HPV-18 E6 Oncoprotein and Its Spliced Isoform E6*I Regulate the Wnt/β-Catenin Cell Signaling Pathway through the TCF-4 Transcriptional Factor

 ,
,  , , , , ,
, , , , ,
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
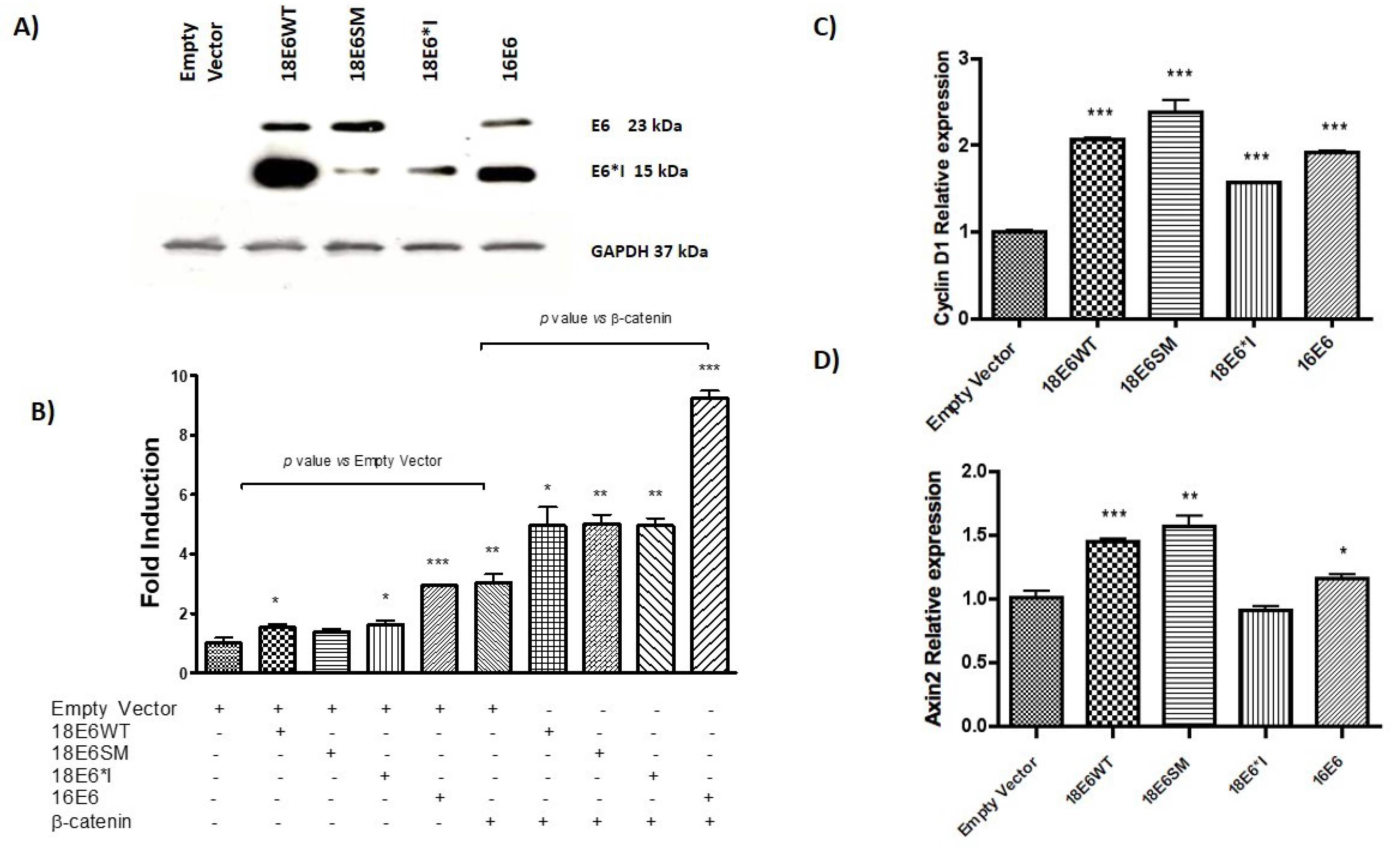
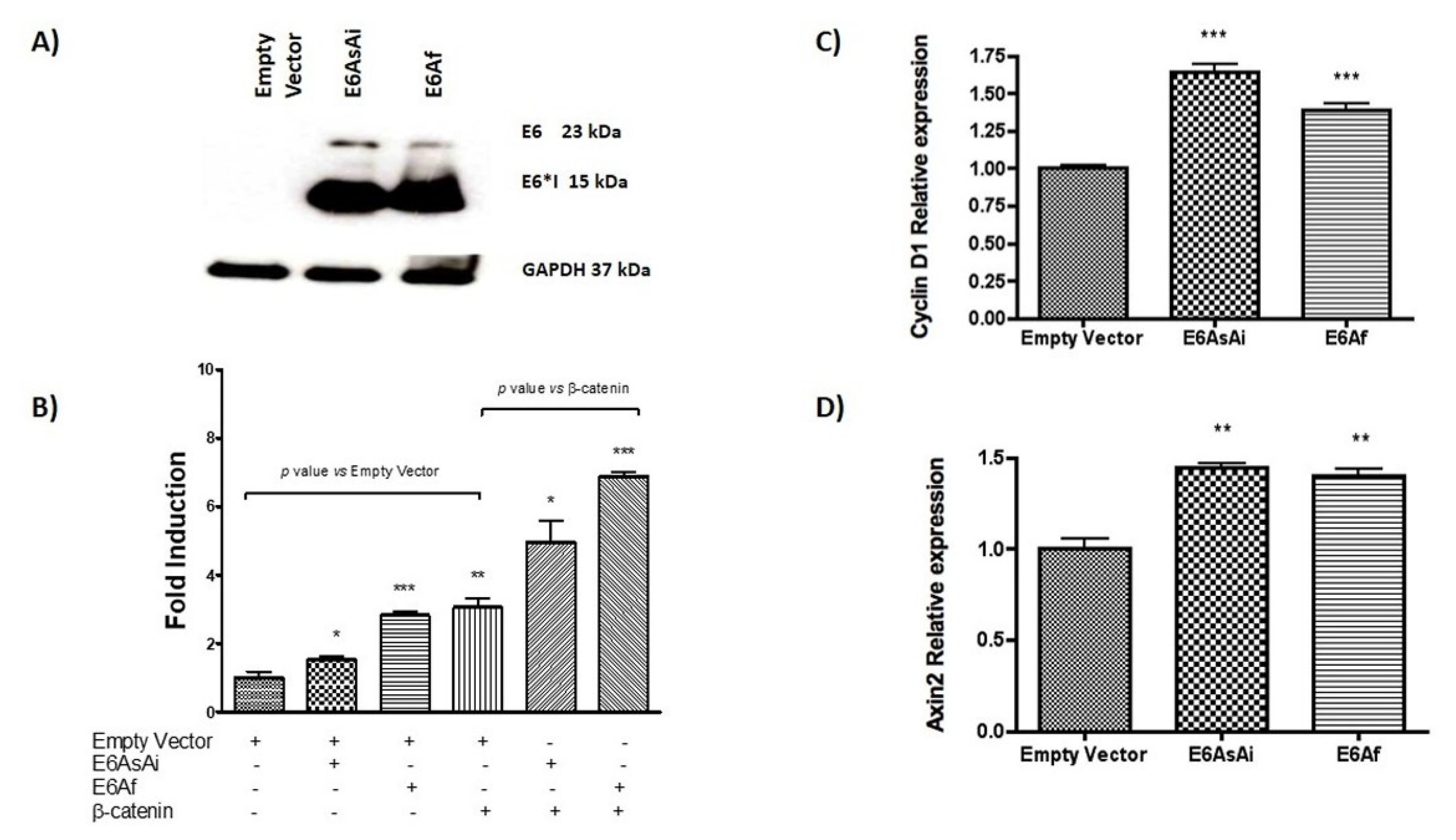
2.1. HPV-18 E6 and E6*I Proteins Enhance β-Catenin/TCF-4 Transcription
