Comparative RNA-Seq Analysis of High- and Low-Oil Yellow Horn During Embryonic Development



Abstract
1. Introduction
2. Results
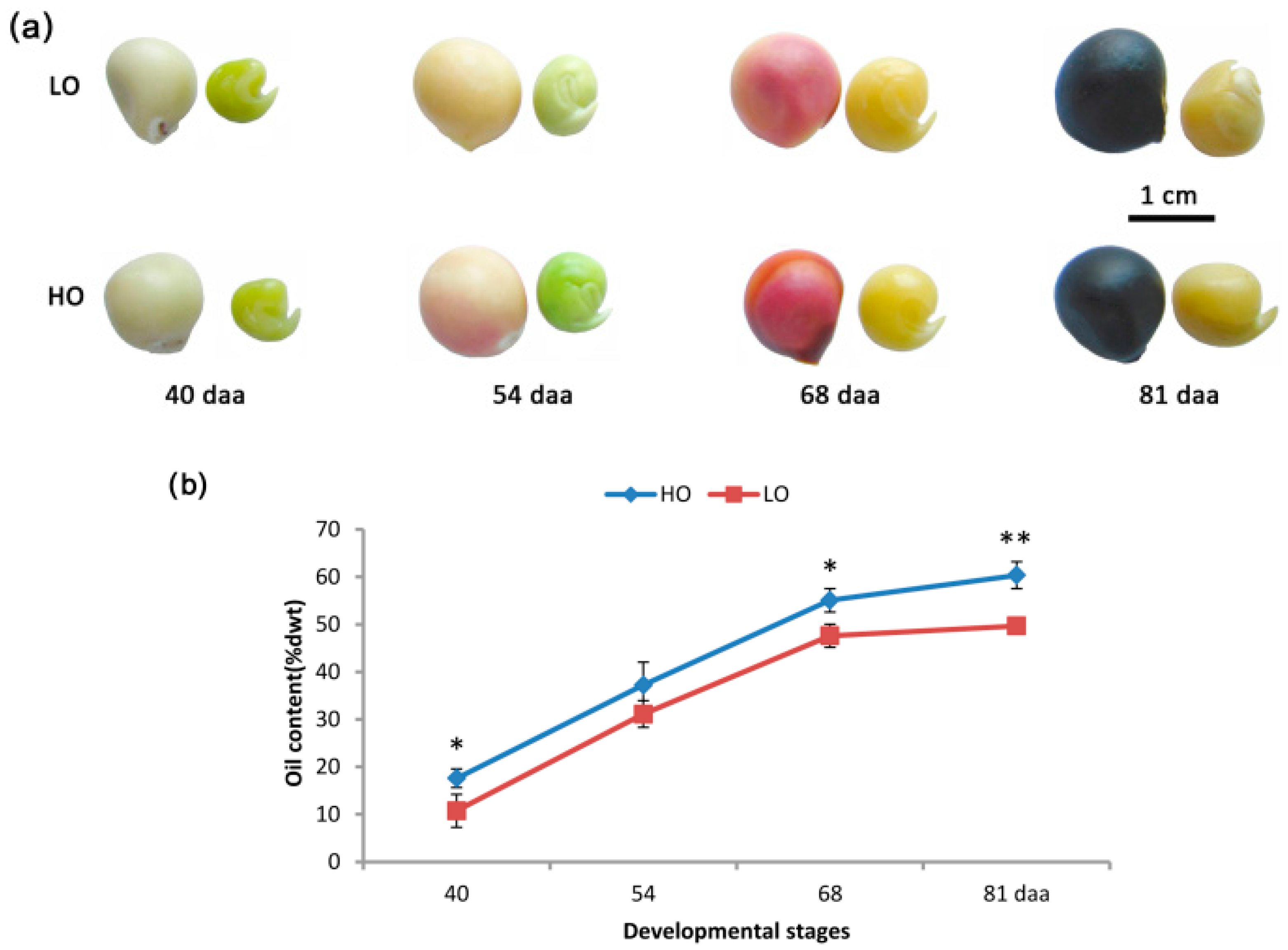
2.1. Characterizations of Oil Accumulation in the Two Yellow Horn Lines
2.2. Sequencing Statistics and Assembly
2.3. Function Annotation of Transcriptome Genes
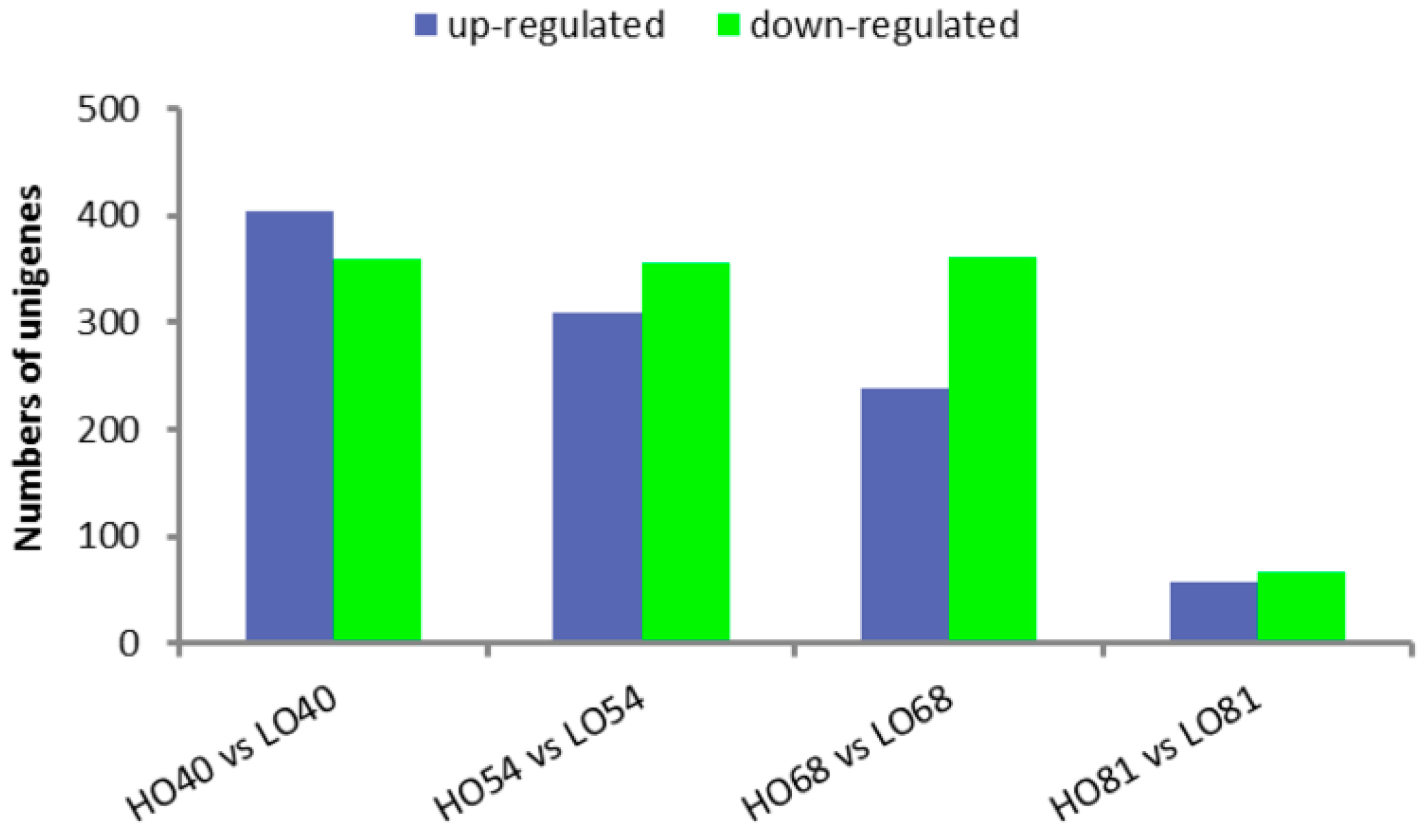
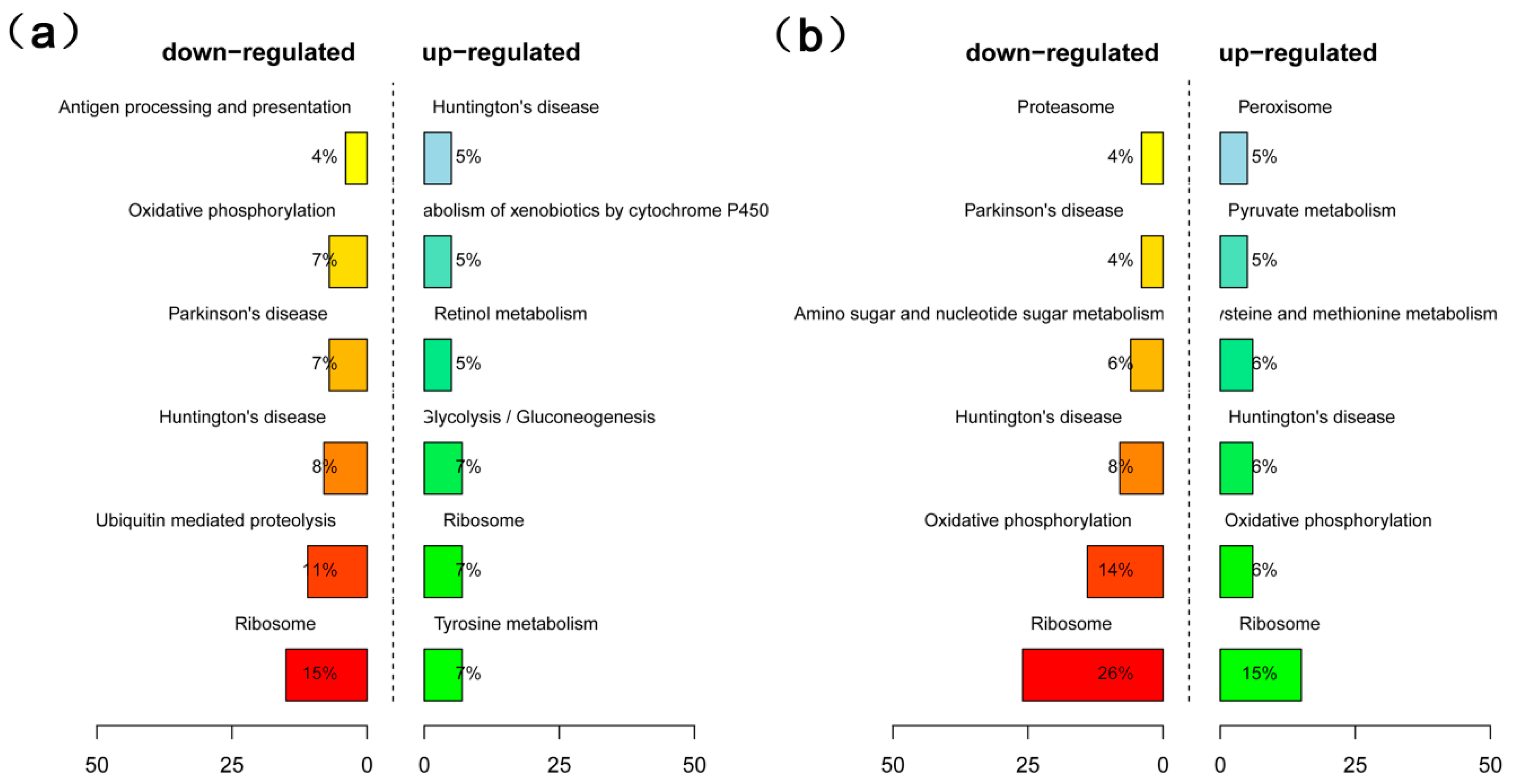
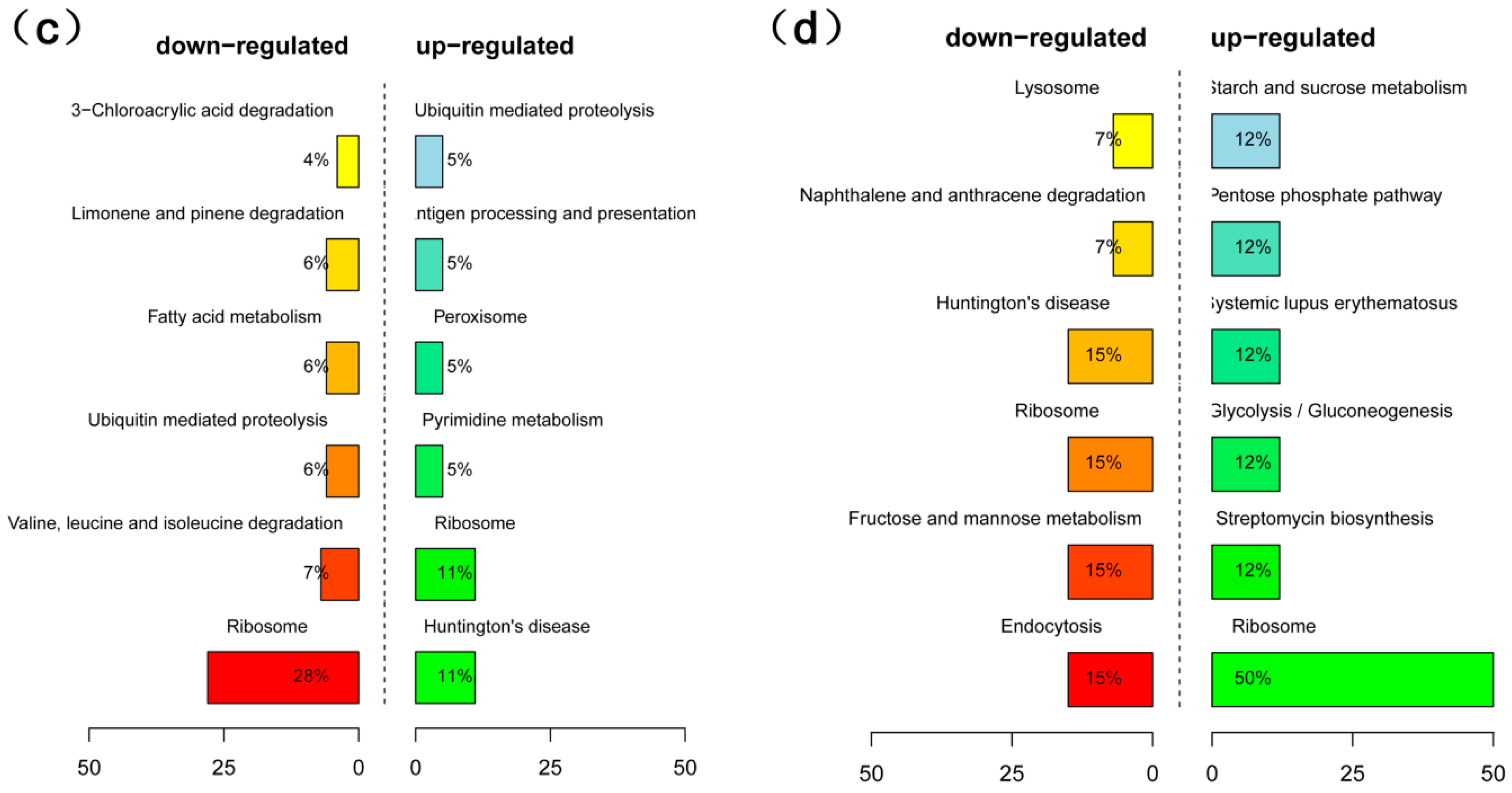
2.4. Analysis of Differentially Expressed Genes (DEGs) in High- and Low-Oil Yellow Horn During Embryonic Development
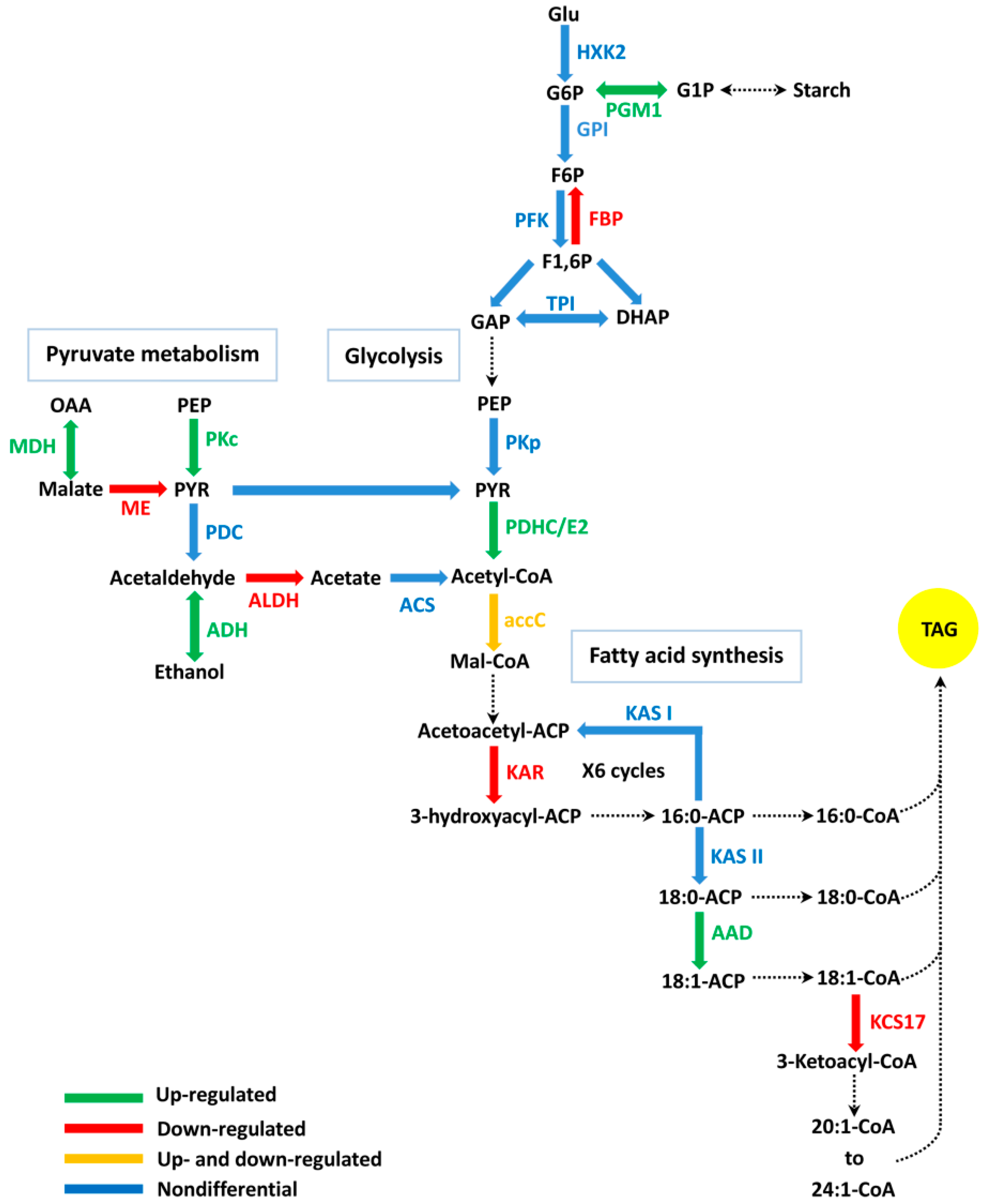
2.5. DEGs Related to FA (Fatty Acid) Biosynthesis
2.6. Differentially Expressed Transcription Factors (TFs)
2.7. DEGs Related to Glycolysis/Gluconeogenesis and Pyruvate Metabolic Pathways
2.8. qPCR Validation of DEGs
3. Discussion
3.1. The Function of FA Biosynthesis on Oil Accumulation
3.2. Role of Transcriptional Regulation in Oil Accumulation
3.3. The Relation Between Carbon Metabolism and Oil Accumulation
4. Materials and Methods
4.1. Plant Material
4.2. Oil Content Analysis
4.3. RNA-Seq Library Preparation and Sequencing
4.4. Quality Control
4.5. De novo Assembly and Annotation
4.6. Reads Mapping to the Reference Genome
4.7. Quantification of Gene Expression Level and Differential Expression Analysis
4.8. GO and KEGG Enrichment Analysis of DEGs
4.9. Quantitative Real-Time PCR (qPCR) Validation
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| GO | Gene Ontology |
| KOG | euKaryotic Ortholog Groups |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| HO | High-oil |
| LO | Low-oil |
| daa | Days after anthesis |
| DEGs | Differentially expressed genes |
| accC | Acetyl-CoA carboxylase/biotin carboxylase |
| FA | Fatty acid |
| KAR | 3-Ketoacyl ACP reductase |
| KAS | 3-Ketoacyl-ACP synthase |
| HAD | Hydroxyacyl-ACP dehydrogenase |
| EAR | Enoyl-ACP reductase |
| AAD | Acyl-ACP desaturase |
| KCS17 | 3-Ketoacyl-CoA synthase 17 |
