The Reelin Receptors Apolipoprotein E receptor 2 (ApoER2) and VLDL Receptor


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
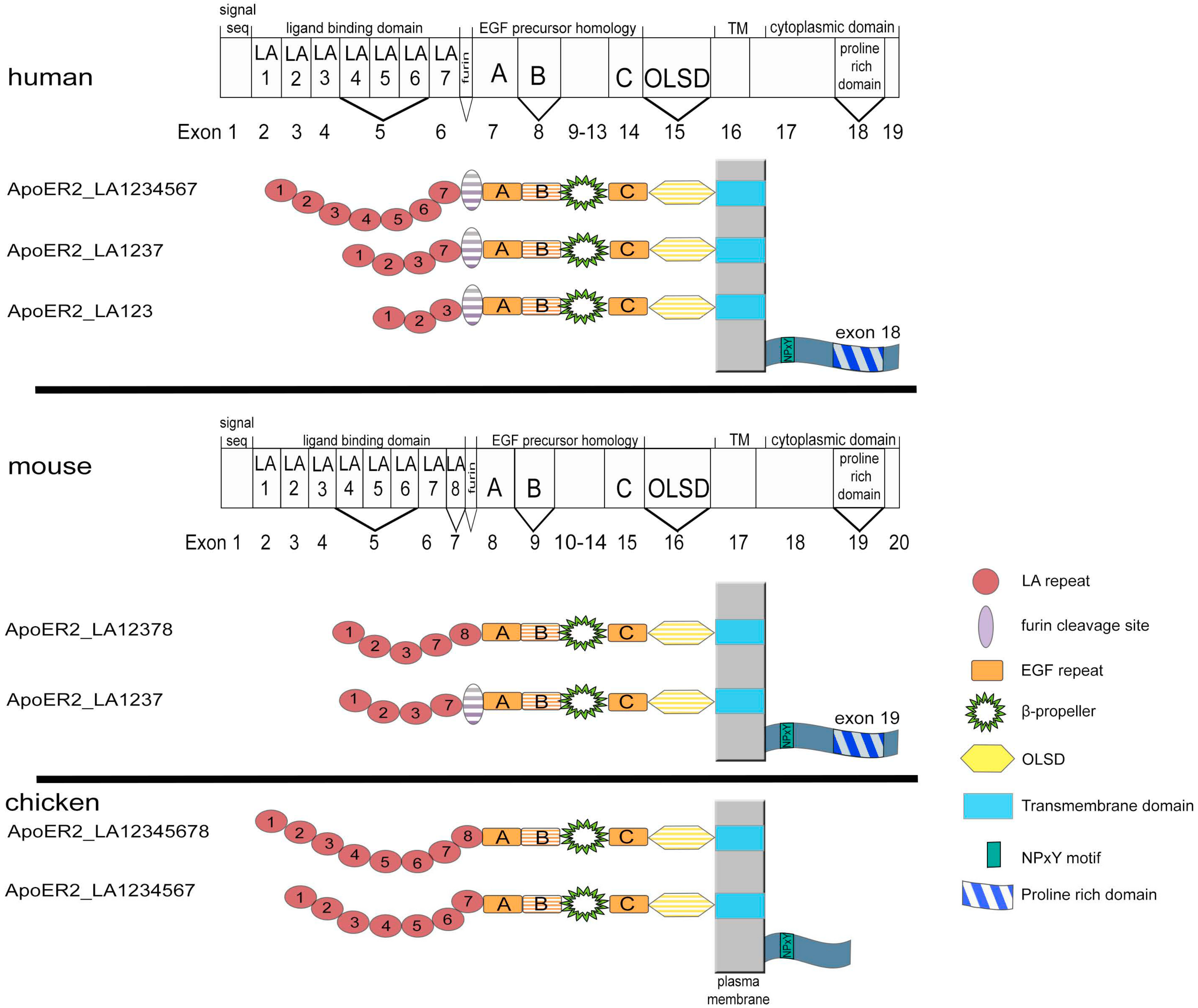
2. Structure and Expression of ApoER2
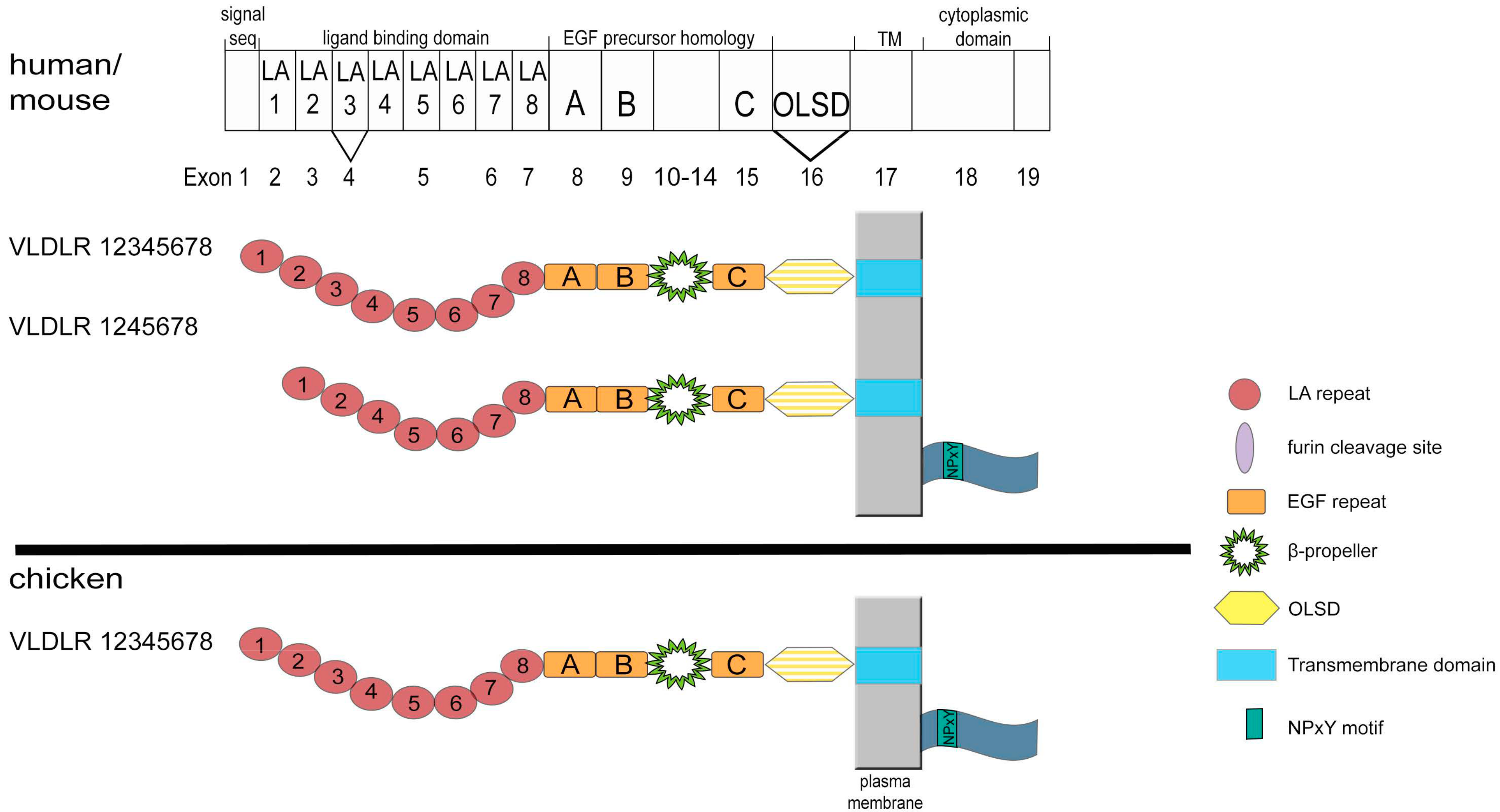
3. Structure and Expression of VLDLR
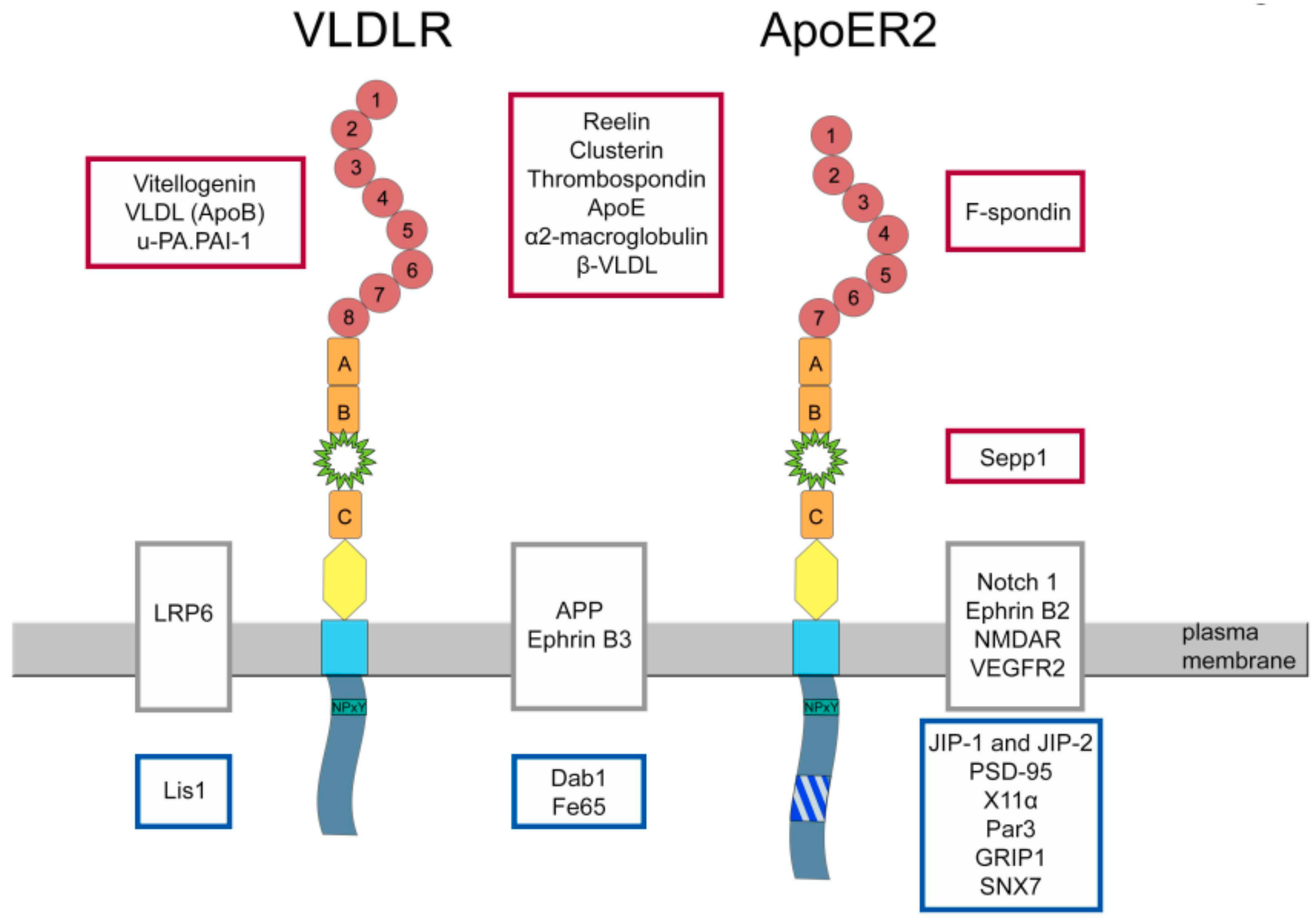
4. Functions of ApoER2
5. Proteins Interacting with ApoER2
6. Functions of VLDLR
7. Reelin, ApoER2, and VLDLR in Alzheimer’s Disease
8. Clusterin
9. Clusterin in Alzheimer’s Disease
10. Outlook
Funding
Conflicts of Interest
References
- Tissir, F.; Goffinet, A.M. Reelin and brain development. Nat. Rev. Neurosci. 2003, 4, 496–505. [Google Scholar] [CrossRef] [PubMed]
- Trommsdorff, M.; Gotthardt, M.; Hiesberger, T.; Shelton, J.; Stockinger, W.; Nimpf, J.; Hammer, R.E.; Richardson, J.A.; Herz, J. Reeler/disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2. Cell 1999, 97, 689–701. [Google Scholar] [CrossRef]
- Kim, D.-H.; Iijima, H.; Goto, K.; Sakai, J.; Ishii, H.; Kim, H.-J.; Suzuki, H.; Kondo, H.; Saeki, S.; Yamamoto, T. Human apolipoprotein E receptor 2. A novel lipoprotein receptor of the low density lipoprotein receptor family predominantly expressed in brain. J. Biol. Chem. 1996, 271, 8373–8380. [Google Scholar] [CrossRef] [PubMed]
- Novak, S.; Hiesberger, T.; Schneider, W.J.; Nimpf, J. A new low density lipoprotein receptor homologue with 8 ligand binding repeats in brain of chicken and mouse. J. Biol. Chem. 1996, 271, 11732–11736. [Google Scholar] [CrossRef] [PubMed]
- Reddy, S.S.; Connor, T.E.; Weeber, E.J.; Rebeck, W. Similarities and differences in structure, expression, and functions of VLDLR and ApoER2. Mol. Neurodegener. 2011, 6, 30. [Google Scholar] [CrossRef] [PubMed]
- Pasten, C.; Cerda, J.; Jausoro, I.; Court, F.A.; Caceres, A.; Marzolo, M.P. ApoER2 and Reelin are expressed in regenerating peripheral nerve and regulate schwann cell migration by activating the R0061c1 GEF protein, tiam1. Mol. Cell. Neurosci. 2015, 69, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Clatworthy, A.E.; Stockinger, W.; Christie, R.H.; Schneider, W.J.; Nimpf, J.; Hyman, B.T.; Rebeck, G.W. Expression and alternate splicing of apolipoprotein E receptor 2 in brain. Neuroscience 1999, 90, 903–911. [Google Scholar] [CrossRef]
- Perez-Martinez, F.J.; Luque-Rio, A.; Sakakibara, A.; Hattori, M.; Miyata, T.; Luque, J.M. Reelin-dependent ApoER2 downregulation uncouples newborn neurons from progenitor cells. Biol. Open 2012, 1, 1258–1263. [Google Scholar] [CrossRef] [PubMed]
- Lutters, B.C.; Derksen, R.H.; Tekelenburg, W.L.; Lenting, P.J.; Arnout, J.; de Groot, P.G. Dimers of β2-glycoprotein I increase platelet deposition to collagen via interaction with phospholipids and the apolipoprotein E receptor 2’. J. Biol. Chem. 2003, 278, 33831–33838. [Google Scholar] [CrossRef] [PubMed]
- Hirota, Y.; Kubo, K.; Katayama, K.; Honda, T.; Fujino, T.; Yamamoto, T.T.; Nakajima, K. Reelin receptors ApoER2 and VLDLR are expressed in distinct spatiotemporal patterns in developing mouse cerebral cortex. J. Comp. Neurol. 2015, 523, 463–478. [Google Scholar] [CrossRef] [PubMed]
- Perez-Garcia, C.G.; Tissir, F.; Goffinet, A.M.; Meyer, G. Reelin receptors in developing laminated brain structures of mouse and human. Eur. J. Neurosci. 2004, 20, 2827–2832. [Google Scholar] [CrossRef] [PubMed]
- Hack, I.; Hellwig, S.; Junghans, D.; Brunne, B.; Bock, H.H.; Zhao, S.; Frotscher, M. Divergent roles of ApoER2 and VLDLR in the migration of cortical neurons. Development 2007, 134, 3883–3891. [Google Scholar] [CrossRef] [PubMed]
- Brandes, C.; Novak, S.; Stockinger, W.; Herz, J.; Schneider, W.J.; Nimpf, J. Avian and murine LR8B and human apolipoprotein E receptor 2: Differentially spliced products from corresponding genes. Genomics 1997, 42, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-J.; Kim, D.-H.; Magoori, K.; Saeki, S.; Yamamoto, T.T. Evolution of the apolipoprotein E receptor 2 gene by exon loss. J. Biochem. Tokyo 1998, 124, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Brandes, C.; Kahr, L.; Stockinger, W.; Hiesberger, T.; Schneider, W.J.; Nimpf, J. Alternative splicing in the ligand binding domain of mouse ApoE receptor-2 produces receptor variants binding reelin but not α2-macroglobulin. J. Biol. Chem. 2001, 276, 22160–22169. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-H.; Magoori, K.; Inoue, T.R.; Mao, C.C.; Kim, H.-J.; Suzuki, H.; Fujita, T.; Endo, Y.; Saeki, S.; Yamamoto, T.T. Exon/intron organization, chromosome localization, alternative splicing, and transcription units of the human apolipoprotein E receptor 2. J. Biol. Chem. 1997, 272, 8498–8504. [Google Scholar] [CrossRef] [PubMed]
- Myant, N.B. Reelin and apolipoprotein E receptor 2 in the embryonic and mature brain: Effects of an evolutionary change in the ApoER2 gene. Proc. Biol. Sci. 2010, 277, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Koch, S.; Strasser, V.; Hauser, C.; Fasching, D.; Brandes, C.; Bajari, T.M.; Schneider, W.J.; Nimpf, J. A secreted soluble form of ApoE receptor 2 acts as a dominant-negative receptor and inhibits Reelin signaling. EMBO J. 2002, 21, 5996–6004. [Google Scholar] [CrossRef] [PubMed]
- May, P.; Bock, H.H.; Nimpf, J.; Herz, J. Differential glycosylation regulates processing of lipoprotein receptors by gamma-secretase. J. Biol. Chem. 2003, 278, 37386–37392. [Google Scholar] [CrossRef] [PubMed]
- Hibi, T.; Mizutani, M.; Baba, A.; Hattori, M. Splicing variations in the ligand-binding domain of ApoER2 results in functional differences in the binding properties to Reelin. Neurosci. Res. 2009, 63, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Gotthardt, M.; Trommsdorff, M.; Nevitt, M.F.; Shelton, J.; Richardson, J.A.; Stockinger, W.; Nimpf, J.; Herz, J. Interactions of the low density lipoprotein receptor gene family with cytosolic adaptor and scaffold proteins suggest diverse biological functions in cellular communication and signal transduction. J. Biol. Chem. 2000, 275, 25616–25624. [Google Scholar] [CrossRef] [PubMed]