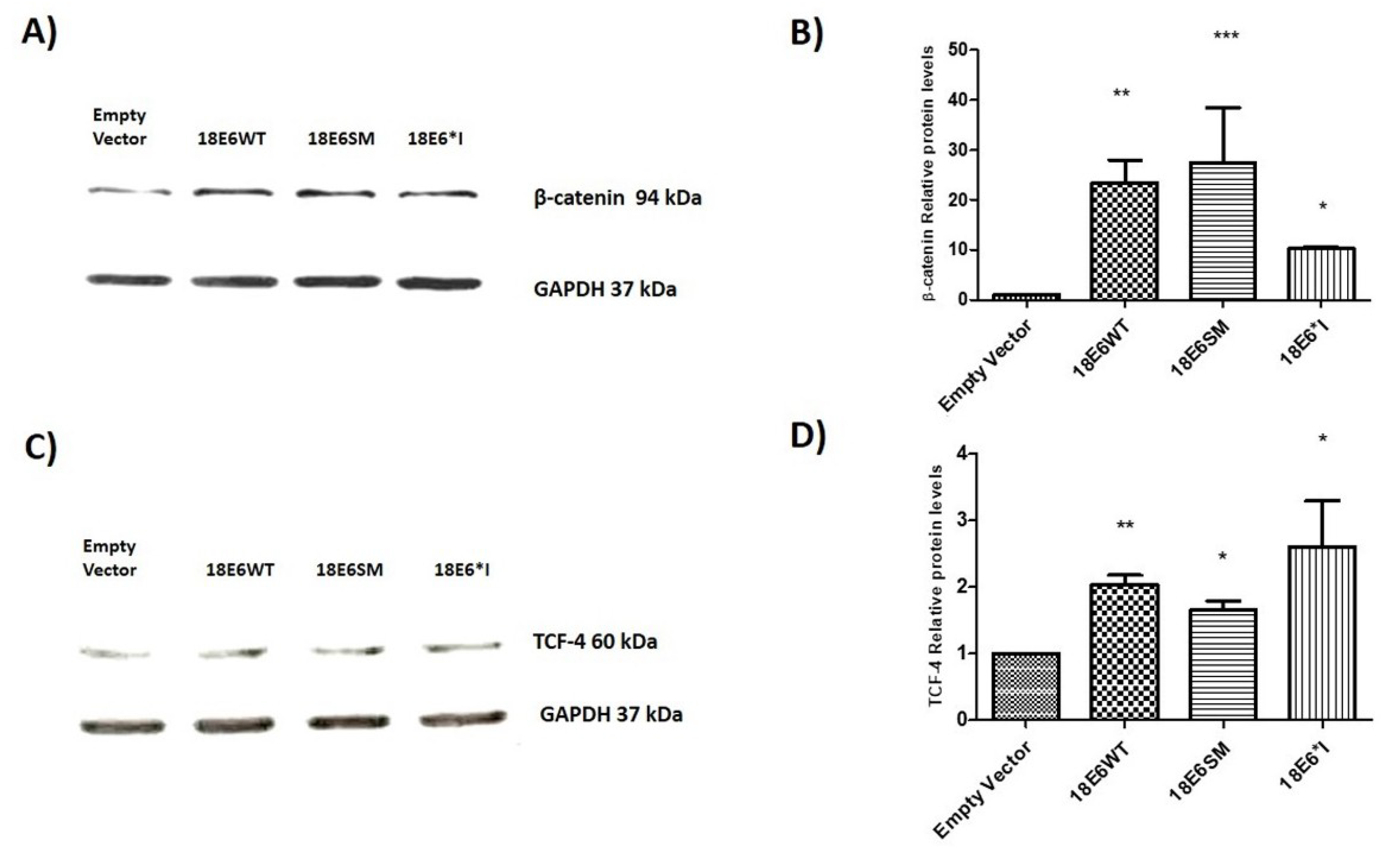
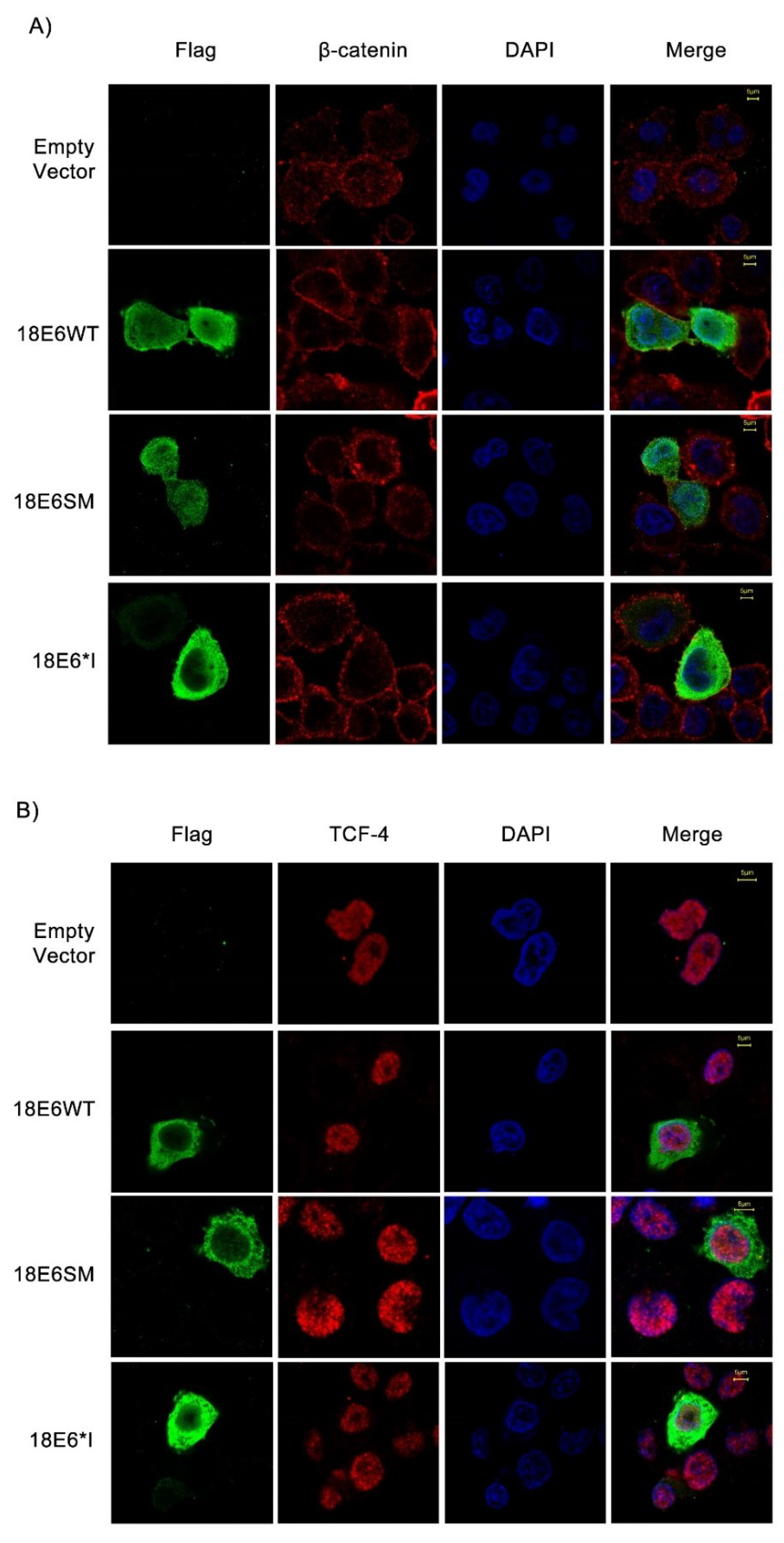
2.2. E6 Proteins Increase β-Catenin and TCF-4 Protein Levels, But Do Not Alter Their Subcellular Localization
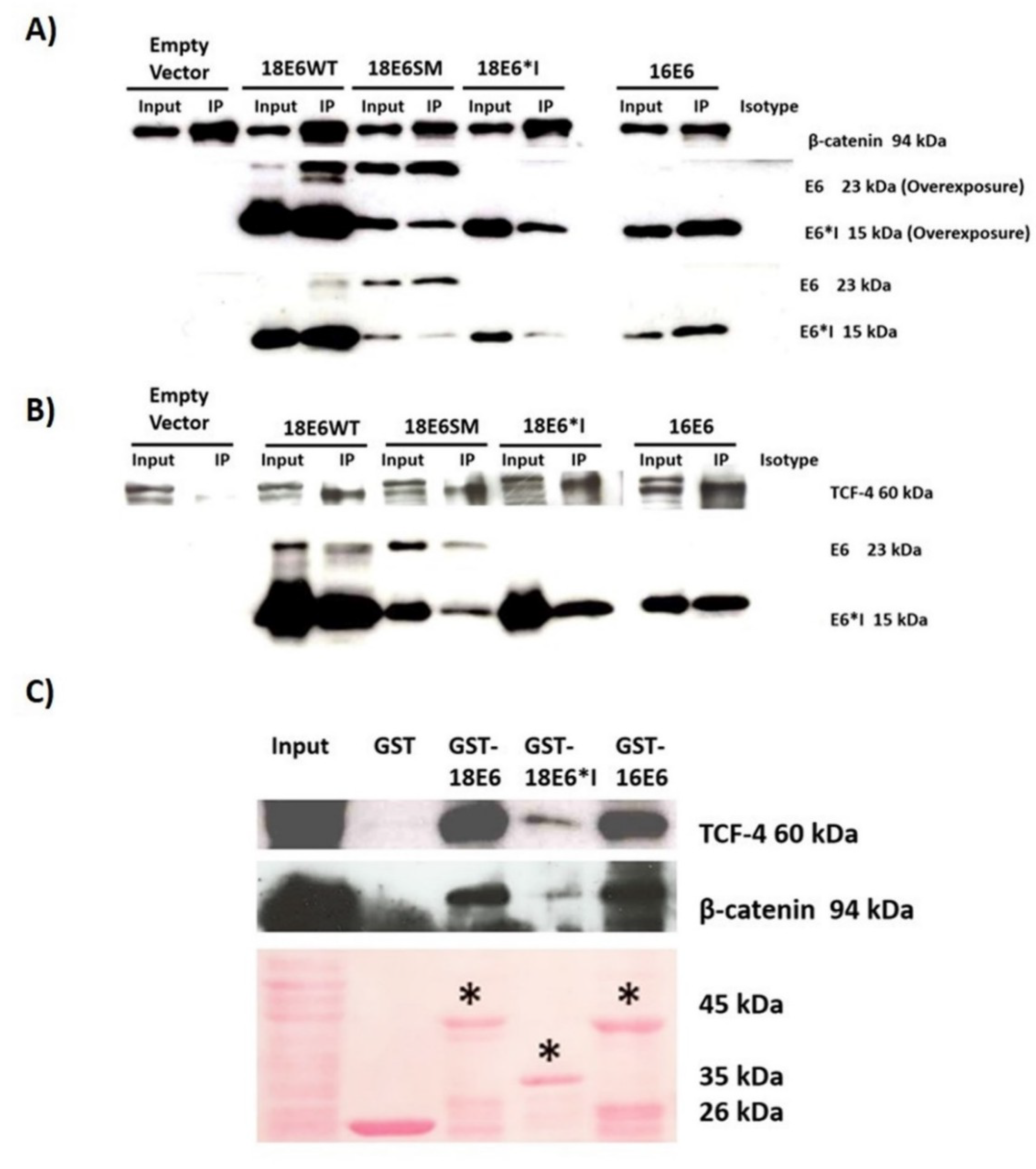
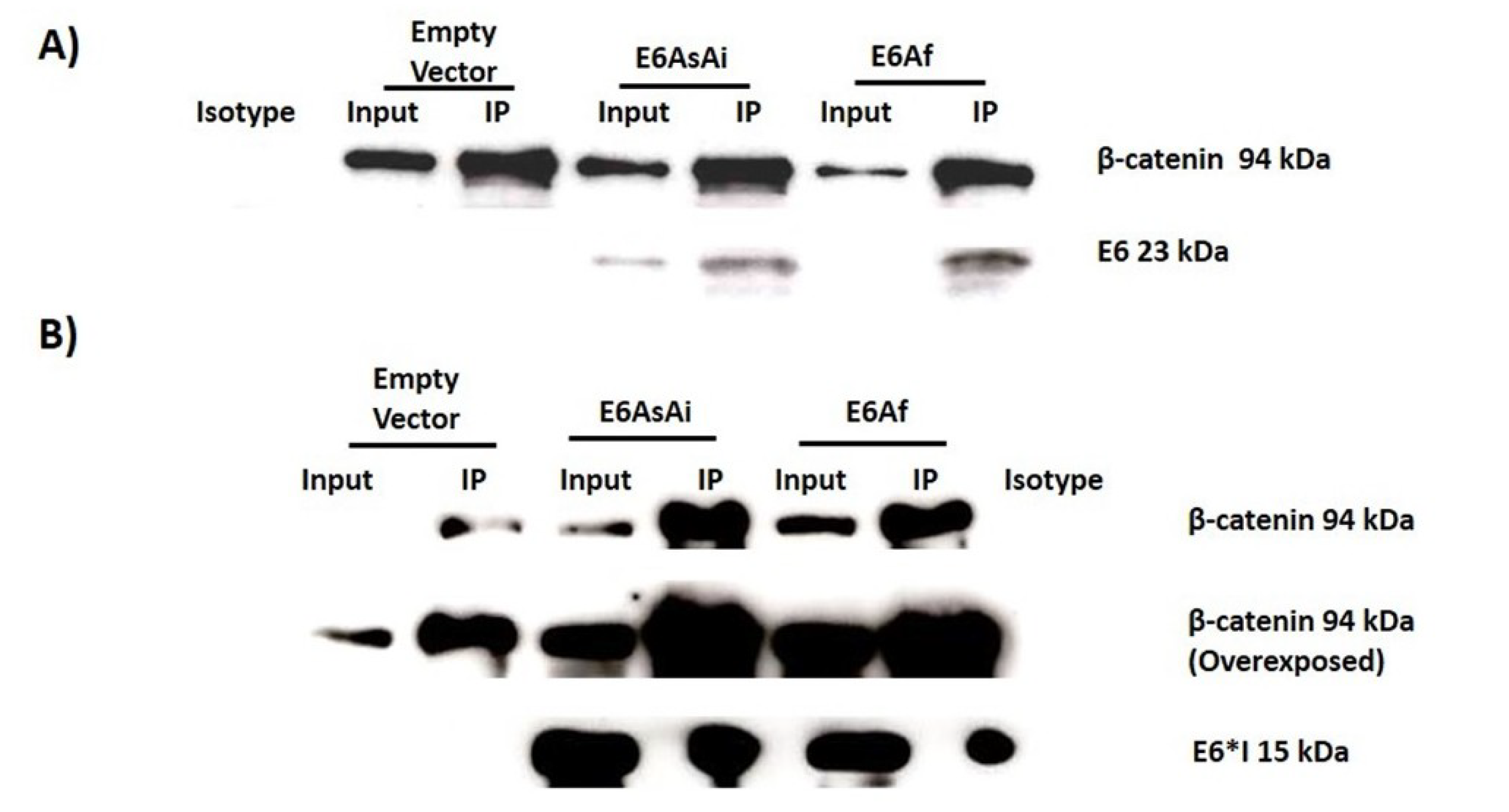
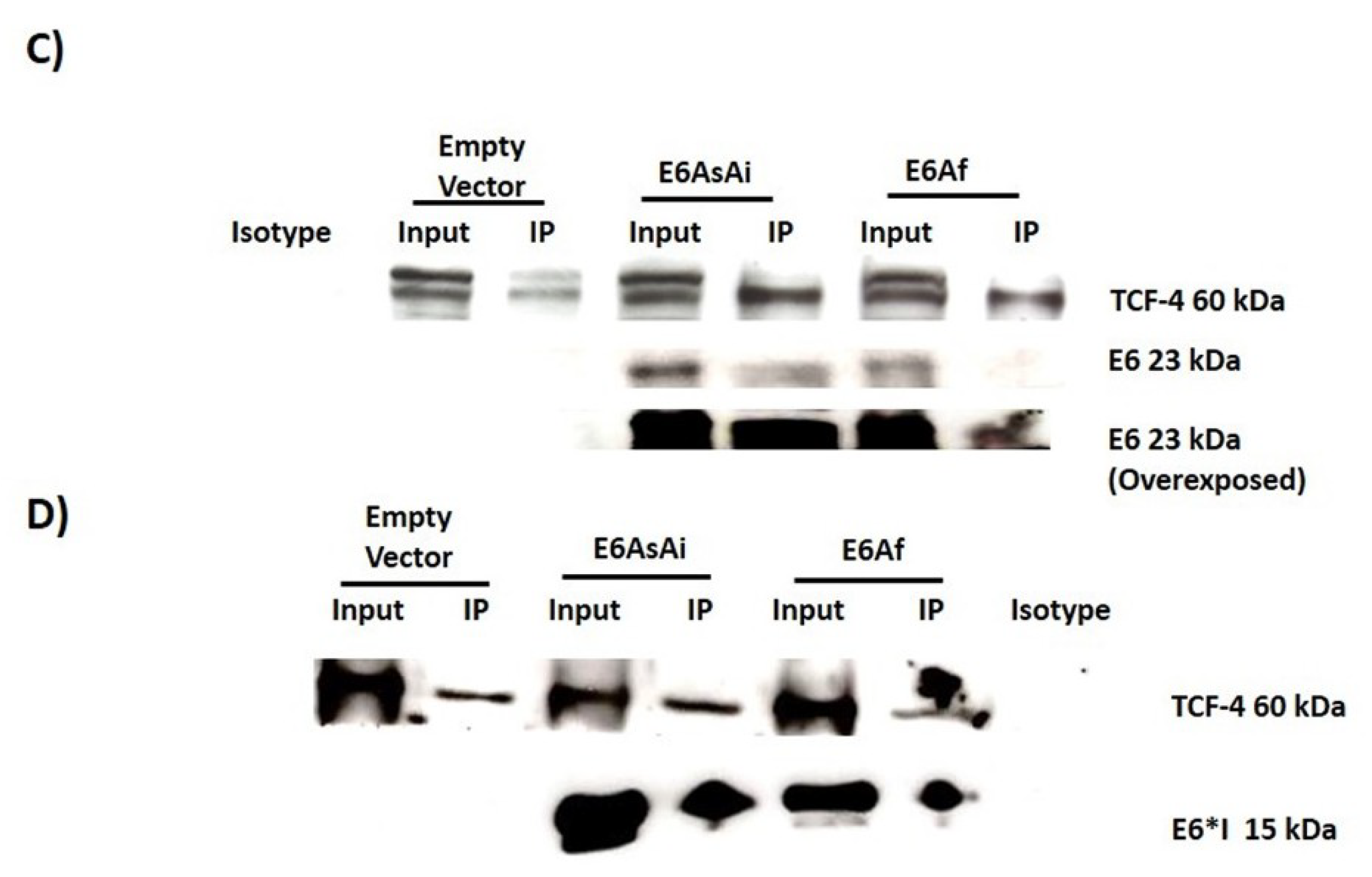
2.3. E6 Proteins Interact with the Wnt Activation Complex In Vivo and In Vitro
