Targeting Oncogenic Signaling in Mutant FLT3 Acute Myeloid Leukemia: The Path to Least Resistance

 , , ,
, , , 

Abstract
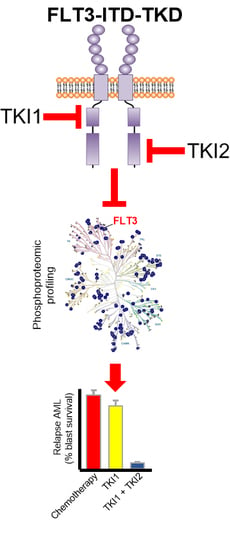
1. Introduction
2. Genomics Underpinning Transformation in AML
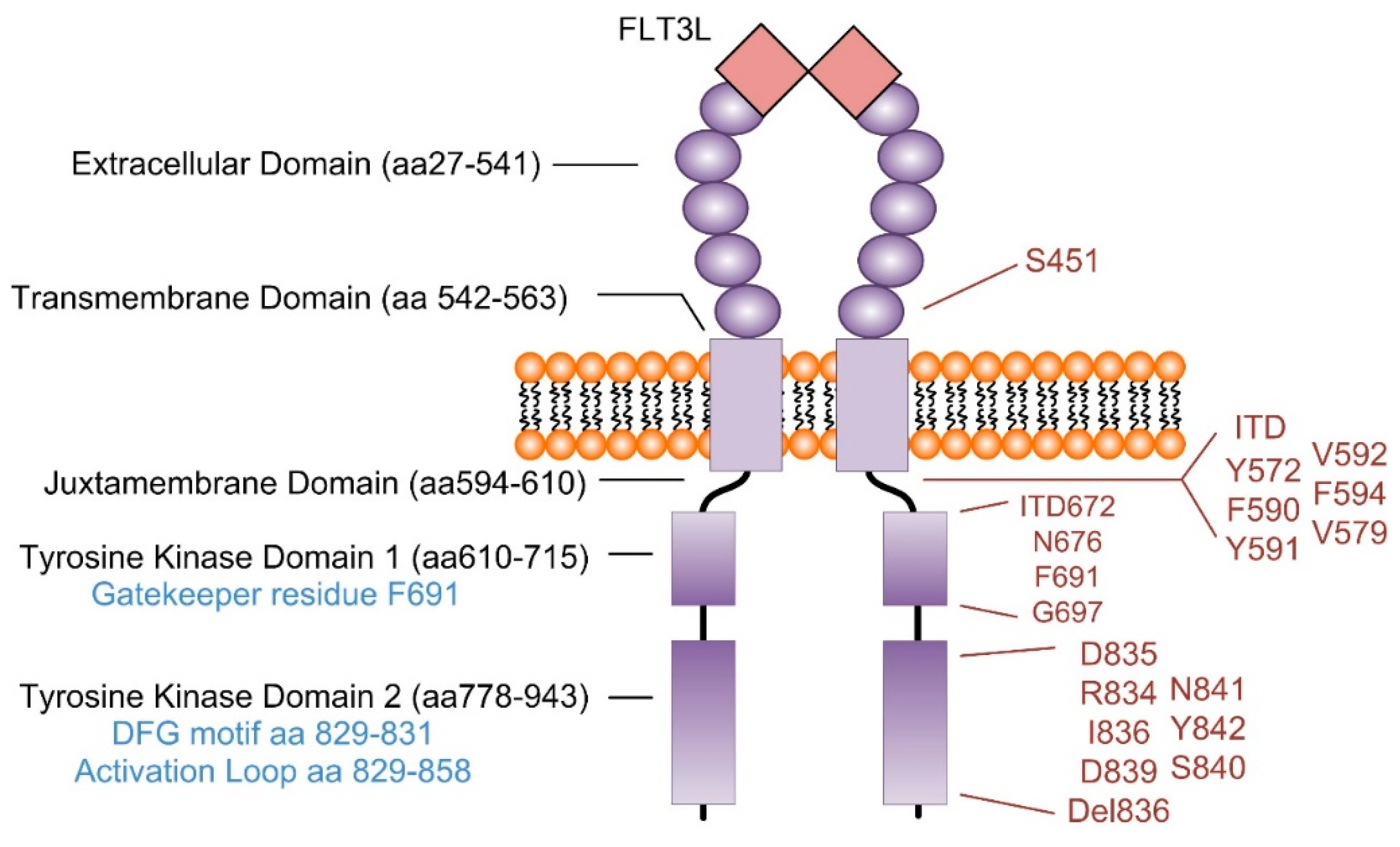
3. FLT3-ITD Mutations Confer a Poor Prognosis in Cytogenetically Normal (CN) AML
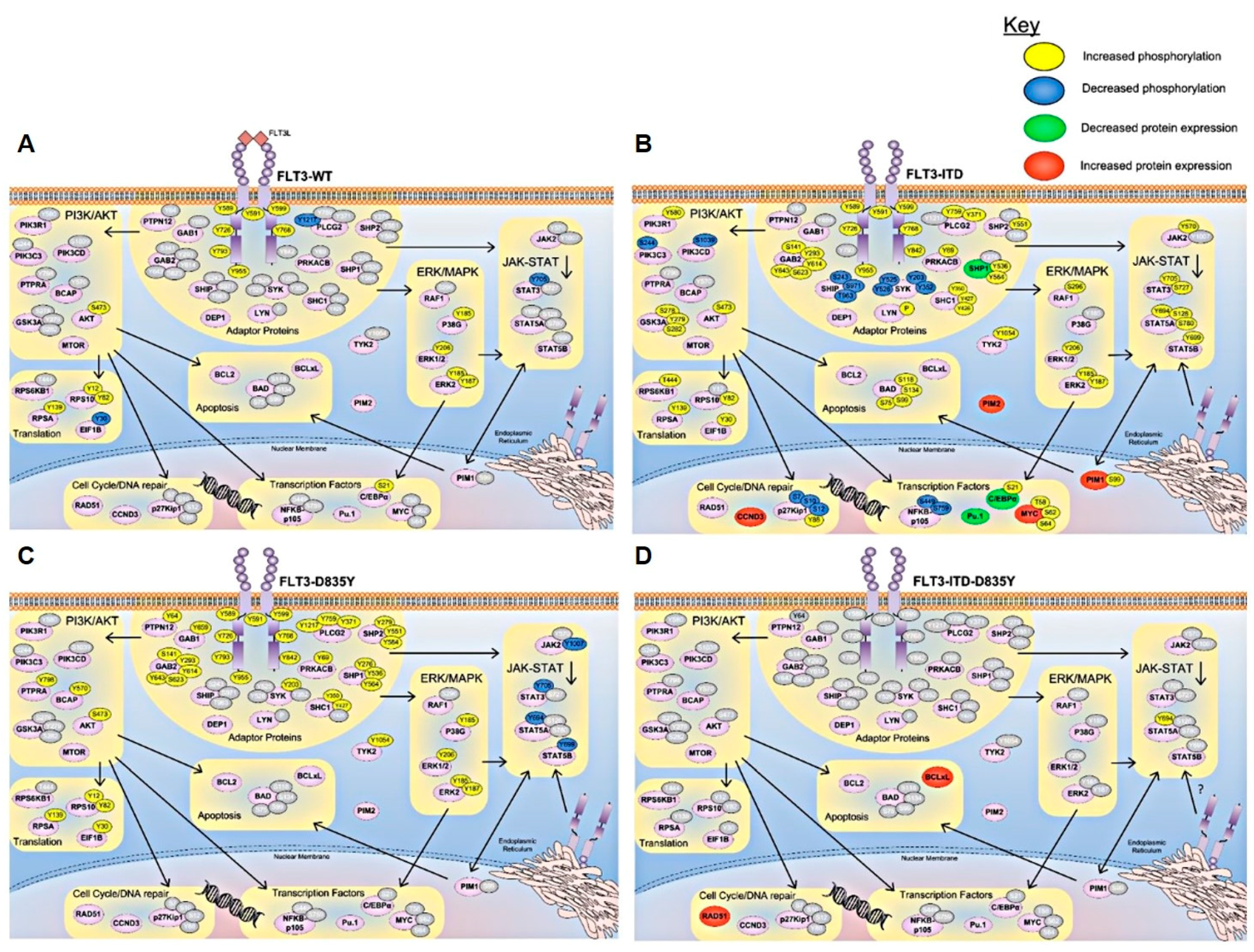
4. Signaling Pathways Regulated in FLT3 Mutant AML
5. Relapse and Resistance is Common in FLT3-ITD AML
6. FLT3 Targeted Therapy
6.1. Tyrosine Kinase Inhibitors in Clinical Development for AML
6.1.1. First-Generation TKIs
6.1.2. Second-Generation TKI
7. Mechanisms of Relapse
7.1. FLT3-Mediated Mechanisms
7.2. Non FLT3-Mediated Mechanisms
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| aa | Amino acid |
| alloHSCT | Allogeneic hematopoietic stem cell transplant |
| AML | Acute myeloid leukemia |
| CK-AML | Complex karyotype AML |
| CML | Chronic myeloid leukemia |
| CN | Cytogenetically normal |
| CR | Complete remission |
| Del | Deletion |
| DFS | Disease-free survival |
| EFS | Event-free survival |
| FLT3 | FMS-like tyrosine kinase 3 |
| FLT3L | FLT3 ligand |
| FLT3-mut | FLT3 mutation |
| HSC | Hematopoietic stem/progenitor cell |
| IL-3 | Interleukin-3 |
| Indels | Small insertions or deletions |
| ITD | Internal tandem duplication |
| LI-C | Leukemia-inducing cells |
| LSCs | Leukemic stem cells |
| MDS | Myelodysplastic syndrome |
| MLL-PTD | MLL-partial tandem duplication |
| MRD | Minimal residual disease |
| OS | Overall survival |
| PDGF | Platelet-derived growth factor |
| PR | Partial remission |
| RFS | Relapse-free survival |
| RTK | Receptor tyrosine kinase |
| TK | Tyrosine kinase |
| TKD | Tyrosine kinase domain |
| TKIs | Tyrosine kinase inhibitors |
References
- Bozic, I.; Antal, T.; Ohtsuki, H.; Carter, H.; Kim, D.; Chen, S.; Karchin, R.; Kinzler, K.W.; Vogelstein, B.; Nowak, M.A. Accumulation of driver and passenger mutations during tumor progression. Proc. Natl. Acad. Sci. USA 2010, 107, 18545–18550. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.M.; Grant, S.; Saleiro, D.; Crispino, J.D.; Hijiya, N.; Giles, F.; Platanias, L.; Eklund, E.A. Targeting novel signaling pathways for resistant acute myeloid leukemia. Mol. Genet. Metab. 2015, 114, 397–402. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute, DCCPS, Surveillance Research Program; Surveillance, Epidemiology, and End Results (SEER) Program Research Data (1973–2015). 2015. Available online: https://www.seer.cancer.gov (accessed on 15 May 2018).