- Stockinger, W.; Brandes, C.; Fasching, D.; Hermann, M.; Gotthardt, M.; Herz, J.; Schneider, W.J.; Nimpf, J. The reelin receptor ApoER2 recruits JNK-interacting proteins-1 and -2. J. Biol. Chem. 2000, 275, 25625–25632. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.; Duit, S.; Jalonen, P.; Out, R.; Scheer, L.; Sorrentino, V.; Boyadjian, R.; Rodenburg, K.W.; Foley, E.; Korhonen, L.; et al. The E3 ubiquitin ligase idol induces the degradation of the low density lipoprotein receptor family members VLDLR and ApoER2. J. Biol. Chem. 2010, 285, 19720–19726. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Marosi, M.; Choi, J.; Achiro, J.M.; Kim, S.; Li, S.; Otis, K.; Martin, K.C.; Portera-Cailliau, C.; Tontonoz, P. The E3 ubiquitin ligase idol regulates synaptic ApoER2 levels and is important for plasticity and learning. eLife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Poirier, S.; Mayer, G.; Benjannet, S.; Bergeron, E.; Marcinkiewicz, J.; Nassoury, N.; Mayer, H.; Nimpf, J.; Prat, A.; Seidah, N.G. The proprotein convertase PCSK9 induces the degradation of low density lipoprotein receptor (LDLR) and its closest family members VLDLR and ApoER2. J. Biol. Chem. 2008, 283, 2363–2372. [Google Scholar] [CrossRef] [PubMed]
- Sotelo, P.; Farfan, P.; Benitez, M.L.; Bu, G.; Marzolo, M.P. Sorting nexin 17 regulates ApoER2 recycling and reelin signaling. PLoS ONE 2014, 9, e93672. [Google Scholar] [CrossRef] [PubMed]
- Stockinger, W.; Sailler, B.; Strasser, V.; Recheis, B.; Fasching, D.; Kahr, L.; Schneider, W.J.; Nimpf, J. The PX-domain protein SNX17 interacts with members of the LDL receptor family and modulates endocytosis of the LDL receptor. EMBO J. 2002, 21, 4259–4267. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Kawarabayasi, Y.; Nakai, T.; Sakai, J.; Yamamoto, T. Rabbit very low density lipoprotein receptor: A low density lipoprotein receptor-like protein with distinct ligand specificity. Proc. Natl. Acad. Sci. USA 1992, 89, 9252–9256. [Google Scholar] [CrossRef] [PubMed]
- Oka, K.; Tzung, K.W.; Sullivan, M.; Lindsay, E.; Baldini, A.; Chan, L. Human very-low-density lipoprotein receptor complementary DNA and deduced amino acid sequence and localization of its gene (VLDLR) to chromosome band 9p24 by fluorescence in situ hybridization. Genomics 1994, 20, 298–300. [Google Scholar] [CrossRef] [PubMed]
- Sakai, J.; Hoshino, A.; Takahashi, S.; Miura, Y.; Ishii, H.; Suzuki, H.; Kawarabayasi, Y.; Yamamoto, T. Structure, chromosome location, and expression of the human very low density lipoprotein receptor gene. J. Biol. Chem. 1994, 269, 2173–2182. [Google Scholar] [PubMed]
- Oka, K.; Ishimura-Oka, K.; Chu, M.J.; Sullivan, M.; Krushkal, J.; Li, W.H.; Chan, L. Mouse very-low-density-lipoprotein receptor (VLDLR) cDNA cloning, tissue-specific expression and evolutionary relationship with the low-density-lipoprotein receptor. Eur J. Biochem 1994, 224, 975–982. [Google Scholar] [CrossRef] [PubMed]
- Bujo, H.; Hermann, M.; Kaderli, M.O.; Jacobsen, L.; Sugawara, S.; Nimpf, J.; Yamamoto, T.; Schneider, W.J. Chicken oocyte growth is mediated by an eight ligand binding repeat member of the LDL receptor family. EMBO J. 1994, 13, 5165–5175. [Google Scholar] [CrossRef] [PubMed]
- Nimpf, J.; Schneider, W.J. From cholesterol transport to signal transduction: Low density lipoprotein receptor, very low density lipoprotein receptor, and apolipoprotein E receptor-2. Biochim. Biophys. Acta 2000, 1529, 287–298. [Google Scholar] [CrossRef]
- Tiebel, O.; Oka, K.; Robinson, K.; Sullivan, M.; Martinez, J.; Nakamuta, M.; Ishimura-Oka, K.; Chan, L. Mouse very low-density lipoprotein receptor (VLDLR): Gene structure, tissue-specific expression and dietary and developmental regulation. Atherosclerosis 1999, 145, 239–251. [Google Scholar] [CrossRef]
- Sakai, K.; Tiebel, O.; Ljungberg, M.C.; Sullivan, M.; Lee, H.J.; Terashima, T.; Li, R.; Kobayashi, K.; Lu, H.C.; Chan, L.; et al. A neuronal VLDLR variant lacking the third complement-type repeat exhibits high capacity binding of ApoE containing lipoproteins. Brain Res. 2009, 1276, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Takahashi, Y.; Oka, K.; Ma, J.X. Functional differences of very-low-density lipoprotein receptor splice variants in regulating wnt signaling. Mol. Cell. Biol. 2016, 36, 2645–2654. [Google Scholar] [CrossRef] [PubMed]
- Iijima, H.; Miyazawa, M.; Sakai, J.; Magoori, K.; Ito, M.R.; Suzuki, H.; Nose, M.; Kawarabayasi, Y.; Yamamoto, T.T. Expression and characterization of a very low density lipoprotein receptor variant lacking the O-linked sugar region generated by alternative splicing. J. Biochem. 1998, 124, 747–755. [Google Scholar] [CrossRef] [PubMed]
- Magrane, J.; Casaroli-Marano, R.P.; Reina, M.; Gafvels, M.; Vilaro, S. The role of O-linked sugars in determining the very low density lipoprotein receptor stability or release from the cell. FEBS Lett. 1999, 451, 56–62. [Google Scholar] [CrossRef]
- Gåfvels, M.E.; Paavola, L.G.; Boyd, C.O.; Nolan, P.M.; Wittmaack, F.; Chawla, A.; Lazar, M.A.; Bucan, M.; Angelin, B.; Strauss, J.F. Cloning of a complementary deoxyribonucleic acid encoding the murine homologue of the very low density lipoprotein/apolipoprotein-e receptor: Expression pattern and assignment of the gene to mouse chromosome 19. Endocrinology 1994, 135, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Jokinen, E.V.; Landschulz, K.T.; Wyne, K.L.; Ho, Y.K.; Frykman, P.K.; Hobbs, H.H. Regulation of the very low density lipoprotein receptor by thyroid hormone in rat sceletal muscle. J. Biol. Chem. 1994, 269, 26411–26418. [Google Scholar] [PubMed]