| PKc | Cytosolic pyruvate kinase |
| PDHC/E2 | Pyruvate dehydrogenase complex/E2 component |
| PGM1 | Phosphoglucomutase |
| FBP | Fructose-1,6-bisphosphatase |
| MDH | Malate dehydrogenase |
| ME | NADP-dependent malic enzyme |
| TFs | Transcription factors |
| ALDH | Aldehyde dehydrogenase |
| ADH | Alcohol dehydrogenase |
| PEP | Phosphoenolpyruvate |
| OAA | Oxaloacetate |
| PYR | Pyruvate |
| TAG | Triacylglycerol |
References
- Zhang, S.; Zu, Y.G.; Fu, Y.J.; Luo, M.; Liu, W.; Li, J.; Efferth, T. Supercritical carbon dioxide extraction of seed oil from yellow horn (Xanthoceras sorbifolia Bunge.) and its anti-oxidant activity. Bioresour. Technol. 2010, 101, 2537–2544. [Google Scholar] [CrossRef] [PubMed]
- Ruan, C.J.; Yan, R.; Wang, B.X.; Mopper, S.; Guan, W.K.; Zhang, J. The importance of yellow horn (Xanthoceras sorbifolia) for restoration of arid habitats and production of bioactive seed oils. Ecol. Eng. 2017, 99, 504–512. [Google Scholar] [CrossRef]
- Li, J.; Fu, Y.J.; Qu, X.J.; Wang, W.; Luo, M.; Zhao, C.J.; Zu, Y.G. Biodiesel production from yellow horn (Xanthoceras sorbifolia Bunge.) seed oil using ion exchange resin as heterogeneous catalyst. Bioresour. Technol. 2012, 108, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.H.; Wang, T.T.; Li, Q.Q.; Zhao, N.; Zhang, Y.; Liu, D.; Hu, Q.; Li, F.L. Two novel diacylglycerol acyltransferase genes from Xanthoceras sorbifolia are responsible for its seed oil content. Gene 2013, 527, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.H.; Li, Q.Q.; Wang, T.T.; Hu, Q.; Deng, W.H.; Xia, X.L.; Gao, H.B. XsFAD2 gene encodes the enzyme responsible for the high linoleic acid content in oil accumulated in Xanthoceras sorbifolia seeds. J. Sci. Food Agric. 2014, 94, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Zhang, Y.; Li, Q.Q.; Li, R.F.; Xia, X.L.; Qin, X.W.; Guo, H.H. Identification and expression of a stearoyl-ACP desaturase gene responsible for oleic acid accumulation in Xanthoceras sorbifolia seeds. Plant Physiol. Biochem. 