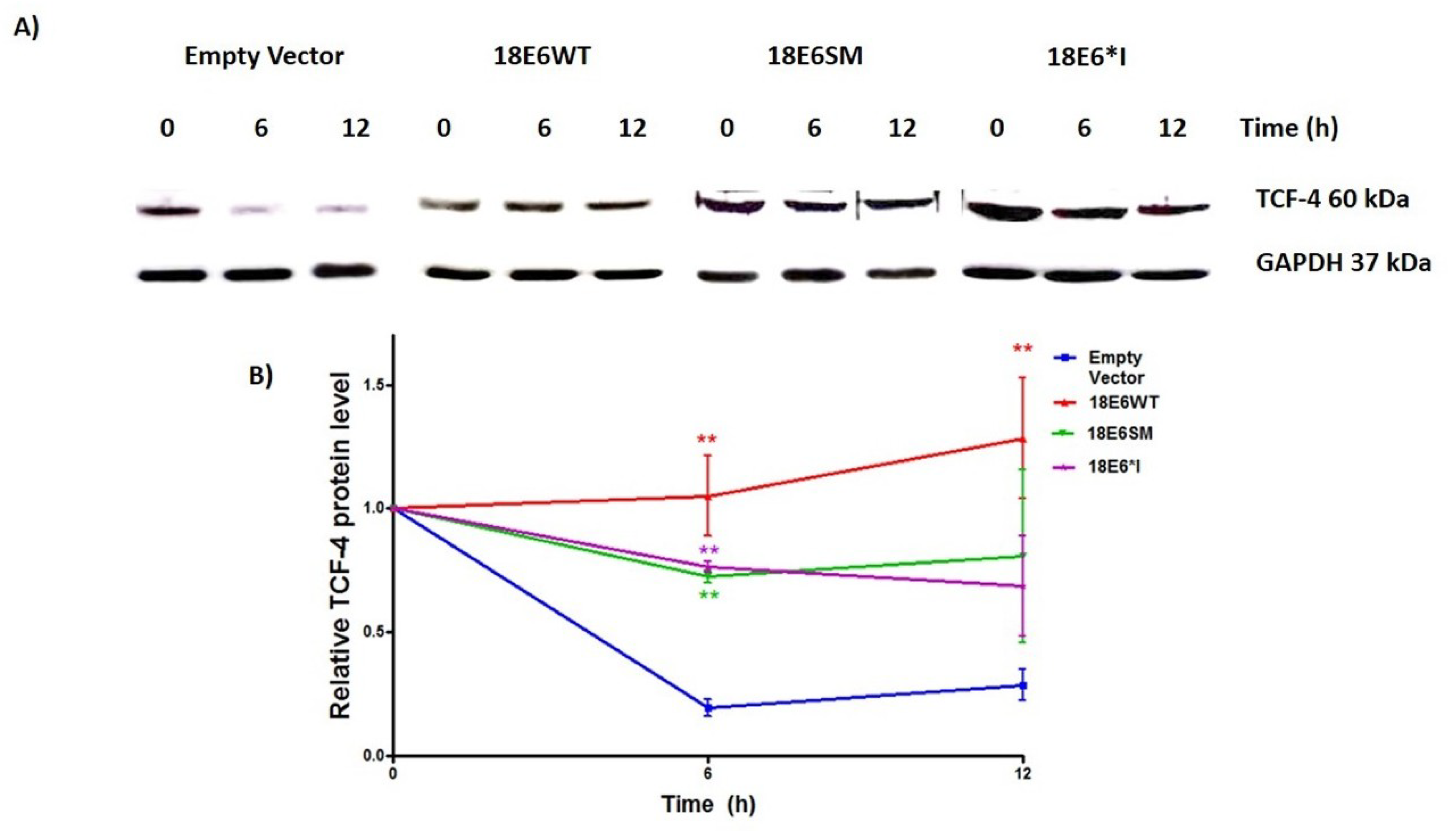
2.4. E6 and E6*I from HPV-18 Increase TCF-4 Protein Stability
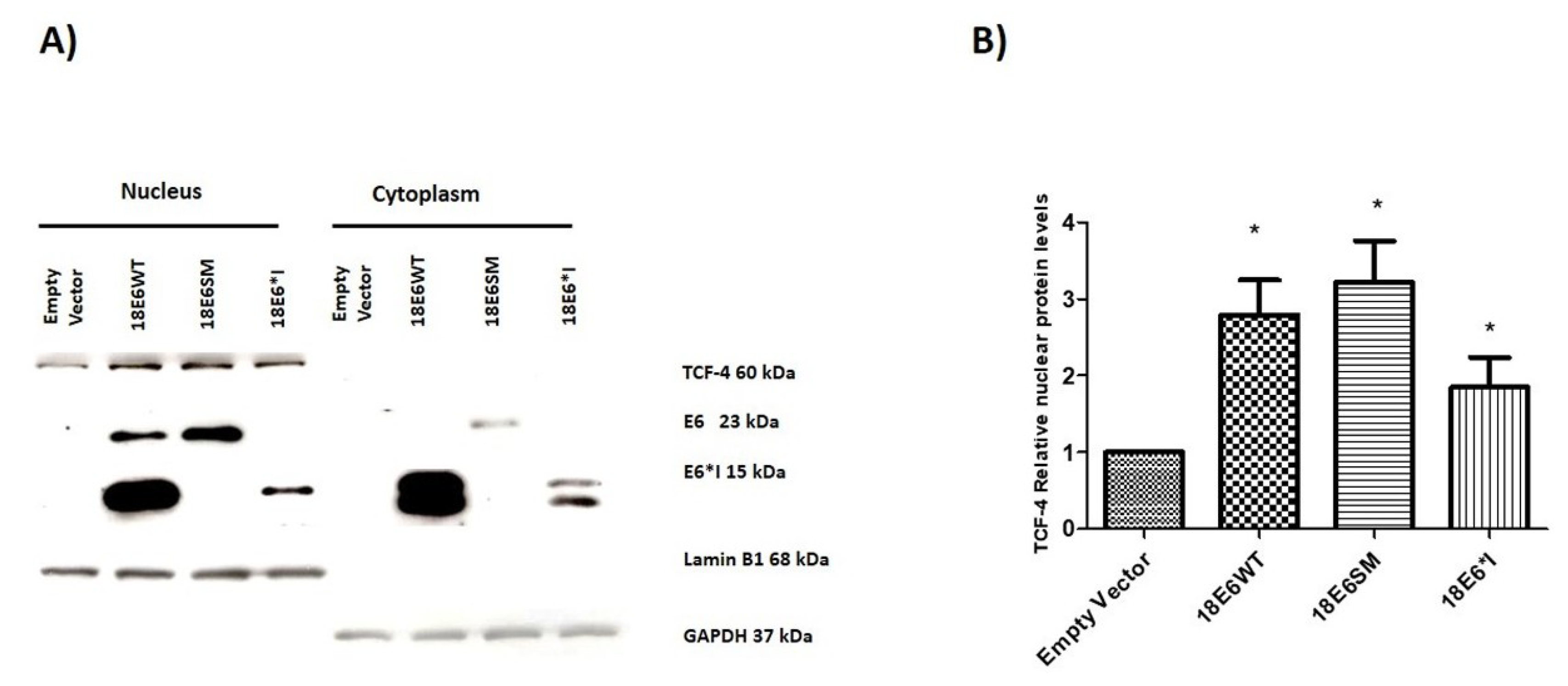
2.5. HPV-18 E6 and E6*I Increase Nuclear TCF-4 Protein Levels
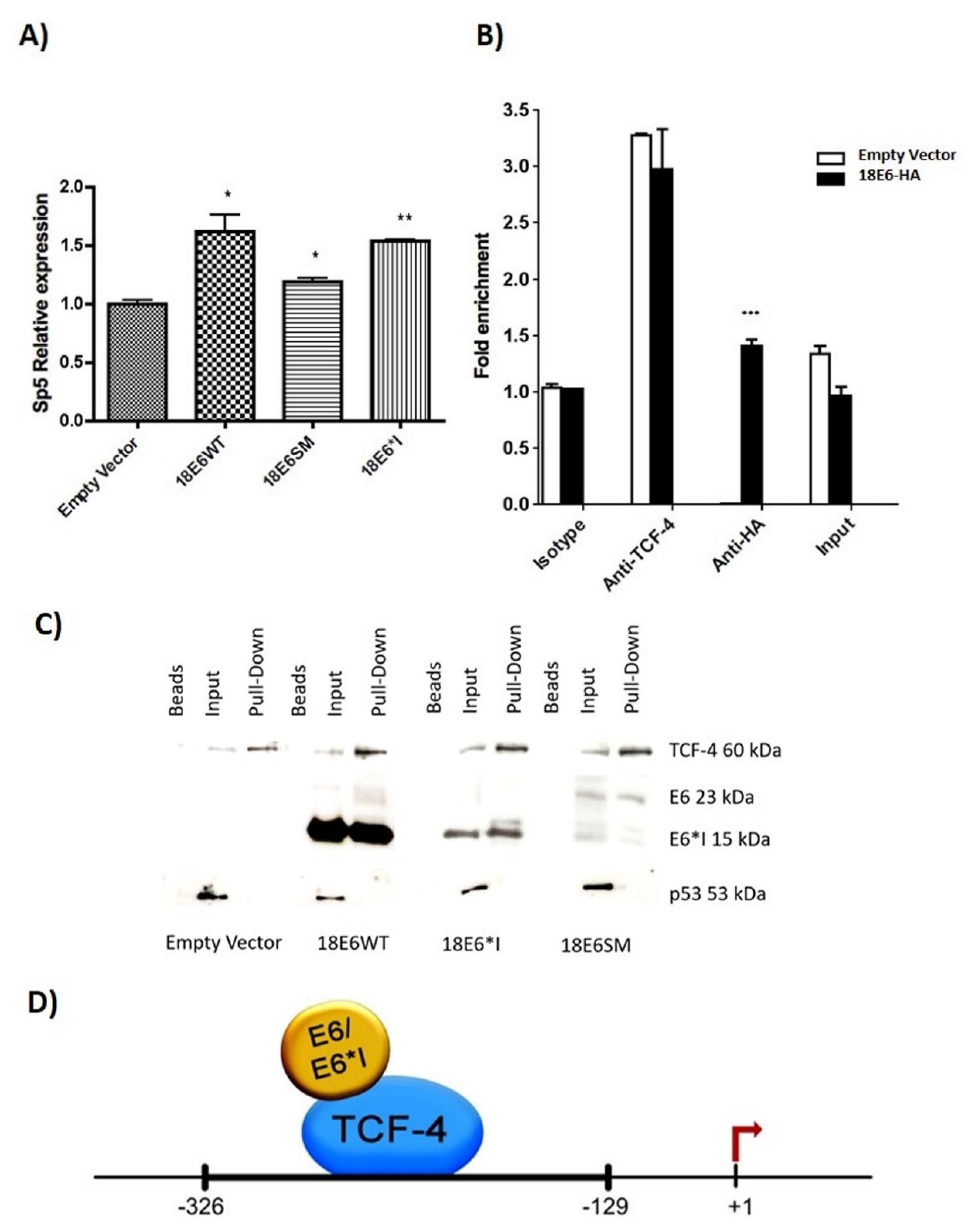
2.6. HPV-18 E6 and E6*I Proteins Bins to a TCF-4 Dependent Promoter In Vivo and In Vitro