- Nakamura, Y.; Yamamoto, M.; Kumamaru, E. Significance of the variant and full-length forms of the very low density lipoprotein receptor in brain. Brain Res. 2001, 922, 209–215. [Google Scholar] [CrossRef]
- Tabata, H.; Nakajima, K. Neurons tend to stop migration and differentiate along the cortical internal plexiform zones in the reelin signal-deficient mice. J. Neurosci. Res. 2002, 69, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Mayer, H.; Duit, S.; Hauser, C.; Schneider, W.J.; Nimpf, J. Reconstitution of the Reelin signaling pathway in fibroblasts demonstrates that Dab1 phosphorylation is independent of receptor localization in lipid rafts. Mol. Cell. Biol. 2006, 26, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Duit, S.; Mayer, H.; Blake, S.M.; Schneider, W.J.; Nimpf, J. Differential functions of ApoER2 and very low density lipoprotein receptor in reelin signaling depend on differential sorting of the receptors. J. Biol. Chem. 2010, 285, 4896–4908. [Google Scholar] [CrossRef] [PubMed]
- Frotscher, M. Role for reelin in stabilizing cortical architecture. Trends Neurosci. 2010, 33, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Ranaivoson, F.M.; von Daake, S.; Comoletti, D. Structural insights into reelin function: Present and future. Front. Cell. Neurosci. 2016, 10, 137. [Google Scholar] [CrossRef] [PubMed]
- Jossin, Y.; Gui, L.; Goffinet, A.M. Processing of reelin by embryonic neurons is important for function in tissue but not in dissociated cultured neurons. J. Neurosci. 2007, 27, 4243–4252. [Google Scholar] [CrossRef] [PubMed]
- Jossin, Y.; Ignatova, N.; Hiesberger, T.; Herz, J.; Lambert de Rouvroit, C.; Goffinet, A.M. The central fragment of reelin, generated by proteolytic processing in vivo, is critical to its function during cortical plate development. J. Neurosci. 2004, 24, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Lambert de Rouvroit, C.; de Bergeyck, V.; Cortvrindt, C.; Bar, I.; Eeckhout, Y.; Goffinet, A.M. Reelin, the extracellular matrix protein deficient in reeler mutant mice, is processed by a metalloproteinase. Exp. Neurol. 1999, 156, 214–217. [Google Scholar] [CrossRef] [PubMed]
- Yasui, N.; Kitago, Y.; Beppu, A.; Kohno, T.; Morishita, S.; Gomi, H.; Nagae, M.; Hattori, M.; Takagi, J. Functional importance of covalent homodimer of reelin protein linked via its central region. J. Biol. Chem. 2011, 286, 35247–35256. [Google Scholar] [CrossRef] [PubMed]
- Andersen, O.M.; Benhayon, D.; Curran, T.; Willnow, T.E. Differential binding of ligands to the apolipoprotein E receptor 2. Biochemistry 2003, 42, 9355–9364. [Google Scholar] [CrossRef] [PubMed]
- Hirai, H.; Yasui, N.; Yamashita, K.; Tabata, S.; Yamamoto, M.; Takagi, J.; Nogi, T. Structural basis for ligand capture and release by the endocytic receptor ApoER2. EMBO Rep. 2017, 18, 982–999. [Google Scholar] [CrossRef] [PubMed]
- Yasui, N.; Nogi, T.; Kitao, T.; Nakano, Y.; Hattori, M.; Takagi, J. Structure of a receptor-binding fragment of reelin and mutational analysis reveal a recognition mechanism similar to endocytic receptors. Proc. Natl. Acad. Sci. USA 2007, 104, 9988–9993. [Google Scholar] [CrossRef] [PubMed]
- D’Arcangelo, G.; Homayoundi, R.; Keshvara, L.; Rice, D.S.; Sheldon, M.; Curran, T. Reelin is a ligand for lipoprotein receptors. Neuron 1999, 24, 471–479. [Google Scholar] [CrossRef]
- Chow, V.W.; Mattson, M.P.; Wong, P.C.; Gleichmann, M. An overview of APP processing enzymes and products. Neuromolecular Med. 2010, 12, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Shih, I.M.; Wang, T.L. Notch signaling, γ-secretase inhibitors, and cancer therapy. Cancer Res. 2007, 67, 1879–1882. [Google Scholar] [CrossRef] [PubMed]
- Hoe, H.S.; Cooper, M.J.; Burns, M.P.; Lewis, P.A.; van der Brug, M.; Chakraborty, G.; Cartagena, C.M.; Pak, D.T.; Cookson, M.R.; Rebeck, G.W. The metalloprotease inhibitor TIMP-3 regulates amyloid precursor protein and apolipoprotein E receptor proteolysis. J. Neurosci. 2007, 27, 10895–10905. [Google Scholar] [CrossRef] [PubMed]
- Balmaceda, V.; Cuchillo-Ibanez, I.; Pujadas, L.; Garcia-Ayllon, M.S.; Saura, C.A.; Nimpf, J.; Soriano, E.; Saez-Valero, J. ApoER2 processing by presenilin-1 modulates reelin expression. FASEB J. 2014, 28, 1543–1554. [Google Scholar] [CrossRef] [PubMed]
- Hoe, H.S.; Tran, T.S.; Matsuoka, Y.; Howell, B.W.; Rebeck, G.W. Dab1 and reelin effects on amyloid precursor protein and ApoE receptor 2 trafficking and processing. J. Biol. Chem. 2006, 281, 35176–35185. [Google Scholar] [CrossRef] [PubMed]
- Telese, F.; Ma, Q.; Perez, P.M.; Notani, D.; Oh, S.; Li, W.; Comoletti, D.; Ohgi, K.A.; Taylor, H.; Rosenfeld, M.G. Lrp8-reelin-regulated neuronal enhancer signature underlying learning and memory formation. Neuron 2015, 86, 696–710. [Google Scholar] [CrossRef] [PubMed]
- Wasser, C.R.; Masiulis, I.; Durakoglugil, M.S.; Lane-Donovan, C.; Xian, X.; Beffert, U.; Agarwala, A.; Hammer, R.E.; Herz, J. Differential splicing and glycosylation of ApoER2 alters synaptic plasticity and fear learning. Sci. Signal. 2014, 7, ra113. [Google Scholar] [CrossRef] [PubMed]
- Blake, S.M.; Strasser, V.; Andrade, N.; Duit, S.; Hofbauer, R.; Schneider, W.J.; Nimpf, J. Thrombospondin-1 binds to ApoER2 and VLDL receptor and functions in postnatal neuronal migration. EMBO. J. 2008, 27, 3069–3080. [Google Scholar] [CrossRef] [PubMed]
- Hoe, H.S.; Wessner, D.; Beffert, U.; Becker, A.G.; Matsuoka, Y.; Rebeck, G.W. F-spondin interaction with the apolipoprotein E receptor ApoER2 affects processing of amyloid precursor protein. Mol. Cell. Biol. 2005, 25, 9259–9268. [Google Scholar] [CrossRef] [PubMed]