2015, 87, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; The, H.F.; Mebus, K.; Ooi, T.E.K.; Kwong, Q.B.; Koo, K.L.; Ong, C.K.; Mayes, S.; Chew, F.T.; Appleton, D.R.; et al. Differential gene expression at different stages of mesocarp development in high- and low-yielding oil palm. BMC Genom. 2017, 18, 470. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.X.; Cheng, H.; Qi, C. Microarray analysis of gene expression in seeds of Brassica napus planted in Nanjing (altitude: 8.9 m), Xining (altitude: 2261.2 m) and Lhasa (altitude: 3658 m) with different oil content. Mol. Biol. Rep. 2009, 36, 2375–2386. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Wang, X.; Zhan, G.; Liu, G.; Hua, W.; Wang, H. Unusually large oilbodies are highly correlated with lower oil content in Brassica napus. Plant Cell Rep. 2009, 28, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Guan, M.; Li, X.; Guan, C. Microarray analysis of differentially expressed genes between Brassica napus strains with high- and low-oleic acid contents. Plant Cell Rep. 2012, 31, 929–943. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.H.; Chen, B.; Yan, X.H.; Wang, L.J.; Zhang, H.F.; Cheng, J.P.; Zhou, X.A.; Sha, A.H.; Shen, H. Identification of differentially expressed genes in soybean seeds differing in oil content. Plant Sci. 2008, 175, 663–673. [Google Scholar] [CrossRef]
- Troncoso-Ponce, M.A.; Garcés, R.; Martínez-Force, E. Glycolytic enzymatic activities in developing seeds involved in the differences between standard and low oil content sunflowers (Helianthus annuus L.). Plant Physiol. Biochem. 2010, 48, 961–965. [Google Scholar] [CrossRef] [PubMed]
- Tsaftaris, A.S.; Scandalios, J.G. Genetic analysis of isocitrate lyase enzyme activity levels in maize lines selected for high or low oil content. J. Hered. 1983, 74, 70–74. [Google Scholar] [CrossRef]