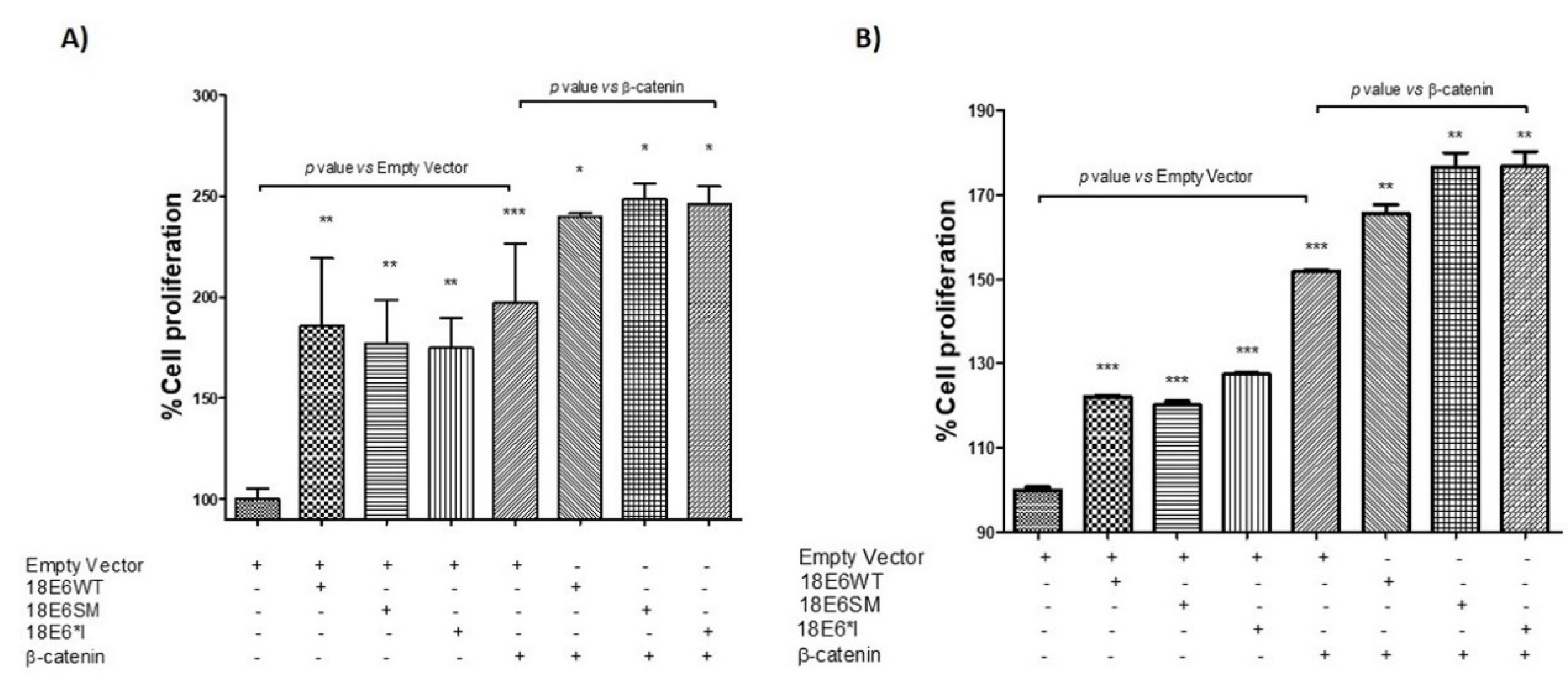
2.7. HPV-18 E6 and E6*I Proteins Induce Cell Proliferation in Cooperation with β-Catenin Overexpression
2.8. HPV-18 E6 Variants Differentially Modulate TCF-4-Mediated Transcription
2.9. HPV-18 Intratype Variants Interact with β-Catenin and TCF-4
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Transfection
4.2. Plasmids
4.3. Luciferase Reporter Activity Assays
4.4. Quantitative Polymerase Chain Reaction (qPCR)
4.5. Western Blotting
4.6. Immunoprecipitation Assay
4.7. Analysis of TCF-4 Stability
4.8. Immunofluorescence Staining and Cell Imaging
4.9. GST- Fusion Protein Purification
4.10. Soluble Cell Fractionation Assay
4.11. DNA Pull-Down Assay
4.12. Chromatin Immunoprecipitation Assay
4.13. Proliferation Assays
4.14. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Clevers, H.; Nusse, R. Wnt/β-catenin signaling and disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R. Wnt signaling. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R. The Wnt Homepage. Available online: http://web.stanford.edu/group/nusselab/cgi-bin/wnt/ (accessed on 9 April 2017).