- Leeb, C.; Eresheim, C.; Nimpf, J. Clusterin is a ligand for apolipoprotein E receptor 2 (ApoER2) and very low density lipoprotein receptor (VLDLR) and signals via the reelin-signaling pathway. J. Biol. Chem. 2014, 289, 4161–4172. [Google Scholar] [CrossRef] [PubMed]
- Hill, K.E.; Zhou, J.; Austin, L.M.; Motley, A.K.; Ham, A.J.; Olson, G.E.; Atkins, J.F.; Gesteland, R.F.; Burk, R.F. The selenium-rich C-terminal domain of mouse selenoprotein P is necessary for the supply of selenium to brain and testis but not for the maintenance of whole body selenium. J. Biol. Chem. 2007, 282, 10972–10980. [Google Scholar] [CrossRef] [PubMed]
- Hill, K.E.; Zhou, J.; McMahan, W.J.; Motley, A.K.; Atkins, J.F.; Gesteland, R.F.; Burk, R.F. Deletion of selenoprotein P alters distribution of selenium in the mouse. J. Biol. Chem. 2003, 278, 13640–13646. [Google Scholar] [CrossRef] [PubMed]
- Burk, R.F.; Hill, K.E.; Olson, G.E.; Weeber, E.J.; Motley, A.K.; Winfrey, V.P.; Austin, L.M. Deletion of apolipoprotein E receptor-2 in mice lowers brain selenium and causes severe neurological dysfunction and death when a low-selenium diet is fed. J. Neurosci. 2007, 27, 6207–6211. [Google Scholar] [CrossRef] [PubMed]
- Olson, G.E.; Winfrey, V.P.; Nagdas, S.K.; Hill, K.E.; Burk, R.F. Apolipoprotein E receptor-2 (ApoER2) mediates selenium uptake from selenoprotein P by the mouse testis. J. Biol. Chem. 2007, 282, 12290–12297. [Google Scholar] [CrossRef] [PubMed]
- Burk, R.F.; Hill, K.E.; Motley, A.K.; Winfrey, V.P.; Kurokawa, S.; Mitchell, S.L.; Zhang, W. Selenoprotein P and apolipoprotein E receptor-2 interact at the blood-brain barrier and also within the brain to maintain an essential selenium pool that protects against neurodegeneration. FASEB J. 2014, 28, 3579–3588. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, S.; Bellinger, F.P.; Hill, K.E.; Burk, R.F.; Berry, M.J. Isoform-specific binding of selenoprotein P to the β-propeller domain of apolipoprotein E receptor 2 mediates selenium supply. J. Biol. Chem. 2014, 289, 9195–9207. [Google Scholar] [CrossRef] [PubMed]
- Bock, H.H.; May, P. Canonical and non-canonical reelin signaling. Front. Cell. Neurosci. 2016, 10, 166. [Google Scholar] [CrossRef] [PubMed]
- Santana, J.; Marzolo, M.P. The functions of reelin in membrane trafficking and cytoskeletal dynamics: Implications for neuronal migration, polarization and differentiation. Biochem. J. 2017, 474, 3137–3165. [Google Scholar] [CrossRef] [PubMed]
- Beffert, U.; Weeber, E.J.; Durudas, A.; Qiu, S.; Masiulis, I.; Sweatt, J.D.; Li, W.P.; Adelmann, G.; Frotscher, M.; Hammer, R.E.; et al. Modulation of synaptic plasticity and memory by reelin involves differential splicing of the lipoprotein receptor ApoER2. Neuron 2005, 47, 567–579. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Beffert, U.; Ertunc, M.; Tang, T.S.; Kavalali, E.T.; Bezprozvanny, I.; Herz, J. Reelin modulates NMDA receptor activity in cortical neurons. J. Neurosci. 2005, 25, 8209–8216. [Google Scholar] [CrossRef] [PubMed]
- Weeber, E.J.; Beffert, U.; Jones, C.; Christian, J.M.; Forster, E.; Sweatt, J.D.; Herz, J. Reelin and ApoE receptors cooperate to enhance hippocampal synaptic plasticity and learning. J. Biol. Chem. 2002, 277, 39944–39952. [Google Scholar] [CrossRef] [PubMed]
- Brai, E.; Marathe, S.; Astori, S.; Fredj, N.B.; Perry, E.; Lamy, C.; Scotti, A.; Alberi, L. Notch1 regulates hippocampal plasticity through interaction with the reelin pathway, glutamatergic transmission and CREB signaling. Front. Cell. Neurosci. 2015, 9, 447. [Google Scholar] [CrossRef] [PubMed]
- Beffert, U.; Nematollah Farsian, F.; Masiulis, I.; Hammer, R.E.; Yoon, S.O.; Giehl, K.M.; Herz, J. ApoE receptor 2 controls neuronal survival in the adult brain. Curr. Biol. 2006, 16, 2446–2452. [Google Scholar] [CrossRef] [PubMed]
- Minami, S.S.; Sung, Y.M.; Dumanis, S.B.; Chi, S.H.; Burns, M.P.; Ann, E.J.; Suzuki, T.; Turner, R.S.; Park, H.S.; Pak, D.T.; et al. The cytoplasmic adaptor protein X11α and extracellular matrix protein reelin regulate ApoE receptor 2 trafficking and cell movement. FASEB J. 2010, 24, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Pfennig, S.; Foss, F.; Bissen, D.; Harde, E.; Treeck, J.C.; Segarra, M.; Acker-Palmer, A. GRIP1 binds to ApoER2 and EphrinB2 to induce activity-dependent AMPA receptor insertion at the synapse. Cell. Rep. 2017, 21, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Bal, M.; Leitz, J.; Reese, A.L.; Ramirez, D.M.; Durakoglugil, M.; Herz, J.; Monteggia, L.M.; Kavalali, E.T. Reelin mobilizes a VAMP7-dependent synaptic vesicle pool and selectively augments spontaneous neurotransmission. Neuron 2013, 80, 934–946. [Google Scholar] [CrossRef] [PubMed]
- Fuentealba, R.A.; Barria, M.I.; Lee, J.; Cam, J.; Araya, C.; Escudero, C.A.; Inestrosa, N.C.; Bronfman, F.C.; Bu, G.; Marzolo, M.P. ApoER2 expression increases Aβ production while decreasing amyloid precursor protein (APP) endocytosis: Possible role in the partitioning of APP into lipid rafts and in the regulation of gamma-secretase activity. Mol. Neurodegener. 2007, 2, 14. [Google Scholar] [CrossRef] [PubMed]
- Senturk, A.; Pfennig, S.; Weiss, A.; Burk, K.; Acker-Palmer, A. Ephrin Bs are essential components of the Reelin pathway to regulate neuronal migration. Nature 2011, 472, 356–360. [Google Scholar] [CrossRef] [PubMed]
- Hoe, H.S.; Pocivavsek, A.; Chakraborty, G.; Fu, Z.; Vicini, S.; Ehlers, M.D.; Rebeck, G.W. Apolipoprotein E receptor 2 interactions with the N-methyl-d-aspartate receptor. J. Biol. Chem. 2006, 281, 3425–3431. [Google Scholar] [CrossRef] [PubMed]
- Segarra, M.; Aburto, M.R.; Cop, F.; Llao-Cid, C.; Hartl, R.; Damm, M.; Bethani, I.; Parrilla, M.; Husainie, D.; Schanzer, A.; et al. Endothelial Dab1 signaling orchestrates neuro-glia-vessel communication in the central nervous system. Science 2018, 361. [Google Scholar] [CrossRef] [PubMed]