- Liu, Z.; Yang, X.; Fu, Y.; Zhang, Y.; Yan, J.; Song, T.; Rocheford, T.; Li, J. Proteomic analysis of early germs with high-oil and normal inbred lines in maize. Mol. Biol. Rep. 2009, 36, 813–821. [Google Scholar] [CrossRef] [PubMed]
- Loei, H.; Lim, J.; Tan, M.; Lim, T.K.; Lin, Q.S.; Chew, F.T.; Kulaveerasingam, H.; Chung, M.C. Proteomic analysis of the oil palm fruit mesocarp reveals elevated oxidative phosphorylation activity is critical for increased storage oil production. J. Proteome Res. 2013, 12, 5096–5109. [Google Scholar] [CrossRef] [PubMed]
- Teh, H.F.; Neoh, B.K.; Hong, M.P.; Low, J.Y.; Ng, T.L.; Ithnin, N.; Thang, Y.M.; Mohamed, M.; Chew, F.T.; Yusof, H.M.; et al. Differential metabolite profiles during fruit development in high-yielding oil palm mesocarp. PLoS ONE 2013, 8, e61344. [Google Scholar] [CrossRef] [PubMed]
- Settlage, S.; Kwanyuen, P.; Wilson, R. Relation between diacylglycerol acyltransferase activity and oil concentration in soybean. J. Am. Oil Chem. Soc. 1998, 75, 775–781. [Google Scholar] [CrossRef]
- Liu, Y.L.; Huang, Z.D.; Ao, Y.; Li, W.; Zhang, Z. Transcriptome analysis of yellow horn (Xanthoceras sorbifolia Bunge): A potential oil-rich seed tree for biodiesel in China. PLoS ONE 2013, 8, e74441. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotech. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Bourgis, F.; Kilaru, A.; Cao, X.; Ngando-Ebongue, G.F.; Drira, N.; Ohlrogge, J.B.; Arondel, V. Comparative transcriptome and metabolite analysis of oil palm and date palm mesocarp that differ dramatically in carbon partitioning. Proc. Natl. Acad. Sci. USA 2011, 108, 12527–12532. [Google Scholar] [CrossRef] [PubMed]
- Troncoso-Ponce, M.A.; Kilaru, A.; Cao, X.; Durrett, T.P.; Fan, J.; Jensen, J.K.; Thrower, N.A.; Pauly, M.; Wilkerson, C.; Ohlrogge, J.B. Comparative deep transcriptional profiling of four developing oilseeds. Plant J. 2011, 68, 1014–1027. [Google Scholar] [CrossRef] [PubMed]
