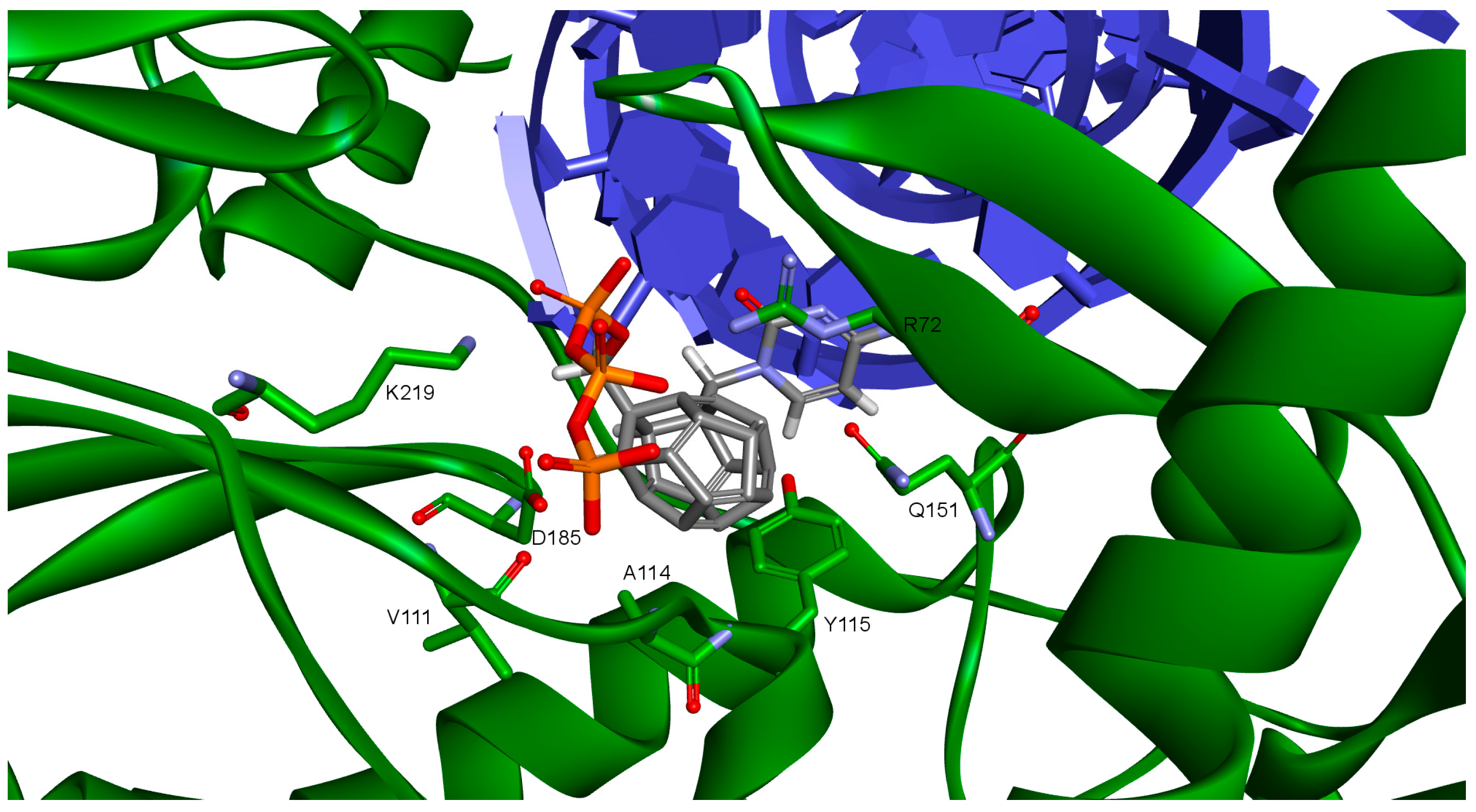
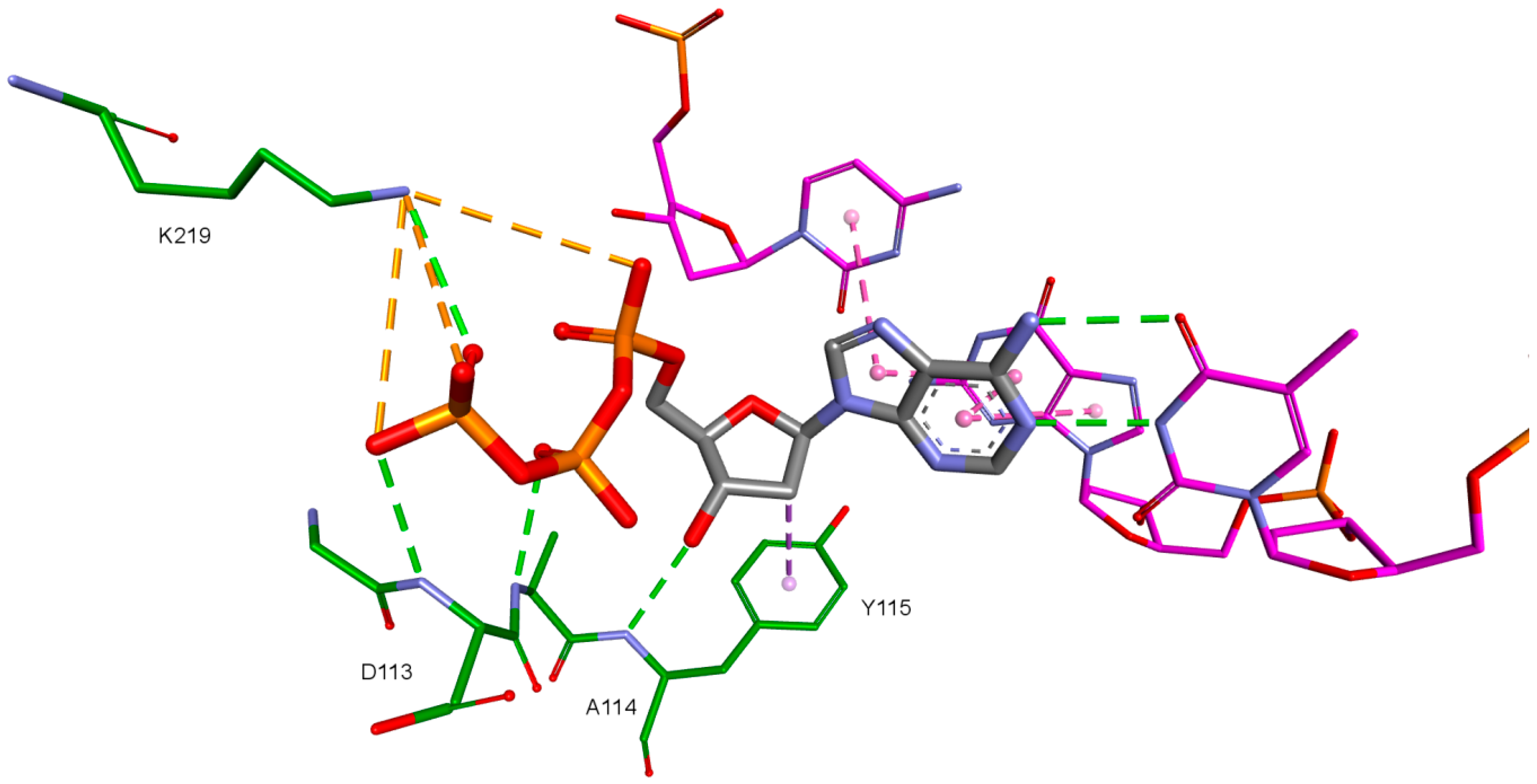
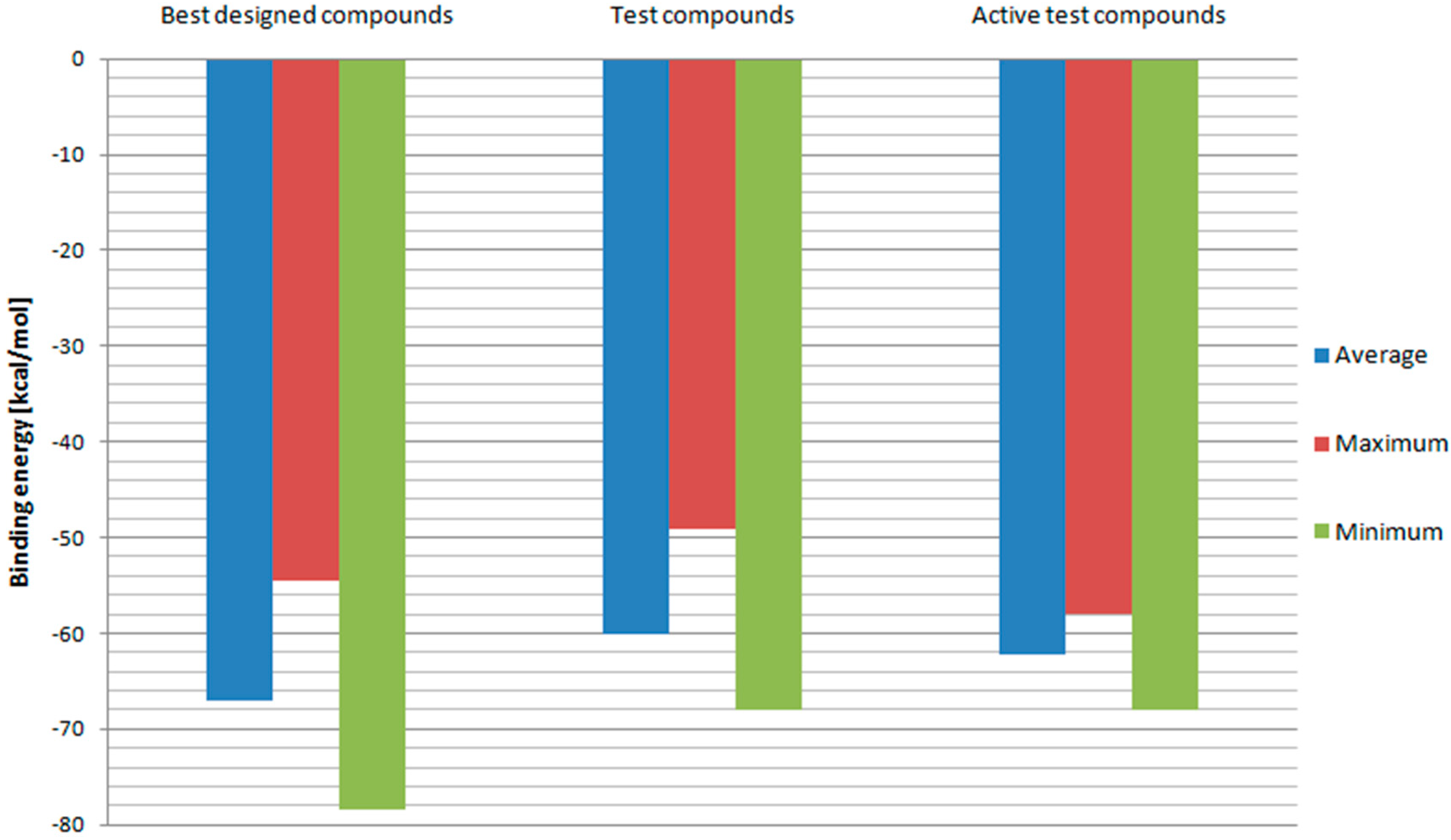
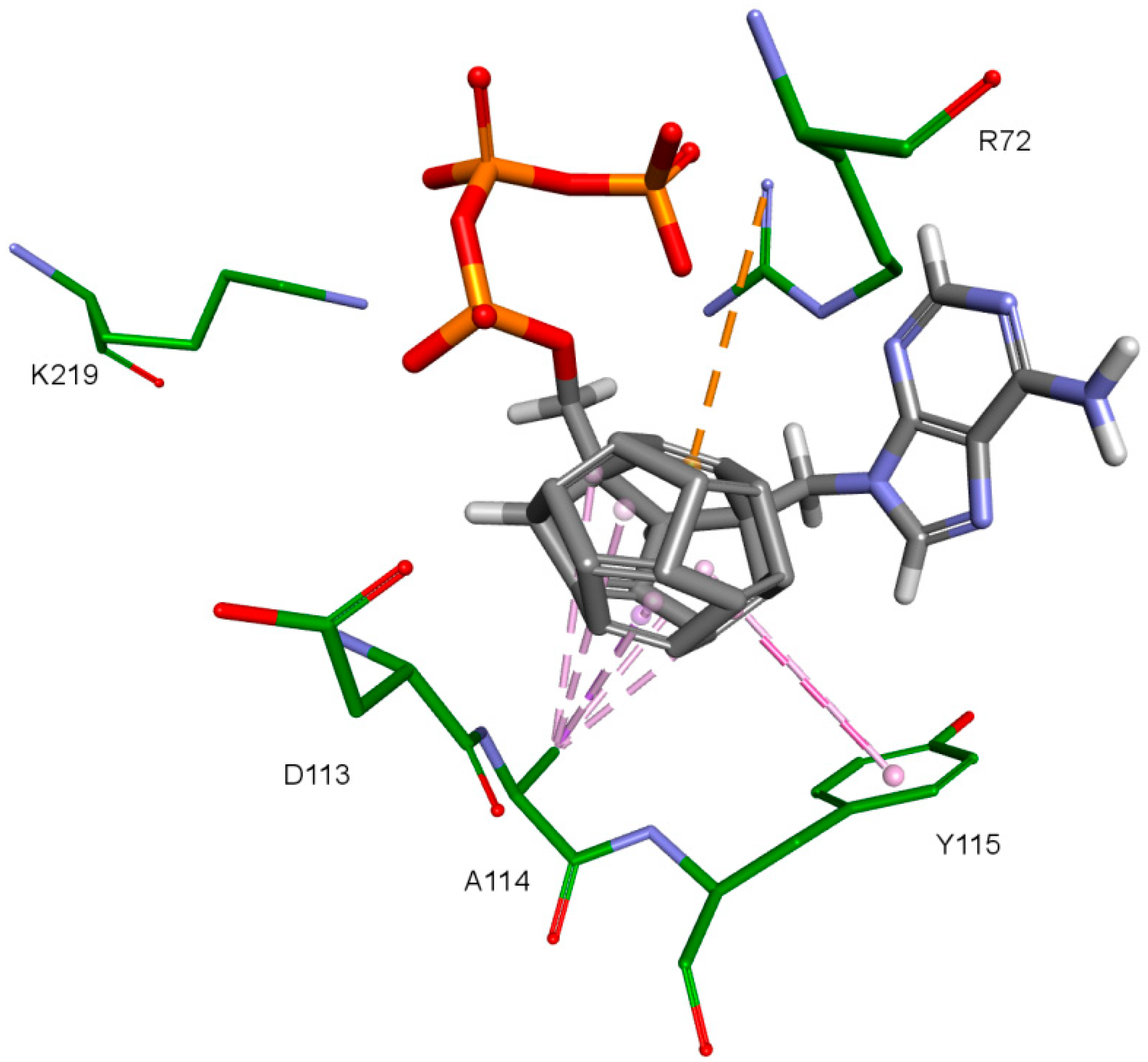
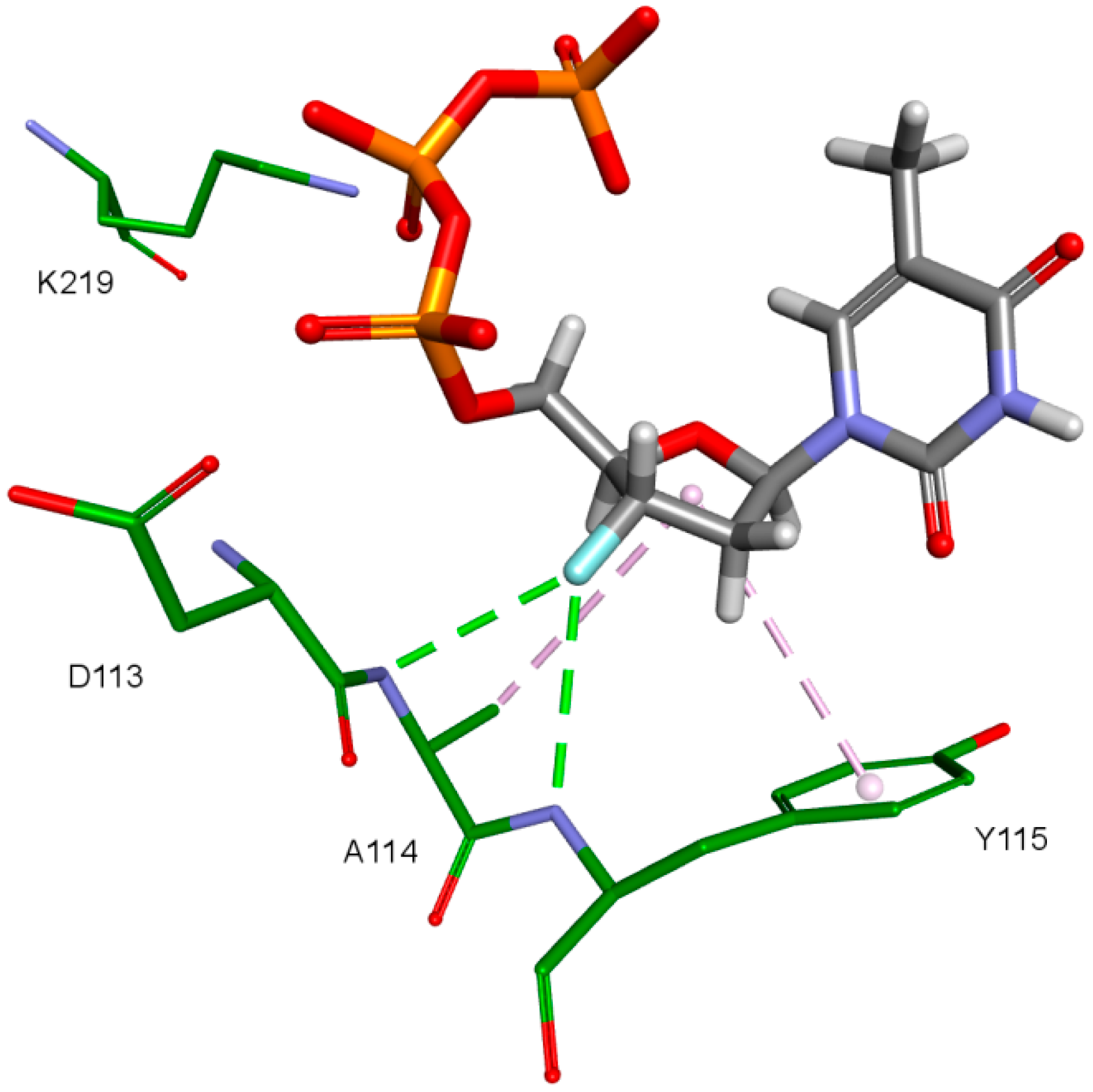
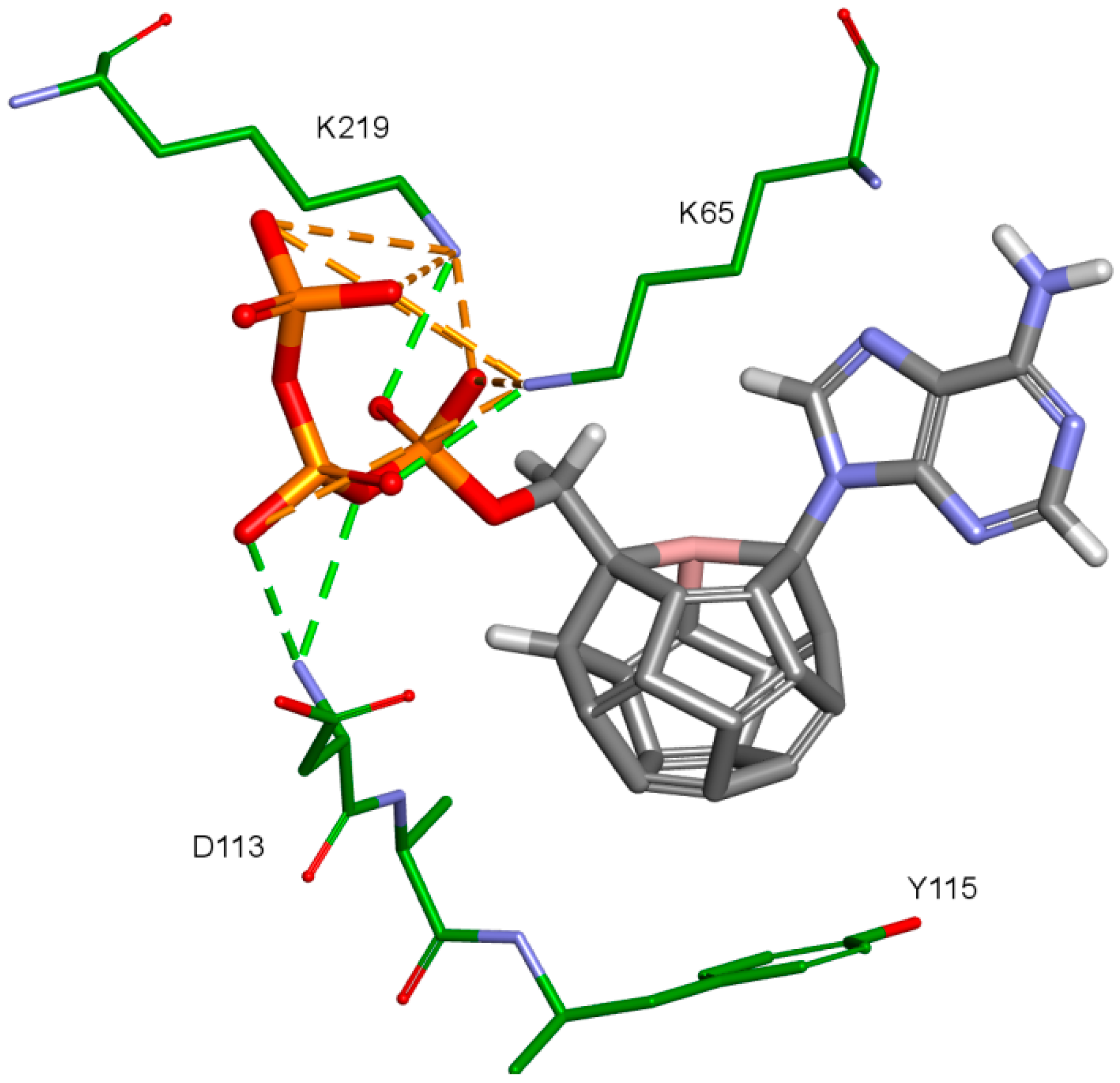