- Dohner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [PubMed]
- Faulk, K.; Gore, L.; Cooper, T. Overview of therapy and strategies for optimizing outcomes in de novo pediatric acute myeloid leukemia. Paediatr. Drugs 2014, 16, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Godley, L.A.; Larson, R.A. Therapy-related myeloid leukemia. Semin. Oncol. 2008, 35, 418–429. [Google Scholar] [CrossRef] [PubMed]
- Welch, J.S.; Ley, T.J.; Link, D.C.; Miller, C.A.; Larson, D.E.; Koboldt, D.C.; Wartman, L.D.; Lamprecht, T.L.; Liu, F.; Xia, J.; et al. The origin and evolution of mutations in acute myeloid leukemia. Cell 2012, 150, 264–278. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research; Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, A.; Hoadley, K.; Triche, T.J., Jr.; Laird, P.W.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [PubMed]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef] [PubMed]
- Hatzimichael, E.; Georgiou, G.; Benetatos, L.; Briasoulis, E. Gene mutations and molecularly targeted therapies in acute myeloid leukemia. Am. J. Blood Res. 2013, 3, 29–51. [Google Scholar] [PubMed]
- Nakao, M.; Yokota, S.; Iwai, T.; Kaneko, H.; Horiike, S.; Kashima, K.; Sonoda, Y.; Fujimoto, T.; Misawa, S. Internal tandem duplication of the flt3 gene found in acute myeloid leukemia. Leukemia 1996, 10, 1911–1918. [Google Scholar] [PubMed]
- Yamamoto, Y.; Kiyoi, H.; Nakano, Y.; Suzuki, R.; Kodera, Y.; Miyawaki, S.; Asou, N.; Kuriyama, K.; Yagasaki, F.; Shimazaki, C.; et al. Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood 2001, 97, 2434–2439. [Google Scholar] [CrossRef] [PubMed]
- Yanada, M.; Matsuo, K.; Suzuki, T.; Kiyoi, H.; Naoe, T. Prognostic significance of FLT3 internal tandem duplication and tyrosine kinase domain mutations for acute myeloid leukemia: A meta-analysis. Leukemia 2005, 19, 1345–1349. [Google Scholar] [CrossRef] [PubMed]
- Thiede, C.; Steudel, C.; Mohr, B.; Schaich, M.; Schakel, U.; Platzbecker, U.; Wermke, M.; Bornhauser, M.; Ritter, M.; Neubauer, A.; et al. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: Association with FAB subtypes and identification of subgroups with poor prognosis. Blood 2002, 99, 4326–4335. [Google Scholar] [CrossRef] [PubMed]
- Whitman, S.P.; Ruppert, A.S.; Radmacher, M.D.; Mrozek, K.; Paschka, P.; Langer, C.; Baldus, C.D.; Wen, J.; Racke, F.; Powell, B.L.; et al. FLT3 D835/I836 mutations are associated with poor disease-free survival and a distinct gene-expression signature among younger adults with de novo cytogenetically normal acute myeloid leukemia lacking FLT3 internal tandem duplications. Blood 2008, 111, 1552–1559. [Google Scholar] [CrossRef] [PubMed]
- Santos, F.P.; Jones, D.; Qiao, W.; Cortes, J.E.; Ravandi, F.; Estey, E.E.; Verma, D.; Kantarjian, H.; Borthakur, G. Prognostic value of FLT3 mutations among different cytogenetic subgroups in acute myeloid leukemia. Cancer 2011, 117, 2145–2155. [Google Scholar] [CrossRef] [PubMed]
- Schlenk, R.F.; Dohner, K.; Krauter, J.; Frohling, S.; Corbacioglu, A.; Bullinger, L.; Habdank, M.; Spath, D.; Morgan, M.; Benner, A.; et al. German-Austrian Acute Myeloid Leukemia Study Group, Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N. Engl. J. Med. 2008, 358, 1909–1918. [Google Scholar] [CrossRef] [PubMed]
- Mead, A.J.; Linch, D.C.; Hills, R.K.; Wheatley, K.; Burnett, A.K.; Gale, R.E. FLT3 tyrosine kinase domain mutations are biologically distinct from and have a significantly more favorable prognosis than FLT3 internal tandem duplications in patients with acute myeloid leukemia. Blood 2007, 110, 1262–1270. [Google Scholar] [CrossRef] [PubMed]
- Mead, A.J.; Gale, R.E.; Hills, R.K.; Gupta, M.; Young, B.D.; Burnett, A.K.; Linch, D.C. Conflicting data on the prognostic significance of FLT3/TKD mutations in acute myeloid leukemia might be related to the incidence of biallelic disease. Blood 2008, 112, 444–445. [Google Scholar] [CrossRef] [PubMed]
- Frohling, S.; Scholl, C.; Levine, R.L.; Loriaux, M.; Boggon, T.J.; Bernard, O.A.; Berger, R.; Dohner, H.; Dohner, K.; Ebert, B.L.; et al. Identification of driver and passenger mutations of FLT3 by high-throughput DNA sequence analysis and functional assessment of candidate alleles. Cancer Cell 2007, 12, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Reindl, C.; Bagrintseva, K.; Vempati, S.; Schnittger, S.; Ellwart, J.W.; Wenig, K.; Hopfner, K.P.; Hiddemann, W.; Spiekermann, K. Point mutations in the juxtamembrane domain of FLT3 define a new class of activating mutations in AML. Blood 2006, 107, 3700–3707. [Google Scholar] [CrossRef] [PubMed]
- Janke, H.; Pastore, F.; Schumacher, D.; Herold, T.; Hopfner, K.P.; Schneider, S.; Berdel, W.E.; Buchner, T.; Woermann, B.J.; Subklewe, M.; et al. Activating FLT3 mutants show distinct gain-of-function phenotypes in vitro and a characteristic signaling pathway profile associated with prognosis in acute myeloid leukemia. PLoS ONE 2014, 9, e89560. [Google Scholar] [CrossRef] [PubMed]
- Breitenbuecher, F.; Markova, B.; Kasper, S.; Carius, B.; Stauder, T.; Bohmer, F.D.; Masson, K.; Ronnstrand, L.; Huber, C.; Kindler, T.; et al. A novel molecular mechanism of primary resistance to FLT3-kinase inhibitors in AML. Blood 2009, 113, 4063–4073. [Google Scholar] [CrossRef] [PubMed]
- Heidel, F.; Solem, F.K.; Breitenbuecher, F.; Lipka, D.B.; Kasper, S.; Thiede, M.H.; Brandts, C.; Serve, H.; Roesel, J.; Giles, F.; et al. Clinical resistance to the kinase inhibitor PKC412 in acute myeloid leukemia by mutation of Asn-676 in the FLT3 tyrosine kinase domain. Blood 2006, 107, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Cools, J.; Mentens, N.; Furet, P.; Fabbro, D.; Clark, J.J.; Griffin, J.D.; Marynen, P.; Gilliland, D.G. Prediction of resistance to small molecule FLT3 inhibitors: Implications for molecularly targeted therapy of acute leukemia. Cancer Res. 2004, 64, 6385–6389. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Singh, A.; Kumari, A.; Pandey, B.; Jamal, S.; Goyal, S.; Sinha, S.; Grover, A. Insight into the inhibitor discrimination by FLT3 F691L. Chem. Biol. Drug Design 2018, 91, 1056–1064. [Google Scholar] [CrossRef] [PubMed]
- Spiekermann, K.; Bagrintseva, K.; Schoch, C.; Haferlach, T.; Hiddemann, W.; Schnittger, S. A new and recurrent activating length mutation in exon 20 of the FLT3 gene in acute myeloid leukemia. Blood 2002, 100, 3423–3425. [Google Scholar] [CrossRef] [PubMed]
- Kindler, T.; Breitenbuecher, F.; Kasper, S.; Estey, E.; Giles, F.; Feldman, E.; Ehninger, G.; Schiller, G.; Klimek, V.; Nimer, S.D.; et al. Identification of a novel activating mutation (Y842C) within the activation loop of FLT3 in patients with acute myeloid leukemia (AML). Blood 2005, 105, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Paez, J.G.; Lee, J.C.; Bo, R.; Stone, R.M.; DeAngelo, D.J.; Galinsky, I.; Wolpin, B.M.; Jonasova, A.; Herman, P.; et al. Identifying and characterizing a novel activating mutation of the FLT3 tyrosine kinase in AML. Blood 2004, 104, 1855–1858. [Google Scholar] [CrossRef] [PubMed]
- Larrosa-Garcia, M.; Baer, M.R. FLT3 Inhibitors in Acute Myeloid Leukemia: Current Status and Future Directions. Mol. Cancer Ther. 2017, 16, 991–1001. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.D.; Zimmerman, E.I.; Wang, Y.D.; Orwick, S.; Zatechka, D.S.; Buaboonnam, J.; Neale, G.A.; Olsen, S.R.; Enemark, E.J.; Shurtleff, S.; et al. Emergence of polyclonal FLT3 tyrosine kinase domain mutations during sequential therapy with sorafenib and sunitinib in FLT3-ITD-positive acute myeloid leukemia. Clin. Cancer Res. 2013, 19, 5758–5768. [Google Scholar] [CrossRef] [PubMed]
- Konig, H.; Levis, M. Targeting FLT3 to treat leukemia. Expert Opin. Ther. Targets 2015, 19, 37–54. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.B.; Nguyen, B.; Li, L.; Brown, P.; Levis, M.; Leahy, D.; Small, D. Mutations of FLT3/ITD confer resistance to multiple tyrosine kinase inhibitors. Leukemia 2013, 27, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Alvarado, Y.; Kantarjian, H.M.; Luthra, R.; Ravandi, F.; Borthakur, G.; Garcia-Manero, G.; Konopleva, M.; Estrov, Z.; Andreeff, M.; Cortes, J.E. Treatment with FLT3 inhibitor in patients with FLT3-mutated acute myeloid leukemia is associated with development of secondary FLT3-tyrosine kinase domain mutations. Cancer 2014, 120, 2142–2149. [Google Scholar] [CrossRef] [PubMed]