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- Deitrick, J.; Pruitt, W.M. Wnt/β Catenin-Mediated Signaling Commonly Altered in Colorectal Cancer. Prog. Mol. Biol. Transl. Sci. 2016, 144, 49–68. [Google Scholar] [CrossRef] [PubMed]
- Takigawa, Y.; Brown, A.M.C. Wnt signaling in liver cancer. Curr. Drug Targets 2008, 9, 1013–1024. [Google Scholar] [CrossRef] [PubMed]
- Khramtsov, A.I.; Khramtsova, G.F.; Tretiakova, M.; Huo, D.; Olopade, O.I.; Goss, K.H. Wnt/beta-catenin pathway activation is enriched in basal-like breast cancers and predicts poor outcome. Am. J. Pathol. 2010, 176, 2911–2920. [Google Scholar] [CrossRef] [PubMed]
- Rampias, T.; Boutati, E.; Pectasides, E.; Sasaki, C.; Kountourakis, P.; Weinberger, P.; Psyrri, A. Activation of Wnt signaling pathway by human papillomavirus E6 and E7 oncogenes in HPV16-positive oropharyngeal squamous carcinoma cells. Mol. Cancer Res. 2010, 8, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Sastre, M.A.; González-Maya, L.; Delgado, R.; Lizano, M.; Tsubaki, G.; Mohar, A.; García-Carrancá, A. Abnormal distribution of E-cadherin and beta-catenin in different histologic types of cancer of the uterine cervix. Gynecol. Oncol. 2005, 97, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Bello, J.O.M.; Nieva, L.O.; Paredes, A.C.; Gonzalez, A.M.F.; Zavaleta, L.R.; Lizano, M. Regulation of the Wnt/β-Catenin Signaling Pathway by Human Papillomavirus E6 and E7 Oncoproteins. Viruses 2015, 7, 4734–4755. [Google Scholar] [CrossRef] [PubMed]
- zur Hausen, H. Papillomaviruses and cancer: From basic studies to clinical application. Nat. Rev. Cancer 2002, 2, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.S.; Lindsay, L.; Hoots, B.; Keys, J.; Franceschi, S.; Winer, R.; Clifford, G.M. Human papillomavirus type distribution in invasive cervical cancer and high-grade cervical lesions: A meta-analysis update. Int. J. cancer 2007, 121, 621–632. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin-Drubin, M.E.; Münger, K. Oncogenic activities of human papillomaviruses. Virus Res. 2009, 143, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Manzo-Merino, J.; Contreras-Paredes, A.; Vázquez-Ulloa, E.; Rocha-Zavaleta, L.; Fuentes-Gonzalez, A.M.; Lizano, M. The role of signaling pathways in cervical cancer and molecular therapeutic targets. Arch. Med. Res. 2014, 45, 525–539. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Tao, M.; McCoy, J.P.; Zheng, Z.-M. The E7 oncoprotein is translated from spliced E6*I transcripts in high-risk human papillomavirus type 16- or type 18-positive cervical cancer cell lines via translation reinitiation. J. Virol. 2006, 80, 4249–4263. [Google Scholar] [CrossRef] [PubMed]
- Shirasawa, H.; Tanzawa, H.; Matsunaga, T.; Simizu, B. Quantitative detection of spliced E6-E7 transcripts of human papillomavirus type 16 in cervical premalignant lesions. Virology 1991, 184, 795–798. [Google Scholar] [CrossRef]
- De la Cruz-Hernández, E.; García-Carrancá, A.; Mohar-Betancourt, A.; Dueñas-González, A.; Contreras-Paredes, A.; Pérez-Cardenas, E.; Herrera-Goepfert, R.; Lizano-Soberón, M. Differential splicing of E6 within human papillomavirus type 18 variants and functional consequences. J. Gen. Virol. 2005, 86, 2459–2468. [Google Scholar] [CrossRef] [PubMed]
- Olmedo-Nieva, L.; Muñoz-Bello, J.; Contreras-Paredes, A.; Lizano, M. The Role of E6 Spliced Isoforms (E6*) in Human Papillomavirus-Induced Carcinogenesis. Viruses 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Al-Shabanah, O.A.; Hafez, M.M.; Hassan, Z.K.; Sayed-Ahmed, M.M.; Abozeed, W.N.; Alsheikh, A.; Al-Rejaie, S.S. Methylation of SFRPs and APC genes in ovarian cancer infected with high risk human papillomavirus. Asian Pac. J. Cancer Prev. 2014, 15, 2719–2725. [Google Scholar] [CrossRef] [PubMed]
- Uren, A.; Fallen, S.; Yuan, H.; Usubütün, A.; Küçükali, T.; Schlegel, R.; Toretsky, J.A. Activation of the canonical Wnt pathway during genital keratinocyte transformation: A model for cervical cancer progression. Cancer Res. 2005, 65, 6199–6206. [Google Scholar] [CrossRef] [PubMed]
- Lichtig, H.; Gilboa, D.A.; Jackman, A.; Gonen, P.; Levav-Cohen, Y.; Haupt, Y.; Sherman, L. HPV16 E6 augments Wnt signaling in an E6AP-dependent manner. Virology 2010, 396, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Sominsky, S.; Kuslansky, Y.; Shapiro, B.; Jackman, A.; Haupt, Y.; Rosin-Arbesfeld, R.; Sherman, L. HPV16 E6 and E6AP differentially cooperate to stimulate or augment Wnt signaling. Virology 2014, 468–470, 510–523. [Google Scholar] [CrossRef] [PubMed]
- Bonilla-Delgado, J.; Bulut, G.; Liu, X.; Cortés-Malagón, E.M.; Schlegel, R.; Flores-Maldonado, C.; Contreras, R.G.; Chung, S.-H.; Lambert, P.F.; Uren, A.; et al. The E6 oncoprotein from HPV16 enhances the canonical Wnt/β-catenin pathway in skin epidermis in vivo. Mol. Cancer Res. 2012, 10, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Nakamura, Y.; Obama, K.; Furukawa, Y. Identification of SP5 as a downstream gene of the beta-catenin/Tcf pathway and its enhanced expression in human colon cancer. Int. J. Oncol. 2005, 27, 1483–1487. [Google Scholar] [PubMed]
- Hatzis, P.; van der Flier, L.G.; van Driel, M.A.; Guryev, V.; Nielsen, F.; Denissov, S.; Nijman, I.J.; Koster, J.; Santo, E.E.; Welboren, W.; et al. Genome-wide pattern of TCF7L2/TCF4 chromatin occupancy in colorectal cancer cells. Mol. Cell. Biol. 2008, 28, 2732–2744. [Google Scholar] [CrossRef] [PubMed]
- TFSiteScan. Available online: http://www.ifti.org/cgi-bin/ifti/Tfsitescan.pl (accessed on 5 August 2018).