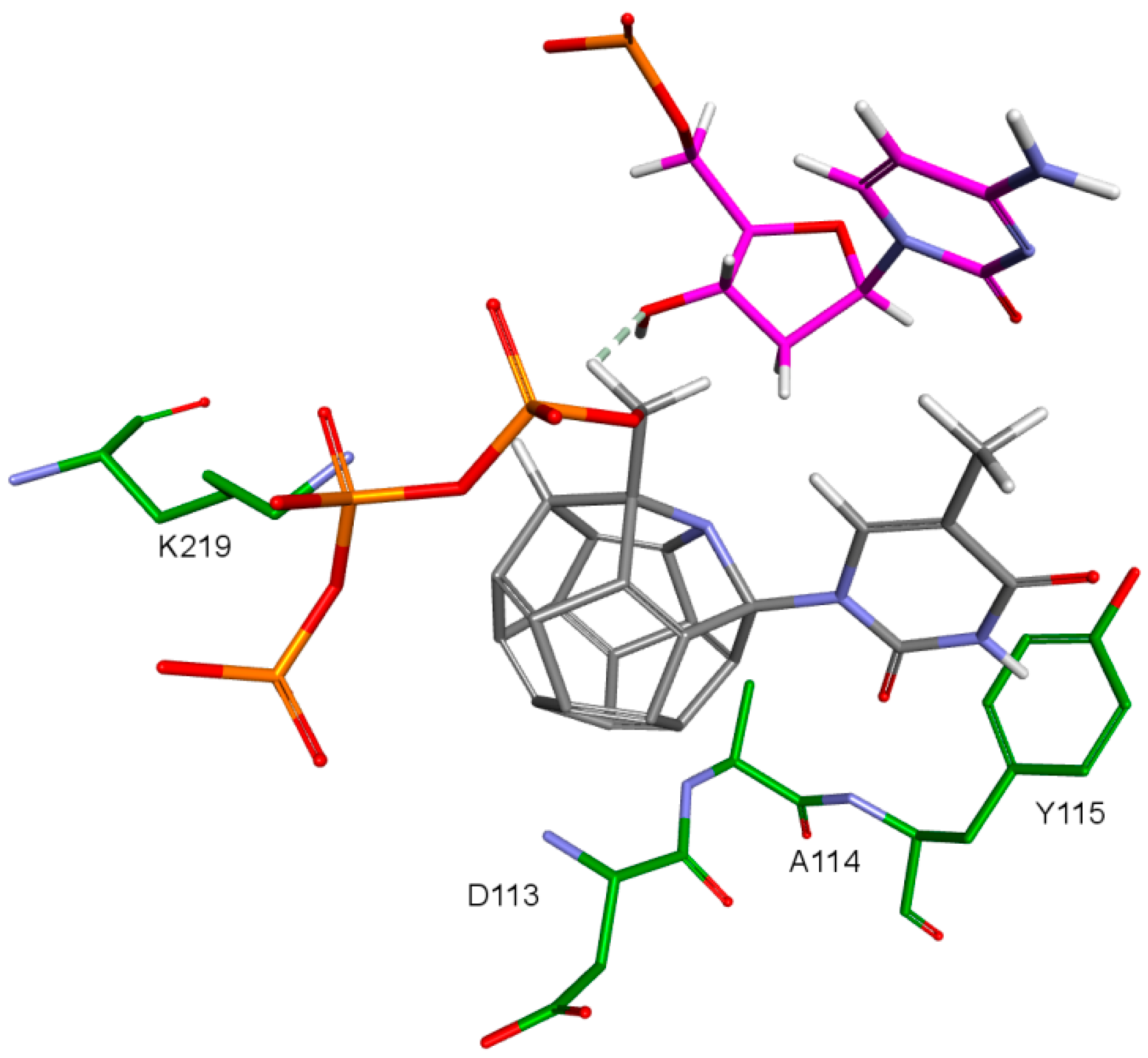
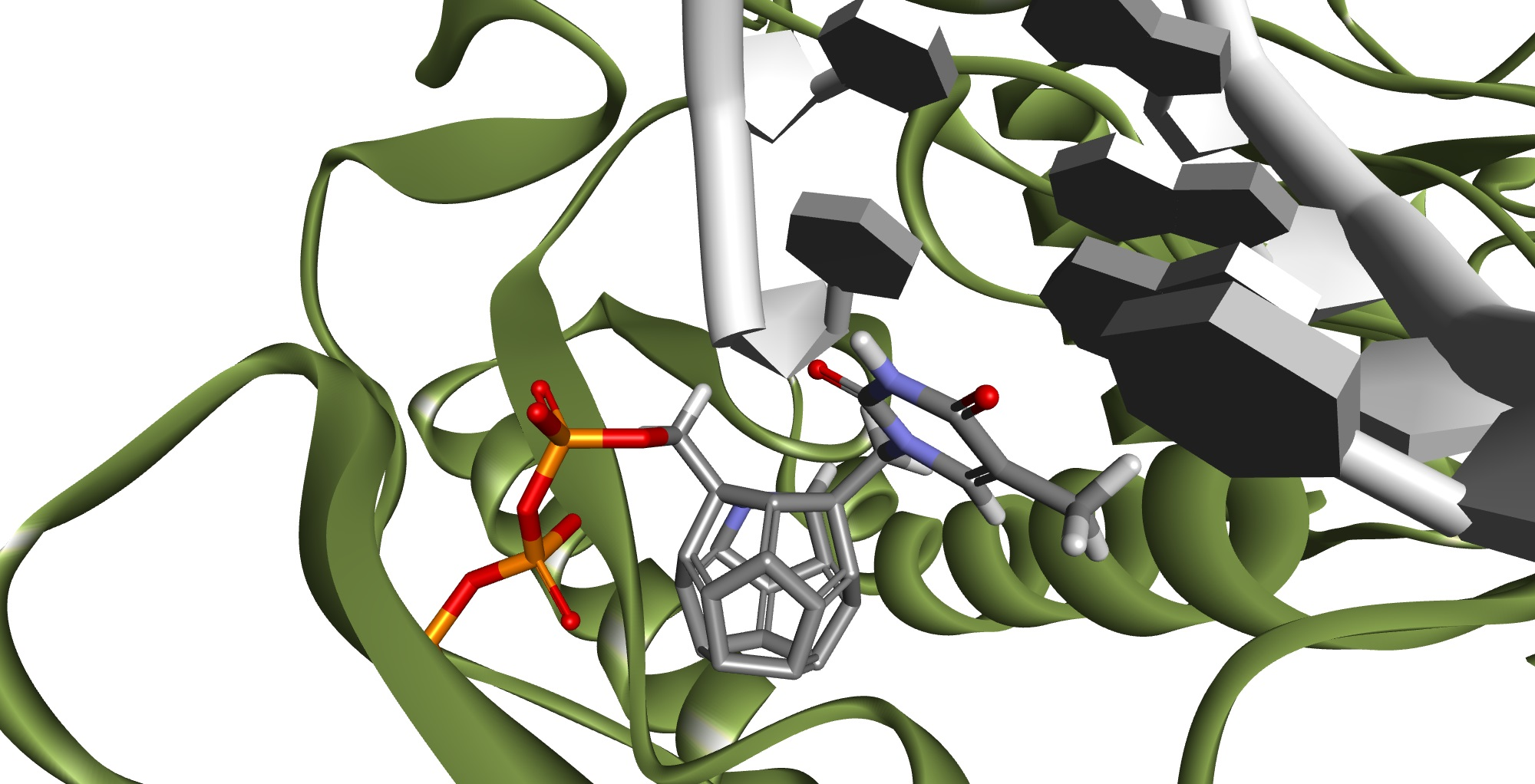
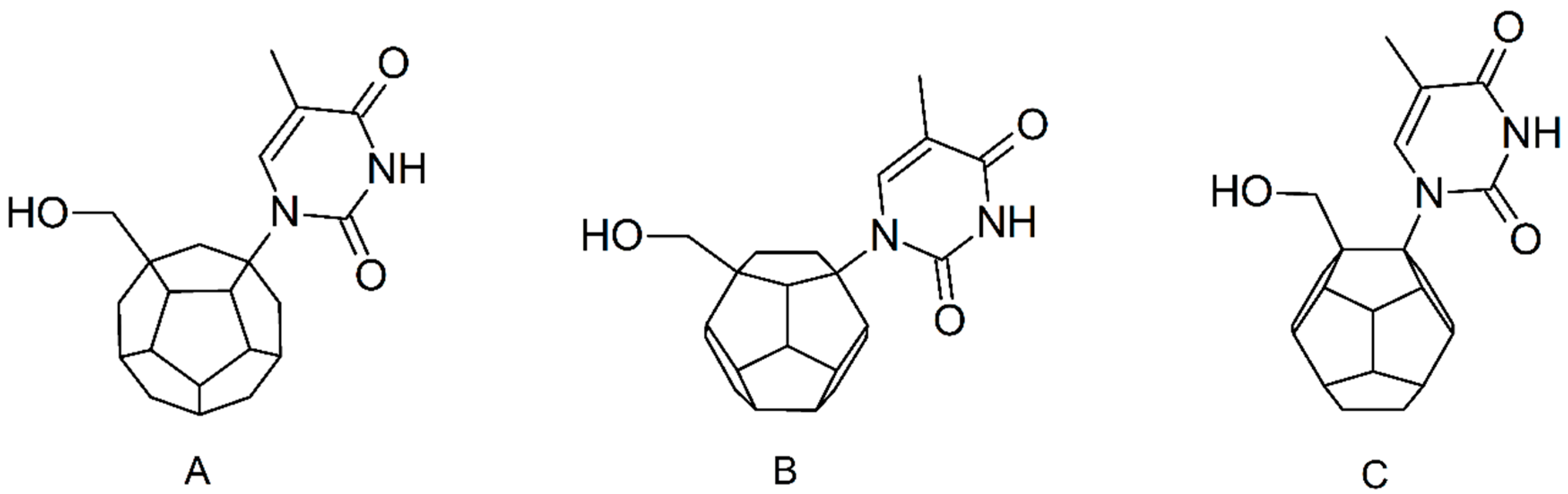
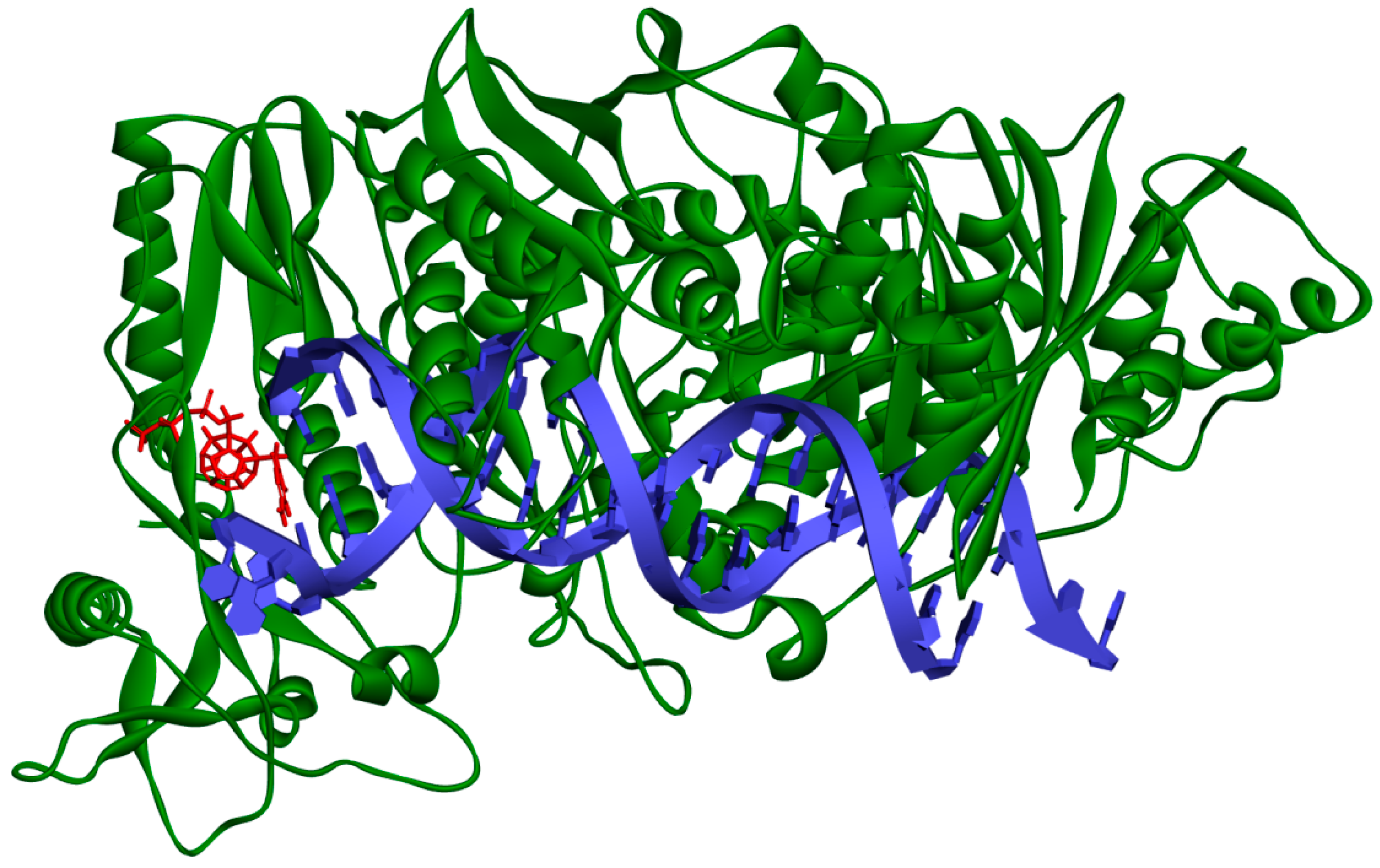
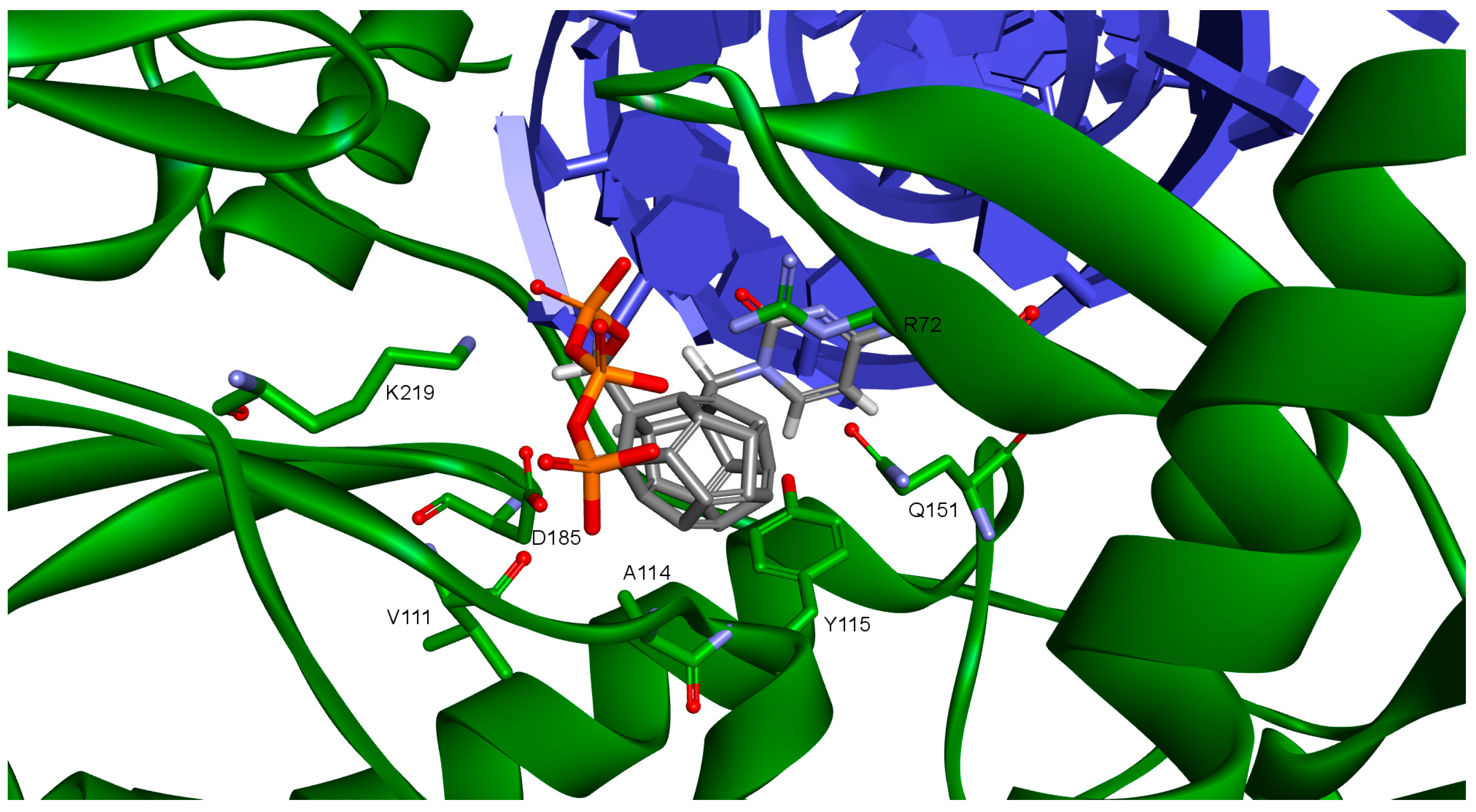
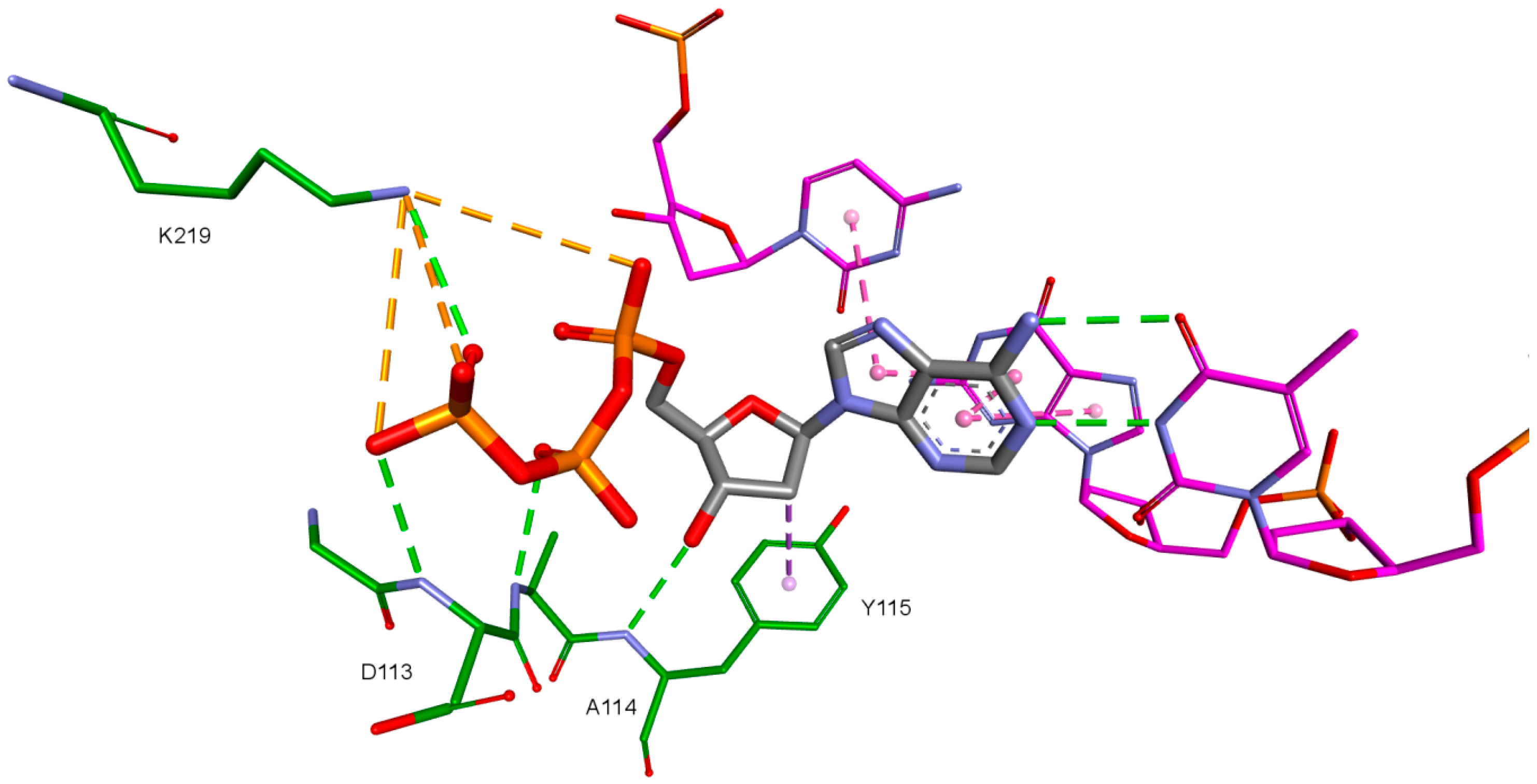