- Venglat, P.; Xiang, D.; Qiu, S.; Stone, S.L.; Tibiche, C.; Cram, D.; Alting-Mees, M.; Nowak, J.; Cloutier, S.; Deyholos, M.; et al. Gene expression analysis of flax seed development. BMC Plant Biol. 2011, 11, 74. [Google Scholar] [CrossRef] [PubMed]
- Dussert, S.; Guerin, C.; Andersson, M.; Joet, T.; Tranbarger, T.J.; Pizot, M.; Sarah, G.; Omore, A.; Durand-Gasselin, T.; Morcillo, F. Comparative transcriptome analysis of three oil palm fruit and seed tissues that differ in oil content and fatty acid composition. Plant Physiol. 2013, 162, 1337–1358. [Google Scholar] [CrossRef] [PubMed]
- Nikolau, B.J.; Ohlrogge, J.B.; Wurtele, E.S. Plant biotin-containing carboxylases. Arch. Biochem. Biophys. 2003, 414, 211–222. [Google Scholar] [CrossRef]
- Andre, C.; Haslam, R.P.; Shanklin, J. Feedback regulation of plastidic acetyl-CoA carboxylase by 18:1-acyl carrier protein in Brassica napus. Proc. Natl. Acad. Sci. USA 2012, 109, 10107–10112. [Google Scholar] [CrossRef] [PubMed]
- Nakkaew, A.; Chotigeat, W.; Eksomtramage, T.; Phongdara, A. Cloning and expression of a plastid-encoded subunit, beta-carboxyltransferase gene (accD) and a nuclear-encoded subunit, biotin carboxylase of acetyl-CoA carboxylase from oil palm (Elaeis guineensis Jacq.). Plant Sci. 2008, 175, 497–504. [Google Scholar] [CrossRef]
- Cui, Y.; Liu, Z.; Zhao, Y.; Wang, Y.; Huang, Y.; Li, L.; Wu, H.; Xu, S.; Hua, J. Overexpression of heteromeric GhACCase subunits enhanced oil accumulation in upland cotton. Plant Mol. Biol. Rep. 2017, 35, 287–297. [Google Scholar] [CrossRef]
- Ohlrogge, J.; Browse, J. Lipid biosynthesis. Plant Cell 1995, 7, 957–970. [Google Scholar] [CrossRef] [PubMed]
- Palaniswamy, S.K.; James, S.; Sun, H.; Lamb, R.S.; Davuluri, R.V.; Grotewold, E. AGRIS and AtRegNet. a platform to link cis-regulatory elements and transcription factors into regulatory networks. Plant Physiol. 2006, 140, 818–829. [Google Scholar] [CrossRef] [PubMed]