- Stone, R.M.; Mandrekar, S.J.; Sanford, B.L.; Laumann, K.; Geyer, S.; Bloomfield, C.D.; Thiede, C.; Prior, T.W.; Dohner, K.; Marcucci, G.; et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N. Engl. J. Med. 2017, 377, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Levis, M. Midostaurin approved for FLT3-mutated AML. Blood 2017, 129, 3403–3406. [Google Scholar] [CrossRef] [PubMed]
- Sekeres, M.A.; Steensma, D.P. Boulevard of broken dreams: Drug approval for older adults with acute myeloid leukemia. J. Clin. Oncol. 2012, 30, 4061–4063. [Google Scholar] [CrossRef] [PubMed]
- Faulkner, S.; Dun, M.D.; Hondermarck, H. Proteogenomics: Emergence and promise. Cell. Mol. Life Sci. 2015, 72, 953–957. [Google Scholar] [CrossRef] [PubMed]
- Murray, H.C.; Dun, M.D.; Verrills, N.M. Harnessing the power of proteomics for identification of oncogenic, druggable signalling pathways in cancer. Expert Opin. Drug Discov. 2017, 12, 431–447. [Google Scholar] [CrossRef] [PubMed]
- Degryse, S.; de Bock, C.E.; Demeyer, S.; Govaerts, I.; Bornschein, S.; Verbeke, D.; Jacobs, K.; Binos, S.; Skerrett-Byrne, D.A.; Murray, H.C.; et al. Mutant JAK3 phosphoproteomic profiling predicts synergism between JAK3 inhibitors and MEK/BCL2 inhibitors for the treatment of T-cell acute lymphoblastic leukemia. Leukemia 2018, 32, 788–800. [Google Scholar] [CrossRef] [PubMed]
- Nixon, B.; Johnston, S.D.; Skerrett-Byrne, D.A.; Anderson, A.L.; Stanger, S.J.; Bromfield, E.G.; Martin, J.H.; Hansbro, P.M.; Dun, M.D. Proteomic profiling of crocodile spermatozoa refutes the tenet that post-testicular maturation is restricted to mammals. Mol. Cell. Proteom. 2018. [Google Scholar] [CrossRef] [PubMed]
- Bagrintseva, K.; Schwab, R.; Kohl, T.M.; Schnittger, S.; Eichenlaub, S.; Ellwart, J.W.; Hiddemann, W.; Spiekermann, K. Mutations in the tyrosine kinase domain of FLT3 define a new molecular mechanism of acquired drug resistance to PTK inhibitors in FLT3-ITD-transformed hematopoietic cells. Blood 2004, 103, 2266–2275. [Google Scholar] [CrossRef] [PubMed]
- Dash, A.; Gilliland, D.G. Molecular genetics of acute myeloid leukaemia. Best Pract. Res. Clin. Haematol. 2001, 14, 49–64. [Google Scholar] [CrossRef] [PubMed]
- Frohling, S.; Scholl, C.; Gilliland, D.G.; Levine, R.L. Genetics of Myeloid Malignancies: Pathogenetic and Clinical Implications. J. Clin. Oncol. 2005, 23, 6285–6295. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Lu, C.; Wang, J.; McLellan, M.D.; Johnson, K.J.; Wendl, M.C.; McMichael, J.F.; Schmidt, H.K.; Yellapantula, V.; Miller, C.A.; et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 2014, 20, 1472–1478. [Google Scholar] [CrossRef] [PubMed]
- Abelson, S.; Collord, G.; Ng, S.W.K.; Weissbrod, O.; Mendelson Cohen, N.; Niemeyer, E.; Barda, N.; Zuzarte, P.C.; Heisler, L.; Sundaravadanam, Y.; et al. Prediction of acute myeloid leukaemia risk in healthy individuals. Nature 2018, 559, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Desai, P.; Mencia-Trinchant, N.; Savenkov, O.; Simon, M.S.; Cheang, G.; Lee, S.; Samuel, M.; Ritchie, E.K.; Guzman, M.L.; Ballman, K.V.; et al. Somatic mutations precede acute myeloid leukemia years before diagnosis. Nat. Med. 2018, 24, 1015–1023. [Google Scholar] [CrossRef] [PubMed]
- Corces-Zimmerman, M.R.; Majeti, R. Pre-leukemic evolution of hematopoietic stem cells: The importance of early mutations in leukemogenesis. Leukemia 2014, 28, 2276–2282. [Google Scholar] [CrossRef] [PubMed]
- Corces, M.R.; Chang, H.Y.; Majeti, R. Preleukemic Hematopoietic Stem Cells in Human Acute Myeloid Leukemia. Front. Oncol. 2017, 7, 263. [Google Scholar] [CrossRef] [PubMed]
- Kelly, L.M.; Gilliland, D.G. Genetics of myeloid leukemias. Annu. Rev. Genom. Hum. Gen. 2002, 3, 179–198. [Google Scholar] [CrossRef] [PubMed]
- Walter, M.J.; Shen, D.; Ding, L.; Shao, J.; Koboldt, D.C.; Chen, K.; Larson, D.E.; McLellan, M.D.; Dooling, D.; Abbott, R.; et al. Clonal architecture of secondary acute myeloid leukemia. N. Engl. J. Med. 2012, 366, 1090–1098. [Google Scholar] [CrossRef] [PubMed]
- Sansregret, L.; Vanhaesebroeck, B.; Swanton, C. Determinants and clinical implications of chromosomal instability in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Meng, C.Y.; Noor, P.J.; Ismail, A.; Ahid, M.F.; Zakaria, Z. Cytogenetic Profile of de novo Acute Myeloid Leukemia Patients in Malaysia. Int. J. Biomed. Sci. 2013, 9, 26–32. [Google Scholar] [PubMed]
- Mrozek, K. Cytogenetic, molecular genetic, and clinical characteristics of acute myeloid leukemia with a complex karyotype. Semin. Oncol. 2008, 35, 365–377. [Google Scholar] [CrossRef] [PubMed]
- Dohner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Buchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [PubMed]
- Warner, J.K.; Wang, J.C.; Hope, K.J.; Jin, L.; Dick, J.E. Concepts of human leukemic development. Oncogene 2004, 23, 7164–7177. [Google Scholar] [CrossRef] [PubMed]
- Grimwade, D.; Hills, R.K.; Moorman, A.V.; Walker, H.; Chatters, S.; Goldstone, A.H.; Wheatley, K.; Harrison, C.J.; Burnett, A.K. National Cancer Research Institute Adult Leukaemia Working; Refinement of cytogenetic classification in acute myeloid leukemia: Determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood 2010, 116, 354–365. [Google Scholar] [PubMed]
- Grimwade, D.; Walker, H.; Oliver, F.; Wheatley, K.; Harrison, C.; Harrison, G.; Rees, J.; Hann, I.; Stevens, R.; Burnett, A.; et al. The importance of diagnostic cytogenetics on outcome in AML: Analysis of 1,612 patients entered into the MRC AML 10 trial. The Medical Research Council Adult and Children′s Leukaemia Working Parties. Blood 1998, 92, 2322–2333. [Google Scholar] [PubMed]
- Schnittger, S.; Schoch, C.; Dugas, M.; Kern, W.; Staib, P.; Wuchter, C.; Loffler, H.; Sauerland, C.M.; Serve, H.; Buchner, T.; et al. Analysis of FLT3 length mutations in 1003 patients with acute myeloid leukemia: Correlation to cytogenetics, FAB subtype, and prognosis in the AMLCG study and usefulness as a marker for the detection of minimal residual disease. Blood 2002, 100, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Grafone, T.; Palmisano, M.; Nicci, C.; Storti, S. An overview on the role of FLT3-tyrosine kinase receptor in acute myeloid leukemia: Biology and treatment. Oncol. Rev. 2012, 6, e8. [Google Scholar] [CrossRef] [PubMed]
- Whitman, S.P.; Maharry, K.; Radmacher, M.D.; Becker, H.; Mrozek, K.; Margeson, D.; Holland, K.B.; Wu, Y.Z.; Schwind, S.; Metzeler, K.H.; et al. FLT3 internal tandem duplication associates with adverse outcome and gene- and microRNA-expression signatures in patients 60 years of age or older with primary cytogenetically normal acute myeloid leukemia: A Cancer and Leukemia Group B study. Blood 2010, 116, 3622–3626. [Google Scholar] [CrossRef] [PubMed]
- Garg, M.; Nagata, Y.; Kanojia, D.; Mayakonda, A.; Yoshida, K.; Keloth, S.H.; Zang, Z.J.; Okuno, Y.; Shiraishi, Y.; Chiba, K.; et al. Profiling of somatic mutations in acute myeloid leukemia with FLT3-ITD at diagnosis and relapse. Blood 2015, 126, 2491–2501. [Google Scholar] [CrossRef] [PubMed]
- Marcucci, G.; Haferlach, T.; Dohner, H. Molecular genetics of adult acute myeloid leukemia: Prognostic and therapeutic implications. J. Clin. Oncol. 2011, 29, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Markova, J.; Michkova, P.; Burcˇkova, K.; Brˇezinova, J.; Michalova, K.; Dohnalova, A.; Maaloufova, J.S.; Soukup, P.; Vıxtek, A.; Cetkovsky, P.; et al. Prognostic impact of DNMT3A mutations in patients with intermediate cytogenetic risk profile acute myeloid leukemia. Eur. J. Haematol. 2011, 88, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Meyer, S.E.; Qin, T.; Muench, D.E.; Masuda, K.; Venkatasubramanian, M.; Orr, E.; Suarez, L.; Gore, S.D.; Delwel, R.; Paietta, E.; et al. DNMT3A Haploinsufficiency Transforms FLT3ITD Myeloproliferative Disease into a Rapid, Spontaneous, and Fully Penetrant Acute Myeloid Leukemia. Cancer Discov. 2016, 6, 501–515. [Google Scholar] [CrossRef] [PubMed]