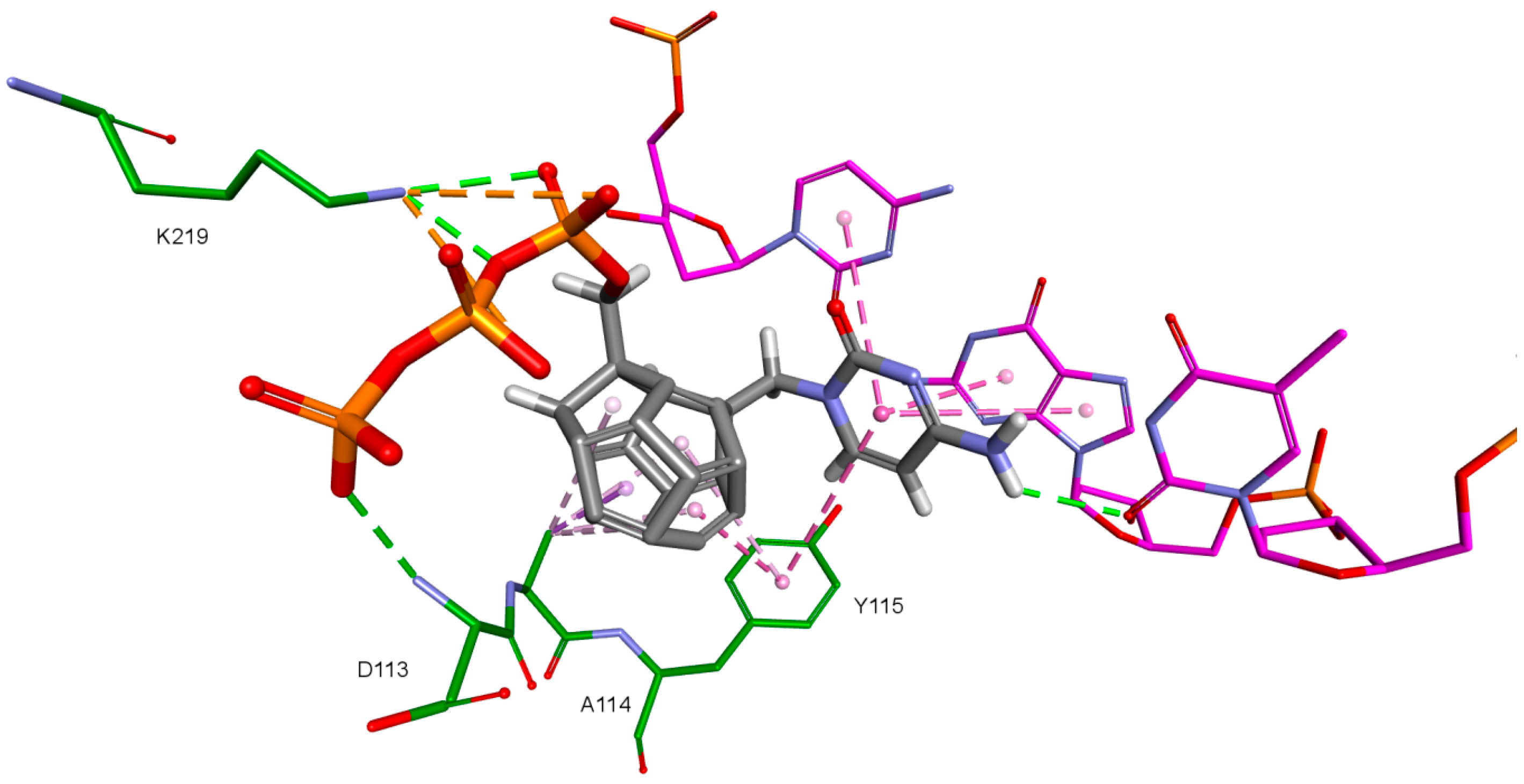
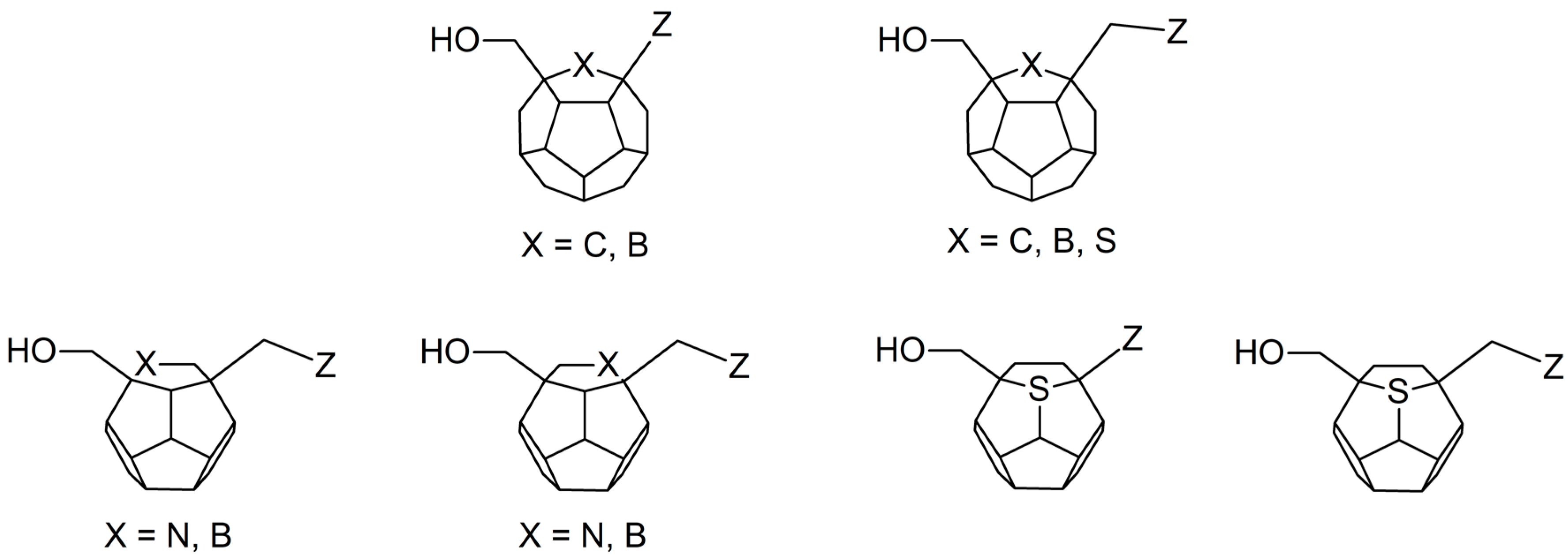
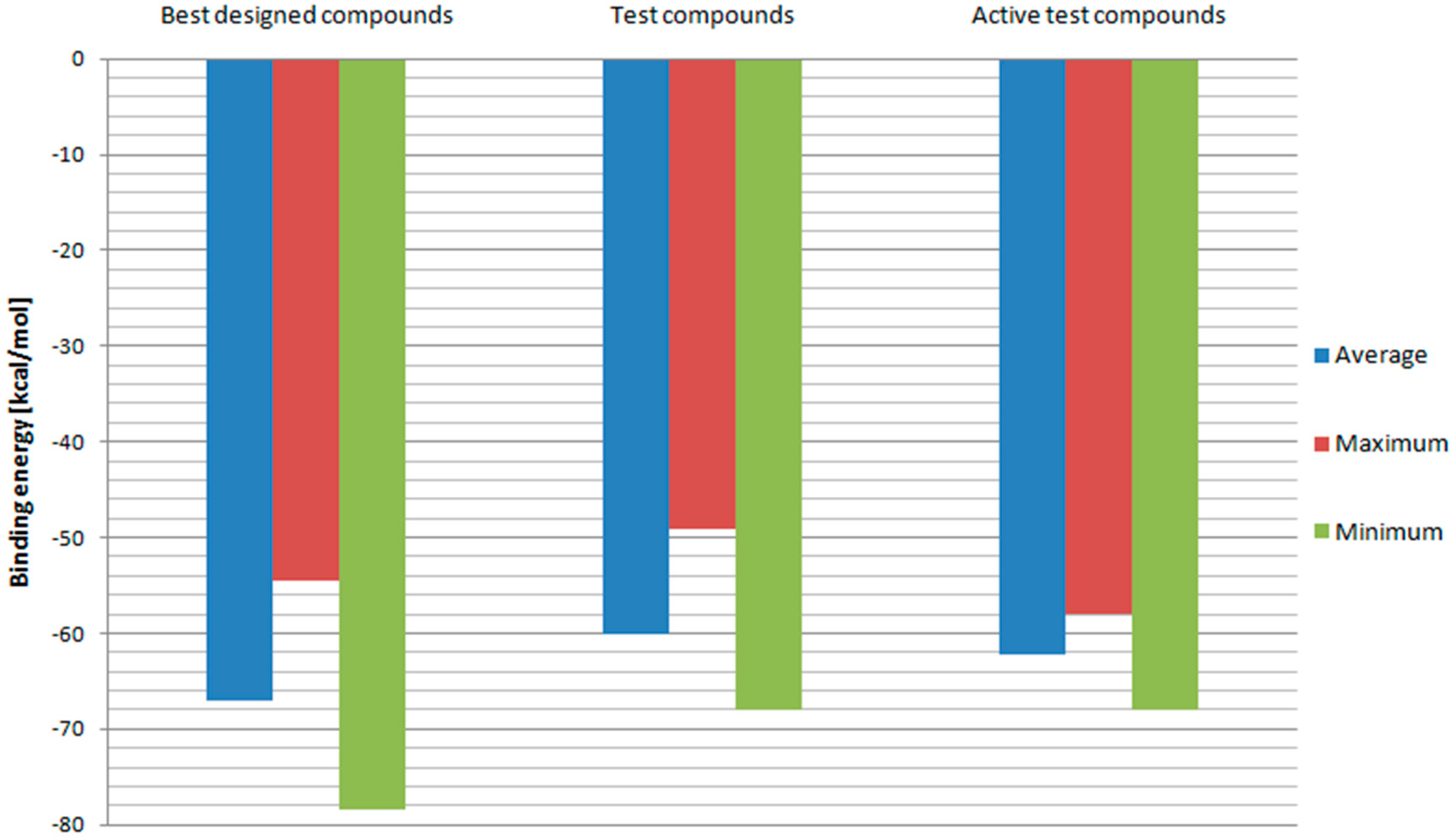
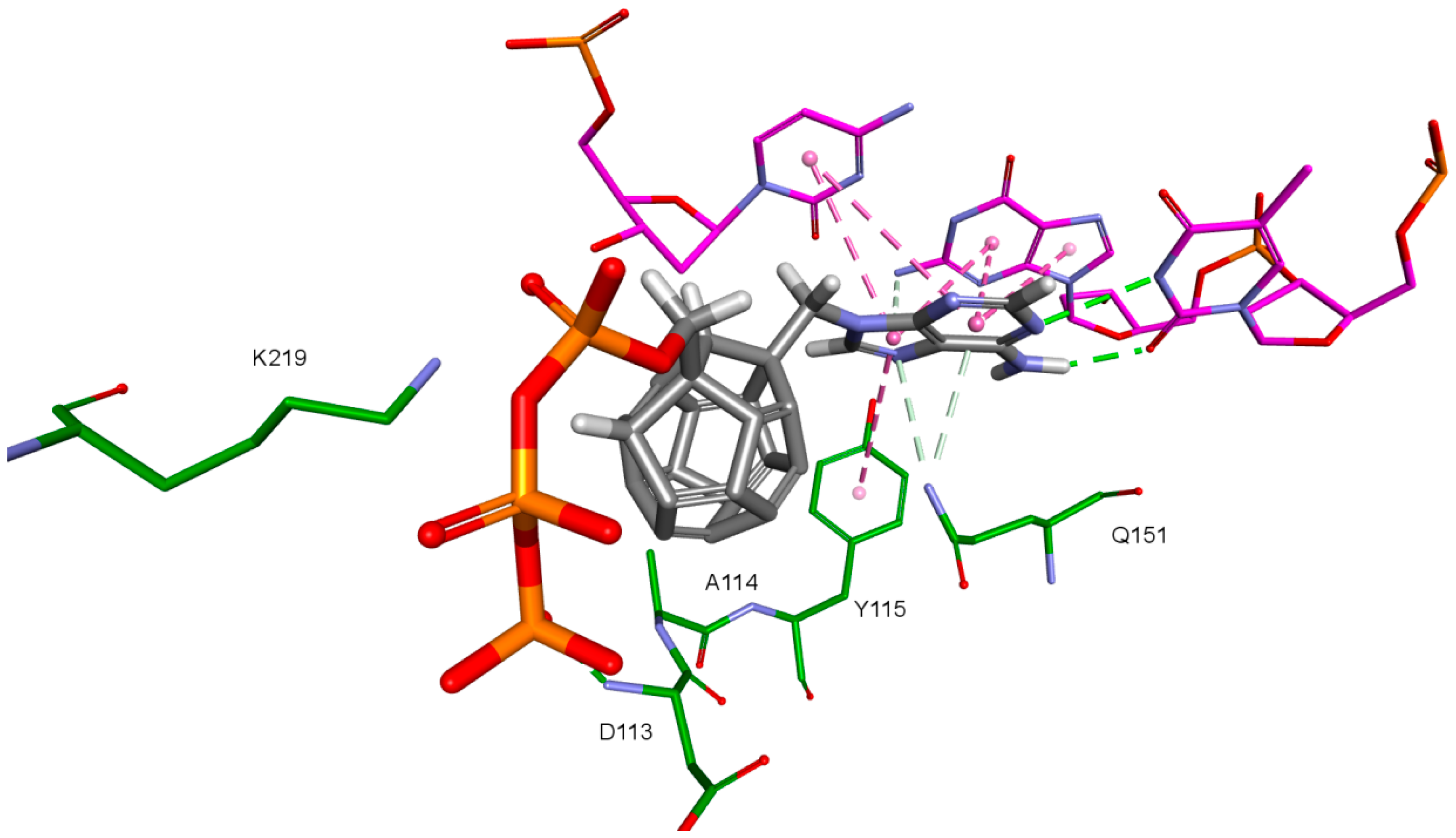
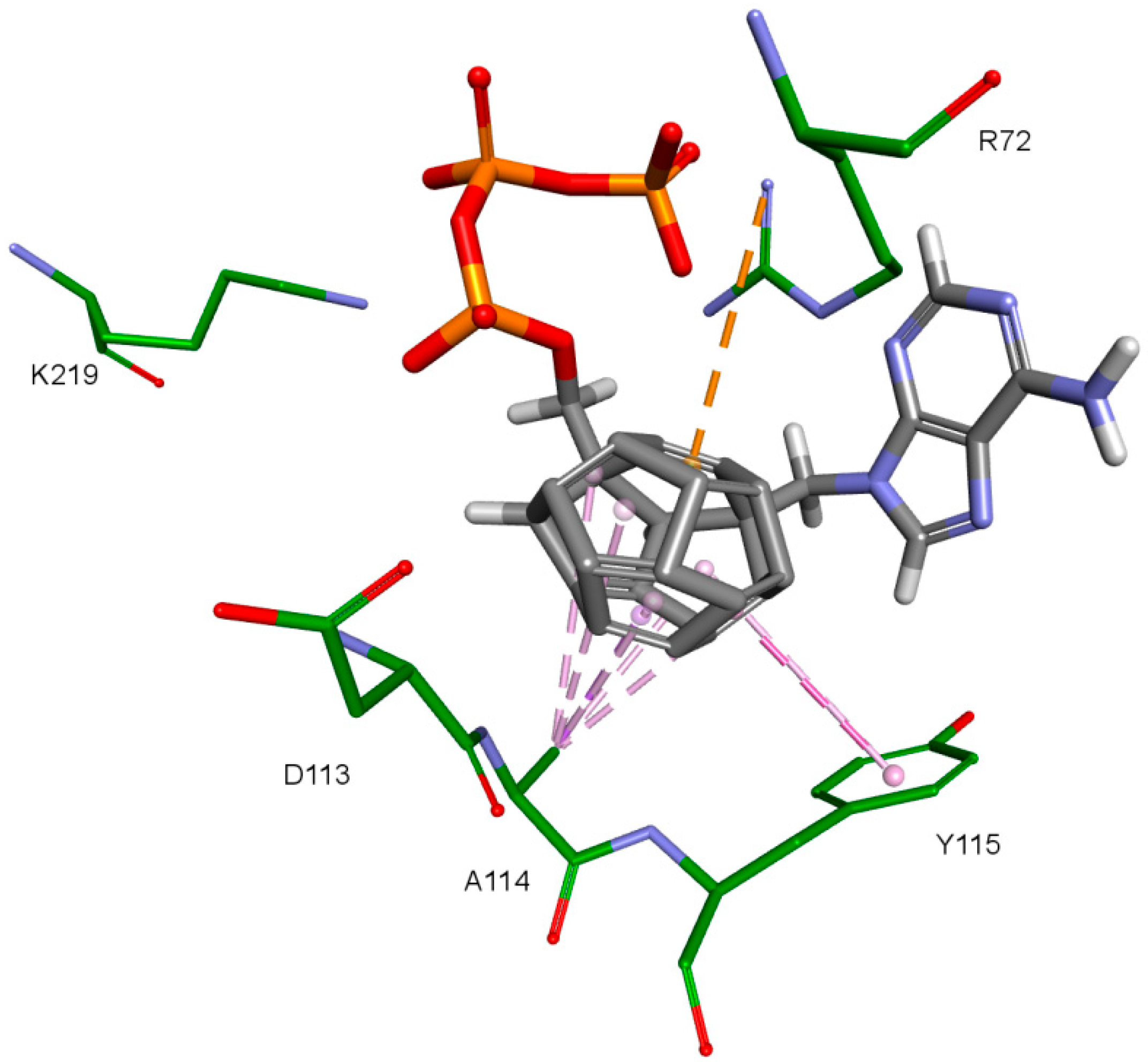
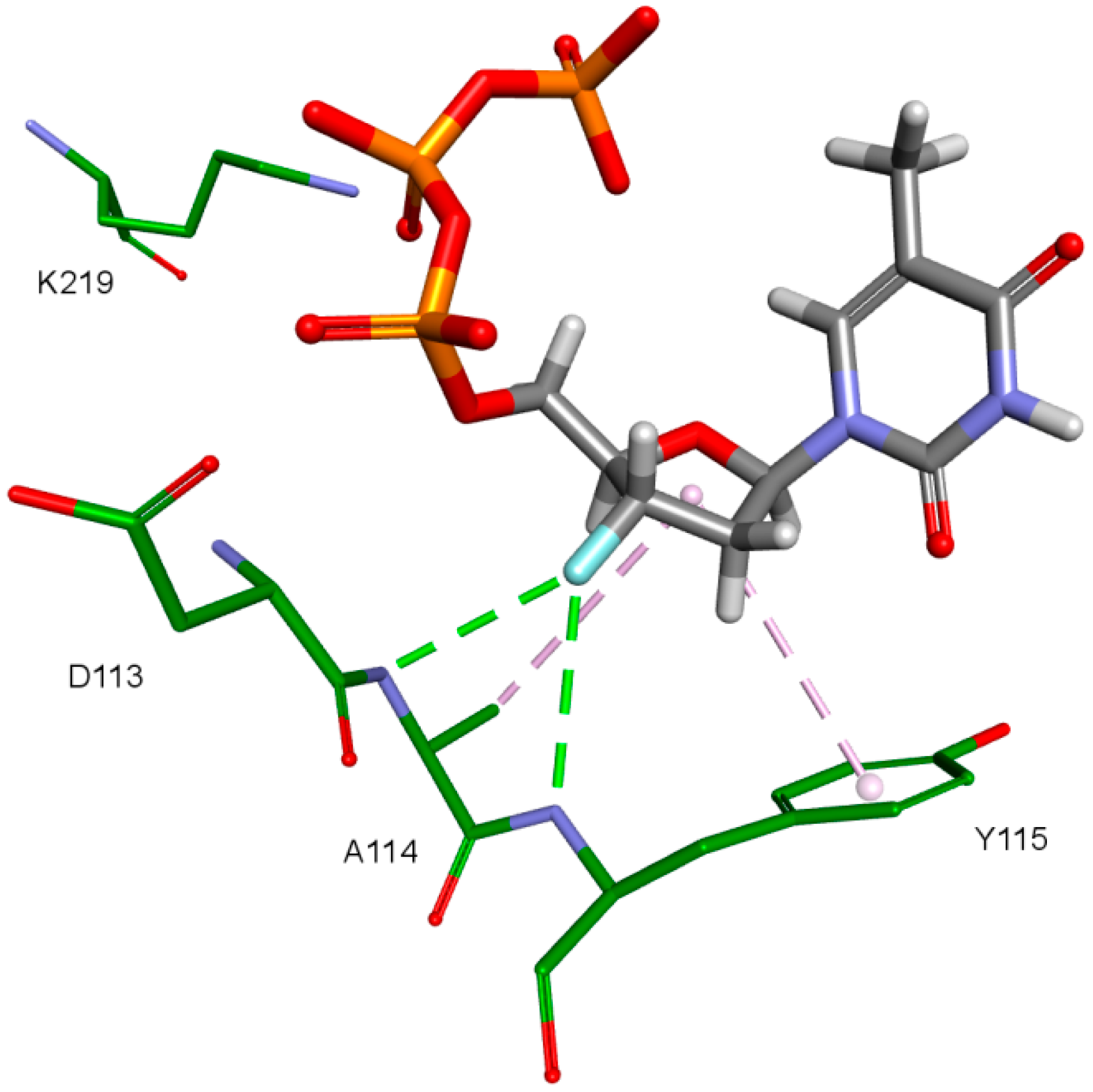