- Niu, J.; Chen, Y.; An, J.; Hou, X.; Cai, J.; Wang, J.; Zhang, Z.; Lin, S. Integrated transcriptome sequencing and dynamic analysis reveal carbon source partitioning between terpenoid and oil accumulation in developing Lindera glauca fruits. Sci. Rep. 2015, 5, 15017. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Q.; Lu, X.; Zhao, F.Y.; Li, Q.T.; Niu, S.L.; Wei, W.; Zhang, W.K.; Ma, B.; Chen, S.Y.; Zhang, J.S. Soybean GmDREBL increases lipid content in seeds of transgenic Arabidopsis. Sci. Rep. 2016, 6, 34307. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Kong, Q.; Arondel, V.; Kilaru, A.; Bates, P.D.; Thrower, N.A.; Benning, C.; Ohlrogge, J.B. WRINKLED1, a ubiquitous regulator in oil accumulating tissues from Arabidopsis embryos to oil palm mesocarp. PLoS ONE 2013, 8, e68887. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Wang, H.H.; McCarty, D.R. Repression of the LEAFY COTYLEDON 1/B3 Regulatory Network in Plant Embryo Development by VP1/ABSCISIC ACID INSENSITIVE 3-LIKE B3 Genes. Plant Physiol. 2007, 143, 902–911. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; McCarty, D.R. Functional symmetry of the B3 network controlling seed development. Curr. Opin. Plant Biol. 2008, 11, 548–553. [Google Scholar] [CrossRef] [PubMed]
- Veerappan, V.; Chen, N.; Reichert, A.I.; Allen, R.D. HSI2/VAL1 PHD-like domain promotes H3K27 trimethylation to repress the expression of seed maturation genes and complex transgenes in Arabidopsis seedlings. BMC Plant Biol. 2014, 14, 293. [Google Scholar] [CrossRef] [PubMed]
- Vanhercke, T.; El Tahchy, A.; Liu, Q.; Zhou, X.R.; Shrestha, P.; Divi, U.K.; Ral, J.P.; Mansour, M.P.; Nichols, P.D.; James, C.N.; et al. Metabolic engineering of biomass for high energy density: Oilseed-like triacylglycerol yields from plant leaves. Plant Biotechnol. J. 2014, 12, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.U.; Lee, K.R.; Jung, S.J.; Shin, H.A.; Go, Y.S.; Suh, M.C.; Kim, J.B. Senescence-inducible LEC2 enhances triacylglycerol accumulation in leaves without negatively affecting plant growth. Plant Biotechnol. J. 2015, 13, 1346–1359. [Google Scholar] [CrossRef] [PubMed]