- Loghavi, S.; Zuo, Z.; Ravandi, F.; Kantarjian, H.M.; Bueso-Ramos, C.; Zhang, L.; Singh, R.R.; Patel, K.P.; Medeiros, L.J.; Stingo, F.; et al. Clinical features of De Novo acute myeloidleukemia with concurrent DNMT3A, FLT3 and NPM1 mutations. J. Hematol. Oncol. 2014, 7, 74. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Liu, Y.; Hou, G.; Wang, L.; Na, L.; Xu, Y.; Xu, Y.; Wang, X.; Xuan, Z.; Jing, Y.; Li, H.; et al. Mutational spectrum and risk stratification of intermediate-risk acute myeloid leukemia patients based on next-generation sequencing. Oncotarget 2016, 7, 32065–32078. [Google Scholar] [CrossRef] [PubMed]
- Zorko, N.A.; Bernot, K.M.; Whitman, S.P.; Siebenaler, R.F.; Ahmed, E.H.; Marcucci, G.G.; Yanes, D.A.; McConnell, K.K.; Mao, C.; Kalu, C.; et al. Mll partial tandem duplication and Flt3 internal tandem duplication in a double knock-in mouse recapitulates features of counterpart human acute myeloid leukemias. Blood 2012, 120, 1130–1136. [Google Scholar] [CrossRef] [PubMed]
- Wakita1, S.; Yamaguchi1, H.; Omori, I.; Terada, K.; Ueda, T.; Manabe, E.; Kurosawa, S.; Iida, S.; Ibaraki, T.; Sato, Y.; et al. Mutations of the epigenetics-modifying gene (DNMT3a, TET2, IDH1/2) at diagnosis may induce FLT3-ITD at relapse in de novo acute myeloid leukemia. Leukemia 2013, 27, 1044–1052. [Google Scholar] [CrossRef] [PubMed]
- Abu-Duhier, F.M.; Goodeve, A.C.; Wilson, G.A.; Care, R.S.; Peake, I.R.; Reilly, J.T. Identification of novel FLT-3 Asp835 mutations in adult acute myeloid leukaemia. Br. J. Haematol. 2001, 113, 983–988. [Google Scholar] [CrossRef] [PubMed]
- Bacher, U.; Haferlach, C.; Kern, W.; Haferlach, T.; Schnittger, S. Prognostic relevance of FLT3-TKD mutations in AML: The combination matters—An analysis of 3082 patients. Blood 2008, 111, 2527–2537. [Google Scholar] [CrossRef] [PubMed]
- Bagrintseva, K.; Geisenhof, S.; Kern, R.; Eichenlaub, S.; Reindl, C.; Ellwart, J.W.; Hiddemann, W.; Spiekermann, K. FLT3-ITD-TKD dual mutants associated with AML confer resistance to FLT3 PTK inhibitors and cytotoxic agents by overexpression of Bcl-x(L). Blood 2005, 105, 3679–3685. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.C.; Wang, Q.; Chin, C.S.; Salerno, S.; Damon, L.E.; Levis, M.J.; Perl, A.E.; Travers, K.J.; Wang, S.; Hunt, J.P.; et al. Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature 2012, 485, 260–263. [Google Scholar] [CrossRef] [PubMed]
- Levis, M. FLT3 mutations in acute myeloid leukemia: What is the best approach in 2013? Hematol. ASH Educ. Prog. 2013, 2013, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Stirewalt, D.L.; Radich, J.P. The role of FLT3 in haematopoietic malignancies. Nat. Rev. Cancer 2003, 3, 650–665. [Google Scholar] [CrossRef] [PubMed]
- Verstraete, K.; Savvides, S.N. Extracellular assembly and activation principles of oncogenic class III receptor tyrosine kinases. Nat. Rev. Cancer 2012, 12, 753–766. [Google Scholar] [CrossRef] [PubMed]
- Griffith, J.; Black, J.; Faerman, C.; Swenson, L.; Wynn, M.; Lu, F.; Lippke, J.; Saxena, K. The structural basis for autoinhibition of FLT3 by the juxtamembrane domain. Mol. Cell 2004, 13, 169–178. [Google Scholar] [CrossRef]
- Weiss, A.; Schlessinger, J. Switching signals on or off by receptor dimerization. Cell 1998, 94, 277–280. [Google Scholar] [CrossRef]
- Rottapel, R.; Turck, C.W.; Casteran, N.; Liu, X.; Birnbaum, D.; Pawson, T.; Dubreuil, P. Substrate specificities and identification of a putative binding site for PI3K in the carboxy tail of the murine Flt3 receptor tyrosine kinase. Oncogene 1994, 9, 1755–1765. [Google Scholar] [PubMed]
- Heiss, E.; Masson, K.; Sundberg, C.; Pedersen, M.; Sun, J.; Bengtsson, S.; Ronnstrand, L. Identification of Y589 and Y599 in the juxtamembrane domain of Flt3 as ligand-induced autophosphorylation sites involved in binding of Src family kinases and the protein tyrosine phosphatase SHP2. Blood 2006, 108, 1542–1550. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Mantel, C.; Broxmeyer, H.E. Flt3 signaling involves tyrosyl-phosphorylation of SHP-2 and SHIP and their association with Grb2 and Shc in Baf3/Flt3 cells. J. Leukoc. Biol. 1999, 65, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Fukuda, S.; Lee, Y.; Hangoc, G.; Cooper, S.; Spolski, R.; Leonard, W.J.; Broxmeyer, H.E. Essential role of signal transducer and activator of transcription (Stat)5a but not Stat5b for Flt3-dependent signaling. J. Exp. Med. 2000, 192, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Broxmeyer, H.E. Flt3 ligand induces tyrosine phosphorylation of gab1 and gab2 and their association with shp-2, grb2, and PI3 kinase. Biochem. Biophys. Res. Commun. 2000, 277, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Rosnet, O.; Schiff, C.; Pebusque, M.-J.; Marchetto, S.; Tonnelle, C.; Toiron, Y.; Birg, F.; Birnbaum, D. Human FLT3/FLK2 Gene: CDNA Cloning and Expression in Hematopoietic Cells. Blood J. 1993, 82, 1110–1119. [Google Scholar]
- Gabbianelli, M.; Pelosi, E.; Montesoro, E.; Valtieri, M.; Luchetti, L.; Samoggia, P.; Vitelli, L.; Barberi, T.; Testa, U.; Lyman, S. Multi-level effects of flt3 ligand on human hematopoiesis: Expansion of putative stem cells and proliferation of granulomonocytic progenitors/monocytic precursors. Blood 1995, 86, 1661–1670. [Google Scholar] [PubMed]
- Ratajczak, M.Z.; Ratajczak, J.; Ford, J.; Kregenow, R.; Marlicz, W.; Gewirtz, A.M. FLT3/FLK-2 (STK-1) Ligand does not stimulate human megakaryopoiesis in vitro. Stem Cells 1996, 14, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Turner, A.M.; Lin, N.L.; Issarachai, S.; Lyman, S.D.; Broudy, V.C. FLT3 receptor expression on the surface of normal and malignant human hematopoietic cells. Blood 1996, 88, 3383–3390. [Google Scholar] [PubMed]
- Carow, C.E.; Levenstein, M.; Kaufmann, S.H.; Chen, J.; Amin, S.; Rockwell, P.; Witte, L.; Borowitz, M.J.; Civin, C.I.; Small, D. Expression of the hematopoietic growth factor receptor FLT3 (STK-1/Flk2) in human leukemias. Blood 1996, 87, 1089–1096. [Google Scholar] [PubMed]
- Kiyoi, H.; Towatari, M.; Yokota, S.; Hamaguchi, M.; Ohno, R.; Saito, H.; Naoe, T. Internal tandem duplication of the FLT3 gene is a novel modality of elongation mutation which causes constitutive activation of the product. Leukemia 1998, 12, 1333–1337. [Google Scholar] [CrossRef] [PubMed]
- Kiyoi, H.; Ohno, R.; Ueda, R.; Saito, H.; Naoe, T. Mechanism of constitutive activation of FLT3 with internal tandem duplication in the juxtamembrane domain. Oncogene 2002, 21, 2555–2563. [Google Scholar] [CrossRef] [PubMed]
- Kelly, L.M.; Liu, Q.; Kutok, J.L.; Williams, I.R.; Boulton, C.L.; Gilliland, D.G. FLT3 internal tandem duplication mutations associated with human acute myeloid leukemias induce myeloproliferative disease in a murine bone marrow transplant model. Blood 2002, 99, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Piloto, O.; Nguyen, H.B.; Greenberg, K.; Takamiya, K.; Racke, F.; Huso, D.; Small, D. Knock-in of an internal tandem duplication mutation into murine FLT3 confers myeloproliferative disease in a mouse model. Blood 2008, 111, 3849–3858. [Google Scholar] [CrossRef] [PubMed]
- Chu, S.H.; Heiser, D.; Li, L.; Kaplan, I.; Collector, M.; Huso, D.; Sharkis, S.J.; Civin, C.; Small, D. FLT3-ITD knockin impairs hematopoietic stem cell quiescence/homeostasis, leading to myeloproliferative neoplasm. Cell Stem Cell 2012, 11, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Kelly, L.M.; Kutok, J.L.; Williams, I.R.; Boulton, C.L.; Amaral, S.M.; Curley, D.P.; Ley, T.J.; Gilliland, D.G. PML/RARalpha and FLT3-ITD induce an APL-like disease in a mouse model. Proc. Natl. Acad. Sci. USA 2002, 99, 8283–8288. [Google Scholar] [CrossRef] [PubMed]
- Greenblatt, S.; Li, L.; Slape, C.; Nguyen, B.; Novak, R.; Duffield, A.; Huso, D.; Desiderio, S.; Borowitz, M.J.; Aplan, P.; et al. Knock-in of a FLT3/ITD mutation cooperates with a NUP98-HOXD13 fusion to generate acute myeloid leukemia in a mouse model. Blood 2012, 119, 2883–2894. [Google Scholar] [CrossRef] [PubMed]
- Poitras, J.L.; Heiser, D.; Li, L.; Nguyen, B.; Nagai, K.; Duffield, A.S.; Gamper, C.; Small, D. Dnmt3a deletion cooperates with the Flt3/ITD mutation to drive leukemogenesis in a murine model. Oncotarget 2016, 7, 69124–69135. [Google Scholar] [CrossRef] [PubMed]