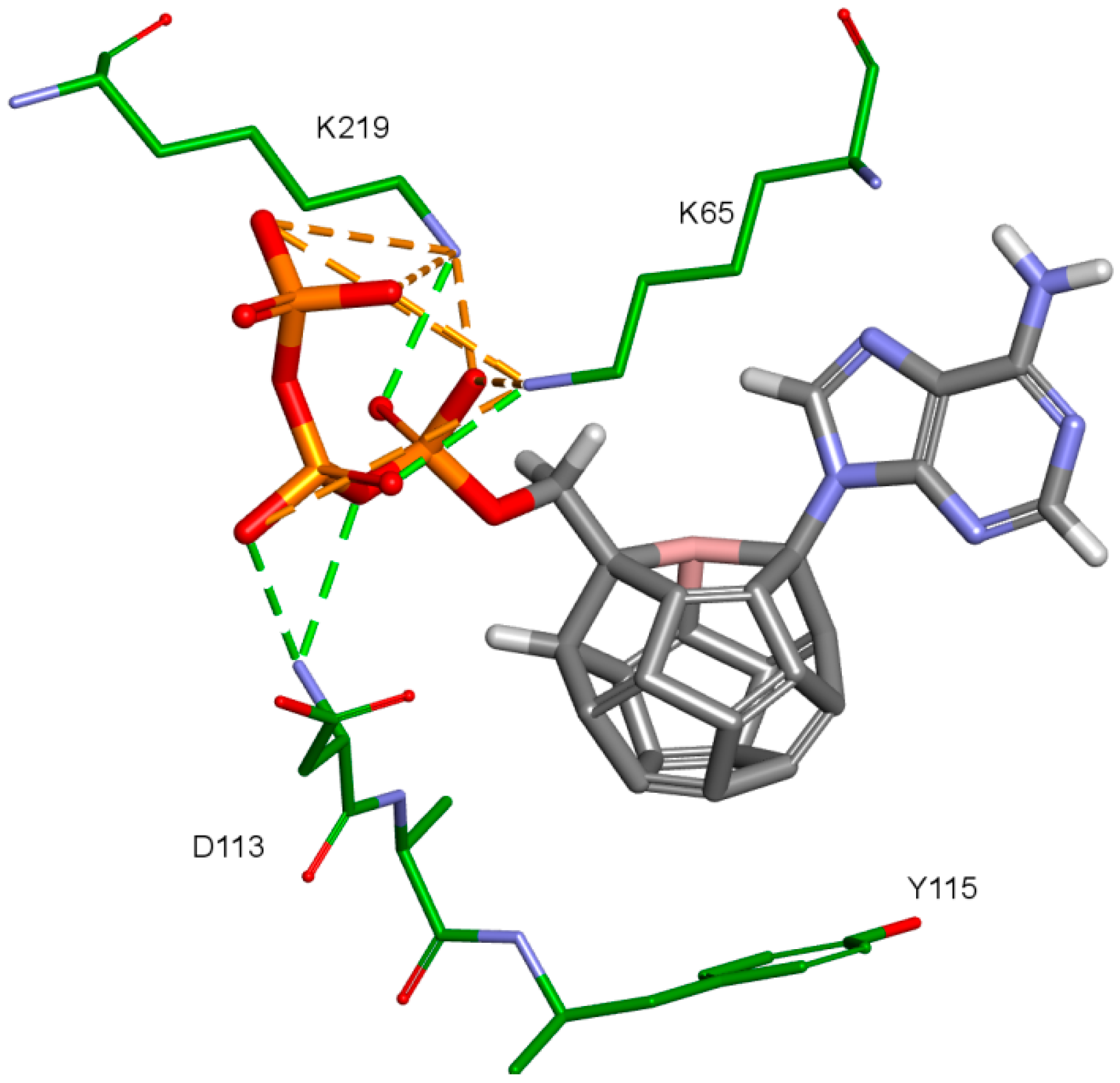
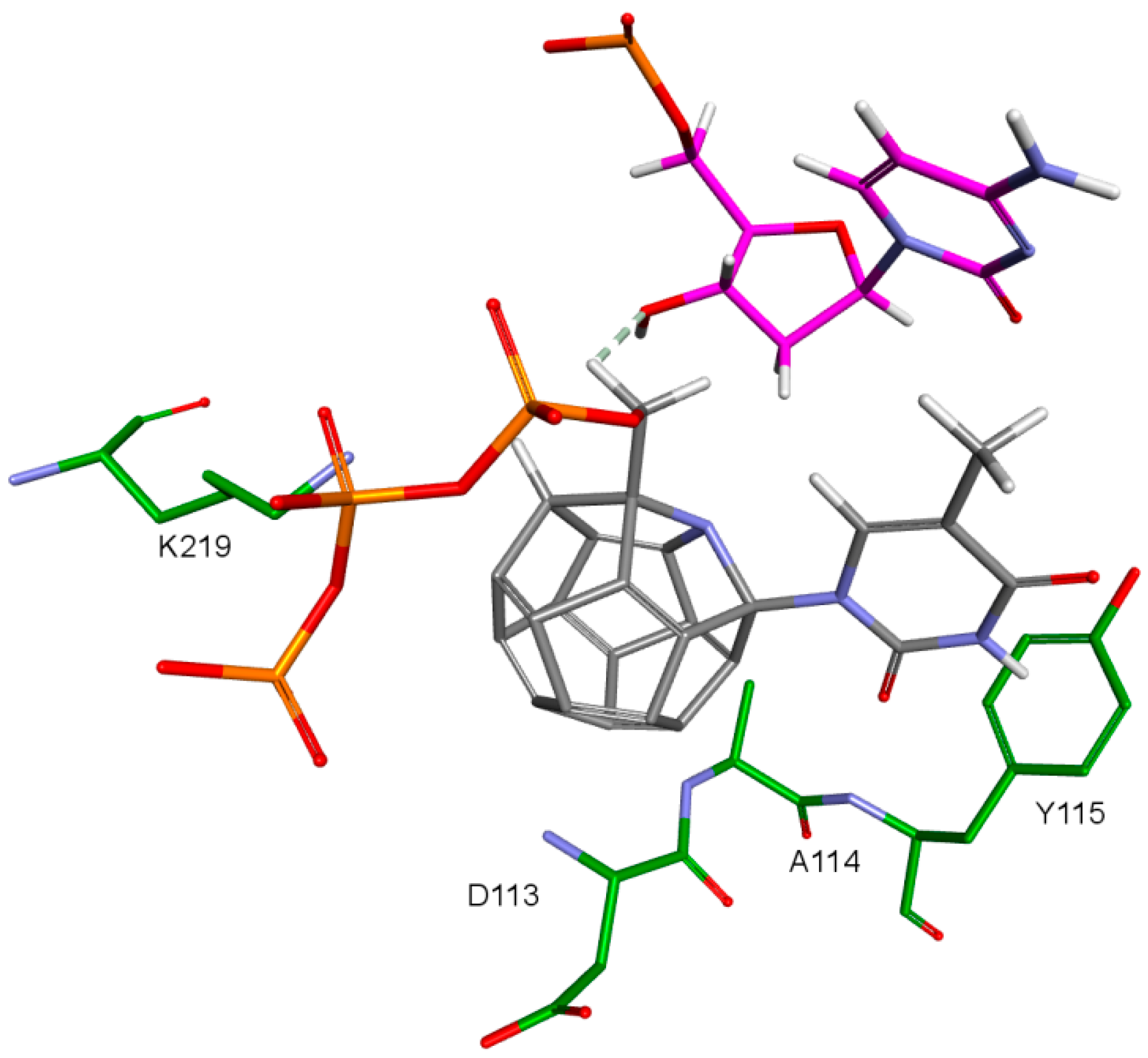
2. Results and Discussion
- 3KK2: 6, 78, 2, 25, 102, 106, 174, 28, 7, 107, 83, 48
- Model 2: 29, 47, 5, 83, 66, 78, 82, 30, 4, 94, 144, 100
- Model 3: 6, 78, 102, 84, 60, 114, 72, 107, 76, 96, 105, 83
- 3V4I: 18, 3, 27, 25, 132, 40, 39.
3. Materials and Methods
3.1. Ligands’ Preparation
3.2. RT Models’ Preparation
3.3. Model Validation and Selection of Scoring Functions
3.4. Preparation of New RT Models
3.5. Docking
3.6. Binding Free Energy Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| HIV | Human immunodeficiency virus |
| AIDS | Acquired Immunodeficiency Syndrome |
| RT | Reverse transcriptase |
| NRTI | Nucleoside/nucleotide RT inhibitor |
| NNRTI | Non-nucleoside RT inhibitor |
References
- World Health Organization. HIV/AIDS Fact Sheet N°360. Available online: http://www.who.int/mediacentre/factsheets/fs360/en/ (accessed on 14 April 2016).