- Boycott, K.M.; Flavelle, S.; Bureau, A.; Glass, H.C.; Fujiwara, T.M.; Wirrell, E.; Davey, K.; Chudley, A.E.; Scott, J.N.; McLeod, D.R.; et al. Homozygous deletion of the very low density lipoprotein receptor gene causes autosomal recessive cerebellar hypoplasia with cerebral gyral simplification. Am. J. Hum. Genet. 2005, 77, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Moheb, L.A.; Tzschach, A.; Garshasbi, M.; Kahrizi, K.; Darvish, H.; Heshmati, Y.; Kordi, A.; Najmabadi, H.; Ropers, H.H.; Kuss, A.W. Identification of a nonsense mutation in the very low-density lipoprotein receptor gene (VLDLR) in an iranian family with dysequilibrium syndrome. Eur. J. Hum. Genet. 2008, 16, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Turkmen, S.; Hoffmann, K.; Demirhan, O.; Aruoba, D.; Humphrey, N.; Mundlos, S. Cerebellar hypoplasia, with quadrupedal locomotion, caused by mutations in the very low-density lipoprotein receptor gene. Eur. J. Hum. Genet. 2008, 16, 1070–1074. [Google Scholar] [CrossRef] [PubMed]
- Ozcelik, T.; Akarsu, N.; Uz, E.; Caglayan, S.; Gulsuner, S.; Onat, O.E.; Tan, M.; Tan, U. Mutations in the very low-density lipoprotein receptor VLDLR cause cerebellar hypoplasia and quadrupedal locomotion in humans. Proc. Natl. Acad. Sci. USA 2008, 105, 4232–4236. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, J.; Kouiavskaia, D.; Migliorini, M.; Robinson, S.; Saenko, E.L.; Gorlatova, N.; Li, D.; Lawrence, D.; Hyman, B.T.; Weisgraber, K.H.; et al. The ApoE isoform binding properties of the VLDL receptor reveal marked differences from LRP and the LDL receptor. J. Lipid Res. 2005, 46, 1721–1731. [Google Scholar] [CrossRef] [PubMed]
- Frykman, P.K.; Brown, M.S.; Yamamoto, T.; Goldstein, J.L.; Herz, J. Normal plasma lipoproteins and fertility in gene-targeted mice homozygous for a disruption in the gene encoding very low density lipoprotein receptor. Proc. Natl. Acad. Sci. USA 1995, 92, 8453–8457. [Google Scholar] [CrossRef] [PubMed]
- Tacken, P.J.; Teusink, B.; Jong, M.C.; Harats, D.; Havekes, L.M.; Dijk, K.W.v.; Hofker, M.H. LDL receptor deficiency unmasks altered VLDL triglyceride metabolism in VLDL receptor transgenic and knock out mice. J. Lipid Res. 2000, 41, 2055–2062. [Google Scholar] [PubMed]
- Goudriaan, J.R.; Tacken, P.J.; Dahlmans, V.E.; Gijbels, M.J.; van Dijk, K.W.; Havekes, L.M.; Jong, M.C. Protection from obesity in mice lacking the VLDL receptor. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1488–1493. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Oka, K.; Forte, T.; Ishida, B.; Teng, B.; Ishimura-Oka, K.; Nakamuta, M.; Chan, L. Reversal of hypercholesterolemia in low density lipoprotein receptor knock out mice by adenovirus-mediated gene transfer of the very low density lipoprotein receptor. J. Biol. Chem. 1996, 271, 6852–6860. [Google Scholar] [CrossRef] [PubMed]
- Kozarsky, K.F.; Jooss, K.; Donahee, M.; Strauss, J.F., III; Wilson, J.M. Effective treatment of familial hypercholesterolaemia in the mouse model using adenovirus-mediated transfer of the VLDL receptor gene. Nat. Genet. 1996, 13, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Heegaard, C.W.; Simonson, A.C.W.; Oka, K.; Kjøller, L.; Christensen, A.; Madsen, B.; Ellgaard, L.; Chan, L.; Andreasen, P.A. Very low density lipoprotein receptor binds and mediates endocytosis of urokinase-type plasminogen activator-type-1 plasminogen activator inhibitor complex. J. Biol. Chem. 1995, 270, 20855–20861. [Google Scholar] [CrossRef] [PubMed]
- Mikhailenko, I.; Krylov, D.; Argraves, K.M.; Roberts, D.D.; Liau, G.; Strickland, D.K. Cellular internalization and degradation of thrombospondin-1 is mediated by the amino-terminal heparin binding domain (HBD). High affinity interaction of dimeric HBD with the low density lipoprotein receptor-related protein. J. Biol. Chem. 1997, 272, 6784–6791. [Google Scholar] [CrossRef] [PubMed]
- Stifani, S.; Barber, D.L.; Nimpf, J.; Schneider, W.J. A single chicken oocyte plasma membrane protein mediates uptake of very low density lipoprotein and vitellogenin. Proc. Natl. Acad. Sci. USA 1990, 87, 1955–1959. [Google Scholar] [CrossRef] [PubMed]
- Nimpf, J.; Schneider, W.J. The VLDL receptor: An LDL receptor relative with eight ligand binding repeats, LR8. Atherosclerosis 1998, 141, 191–202. [Google Scholar] [CrossRef]
- Bujo, H.; Yamamoto, T.; Hayashi, K.; Hermann, M.; Nimpf, J.; Schneider, W.J. Mutant oocytic low density lipoprotein receptor gene family member causes atherosclerosis and female sterility. Proc. Natl. Acad. Sci. USA 1995, 92, 9905–9909. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H.; Nusse, R. Wnt/β-catenin signaling and disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef] [PubMed]
- Arnes, M.; Casas Tinto, S. Aberrant Wnt signaling: A special focus in CNS diseases. J. Neurogenet. 2017, 31, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Hu, Y.; Lu, K.; Flannery, J.G.; Ma, J.X. Very low density lipoprotein receptor, a negative regulator of the Wnt signaling pathway and choroidal neovascularization. J. Biol. Chem. 2007, 282, 34420–34428. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Shin, Y.; Cheng, R.; Park, K.; Hu, Y.; McBride, J.; He, X.; Takahashi, Y.; Ma, J.X. Receptor heterodimerization as a novel mechanism for the regulation of Wnt/β-catenin signaling. J. Cell. Sci. 2014, 127, 4857–4869. [Google Scholar] [CrossRef] [PubMed]
- Hoe, H.S.; Magill, L.A.; Guenette, S.; Fu, Z.; Vicini, S.; Rebeck, G.W. Fe65 interaction with the ApoE receptor ApoER2. J. Biol. Chem. 2006, 281, 24521–24530. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Assadi, A.H.; McNeil, R.S.; Beffert, U.; Wynshaw-Boris, A.; Herz, J.; Clark, G.D.; D’Arcangelo, G. The Pafah1b complex interacts with the reelin receptor VLDLR. PLoS ONE 2007, 2, e252. [Google Scholar] [CrossRef] [PubMed]