- Kang, F.; Rawsthorne, S. Metabolism of glucose-6-phosphate and utilization of multiple metabolites for fatty acid synthesis by plastids from developing oilseed rape embryos. Planta 1996, 199, 321–327. [Google Scholar] [CrossRef]
- Alonso, A.P.; Goffman, F.D.; Ohlrogge, J.B.; Shachar-Hill, Y. Carbon conversion efficiency and central metabolic fluxes in developing sunflower (Helianthus annuus L.) embryos. Plant J. 2007, 52, 296–308. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Behal, R.; Oliver, D.J. Disruption of plE2, the gene for the E2 subunit of the plastid pyruvate dehydrogenase complex, in Arabidopsis causes an early embryo lethal phenotype. Plant Mol. Biol. 2003, 52, 865–872. [Google Scholar] [CrossRef] [PubMed]
- Li, R.J.; Wang, H.Z.; Mao, H.; Lu, Y.T.; Hua, W. Identification of differentially expressed genes in seeds of two near-isogenic Brassica napus lines with different oil content. Planta 2006, 224, 952–962. [Google Scholar] [CrossRef] [PubMed]
- Baud, S.; Lepiniec, L. Physiological and developmental regulation of seed oil production. Prog. Lipid Res. 2010, 49, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Shockey, J.M.; Gidda, S.K.; Chapital, D.C.; Kuan, J.C.; Dhanoa, P.K.; Bland, J.M.; Rothstein, S.J.; Mullen, R.T.; Dyer, J.M. Tung tree DGAT1 and DGAT2 have nonredundant functions in triacylglycerol biosynthesis and are localized to different subdomains of the endoplasmic reticulum. Plant Cell 2006, 18, 2294–2313. [Google Scholar] [CrossRef] [PubMed]
- FastQC. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 3 May 2012).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. Embnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- fqtrim. Available online: http://ccb.jhu.edu/software/fqtrim/index.shtml (accessed on 27 February 2018).