- Grundler, R.; Miething, C.; Thiede, C.; Peschel, C.; Duyster, J. FLT3-ITD and tyrosine kinase domain mutants induce 2 distinct phenotypes in a murine bone marrow transplantation model. Blood 2005, 105, 4792–4799. [Google Scholar] [CrossRef] [PubMed]
- Bailey, E.; Li, L.; Duffield, A.S.; Ma, H.S.; Huso, D.L.; Small, D. FLT3/D835Y mutation knock-in mice display less aggressive disease compared with FLT3/internal tandem duplication (ITD) mice. Proc. Natl. Acad. Sci. USA 2013, 110, 21113–21118. [Google Scholar] [CrossRef] [PubMed]
- Razumovskaya, E.; Masson, K.; Khan, R.; Bengtsson, S.; Ronnstrand, L. Oncogenic Flt3 receptors display different specificity and kinetics of autophosphorylation. Exp. Hematol. 2009, 37, 979–989. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Askenazi, M.; Jiang, J.; Luckey, C.J.; Griffin, J.D.; Marto, J.A. A robust error model for iTRAQ quantification reveals divergent signaling between oncogenic FLT3 mutants in acute myeloid leukemia. Mol. Cell. Proteom. 2010, 9, 780–790. [Google Scholar] [CrossRef] [PubMed]
- Puissant, A.; Fenouille, N.; Alexe, G.; Pikman, Y.; Bassil, C.F.; Mehta, S.; Du, J.; Kazi, J.U.; Luciano, F.; Ronnstrand, L.; et al. SYK is a critical regulator of FLT3 in acute myeloid leukemia. Cancer Cell 2014, 25, 226–242. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, C.; Olsen, J.V.; Brandts, C.; Cox, J.; Reddy, P.N.; Bohmer, F.D.; Gerke, V.; Schmidt-Arras, D.E.; Berdel, W.E.; Muller-Tidow, C.; et al. Mislocalized activation of oncogenic RTKs switches downstream signaling outcomes. Mol. Cell 2009, 36, 326–339. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.T.; Baird, K.; Ahn, J.Y.; Meltzer, P.; Lilly, M.; Levis, M.; Small, D. Pim-1 is up-regulated by constitutively activated FLT3 and plays a role in FLT3-mediated cell survival. Blood 2005, 105, 1759–1767. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, C.; Schwable, J.; Brandts, C.; Tickenbrock, L.; Sargin, B.; Kindler, T.; Fischer, T.; Berdel, W.E.; Muller-Tidow, C.; Serve, H. AML-associated Flt3 kinase domain mutations show signal transduction differences compared with Flt3 ITD mutations. Blood 2005, 106, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Mizuki, M.; Schwable, J.; Steur, C.; Choudhary, C.; Agrawal, S.; Sargin, B.; Steffen, B.; Matsumura, I.; Kanakura, Y.; Bohmer, F.D.; et al. Suppression of myeloid transcription factors and induction of STAT response genes by AML-specific Flt3 mutations. Blood 2003, 101, 3164–3173. [Google Scholar] [CrossRef] [PubMed]
- Radomska, H.S.; Basseres, D.S.; Zheng, R.; Zhang, P.; Dayaram, T.; Yamamoto, Y.; Sternberg, D.W.; Lokker, N.; Giese, N.A.; Bohlander, S.K.; et al. Block of C/EBP alpha function by phosphorylation in acute myeloid leukemia with FLT3 activating mutations. J. Exp. Med. 2006, 203, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Peschel, I.; Podmirseg, S.R.; Taschler, M.; Duyster, J.; Gotze, K.S.; Sill, H.; Nachbaur, D.; Jakel, H.; Hengst, L. FLT3 and FLT3-ITD phosphorylate and inactivate the cyclin-dependent kinase inhibitor p27(Kip1) in acute myeloid leukemia. Haematologica 2017, 102, 1378–1389. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.T.; Levis, M.; Small, D. Constitutively activated FLT3 phosphorylates BAD partially through pim-1. Br. J. Haematol. 2006, 134, 500–509. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Levis, M.; Brown, P.; Kim, K.T.; Allebach, J.; Small, D. FLT3/ITD mutation signaling includes suppression of SHP-1. J. Biol. Chem. 2005, 280, 5361–5369. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, M.; Hayakawa, F.; Miyata, Y.; Watamoto, K.; Emi, N.; Abe, A.; Kiyoi, H.; Towatari, M.; Naoe, T. Lyn is an important component of the signal transduction pathway specific to FLT3/ITD and can be a therapeutic target in the treatment of AML with FLT3/ITD. Leukemia 2007, 21, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Arras, D.; Bohmer, S.A.; Koch, S.; Muller, J.P.; Blei, L.; Cornils, H.; Bauer, R.; Korasikha, S.; Thiede, C.; Bohmer, F.D. Anchoring of FLT3 in the endoplasmic reticulum alters signaling quality. Blood 2009, 113, 3568–3576. [Google Scholar] [CrossRef] [PubMed]
- Klingmuller, U.; Lorenz, U.; Cantley, L.C.; Neel, B.G.; Lodish, H.F. Specific recruitment of SH-PTP1 to the erythropoietin receptor causes inactivation of JAK2 and termination of proliferative signals. Cell 1995, 80, 729–738. [Google Scholar] [CrossRef]
- Weisberg, E.; Liu, Q.; Nelson, E.; Kung, A.L.; Christie, A.L.; Bronson, R.; Sattler, M.; Sanda, T.; Zhao, Z.; Hur, W.; et al. Using combination therapy to override stromal-mediated chemoresistance in mutant FLT3-positive AML: Synergism between FLT3 inhibitors, dasatinib/multi-targeted inhibitors and JAK inhibitors. Leukemia 2012, 26, 2233–2244. [Google Scholar] [CrossRef] [PubMed]
- Wiernik, P.H.; Banks, P.L.; Case, D.C., Jr.; Arlin, Z.A.; Periman, P.O.; Todd, M.B.; Ritch, P.S.; Enck, R.E.; Weitberg, A.B. Cytarabine plus idarubicin or daunorubicin as induction and consolidation therapy for previously untreated adult patients with acute myeloid leukemia. Blood 1992, 79, 313–319. [Google Scholar] [PubMed]
- Mayer, R.J.; Davis, R.B.; Schiffer, C.A.; Berg, D.T.; Powell, B.L.; Schulman, P.; Omura, G.A.; Moore, J.O.; McIntyre, O.R.; Frei, E., 3rd. Intensive postremission chemotherapy in adults with acute myeloid leukemia. Cancer and Leukemia Group B. N. Engl. J. Med. 1994, 331, 896–903. [Google Scholar] [CrossRef] [PubMed]
- Bloomfield, C.D.; Lawrence, D.; Byrd, J.C.; Carroll, A.; Pettenati, M.J.; Tantravahi, R.; Patil, S.R.; Davey, F.R.; Berg, D.T.; Schiffer, C.A.; et al. Frequency of prolonged remission duration after high-dose cytarabine intensification in acute myeloid leukemia varies by cytogenetic subtype. Cancer Res. 1998, 58, 4173–4179. [Google Scholar] [PubMed]
- Byrd, J.C.; Ruppert, A.S.; Mrozek, K.; Carroll, A.J.; Edwards, C.G.; Arthur, D.C.; Pettenati, M.J.; Stamberg, J.; Koduru, P.R.; Moore, J.O.; et al. Repetitive cycles of high-dose cytarabine benefit patients with acute myeloid leukemia and inv(16)(p13q22) or t(16;16)(p13;q22): Results from CALGB 8461. J. Clin. Oncol. 2004, 22, 1087–1094. [Google Scholar] [CrossRef] [PubMed]
- Cornelissen, J.J.; van Putten, W.L.; Verdonck, L.F.; Theobald, M.; Jacky, E.; Daenen, S.M.; van Marwijk Kooy, M.; Wijermans, P.; Schouten, H.; Huijgens, P.C.; et al. Results of a HOVON/SAKK donor versus no-donor analysis of myeloablative HLA-identical sibling stem cell transplantation in first remission acute myeloid leukemia in young and middle-aged adults: Benefits for whom? Blood 2007, 109, 3658–3666. [Google Scholar] [CrossRef] [PubMed]
- Ho, A.D.; Schetelig, J.; Bochtler, T.; Schaich, M.; Schafer-Eckart, K.; Hanel, M.; Rosler, W.; Einsele, H.; Kaufmann, M.; Serve, H.; et al. Allogeneic Stem Cell Transplantation Improves Survival in Patients with Acute Myeloid Leukemia Characterized by a High Allelic Ratio of Mutant FLT3-ITD. Biol. Blood Marrow Transpl. 2016, 22, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Byrd, J.C.; Mrozek, K.; Dodge, R.K.; Carroll, A.J.; Edwards, C.G.; Arthur, D.C.; Pettenati, M.J.; Patil, S.R.; Rao, K.W.; Watson, M.S.; et al. Pretreatment cytogenetic abnormalities are predictive of induction success, cumulative incidence of relapse, and overall survival in adult patients with de novo acute myeloid leukemia: Results from Cancer and Leukemia Group B (CALGB 8461). Blood 2002, 100, 4325–4336. [Google Scholar] [CrossRef] [PubMed]
- Breems, D.A.; Van Putten, W.L.; Huijgens, P.C.; Ossenkoppele, G.J.; Verhoef, G.E.; Verdonck, L.F.; Vellenga, E.; De Greef, G.E.; Jacky, E.; Van der Lelie, J.; et al. Prognostic index for adult patients with acute myeloid leukemia in first relapse. J. Clin. Oncol. 2005, 23, 1969–1978. [Google Scholar] [CrossRef] [PubMed]