- Alcantara, S.; Ruiz, M.; D’Arcangelo, G.; Ezan, F.; de Lecea, L.; Curran, T.; Sotelo, C.; Soriano, E. Regional and cellular patterns of reelin mRNA expression in the forebrain of the developing and adult mouse. J. Neurosci. 1998, 18, 7779–7799. [Google Scholar] [CrossRef] [PubMed]
- Doehner, J.; Knuesel, I. Reelin-mediated signaling during normal and pathological forms of aging. Aging Dis. 2010, 1, 12–29. [Google Scholar] [PubMed]
- Herring, A.; Donath, A.; Steiner, K.M.; Widera, M.P.; Hamzehian, S.; Kanakis, D.; Kolble, K.; ElAli, A.; Hermann, D.M.; Paulus, W.; et al. Reelin depletion is an early phenomenon of Alzheimer’s pathology. J. Alzheimers Dis. 2012, 30, 963–979. [Google Scholar] [CrossRef] [PubMed]
- Holtzman, D.M.; Morris, J.C.; Goate, A.M. Alzheimer’s disease: The challenge of the second century. Sci. Transl. Med. 2011, 3, 77sr71. [Google Scholar] [CrossRef] [PubMed]
- Durakoglugil, M.S.; Chen, Y.; White, C.L.; Kavalali, E.T.; Herz, J. Reelin signaling antagonizes β-amyloid at the synapse. Proc. Natl. Acad. Sci. USA 2009, 106, 15938–15943. [Google Scholar] [CrossRef] [PubMed]
- Mota, S.I.; Ferreira, I.L.; Valero, J.; Ferreiro, E.; Carvalho, A.L.; Oliveira, C.R.; Rego, A.C. Impaired Src signaling and post-synaptic actin polymerization in Alzheimer’s disease mice hippocampus—Linking NMDA receptors and the reelin pathway. Exp. Neurol. 2014, 261, 698–709. [Google Scholar] [CrossRef] [PubMed]
- Ohkubo, N.; Lee, Y.D.; Morishima, A.; Terashima, T.; Kikkawa, S.; Tohyama, M.; Sakanaka, M.; Tanaka, J.; Maeda, N.; Vitek, M.P.; et al. Apolipoprotein E and reelin ligands modulate tau phosphorylation through an apolipoprotein e receptor/disabled-1/glycogen synthase kinase-3β cascade. FASEB J. 2003, 17, 295–297. [Google Scholar] [CrossRef] [PubMed]
- Kocherhans, S.; Madhusudan, A.; Doehner, J.; Breu, K.S.; Nitsch, R.M.; Fritschy, J.M.; Knuesel, I. Reduced reelin expression accelerates amyloid-β plaque formation and tau pathology in transgenic Alzheimer’s disease mice. J. Neurosci. 2010, 30, 9228–9240. [Google Scholar] [CrossRef] [PubMed]
- Lane-Donovan, C.; Philips, G.T.; Wasser, C.R.; Durakoglugil, M.S.; Masiulis, I.; Upadhaya, A.; Pohlkamp, T.; Coskun, C.; Kotti, T.; Steller, L.; et al. Reelin protects against amyloid β toxicity in vivo. Sci. Signal. 2015, 8, ra67. [Google Scholar] [CrossRef] [PubMed]
- Pujadas, L.; Rossi, D.; Andres, R.; Teixeira, C.M.; Serra-Vidal, B.; Parcerisas, A.; Maldonado, R.; Giralt, E.; Carulla, N.; Soriano, E. Reelin delays amyloid-β fibril formation and rescues cognitive deficits in a model of Alzheimer’s disease. Nat. Commun. 2014, 5, 3443. [Google Scholar] [CrossRef] [PubMed]
- Saez-Valero, J.; Costell, M.; Sjogren, M.; Andreasen, N.; Blennow, K.; Luque, J.M. Altered levels of cerebrospinal fluid reelin in frontotemporal dementia and Alzheimer’s disease. J. Neurosci. Res. 2003, 72, 132–136. [Google Scholar] [CrossRef] [PubMed]
- Cuchillo-Ibanez, I.; Mata-Balaguer, T.; Balmaceda, V.; Arranz, J.J.; Nimpf, J.; Saez-Valero, J. The β-amyloid peptide compromises reelin signaling in Alzheimer’s disease. Sci. Rep. 2016, 6, 31646. [Google Scholar] [CrossRef] [PubMed]
- Cuchillo-Ibanez, I.; Balmaceda, V.; Mata-Balaguer, T.; Lopez-Font, I.; Saez-Valero, J. Reelin in Alzheimer’s disease, increased levels but impaired signaling: When more is less. J. Alzheimers Dis. 2016, 52, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Cuchillo-Ibanez, I.; Balmaceda, V.; Botella-Lopez, A.; Rabano, A.; Avila, J.; Saez-Valero, J. β-amyloid impairs reelin signaling. PLoS ONE 2013, 8, e72297. [Google Scholar] [CrossRef] [PubMed]
- Madhusudan, A.; Sidler, C.; Knuesel, I. Accumulation of reelin-positive plaques is accompanied by a decline in basal forebrain projection neurons during normal aging. Eur. J. Neurosci. 2009, 30, 1064–1076. [Google Scholar] [CrossRef] [PubMed]
- Knuesel, I.; Nyffeler, M.; Mormede, C.; Muhia, M.; Meyer, U.; Pietropaolo, S.; Yee, B.K.; Pryce, C.R.; LaFerla, F.M.; Marighetto, A.; et al. Age-related accumulation of reelin in amyloid-like deposits. Neurobiol. Aging 2009, 30, 697–716. [Google Scholar] [CrossRef] [PubMed]
- Krstic, D.; Rodriguez, M.; Knuesel, I. Regulated proteolytic processing of reelin through interplay of tissue plasminogen activator (tPA), ADAMTS-4, ADAMTS-5, and their modulators. PLoS ONE 2012, 7, e47793. [Google Scholar] [CrossRef] [PubMed]
- Botella-Lopez, A.; Burgaya, F.; Gavin, R.; Garcia-Ayllon, M.S.; Gomez-Tortosa, E.; Pena-Casanova, J.; Urena, J.M.; Del Rio, J.A.; Blesa, R.; Soriano, E.; et al. Reelin expression and glycosylation patterns are altered in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2006, 103, 5573–5578. [Google Scholar] [CrossRef] [PubMed]
- Botella-Lopez, A.; Cuchillo-Ibanez, I.; Cotrufo, T.; Mok, S.S.; Li, Q.X.; Barquero, M.S.; Dierssen, M.; Soriano, E.; Saez-Valero, J. β-amyloid controls altered reelin expression and processing in Alzheimer’s disease. Neurobiol. Dis. 2010, 37, 682–691. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, S.I.; Rosse, R.B.; Deutsch, L.H. Faulty regulation of tau phosphorylation by the reelin signal transduction pathway is a potential mechanism of pathogenesis and therapeutic target in Alzheimer’s disease. Eur. Neuropsychopharmacol. 2006, 16, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Kramer, P.L.; Xu, H.; Woltjer, R.L.; Westaway, S.K.; Clark, D.; Erten-Lyons, D.; Kaye, J.A.; Welsh-Bohmer, K.A.; Troncoso, J.C.; Markesbery, W.R.; et al. Alzheimer disease pathology in cognitively healthy elderly: A genome-wide study. Neurobiol. Aging 2011, 32, 2113–2122. [Google Scholar] [CrossRef] [PubMed]