- Lizano, M.; Berumen, J.; Guido, M.C.; Casas, L.; García-Carrancá, A. Association between human papillomavirus type 18 variants and histopathology of cervical cancer. J. Natl. Cancer Inst. 1997, 89, 1227–1231. [Google Scholar] [CrossRef] [PubMed]
- Lizano, M.; De la Cruz-Hernández, E.; Carrillo-García, A.; García-Carrancá, A.; Ponce de Leon-Rosales, S.; Dueñas-González, A.; Hernández-Hernández, D.M.; Mohar, A. Distribution of HPV16 and 18 intratypic variants in normal cytology, intraepithelial lesions, and cervical cancer in a Mexican population. Gynecol. Oncol. 2006, 102, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Contreras-Paredes, A.; De la Cruz-Hernández, E.; Martínez-Ramírez, I.; Dueñas-González, A.; Lizano, M. E6 variants of human papillomavirus 18 differentially modulate the protein kinase B/phosphatidylinositol 3-kinase (akt/PI3K) signaling pathway. Virology 2009, 383, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Moody, C.A.; Laimins, L.A. Human papillomavirus oncoproteins: Pathways to transformation. Nat. Rev. Cancer 2010, 10, 550–560. [Google Scholar] [CrossRef] [PubMed]
- Pim, D.; Massimi, P.; Banks, L. Alternatively spliced HPV-18 E6* protein inhibits E6 mediated degradation of p53 and suppresses transformed cell growth. Oncogene 1997, 15, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Pim, D.; Tomaic, V.; Banks, L. The human papillomavirus (HPV) E6* proteins from high-risk, mucosal HPVs can direct degradation of cellular proteins in the absence of full-length E6 protein. J. Virol. 2009, 83, 9863–9874. [Google Scholar] [CrossRef] [PubMed]
- Manzo-Merino, J.; Massimi, P.; Lizano, M.; Banks, L. The human papillomavirus (HPV) E6 oncoproteins promotes nuclear localization of active caspase 8. Virology 2014, 450–451, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Evans, W.; Filippova, M.; Filippov, V.; Bashkirova, S.; Zhang, G.; Reeves, M.E.; Duerksen-Hughes, P. Overexpression of HPV16 E6* Alters β-Integrin and Mitochondrial Dysfunction Pathways in Cervical Cancer Cells. Cancer Genomics Proteomics 2016, 13, 259–273. [Google Scholar] [PubMed]
- Tungteakkhun, S.S.; Filippova, M.; Fodor, N.; Duerksen-Hughes, P.J. The full-length isoform of human papillomavirus 16 E6 and its splice variant E6* bind to different sites on the procaspase 8 death effector domain. J. Virol. 2010, 84, 1453–1463. [Google Scholar] [CrossRef] [PubMed]
- Filippova, M.; Johnson, M.M.; Bautista, M.; Filippov, V.; Fodor, N.; Tungteakkhun, S.S.; Williams, K.; Duerksen-Hughes, P.J. The large and small isoforms of human papillomavirus type 16 E6 bind to and differentially affect procaspase 8 stability and activity. J. Virol. 2007, 81, 4116–4129. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Solano, M.; Meza-Canales, I.D.; Torres-Reyes, L.A.; Alvarez-Zavala, M.; Alvarado-Ruíz, L.; Rincon-Orozco, B.; Garcia-Chagollan, M.; Ochoa-Hernández, A.B.; Ortiz-Lazareno, P.C.; Rösl, F.; et al. Expression of WNT genes in cervical cancer-derived cells: Implication of WNT7A in cell proliferation and migration. Exp. Cell Res. 2015, 335, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Kuslansky, Y.; Sominsky, S.; Jackman, A.; Gamell, C.; Monahan, B.J.; Haupt, Y.; Rosin-Arbesfeld, R.; Sherman, L. Ubiquitin ligase E6AP mediates nonproteolytic polyubiquitylation of β-catenin independent of the E6 oncoprotein. J. Gen. Virol. 2016, 97, 3313–3330. [Google Scholar] [CrossRef] [PubMed]
- Koch, A.; Hrychyk, A.; Hartmann, W.; Waha, A.; Mikeska, T.; Waha, A.; Schüller, U.; Sörensen, N.; Berthold, F.; Goodyer, C.G.; et al. Mutations of the Wnt antagonist AXIN2 (Conductin) result in TCF-dependent transcription in medulloblastomas. Int. J. cancer 2007, 121, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Dong, X.; Mai, M.; Seelan, R.S.; Taniguchi, K.; Krishnadath, K.K.; Halling, K.C.; Cunningham, J.M.; Boardman, L.A.; Qian, C.; et al. Mutations in AXIN2 cause colorectal cancer with defective mismatch repair by activating beta-catenin/TCF signalling. Nat. Genet. 2000, 26, 146–147. [Google Scholar] [CrossRef] [PubMed]
- Leung, J.Y.; Kolligs, F.T.; Wu, R.; Zhai, Y.; Kuick, R.; Hanash, S.; Cho, K.R.; Fearon, E.R. Activation of AXIN2 expression by beta-catenin-T. cell factor. A feedback repressor pathway regulating Wnt signaling. J. Biol. Chem. 2002, 277, 21657–21665. [Google Scholar] [CrossRef] [PubMed]
- Lustig, B.; Jerchow, B.; Sachs, M.; Weiler, S.; Pietsch, T.; Karsten, U.; van de Wetering, M.; Clevers, H.; Schlag, P.M.; Birchmeier, W.; et al. Negative feedback loop of Wnt signaling through upregulation of conductin/axin2 in colorectal and liver tumors. Mol. Cell. Biol. 2002, 22, 1184–1193. [Google Scholar] [CrossRef] [PubMed]
- Shitashige, M.; Hirohashi, S.; Yamada, T. Wnt signaling inside the nucleus. Cancer Sci. 2008, 99, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Pang, T.; Gao, M.; Kang, H.; Ding, W.; Sun, X.; Zhao, Y.; Zhu, W.; Tang, X.; Yao, Y.; et al. HPV E6 induces eIF4E transcription to promote the proliferation and migration of cervical cancer. FEBS Lett. 2013, 587, 690–697. [Google Scholar] [CrossRef] [PubMed]
- Ben, W.; Yang, Y.; Yuan, J.; Sun, J.; Huang, M.; Zhang, D.; Zheng, J. Human papillomavirus 16 E6 modulates the expression of host microRNAs in cervical cancer. Taiwan. J. Obstet. Gynecol. 2015, 54, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Lizano, M.; Berumen, J.; García-Carrancá, A. HPV-related carcinogenesis: Basic concepts, viral types and variants. Arch. Med. Res. 2009, 40, 428–434. [Google Scholar] [CrossRef] [PubMed]
- Burk, R.D.; Harari, A.; Chen, Z. Human papillomavirus genome variants. Virology 2013, 445, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Zehbe, I.; Lichtig, H.; Westerback, A.; Lambert, P.F.; Tommasino, M.; Sherman, L. Rare human papillomavirus 16 E6 variants reveal significant oncogenic potential. Mol. Cancer 2011, 10. [Google Scholar] [CrossRef] [PubMed]











© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muñoz-Bello, J.O.; Olmedo-Nieva, L.; Castro-Muñoz, L.J.; Manzo-Merino, J.; Contreras-Paredes, A.; González-Espinosa, C.; López-Saavedra, A.; Lizano, M. HPV-18 E6 Oncoprotein and Its Spliced Isoform E6*I Regulate the Wnt/β-Catenin Cell Signaling Pathway through the TCF-4 Transcriptional Factor. Int. J. Mol. Sci. 2018, 19, 3153. https://doi.org/10.3390/ijms19103153
Muñoz-Bello JO, Olmedo-Nieva L, Castro-Muñoz LJ, Manzo-Merino J, Contreras-Paredes A, González-Espinosa C, López-Saavedra A, Lizano M. HPV-18 E6 Oncoprotein and Its Spliced Isoform E6*I Regulate the Wnt/β-Catenin Cell Signaling Pathway through the TCF-4 Transcriptional Factor. International Journal of Molecular Sciences. 2018; 19(10):3153. https://doi.org/10.3390/ijms19103153
Chicago/Turabian StyleMuñoz-Bello, J. Omar, Leslie Olmedo-Nieva, Leonardo Josué Castro-Muñoz, Joaquín Manzo-Merino, Adriana Contreras-Paredes, Claudia González-Espinosa, Alejandro López-Saavedra, and Marcela Lizano. 2018. "HPV-18 E6 Oncoprotein and Its Spliced Isoform E6*I Regulate the Wnt/β-Catenin Cell Signaling Pathway through the TCF-4 Transcriptional Factor" International Journal of Molecular Sciences 19, no. 10: 3153. https://doi.org/10.3390/ijms19103153
APA StyleMuñoz-Bello, J. O., Olmedo-Nieva, L., Castro-Muñoz, L. J., Manzo-Merino, J., Contreras-Paredes, A., González-Espinosa, C., López-Saavedra, A., & Lizano, M. (2018). HPV-18 E6 Oncoprotein and Its Spliced Isoform E6*I Regulate the Wnt/β-Catenin Cell Signaling Pathway through the TCF-4 Transcriptional Factor. International Journal of Molecular Sciences, 19(10), 3153. https://doi.org/10.3390/ijms19103153