- Parkin, B.; Ouillette, P.; Li, Y.; Keller, J.; Lam, C.; Roulston, D.; Li, C.; Shedden, K.; Malek, S.N. Clonal evolution and devolution after chemotherapy in adult acute myelogenous leukemia. Blood 2013, 121, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Shlush, L.I.; Mitchell, A.; Heisler, L.; Abelson, S.; Ng, S.W.K.; Trotman-Grant, A.; Medeiros, J.J.F.; Rao-Bhatia, A.; Jaciw-Zurakowsky, I.; Marke, R.; et al. Tracing the origins of relapse in acute myeloid leukaemia to stem cells. Nature 2017, 547, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Hope, K.J.; Jin, L.; Dick, J.E. Acute myeloid leukemia originates from a hierarchy of leukemic stem cell classes that differ in self-renewal capacity. Nat. Immunol. 2004, 5, 738–743. [Google Scholar] [CrossRef] [PubMed]
- Mosna, F.; Capelli, D.; Gottardi, M. Minimal Residual Disease in Acute Myeloid Leukemia: Still a Work in Progress? J. Clin. Med. 2017, 6, 57. [Google Scholar] [CrossRef] [PubMed]
- Van Rhenen, A.; Moshaver, B.; Kelder, A.; Feller, N.; Nieuwint, A.W.; Zweegman, S.; Ossenkoppele, G.J.; Schuurhuis, G.J. Aberrant marker expression patterns on the CD34+CD38- stem cell compartment in acute myeloid leukemia allows to distinguish the malignant from the normal stem cell compartment both at diagnosis and in remission. Leukemia 2007, 21, 1700–1707. [Google Scholar] [CrossRef] [PubMed]
- Hackl, H.; Astanina, K.; Wieser, R. Molecular and genetic alterations associated with therapy resistance and relapse of acute myeloid leukemia. J. Hematol. Oncol. 2017, 10, 51. [Google Scholar] [CrossRef] [PubMed]
- Al-Mawali, A.; Gillis, D.; Lewis, I. Immunoprofiling of leukemic stem cells CD34+/CD38-/CD123+ delineate FLT3/ITD-positive clones. J. Hematol. Oncol. 2016, 9, 61. [Google Scholar] [CrossRef] [PubMed]
- Angelini, D.F.; Ottone, T.; Guerrera, G.; Lavorgna, S.; Cittadini, M.; Buccisano, F.; De Bardi, M.; Gargano, F.; Maurillo, L.; Divona, M.; et al. A Leukemia-Associated CD34/CD123/CD25/CD99+ Immunophenotype Identifies FLT3-Mutated Clones in Acute Myeloid Leukemia. Clin. Cancer Res. 2015, 21, 3977–3985. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.W.; Mitchell, A.; Kennedy, J.A.; Chen, W.C.; McLeod, J.; Ibrahimova, N.; Arruda, A.; Popescu, A.; Gupta, V.; Schimmer, A.D.; et al. A 17-gene stemness score for rapid determination of risk in acute leukaemia. Nature 2016, 540, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Richard-Carpentier, G.; Sauvageau, G. Bringing a Leukemic Stem Cell Gene Signature into Clinics: Are We There Yet? Cell Stem Cell 2017, 20, 300–301. [Google Scholar] [CrossRef] [PubMed]
- Al-Mawali, A.; Pinto, A.D.; Al-Zadjali, S. CD34+CD38-CD123+ Cells Are Present in Virtually All Acute Myeloid Leukaemia Blasts: A Promising Single Unique Phenotype for Minimal Residual Disease Detection. Acta Haematol. 2017, 138, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Benicio, M.T.L.; Scheucher, P.S.; Garcia, A.B.; Falcao, R.P.; Rego, E.M. Characterization of leukemic stem cells in AML cell lines using ALDH staining. Blood 2013, 122, 5409. [Google Scholar]
- Johnson, J.R.; Bross, P.; Cohen, M.; Rothmann, M.; Chen, G.; Zajicek, A.; Gobburu, J.; Rahman, A.; Staten, A.; Pazdur, R. Approval summary: Imatinib mesylate capsules for treatment of adult patients with newly diagnosed philadelphia chromosome-positive chronic myelogenous leukemia in chronic phase. Clin. Cancer Res. 2003, 9, 1972–1979. [Google Scholar] [PubMed]
- Chen, Y.; Wang, H.; Kantarjian, H.; Cortes, J. Trends in chronic myeloid leukemia incidence and survival in the United States from 1975 to 2009. Leuk. Lymph. 2013, 54, 1411–1417. [Google Scholar] [CrossRef] [PubMed]
- Hehlmann, R.; Lauseker, M.; Saussele, S.; Pfirrmann, M.; Krause, S.; Kolb, H.J.; Neubauer, A.; Hossfeld, D.K.; Nerl, C.; Gratwohl, A.; et al. Assessment of imatinib as first-line treatment of chronic myeloid leukemia: 10-year survival results of the randomized CML study IV and impact of non-CML determinants. Leukemia 2017, 31, 2398–2406. [Google Scholar] [CrossRef] [PubMed]
- Grundler, R.; Thiede, C.; Miething, C.; Steudel, C.; Peschel, C.; Duyster, J. Sensitivity toward tyrosine kinase inhibitors varies between different activating mutations of the FLT3 receptor. Blood 2003, 102, 646–651. [Google Scholar] [CrossRef] [PubMed]
- Eid, S.; Turk, S.; Volkamer, A.; Rippmann, F.; Fulle, S. KinMap: A web-based tool for interactive navigation through human kinome data. BMC Bioinf. 2017, 18, 16. [Google Scholar] [CrossRef] [PubMed]
- Klaeger, S.; Heinzlmeir, S.; Wilhelm, M.; Polzer, H.; Vick, B.; Koenig, P.A.; Reinecke, M.; Ruprecht, B.; Petzoldt, S.; Meng, C.; et al. The target landscape of clinical kinase drugs. Science 2017, 358, eaan4368. [Google Scholar] [CrossRef] [PubMed]
- Karaman, M.W.; Herrgard, S.; Treiber, D.K.; Gallant, P.; Atteridge, C.E.; Campbell, B.T.; Chan, K.W.; Ciceri, P.; Davis, M.I.; Edeen, P.T.; et al. A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2008, 26, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.I.; Hunt, J.P.; Herrgard, S.; Ciceri, P.; Wodicka, L.M.; Pallares, G.; Hocker, M.; Treiber, D.K.; Zarrinkar, P.P. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. [Google Scholar] [CrossRef] [PubMed]
- Von Bubnoff, N.; Engh, R.A.; Aberg, E.; Sanger, J.; Peschel, C.; Duyster, J. FMS-like tyrosine kinase 3-internal tandem duplication tyrosine kinase inhibitors display a nonoverlapping profile of resistance mutations in vitro. Cancer Res. 2009, 69, 3032–3041. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.C.; Lin, K.; Stecula, A.; Sali, A.; Shah, N.P. FLT3 D835 mutations confer differential resistance to type II FLT3 inhibitors. Leukemia 2015, 29, 2390–2392. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.S.; Faisal, A.; Gonzalez de Castro, D.; Bavetsias, V.; Sun, C.; Atrash, B.; Valenti, M.; de Haven Brandon, A.; Avery, S.; Mair, D.; et al. Selective FLT3 inhibition of FLT3-ITD+ acute myeloid leukaemia resulting in secondary D835Y mutation: A model for emerging clinical resistance patterns. Leukemia 2012, 26, 1462–1470. [Google Scholar] [CrossRef] [PubMed]
- McMahon, C.M.; Canaani, J.; Rea, B.; Sargent, R.L.; Morrissette, J.J.D.; Lieberman, D.B.; Watt, C.; Schwartz, G.W.; Faryabi, R.B.; Ferng, T.T.; et al. Mechanisms of Acquired Resistance to Gilteritinib Therapy in Relapsed and Refractory FLT3 -Mutated Acute Myeloid Leukemia. Blood 2017, 130 (Suppl. 1), 295. [Google Scholar]
- Lee, L.Y.; Hernandez, D.; Rajkhowa, T.; Smith, S.C.; Raman, J.R.; Nguyen, B.; Small, D.; Levis, M. Preclinical studies of gilteritinib, a next-generation FLT3 inhibitor. Blood 2017, 129, 257–260. [Google Scholar] [CrossRef] [PubMed]
- Galanis, A.; Ma, H.; Rajkhowa, T.; Ramachandran, A.; Small, D.; Cortes, J.; Levis, M. Crenolanib is a potent inhibitor of FLT3 with activity against resistance-conferring point mutants. Blood 2014, 123, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, E.; Boulton, C.; Kelly, L.M.; Manley, P.; Fabbro, D.; Meyer, T.; Gilliland, D.G.; Griffin, J.D. Inhibition of mutant FLT3 receptors in leukemia cells by the small molecule tyrosine kinase inhibitor PKC412. Cancer Cell 2002, 1, 433–443. [Google Scholar] [CrossRef]
- Warkentin, A.A.; Lopez, M.S.; Lasater, E.A.; Lin, K.; He, B.L.; Leung, A.Y.; Smith, C.C.; Shah, N.P.; Shokat, K.M. Overcoming myelosuppression due to synthetic lethal toxicity for FLT3-targeted acute myeloid leukemia therapy. ELife 2014, 3, e03445. [Google Scholar] [CrossRef] [PubMed]
- Stone, R.M.; DeAngelo, D.J.; Klimek, V.; Galinsky, I.; Estey, E.; Nimer, S.D.; Grandin, W.; Lebwohl, D.; Wang, Y.; Cohen, P.; et al. Patients with acute myeloid leukemia and an activating mutation in FLT3 respond to a small-molecule FLT3 tyrosine kinase inhibitor, PKC412. Blood 2005, 105, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Fischer, T.; Stone, R.M.; Deangelo, D.J.; Galinsky, I.; Estey, E.; Lanza, C.; Fox, E.; Ehninger, G.; Feldman, E.J.; Schiller, G.J.; et al. Phase IIB trial of oral Midostaurin (PKC412), the FMS-like tyrosine kinase 3 receptor (FLT3) and multi-targeted kinase inhibitor, in patients with acute myeloid leukemia and high-risk myelodysplastic syndrome with either wild-type or mutated FLT3. J. Clin. Oncol. 2010, 28, 4339–4345. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, B.; Williams, A.B.; Young, D.J.; Ma, H.; Li, L.; Levis, M.; Brown, P.; Small, D. FLT3 activating mutations display differential sensitivity to multiple tyrosine kinase inhibitors. Oncotarget 2017, 8, 10931–10944. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.J.; Cools, J.; Curley, D.P.; Yu, J.C.; Lokker, N.A.; Giese, N.A.; Gilliland, D.G. Variable sensitivity of FLT3 activation loop mutations to the small molecule tyrosine kinase inhibitor MLN518. Blood 2004, 104, 2867–2872. [Google Scholar] [CrossRef] [PubMed]