- Ohkubo, N.; Vitek, M.P.; Morishima, A.; Suzuki, Y.; Miki, T.; Maeda, N.; Mitsuda, N. Reelin signals survival through Src-family kinases that inactivate bad activity. J. Neurochem. 2007, 103, 820–830. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Liu, C.C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein e and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Durakoglugil, M.S.; Xian, X.; Herz, J. ApoE4 reduces glutamate receptor function and synaptic plasticity by selectively impairing ApoE receptor recycling. Proc. Natl. Acad. Sci. USA 2010, 107, 12011–12016. [Google Scholar] [CrossRef] [PubMed]
- Deane, R.; Sagare, A.; Hamm, K.; Parisi, M.; Lane, S.; Finn, M.B.; Holtzman, D.M.; Zlokovic, B.V. ApoE isoform-specific disruption of amyloid β peptide clearance from mouse brain. J. Clin. Investig. 2008, 118, 4002–4013. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.L.; Ng, H.K.; Baum, L.; Pang, J.C.; Chiu, H.F.; Woo, J.; Tang, N.L.; Lam, L.C. Low-density lipoprotein receptor-related protein 8 (apolipoprotein E receptor 2) gene polymorphisms in Alzheimer’s disease. Neurosci. Lett. 2002, 332, 216–218. [Google Scholar] [CrossRef]
- Wang, W.; Moerman-Herzog, A.M.; Slaton, A.; Barger, S.W. Presenilin 1 mutations influence processing and trafficking of the ApoE receptor ApoER2. Neurobiol. Aging 2017, 49, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Mata-Balaguer, T.; Cuchillo-Ibanez, I.; Calero, M.; Ferrer, I.; Saez-Valero, J. Decreased generation of C-terminal fragments of ApoER2 and increased reelin expression in Alzheimer’s disease. FASEB J. 2018, 32, 3536–3546. [Google Scholar] [CrossRef] [PubMed]
- Hoe, H.S.; Rebeck, G.W. Regulation of ApoE receptor proteolysis by ligand binding. Brain Res. Mol. Brain Res. 2005, 137, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Hinrich, A.J.; Jodelka, F.M.; Chang, J.L.; Brutman, D.; Bruno, A.M.; Briggs, C.A.; James, B.D.; Stutzmann, G.E.; Bennett, D.A.; Miller, S.A.; et al. Therapeutic correction of ApoER2 splicing in Alzheimer’s disease mice using antisense oligonucleotides. EMBO Mol. Med. 2016, 8, 328–345. [Google Scholar] [CrossRef] [PubMed]
- Helbecque, N.; Amouyel, P. Very low density lipoprotein receptor in Alzheimer disease. Microsc. Res. Tech. 2000, 50, 273–277. [Google Scholar] [CrossRef]
- Helbecque, N.; Berr, C.; Cottel, D.; Fromentin-David, I.; Sazdovitch, V.; Ricolfi, F.; Ducimetiere, P.; Di Menza, C.; Amouyel, P. VLDL receptor polymorphism, cognitive impairment, and dementia. Neurology 2001, 56, 1183–1188. [Google Scholar] [CrossRef] [PubMed]
- Okuizumi, K.; Onodera, O.; Namba, Y.; Ikeda, K.; Yamamoto, T.; Seki, K.; Ueki, A.; Nanko, S.; Tanaka, H.; Takahashi, H.; et al. Genetic association of the very low density lipoprotein (VLDL) receptor gene with sporadic Alzheimer’s disease. Nat. Genet. 1995, 11, 207–209. [Google Scholar] [CrossRef] [PubMed]
- Christie, R.H.; Chung, H.; Rebeck, G.W.; Strickland, D.; Hyman, B.T. Expression of the very low-density lipoprotein receptor (VLDL-R), an apolipoprotein-e receptor, in the central nervous system and in Alzheimer’s disease. J. Neuropathol. Exp. Neurol. 1996, 55, 491–498. [Google Scholar] [CrossRef] [PubMed]
- De Silva, H.V.; Harmony, J.A.; Stuart, W.D.; Gil, C.M.; Robbins, J. Apolipoprotein j: Structure and tissue distribution. Biochemistry 1990, 29, 5380–5389. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.E.; Jomary, C. Clusterin. Int. J. Biochem. Cell. Biol. 2002, 34, 427–431. [Google Scholar] [CrossRef]
- Kapron, J.T.; Hilliard, G.M.; Lakins, J.N.; Tenniswood, M.P.; West, K.A.; Carr, S.A.; Crabb, J.W. Identification and characterization of glycosylation sites in human serum clusterin. Protein Sci. 1997, 6, 2120–2133. [Google Scholar] [CrossRef] [PubMed]
- Murphy, B.F.; Kirszbaum, L.; Walker, I.D.; d’Apice, A.J. SP-40,40, a newly identified normal human serum protein found in the SC5b-9 complex of complement and in the immune deposits in glomerulonephritis. J. Clin. Investig. 1988, 81, 1858–1864. [Google Scholar] [CrossRef] [PubMed]
- De Silva, H.V.; Stuart, W.D.; Duvic, C.R.; Wetterau, J.R.; Ray, M.J.; Ferguson, D.G.; Albers, H.W.; Smith, W.R.; Harmony, J.A. A 70-kDa apolipoprotein designated ApoJ is a marker for subclasses of human plasma high density lipoproteins. J. Biol. Chem. 1990, 265, 13240–13247. [Google Scholar] [PubMed]
- Trougakos, I.P.; Djeu, J.Y.; Gonos, E.S.; Boothman, D.A. Advances and challenges in basic and translational research on clusterin. Cancer Res. 2009, 69, 403–406. [Google Scholar] [CrossRef] [PubMed]
- Trougakos, I.P.; Gonos, E.S. Regulation of clusterin/apolipoprotein j, a functional homologue to the small heat shock proteins, by oxidative stress in ageing and age-related diseases. Free Radic. Res. 2006, 40, 1324–1334. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.R.; Easterbrook-Smith, S.B. Clusterin is a secreted mammalian chaperone. Trends Biochem. Sci. 2000, 25, 95–98. [Google Scholar] [CrossRef]
- Wyatt, A.; Yerbury, J.; Poon, S.; Dabbs, R.; Wilson, M. The chaperone action of clusterin and its putative role in quality control of extracellular protein folding. Adv. Cancer Res. 2009, 104, 89–114. [Google Scholar] [PubMed]
- Wyatt, A.R.; Yerbury, J.J.; Berghofer, P.; Greguric, I.; Katsifis, A.; Dobson, C.M.; Wilson, M.R. Clusterin facilitates in vivo clearance of extracellular misfolded proteins. Cell. Mol. Life Sci. 2011, 68, 3919–3931. [Google Scholar] [CrossRef] [PubMed]
- Karch, C.M.; Cruchaga, C.; Goate, A.M. Alzheimer’s disease genetics: From the bench to the clinic. Neuron 2014, 83, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; DeStafano, A.L.; Bis, J.C.; Beecham, G.W.; Grenier-Boley, B.; et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.C.; Heath, S.; Even, G.; Campion, D.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Zelenika, D.; Bullido, M.J.; Tavernier, B.; et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1094–1099. [Google Scholar] [CrossRef] [PubMed]