- Barre-Sinoussi, F.; Chermann, J.C.; Rey, F.; Nugeyre, M.T.; Chamaret, S.; Gruest, J.; Dauguet, C.; Axler-Blin, C.; Vezinet-Brun, F.; Rouzioux, C.; et al. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science 1983, 220, 868–871. [Google Scholar] [CrossRef] [PubMed]
- Waterson, A.P. Acquired immune deficiency syndrome. Br. Med. J. 1983, 286, 743–746. [Google Scholar] [CrossRef]
- Furman, P.A.; Fyfe, J.A.; St Clair, M.H.; Weinhold, K.; Rideout, J.L.; Freeman, G.A.; Lehrman, S.N.; Bolognesi, D.P.; Broder, S.; Mitsuya, H. Phosphorylation of 3′-azido-3′-deoxythymidine and selective interaction of the 5′-triphosphate with human immunodeficiency virus reverse transcriptase. Proc. Natl. Acad. Sci. USA 1986, 83, 8333–8337. [Google Scholar] [CrossRef] [PubMed]
- Van Rompay, A.R.; Johansson, M.; Karlsson, A. Phosphorylation of nucleosides and nucleoside analogs by mammalian nucleoside monophosphate kinases. Pharmacol. Ther. 2000, 87, 189–198. [Google Scholar] [CrossRef]
- Tapia, A.; Villanueva, C.; Peón-Escalante, R.; Quintal, R.; Medina, J.; Peñuñuri, F.; Avilés, F. The bond force constant and bulk modulus of small fullerenes using density functional theory and finite element analysis. J. Mol. Model. 2015, 21, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.H.; DeCamp, D.L.; Sijbesma, R.P.; Srdanov, G.; Wudl, F.; Kenyon, G.L. Inhibition of the HIV-1 protease by fullerene derivatives: Model building studies and experimental verification. J. Am. Chem. Soc. 1993, 115, 6506–6509. [Google Scholar] [CrossRef]
- Sijbesma, R.; Srdanov, G.; Wudl, F.; Castoro, J.A.; Wilkins, C.; Friedman, S.H.; DeCamp, D.L.; Kenyon, G.L. Synthesis of a fullerene derivative for the inhibition of HIV enzymes. J. Am. Chem. Soc. 1993, 115, 6510–6512. [Google Scholar] [CrossRef]
- Calvaresi, M.; Zerbetto, F.; Ciamician, C.G.; Bologna, U.; Selmi, V.F. Baiting proteins with C 60. ACS Nano 2010, 4, 2283–2299. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.H.; Ganapathi, P.S.; Rubin, Y.; Kenyon, G.L. Optimizing the binding of fullerene inhibitors of the HIV-1 protease through predicted increases in hydrophobic desolvation. J. Med. Chem. 1998, 41, 2424–2429. [Google Scholar] [CrossRef] [PubMed]
- Mashino, T.; Shimotohno, K.; Ikegami, N.; Nishikawa, D.; Okuda, K.; Takahashi, K.; Nakamura, S.; Mochizuki, M. Human immunodeficiency virus-reverse transcriptase inhibition and hepatitis C virus RNA-dependent RNA polymerase inhibition activities of fullerene derivatives. Bioorg. Med. Chem. Lett. 2005, 15, 1107–1109. [Google Scholar] [CrossRef] [PubMed]
- Yasuno, T.; Ohe, T.; Takahashi, K.; Nakamura, S.; Mashino, T. The human immunodeficiency virus-reverse transcriptase inhibition activity of novel pyridine/pyridinium-type fullerene derivatives. Bioorg. Med. Chem. Lett. 2015, 25, 3226–3229. [Google Scholar] [CrossRef] [PubMed]
- Hahn, M. Receptor Surface Models. 1. Definition and Construction. J. Med. Chem. 1995, 38, 2080–2090. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Das, K.; Martinez, S.E.; Bauman, J.D.; Arnold, E. HIV-1 reverse transcriptase complex with DNA and nevirapine reveals non-nucleoside inhibition mechanism. Nat. Struct. Mol. Biol. 2012, 19, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Huang, H. Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: Implications for drug resistance. Science 1998, 282, 1669–1675. [Google Scholar] [CrossRef] [PubMed]
- Tuske, S.; Sarafianos, S.G.; Clark, A.D.; Ding, J.; Naeger, L.K.; White, K.L.; Miller, M.D.; Gibbs, C.S.; Boyer, P.L.; Clark, P.; et al. Structures of HIV-1 RT-DNA complexes before and after incorporation of the anti-AIDS drug tenofovir. Nat. Struct. Mol. Biol. 2004, 11, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Lansdon, E.B.; Samuel, D.; Lagpacan, L.; Brendza, K.M.; White, K.L.; Hung, M.; Liu, X.; Boojamra, C.G.; Mackman, R.L.; Cihlar, T.; et al. Visualizing the molecular interactions of a nucleotide analog, GS-9148, with HIV-1 reverse transcriptase-DNA complex. J. Mol. Biol. 2010, 397, 967–978. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R.; Bruccoleri, R.E.; Olafson, B.D.; States, D.J.; Swaminathan, S.; Karplus, M. CHARMM: A program for macromolecular energy minimization and dynamics calculations. J. Comp. Chem. 1983, 4, 187–217. [Google Scholar] [CrossRef]
- Boyle, N.A.; Rajwanshi, V.K.; Prhavc, M.; Wang, G.; Fagan, P.; Chen, F.; Ewing, G.J.; Brooks, J.L.; Hurd, T.; Leeds, J.M.; et al. Synthesis of 2′,3′-Dideoxynucleoside 5′-α-P-Borano-β,γ-(difluoromethylene)triphosphates and their inhibition of HIV-1 reverse transcriptase. J. Med. Chem. 2005, 48, 2695–2700. [Google Scholar] [CrossRef] [PubMed]
- Inhibitory Constant Against HIV-1 Reverse Transcriptase. Available online: https://pubchem.ncbi.nlm.nih.gov/bioassay/238445 (accessed on 6 May 2016).
- Wu, G.; Robertson, D.H.; Brooks, C.L.; Vieth, M. Detailed analysis of grid-based molecular docking: A case study of CDOCKER—A CHARMm-based MD docking algorithm. J. Comput. Chem. 2003, 24, 1549–1562. [Google Scholar] [CrossRef] [PubMed]
- Ponder, J.W.; Richards, F.M. Tertiary templates for proteins. Use of packing criteria in the enumeration of allowed sequences for different structural classes. J. Mol. Biol. 1987, 193, 775–791. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Structure | 3KK2 −PMF | Model 2 −PMF | Model 3 −PMF | 3V4I −PLP1 | Binding Energy [kcal/mol] |
|---|---|---|---|---|---|---|
| 2 |  | 194.64 | 163.72 | 162.48 | 117.20 | −65.91 |
| 3 | 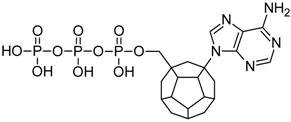 | 165.14 | 179.73 | 165.96 | 130.55 | −69.63 |
| 4 | 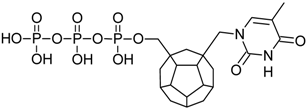 | 189.20 | 191.35 | 169.64 | 115.46 | −71.25 |
| 5 | 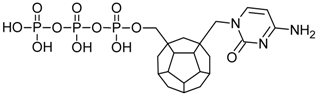 | 185.71 | 199.49 | 164.67 | 116.01 | −67.27 |
| 6 | 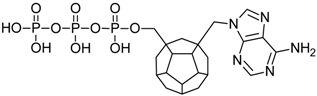 | 197.88 | 173.01 | 185.19 | 114.23 | −70.24 |
| 18 | 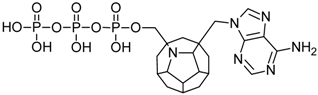 | 174.92 | 163.39 | 149.00 | 133.72 | −66.40 |
| 29 |  | 181.57 | 208.45 | 166.09 | 113.58 | −72.40 |
| 47 |  | 186.98 | 203.10 | 162.20 | 92.20 | −72.27 |
| 72 | 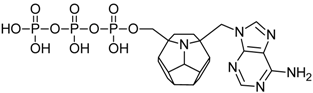 | 168.74 | 187.06 | 174.43 | 120.65 | −76.76 |
| 76 | 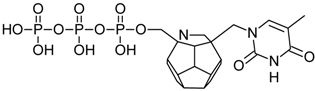 | 162.71 | 185.88 | 173.42 | 86.63 | −71.85 |
| 78 | 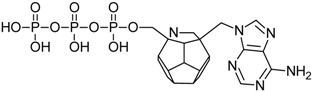 | 197.16 | 193.67 | 183.40 | 95.51 | −68.37 |
| 82 | 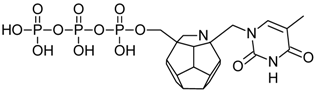 | 185.56 | 192.94 | 167.86 | 133.56 | −73.34 |
| 83 | 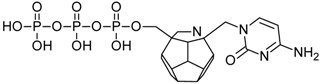 | 189.50 | 198.07 | 172.67 | 78.95 | −65.76 |
| 84 |  | 175.09 | 164.56 | 177.27 | 103.63 | −70.90 |
| 94 | 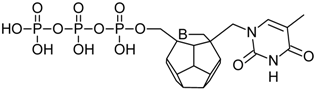 | 184.85 | 191.20 | 168.90 | 97.54 | −66.48 |
| 96 | 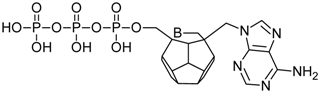 | 167.58 | 205.23 | 173.28 | 104.58 | −73.08 |
| 100 |  | 183.42 | 190.17 | 166.22 | 106.16 | −68.28 |
| 102 |  | 190.89 | 188.32 | 179.27 | 114.29 | −78.33 |
| 106 | 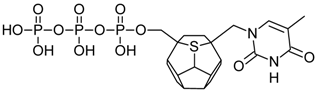 | 190.73 | 174.12 | 168.01 | 101.99 | −72.28 |
| 114 |  | 171.32 | 186.77 | 175.31 | 127.41 | −74.59 |
| 144 |  | 181.78 | 190.96 | 158.44 | 106.51 | −67.94 |
| PDB ID | Ligand | Resolution [Å] |
|---|---|---|
| 1RTD | dTTP | 3.2 |
| 1T05 | Tenofovir diphosphate | 3.0 |
| 3KJV | - | 3.1 |
| 3KK1 | GS-9148 diphosphate | 2.7 |
| 3KK2 | dATP | 2.9 |
| 3V4I | AZTTP | 2.8 |
| Amino Acid | 3KK2 | Model 1 | Model 2 | Model 3 | Model 4 |
|---|---|---|---|---|---|
| Lys65 | −51.24 | −162.38 | - | −68.90 (40.90%) | - |
| Lys66 | 177.09 | −44.71 | - | - | - |
| Asp67 | 89.42 | 48.67 | - | - | - |
| Arg72 | 178.95 | −169.77 | - | −67.60 (46.30%) | - |
| Asp110 | −175.07 | −172.63 | −171.83 | - | - |
| Val111 | 167.86 | 179.67 | 179.75 | - | - |
| Asp113 | −81.13 | 61.08 | −57.58 | - | - |
| Gln151 | −64.55 | −59.13 | - | - | −174.60 (21.10%) |
| Asp185 | 75.22 | 62.93 | 61.09 | - | −68.30 (47.70%) |
| Lys219 | −141.50 | −117.40 | −118.25 | - | −68.90 (40.90%) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dąbrowska, A.; Pieńko, T.; Taciak, P.; Wiktorska, K.; Chilmonczyk, Z.; Mazurek, A.P.; Stasiulewicz, A. Fullerene Derivatives of Nucleoside HIV Reverse Transcriptase Inhibitors—In Silico Activity Prediction. Int. J. Mol. Sci. 2018, 19, 3231. https://doi.org/10.3390/ijms19103231
Dąbrowska A, Pieńko T, Taciak P, Wiktorska K, Chilmonczyk Z, Mazurek AP, Stasiulewicz A. Fullerene Derivatives of Nucleoside HIV Reverse Transcriptase Inhibitors—In Silico Activity Prediction. International Journal of Molecular Sciences. 2018; 19(10):3231. https://doi.org/10.3390/ijms19103231
Chicago/Turabian StyleDąbrowska, Aleksandra, Tomasz Pieńko, Przemysław Taciak, Katarzyna Wiktorska, Zdzisław Chilmonczyk, Aleksander P. Mazurek, and Adam Stasiulewicz. 2018. "Fullerene Derivatives of Nucleoside HIV Reverse Transcriptase Inhibitors—In Silico Activity Prediction" International Journal of Molecular Sciences 19, no. 10: 3231. https://doi.org/10.3390/ijms19103231
APA StyleDąbrowska, A., Pieńko, T., Taciak, P., Wiktorska, K., Chilmonczyk, Z., Mazurek, A. P., & Stasiulewicz, A. (2018). Fullerene Derivatives of Nucleoside HIV Reverse Transcriptase Inhibitors—In Silico Activity Prediction. International Journal of Molecular Sciences, 19(10), 3231. https://doi.org/10.3390/ijms19103231