- Barry, E.V.; Clark, J.J.; Cools, J.; Roesel, J.; Gilliland, D.G. Uniform sensitivity of FLT3 activation loop mutants to the tyrosine kinase inhibitor midostaurin. Blood 2007, 110, 4476–4479. [Google Scholar] [CrossRef] [PubMed]
- Yee, K.W.; Schittenhelm, M.; O′Farrell, A.M.; Town, A.R.; McGreevey, L.; Bainbridge, T.; Cherrington, J.M.; Heinrich, M.C. Synergistic effect of SU11248 with cytarabine or daunorubicin on FLT3 ITD-positive leukemic cells. Blood 2004, 104, 4202–4209. [Google Scholar] [CrossRef] [PubMed]
- Kampa-Schittenhelm, K.M.; Heinrich, M.C.; Akmut, F.; Dohner, H.; Dohner, K.; Schittenhelm, M.M. Quizartinib (AC220) is a potent second generation class III tyrosine kinase inhibitor that displays a distinct inhibition profile against mutant-FLT3, -PDGFRA and -KIT isoforms. Mol. Cancer 2013, 12, 19. [Google Scholar] [CrossRef] [PubMed]
- Naqvi, K.; Konopleva, M.; Ravandi, F. Targeted therapies in Acute Myeloid Leukemia: A focus on FLT-3 inhibitors and ABT199. Expert Rev. Hematol. 2017, 10, 863–874. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Konopleva, M.; Shi, Y.X.; McQueen, T.; Harris, D.; Ling, X.; Estrov, Z.; Quintas-Cardama, A.; Small, D.; Cortes, J.; et al. Mutant FLT3: A direct target of sorafenib in acute myelogenous leukemia. J. Nat. Cancer Inst. 2008, 100, 184–198. [Google Scholar] [CrossRef] [PubMed]
- Inaba, H.; Rubnitz, J.E.; Coustan-Smith, E.; Li, L.; Furmanski, B.D.; Mascara, G.P.; Heym, K.M.; Christensen, R.; Onciu, M.; Shurtleff, S.A.; et al. Phase I pharmacokinetic and pharmacodynamic study of the multikinase inhibitor sorafenib in combination with clofarabine and cytarabine in pediatric relapsed/refractory leukemia. J. Clin. Oncol. 2011, 29, 3293–3300. [Google Scholar] [CrossRef] [PubMed]
- Ravandi, F.; Arana Yi, C.; Cortes, J.E.; Levis, M.; Faderl, S.; Garcia-Manero, G.; Jabbour, E.; Konopleva, M.; O′Brien, S.; Estrov, Z.; et al. Final report of phase II study of sorafenib, cytarabine and idarubicin for initial therapy in younger patients with acute myeloid leukemia. Leukemia 2014, 28, 1543–1545. [Google Scholar] [CrossRef] [PubMed]
- Rollig, C.; Serve, H.; Huttmann, A.; Noppeney, R.; Muller-Tidow, C.; Krug, U.; Baldus, C.D.; Brandts, C.H.; Kunzmann, V.; Einsele, H.; et al. Addition of sorafenib versus placebo to standard therapy in patients aged 60 years or younger with newly diagnosed acute myeloid leukaemia (SORAML): A multicentre, phase 2, randomised controlled trial. Lancet Oncol. 2015, 16, 1691–1699. [Google Scholar] [CrossRef]
- Ravandi, F.; Alattar, M.L.; Grunwald, M.R.; Rudek, M.A.; Rajkhowa, T.; Richie, M.A.; Pierce, S.; Daver, N.; Garcia-Manero, G.; Faderl, S.; et al. Phase 2 study of azacytidine plus sorafenib in patients with acute myeloid leukemia and FLT3 internal tandem duplication mutation. Blood 2013, 121, 4655–4662. [Google Scholar] [CrossRef] [PubMed]
- Ohanian, M.; Garcia-Manero, G.; Levis, M.; Jabbour, E.; Daver, N.; Borthakur, G.; Kadia, T.; Pierce, S.; Burger, J.; Richie, M.A.; et al. Sorafenib Combined with 5-azacytidine (AZA) in Older Patients with Untreated FLT3-ITD Mutated Acute Myeloid Leukemia (AML). Am. J. Hematol. 2018, 93, 1136–1141. [Google Scholar] [CrossRef] [PubMed]
- Ohanian, M.; Garcia-Manero, G.; Levis, M.J.; Jabbour, E.; Daver, N.G.; Borthakur, G.; Kadia, T.M.; Brandt, M.; Pierce, S.; Burger, J.A.; et al. Sorafenib plus 5-azacytidine (AZA) in older untreated FLT3-ITD mutated AML. J. Clin. Oncol. 2017, 35 (Suppl. 15), 7029. [Google Scholar] [CrossRef]
- Kancha, R.K.; Grundler, R.; Peschel, C.; Duyster, J. Sensitivity toward sorafenib and sunitinib varies between different activating and drug-resistant FLT3-ITD mutations. Exp. Hematol. 2007, 35, 1522–1526. [Google Scholar] [CrossRef] [PubMed]
- Fiedler, W.; Serve, H.; Dohner, H.; Schwittay, M.; Ottmann, O.G.; O′Farrell, A.M.; Bello, C.L.; Allred, R.; Manning, W.C.; Cherrington, J.M.; et al. A phase 1 study of SU11248 in the treatment of patients with refractory or resistant acute myeloid leukemia (AML) or not amenable to conventional therapy for the disease. Blood 2005, 105, 986–993. [Google Scholar] [CrossRef] [PubMed]
- Levis, M.; Allebach, J.; Tse, K.F.; Zheng, R.; Baldwin, B.R.; Smith, B.D.; Jones-Bolin, S.; Ruggeri, B.; Dionne, C.; Small, D. A FLT3-targeted tyrosine kinase inhibitor is cytotoxic to leukemia cells in vitro and in vivo. Blood 2002, 99, 3885–3891. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.D.; Levis, M.; Beran, M.; Giles, F.; Kantarjian, H.; Berg, K.; Murphy, K.M.; Dauses, T.; Allebach, J.; Small, D. Single-agent CEP-701, a novel FLT3 inhibitor, shows biologic and clinical activity in patients with relapsed or refractory acute myeloid leukemia. Blood 2004, 103, 3669–3676. [Google Scholar] [CrossRef] [PubMed]
- Knapper, S.; Burnett, A.K.; Littlewood, T.; Kell, W.J.; Agrawal, S.; Chopra, R.; Clark, R.; Levis, M.J.; Small, D. A phase 2 trial of the FLT3 inhibitor lestaurtinib (CEP701) as first-line treatment for older patients with acute myeloid leukemia not considered fit for intensive chemotherapy. Blood 2006, 108, 3262–3270. [Google Scholar] [CrossRef] [PubMed]
- Levis, M.; Ravandi, F.; Wang, E.S.; Baer, M.R.; Perl, A.; Coutre, S.; Erba, H.; Stuart, R.K.; Baccarani, M.; Cripe, L.D.; et al. Results from a randomized trial of salvage chemotherapy followed by lestaurtinib for patients with FLT3 mutant AML in first relapse. Blood 2011, 117, 3294–3301. [Google Scholar] [CrossRef] [PubMed]
- Kelly, L.M.; Yu, J.C.; Boulton, C.L.; Apatira, M.; Li, J.; Sullivan, C.M.; Williams, I.; Amaral, S.M.; Curley, D.P.; Duclos, N.; et al. CT53518, a novel selective FLT3 antagonist for the treatment of acute myelogenous leukemia (AML). Cancer Cell 2002, 1, 421–432. [Google Scholar] [CrossRef]
- DeAngelo, D.J.; Stone, R.M.; Heaney, M.L.; Nimer, S.D.; Paquette, R.L.; Klisovic, R.B.; Caligiuri, M.A.; Cooper, M.R.; Lecerf, J.M.; Karol, M.D.; et al. Phase 1 clinical results with tandutinib (MLN518), a novel FLT3 antagonist, in patients with acute myelogenous leukemia or high-risk myelodysplastic syndrome: Safety, pharmacokinetics, and pharmacodynamics. Blood 2006, 108, 3674–3681. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.E.; Kantarjian, H.; Foran, J.M.; Ghirdaladze, D.; Zodelava, M.; Borthakur, G.; Gammon, G.; Trone, D.; Armstrong, R.C.; James, J.; et al. Phase I study of quizartinib administered daily to patients with relapsed or refractory acute myeloid leukemia irrespective of FMS-like tyrosine kinase 3-internal tandem duplication status. J. Clin. Oncol. 2013, 31, 3681–3687. [Google Scholar] [CrossRef] [PubMed]
- Tallman, M.S.; Schiller, G.; Trone, D.; Gammon, G.; Goldberg, S.; Perl, A.E.; Marie, J.-P.; Martinelli, G.; Levis, M. Results of a Phase 2 Randomized, Open-Label, Study Of Lower Doses Of Quizartinib (AC220; ASP2689) In Subjects With FLT3-ITD Positive Relapsed Or Refractory Acute Myeloid Leukemia (AML). Blood 2013, 122, 494. [Google Scholar]
- Cortes, J.; Perl, A.E.; Döhner, H.; Kantarjian, H.; Martinelli, G.; Kovacsovics, T.; Rousselot, P.; Steffen, B.; Dombret, H.; Estey, E.; et al. Quizartinib, an FLT3 inhibitor, as monotherapy in patients with relapsed or refractory acute myeloid leukaemia: An open-label, multicentre, single-arm, phase 2 trial. Lancet Oncol. 2018, 19, 889–903. [Google Scholar] [CrossRef]
- Cortes, J.; Khaled, S.; Martinelli, G.; Perl, A.E.; Ganguly, S.; Russell, N.; Krämer, A.; Dombret, H.; Hogge, D.; Jonas, B.A.; et al. Quizartinib significantly prolongs overall survival in patients with flt3-internal tandem duplication–mutated (mut) relapsed/refractory aml in the phase 3, randomized, controlled quantum-r trial. EHA Learn. Cent. 2018. Available online: http://www.oncoletter.ch/files/cto_layout/Kongressdateien/EHA2018/LB2600.pdf (accessed on 16 October 2018).