- Bettens, K.; Brouwers, N.; Engelborghs, S.; Lambert, J.C.; Rogaeva, E.; Vandenberghe, R.; Le Bastard, N.; Pasquier, F.; Vermeulen, S.; Van Dongen, J.; et al. Both common variations and rare non-synonymous substitutions and small insertion/deletions in CLU are associated with increased Alzheimer risk. Mol. Neurodegener. 2012, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- May, P.C.; Lampert-Etchells, M.; Johnson, S.A.; Poirier, J.; Masters, J.N.; Finch, C.E. Dynamics of gene expression for a hippocampal glycoprotein elevated in Alzheimer’s disease and in response to experimental lesions in rat. Neuron 1990, 5, 831–839. [Google Scholar] [CrossRef]
- McGeer, P.L.; Kawamata, T.; Walker, D.G. Distribution of clusterin in Alzheimer brain tissue. Brain Res. 1992, 579, 337–341. [Google Scholar] [CrossRef]
- Ghiso, J.; Matsubara, E.; Koudinov, A.; Choi Miura, N.H.; Tomita, M.; Wisniewski, T.; Frangione, B. The cerebrospinal-fluid soluble form of Alzheimer’s amyloid β is complexed to sp-40,40 (apolipoprotein j), an inhibitor of the complement membrane-attack complex. Biochem. J. 1993, 293, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Narayan, P.; Orte, A.; Clarke, R.W.; Bolognesi, B.; Hook, S.; Ganzinger, K.A.; Meehan, S.; Wilson, M.R.; Dobson, C.M.; Klenerman, D. The extracellular chaperone clusterin sequesters oligomeric forms of the amyloid-β(1-40) peptide. Nat. Struct. Mol. Biol. 2012, 19, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Cleary, J.P.; Walsh, D.M.; Hofmeister, J.J.; Shankar, G.M.; Kuskowski, M.A.; Selkoe, D.J.; Ashe, K.H. Natural oligomers of the amyloid-β protein specifically disrupt cognitive function. Nat. Neurosci. 2005, 8, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.E.; Bateman, R.J. Decreased clearance of CNS β-amyloid in Alzheimer’s disease. Science 2010, 330, 1774. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.D.; Sagare, A.P.; Friedman, A.E.; Bedi, G.S.; Holtzman, D.M.; Deane, R.; Zlokovic, B.V. Transport pathways for clearance of human Alzheimer’s amyloid β-peptide and apolipoproteins E and J in the mouse central nervous system. J. Cereb. Blood Flow Metab. 2007, 27, 909–918. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V. Cerebrovascular transport of Alzheimer’s amyloid β and apolipoproteins J and E: Possible anti-amyloidogenic role of the blood-brain barrier. Life Sci. 1996, 59, 1483–1497. [Google Scholar] [CrossRef]
- Zlokovic, B.V.; Martel, C.L.; Matsubara, E.; McComb, J.G.; Zheng, G.; McCluskey, R.T.; Frangione, B.; Ghiso, J. Glycoprotein 330/megalin: Probable role in receptor-mediated transport of apolipoprotein j alone and in a complex with Alzheimer disease amyloid β at the blood-brain and blood-cerebrospinal fluid barriers. Proc. Natl. Acad. Sci. USA 1996, 93, 4229–4234. [Google Scholar] [CrossRef] [PubMed]
- DeMattos, R.B.; O’Dell, M.A.; Parsadanian, M.; Taylor, J.W.; Harmony, J.A.; Bales, K.R.; Paul, S.M.; Aronow, B.J.; Holtzman, D.M. Clusterin promotes amyloid plaque formation and is critical for neuritic toxicity in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2002, 99, 10843–10848. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A.R.; Sagare, A.P.; Zlokovic, B.V. Role of clusterin in the brain vascular clearance of amyloid-β. Proc. Natl. Acad. Sci. USA 2017, 114, 8681–8682. [Google Scholar] [CrossRef] [PubMed]
- Wojtas, A.M.; Kang, S.S.; Olley, B.M.; Gatherer, M.; Shinohara, M.; Lozano, P.A.; Liu, C.C.; Kurti, A.; Baker, K.E.; Dickson, D.W.; et al. Loss of clusterin shifts amyloid deposition to the cerebrovasculature via disruption of perivascular drainage pathways. Proc. Natl. Acad. Sci. USA 2017, 114, E6962–E6971. [Google Scholar] [CrossRef] [PubMed]
- Kounnas, M.Z.; Loukinova, E.B.; Stefansson, S.; Harmony, J.A.; Brewer, B.H.; Strickland, D.K.; Argraves, W.S. Identification of glycoprotein 330 as an endocytic receptor for apolipoprotein j/clusterin. J. Biol. Chem. 1995, 270, 13070–13075. [Google Scholar] [CrossRef] [PubMed]
- Bartl, M.M.; Luckenbach, T.; Bergner, O.; Ullrich, O.; Koch-Brandt, C. Multiple receptors mediate apoj-dependent clearance of cellular debris into nonprofessional phagocytes. Exp. Cell. Res. 2001, 271, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Hammad, S.M.; Ranganathan, S.; Loukinova, E.; Twal, W.O.; Argraves, W.S. Interaction of apolipoprotein J-amyloid β-peptide complex with low density lipoprotein receptor-related protein-2/megalin. A mechanism to prevent pathological accumulation of amyloid β-peptide. J. Biol. Chem. 1997, 272, 18644–18649. [Google Scholar] [CrossRef] [PubMed]
- Bajari, T.M.; Strasser, V.; Nimpf, J.; Schneider, W.J. A model for modulation of leptin activity by association with clusterin. FASEB J. 2003, 17, 1505–1507. [Google Scholar] [CrossRef] [PubMed]
- Khialeeva, E.; Carpenter, E.M. Nonneuronal roles for the reelin signaling pathway. Dev. Dyn. 2017, 246, 217–226. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dlugosz, P.; Nimpf, J. The Reelin Receptors Apolipoprotein E receptor 2 (ApoER2) and VLDL Receptor. Int. J. Mol. Sci. 2018, 19, 3090. https://doi.org/10.3390/ijms19103090
Dlugosz P, Nimpf J. The Reelin Receptors Apolipoprotein E receptor 2 (ApoER2) and VLDL Receptor. International Journal of Molecular Sciences. 2018; 19(10):3090. https://doi.org/10.3390/ijms19103090
Chicago/Turabian StyleDlugosz, Paula, and Johannes Nimpf. 2018. "The Reelin Receptors Apolipoprotein E receptor 2 (ApoER2) and VLDL Receptor" International Journal of Molecular Sciences 19, no. 10: 3090. https://doi.org/10.3390/ijms19103090
APA StyleDlugosz, P., & Nimpf, J. (2018). The Reelin Receptors Apolipoprotein E receptor 2 (ApoER2) and VLDL Receptor. International Journal of Molecular Sciences, 19(10), 3090. https://doi.org/10.3390/ijms19103090