- Aoki-Kinoshita, K.F.; Kanehisa, M. Gene annotation and pathway mapping in KEGG. Methods Mol. Biol. 2007, 396, 71–91. [Google Scholar] [CrossRef] [PubMed]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35, W182–W185. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Meth. 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- R: A Language and Environment for Statistical Computing. Available online: https://www.R-project.org (accessed on 23 April 2018).
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Annotated Databases | Number of Annotated Genes | Percentage of Annotated Genes |
|---|---|---|
| Nr | 37,043 | 46.35% |
| SwissProt | 21,450 | 26.84% |
| Pfam | 26,318 | 32.93% |
| KOG | 28,946 | 36.22% |
| GO | 18,936 | 23.70% |
| KEGG | 11,984 | 15.00% |
| Annotated in at least one database | 38,088 | 47.66% |
| Total number of genes | 79,915 | 100% |
| Sample Name | Total No. of Clean Reads | Reads Mapped | Percentage of Mapped Reads (%) |
|---|---|---|---|
| LO40_1 | 54,222,626 | 47,575,480 | 87.74% |
| LO40_2 | 63,290,894 | 53,054,604 | 83.83% |
| LO54_1 | 51,724,078 | 48,476,883 | 93.72% |
| LO54_2 | 59,978,660 | 56,140,038 | 93.60% |
| LO68_1 | 58,729,700 | 53,863,346 | 91.71% |
| LO68_2 | 62,4351,74 | 54,673,650 | 87.57% |
| LO81_1 | 62,510,098 | 57,732,856 | 92.36% |
| LO81_2 | 47,836,828 | 44,564,451 | 93.16% |
| HO40_1 | 53,006,640 | 43,436,912 | 81.95% |
| HO40_2 | 64,749,242 | 54,235,860 | 83.76% |
| HO54_1 | 59,699,584 | 53,801,599 | 90.12% |
| HO54_2 | 61,692,916 | 57,458,341 | 93.14% |
| HO68_1 | 51,553,508 | 47,005,432 | 91.18% |
| HO68_2 | 62,726,966 | 58,733,804 | 93.63% |
| HO81_1 | 56,526,196 | 52,826,069 | 93.45% |
| HO81_2 | 51,719,960 | 47,510,223 | 91.86% |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Ruan, C.; Liu, L.; Du, W.; Bao, A. Comparative RNA-Seq Analysis of High- and Low-Oil Yellow Horn During Embryonic Development. Int. J. Mol. Sci. 2018, 19, 3071. https://doi.org/10.3390/ijms19103071
Wang L, Ruan C, Liu L, Du W, Bao A. Comparative RNA-Seq Analysis of High- and Low-Oil Yellow Horn During Embryonic Development. International Journal of Molecular Sciences. 2018; 19(10):3071. https://doi.org/10.3390/ijms19103071
Chicago/Turabian StyleWang, Li, Chengjiang Ruan, Lingyue Liu, Wei Du, and Aomin Bao. 2018. "Comparative RNA-Seq Analysis of High- and Low-Oil Yellow Horn During Embryonic Development" International Journal of Molecular Sciences 19, no. 10: 3071. https://doi.org/10.3390/ijms19103071
APA StyleWang, L., Ruan, C., Liu, L., Du, W., & Bao, A. (2018). Comparative RNA-Seq Analysis of High- and Low-Oil Yellow Horn During Embryonic Development. International Journal of Molecular Sciences, 19(10), 3071. https://doi.org/10.3390/ijms19103071