- Bowen, D.; Russell, N.; Knapper, S.; Milligan, D.; Hunter, A.E.; Khwaja, A.; Clark, R.E.; Culligan, D.; Clark, H.; Hills, R.K. AC220 (Quizartinib) Can Be Safely Combined with Conventional Chemotherapy In Older Patients With Newly Diagnosed Acute Myeloid Leukaemia: Experience From The AML18 Pilot Trial. Blood 2013, 122, 622. [Google Scholar]
- Park, I.-K.; Mundy-Bosse, B.; Warner, S.L.; Bearss, D.J.; Marcucci, G.; Caligiuri, M.A. The Receptor Tyrosine Kinase Axl Is Required for Resistance to FLT3-Targeted Therapy in Acute Myeloid Leukemia. Blood 2014, 124, 2350. [Google Scholar] [CrossRef] [PubMed]
- Perl, A.E.; Altman, J.K.; Cortes, J.; Smith, C.; Litzow, M.; Baer, M.R.; Claxton, D.; Erba, H.P.; Gill, S.; Goldberg, S.; et al. Selective inhibition of FLT3 by gilteritinib in relapsed or refractory acute myeloid leukaemia: A multicentre, first-in-human, open-label, phase 1–2 study. Lancet Oncol. 2017, 18, 1061–1075. [Google Scholar] [CrossRef]
- Perl, A.E.; Cortes, J.E.; Strickland, S.A.; Ritchie, E.K.; Neubauer, A.; Martinelli, G.; Naoe, T.; Pigneux, A.; Rousselot, P.H.; Röllig, C.; et al. A phase 3, open-label, randomized study of the FLT3 inhibitor gilteritinib versus salvage chemotherapy in adults with first relapse or primary refractory FLT3 mutation-positive acute myeloid leukemia. J. Clin. Oncol. 2016, 34 (Suppl. 15), TPS7072. [Google Scholar] [CrossRef]
- Randhawa, J.K.; Kantarjian, H.M.; Borthakur, G.; Thompson, P.A.; Konopleva, M.; Daver, N.; Pemmaraju, N.; Jabbour, E.; Kadia, T.M.; Estrov, Z.; et al. Results of a Phase II Study of Crenolanib in Relapsed/Refractory Acute Myeloid Leukemia Patients (Pts) with Activating FLT3 Mutations. Blood 2014, 124, 389. [Google Scholar]
- Cortes, J.E.; Kantarjian, H.M.; Kadia, T.M.; Borthakur, G.; Konopleva, M.; Garcia-Manero, G.; Daver, N.G.; Pemmaraju, N.; Jabbour, E.; Estrov, Z.; et al. Crenolanib besylate, a type I pan-FLT3 inhibitor, to demonstrate clinical activity in multiply relapsed FLT3-ITD and D835 AML. J. Clin. Oncol. 2016, 34 (Suppl. 15), 7008. [Google Scholar] [CrossRef]
- Ghiaur, G.; Levis, M. Mechanisms of Resistance to FLT3 Inhibitors and the Role of the Bone Marrow Microenvironment. Hematol. Oncol. Clin. N. Am. 2017, 31, 681–692. [Google Scholar] [CrossRef] [PubMed]
- Chu, S.H.; Small, D. Mechanisms of resistance to FLT3 inhibitors. Drug Resist. Updat. 2009, 12, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Traer, E.; Martinez, J.; Javidi-Sharifi, N.; Agarwal, A.; Dunlap, J.; English, I.; Kovacsovics, T.; Tyner, J.W.; Wong, M.; Druker, B.J. FGF2 from Marrow Microenvironment Promotes Resistance to FLT3 Inhibitors in Acute Myeloid Leukemia. Cancer Res. 2016, 76, 6471–6482. [Google Scholar] [CrossRef] [PubMed]
- Chang, E.; Ganguly, S.; Rajkhowa, T.; Gocke, C.D.; Levis, M.; Konig, H. The combination of FLT3 and DNA methyltransferase inhibition is synergistically cytotoxic to FLT3/ITD acute myeloid leukemia cells. Leukemia 2016, 30, 1025–1032. [Google Scholar] [CrossRef] [PubMed]
- Salih, H.R.; Hofmann, M.; Grosse-Hovest, L.; Nuebling, T.; Bamberg, M.; Aulwurm, S.; Buehring, H.J.; Bethge, W.A.; Kanz, L.; Jung, G. Elimination of Minimal Residual Disease (MRD) In AML Patients With a Novel Fc-Optimized FLT3 Antibody (4G8-SDIEM). Blood 2013, 122, 21. [Google Scholar]
- Piloto, O.; Wright, M.; Brown, P.; Kim, K.T.; Levis, M.; Small, D. Prolonged exposure to FLT3 inhibitors leads to resistance via activation of parallel signaling pathways. Blood 2007, 109, 1643–1652. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Sexauer, A.; Levis, M. Bone marrow stroma-mediated resistance to FLT3 inhibitors in FLT3-ITD AML is mediated by persistent activation of extracellular regulated kinase. Br. J. Haematol. 2014, 164, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Yokota, S.; Kiyoi, H.; Nakao, M.; Iwai, T.; Misawa, S.; Okuda, T.; Sonoda, Y.; Abe, T.; Kahsima, K.; Matsuo, Y.; et al. Internal tandem duplication of the FLT3 gene is preferentially seen in acute myeloid leukemia and myelodysplastic syndrome among various hematological malignancies. A study on a large series of patients and cell lines. Leukemia 1997, 11, 1605–1609. [Google Scholar] [CrossRef] [PubMed]
- Iwai, T.; Yokota, S.; Nakao, M.; Okamoto, T.; Taniwaki, M.; Onodera, N.; Watanabe, A.; Kikuta, A.; Tanaka, A.; Asami, K.; et al. Internal tandem duplication of the FLT3 gene and clinical evaluation in childhood acute myeloid leukemia. The Children′s Cancer and Leukemia Study Group, Japan. Leukemia 1999, 13, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Wei, A.H.; Tiong, I.S. Midostaurin, enasidenib, CPX-351, gemtuzumab ozogomycin and venetoclax bring new hope to AML. Blood 2017, 130, 2469–2474. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FLT3 Inhibitor | Class, Type | Mutation/Amino Acid Change | Tyrosine Kinase Domain |
|---|---|---|---|
| PKC412 (Midostaurin) | First generation; Type 1 | N676K [24] 672E [23] N676D [25,142] F691L, G697R [25] N676I, N676S, F691I [142] | TKD1 |
| Sunitinib | First generation; Type 1 | A627P, F691L [33] D835Y [31] Y842C [33] | TKD1 TKD2 |
| Lestaurtinib | First generation; Type 1 | A627P [33] | TKD1 |
| Sorafenib | First generation; Type 2 | F691L [31,33,142] A627P [33] D835H [31,143] D835V/F/I/del/Y/A [143] F691I, Y842N [142] Y842C [33] | TKD1 TKD2 |
| Tandutinib | First generation; Type 2 | D835Y [144] | TKD2 |
| Quizartinib | Second generation; Type 2 | F691L [31,33,73] A627P [33] D835V/F/I/del/Y [31,73,143] D835H [31] Y842C/H [73] | TKD1 TKD2 |
| Gilteritinib | Second generation, Type 1 | F961L [145,146] | TKD1 |
| Crenolanib | Second generation, Type 1 | F691L [147] | TKD1 |
| FLT3-Mut | Midostaurin | Sorafenib | Sunitinib | Lestaurtinib | Tandutinib | Quizartinib | Crenolanib | Gilteritinib |
|---|---|---|---|---|---|---|---|---|
| FLT3-ITD | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| D835Y | −3.00 | 190.91 | 7.22 | 1.14 | >18.18 | 73.33 | −1.72 | −1.13 |
| D835V | −3.73 | - | 13.75 | - | >18.18 | 19.11 | −3.14 | - |
| ITD-D835Y | 7.33 | 3000.00 | 31.67 | −1.11 | - | 1000.00 | 1.53 | 1.17 |
| ITD-D835V | 1.08 | 780.94 | 3.25 | - | - | 468.42 | 1.44 | - |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Staudt, D.; Murray, H.C.; McLachlan, T.; Alvaro, F.; Enjeti, A.K.; Verrills, N.M.; Dun, M.D. Targeting Oncogenic Signaling in Mutant FLT3 Acute Myeloid Leukemia: The Path to Least Resistance. Int. J. Mol. Sci. 2018, 19, 3198. https://doi.org/10.3390/ijms19103198
Staudt D, Murray HC, McLachlan T, Alvaro F, Enjeti AK, Verrills NM, Dun MD. Targeting Oncogenic Signaling in Mutant FLT3 Acute Myeloid Leukemia: The Path to Least Resistance. International Journal of Molecular Sciences. 2018; 19(10):3198. https://doi.org/10.3390/ijms19103198
Chicago/Turabian StyleStaudt, Dilana, Heather C. Murray, Tabitha McLachlan, Frank Alvaro, Anoop K. Enjeti, Nicole M. Verrills, and Matthew D. Dun. 2018. "Targeting Oncogenic Signaling in Mutant FLT3 Acute Myeloid Leukemia: The Path to Least Resistance" International Journal of Molecular Sciences 19, no. 10: 3198. https://doi.org/10.3390/ijms19103198
APA StyleStaudt, D., Murray, H. C., McLachlan, T., Alvaro, F., Enjeti, A. K., Verrills, N. M., & Dun, M. D. (2018). Targeting Oncogenic Signaling in Mutant FLT3 Acute Myeloid Leukemia: The Path to Least Resistance. International Journal of Molecular Sciences, 19(10), 3198. https://doi.org/10.3390/ijms19103198