Characterization of Apoptosis, Autophagy and Oxidative Stress in Pancreatic Islets Cells and Intestinal Epithelial Cells Isolated from Equine Metabolic Syndrome (EMS) Horses


Abstract

1. Introduction
2. Results
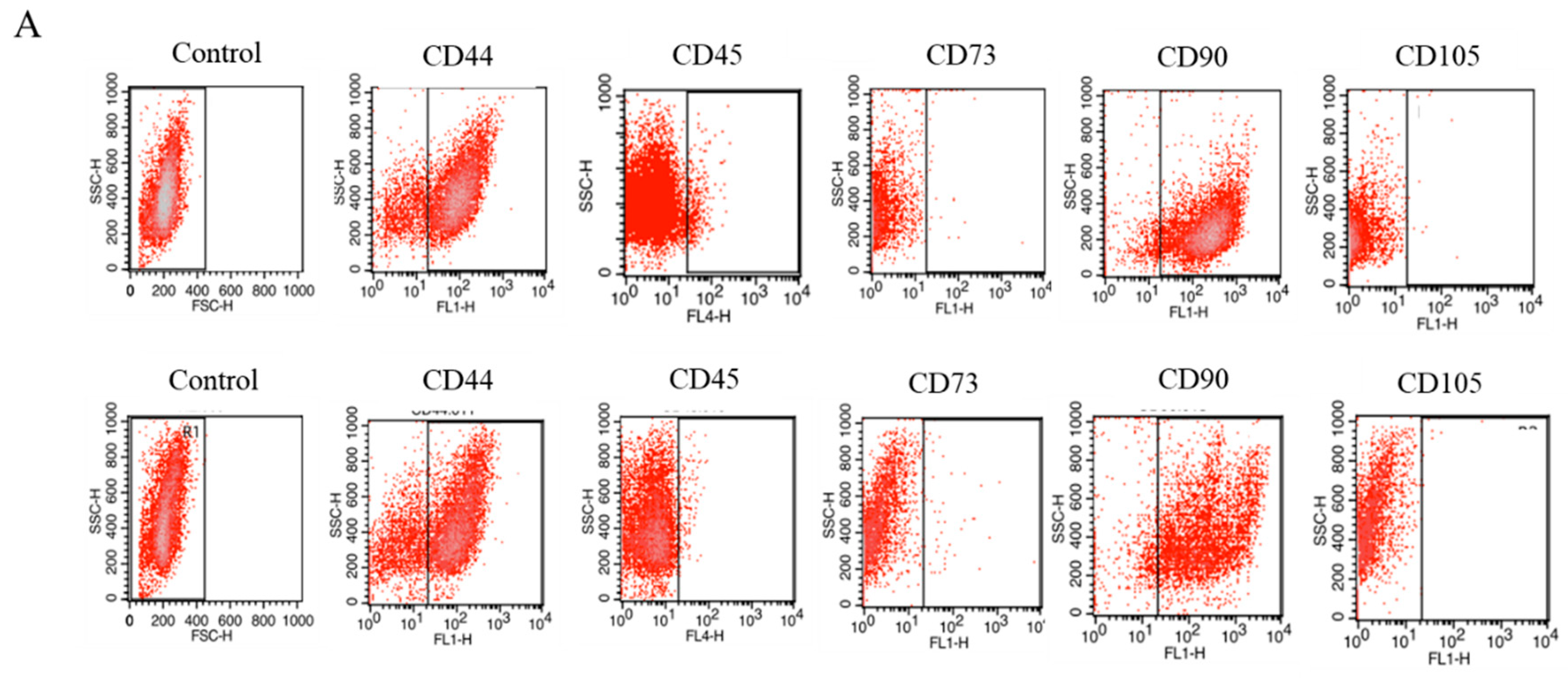
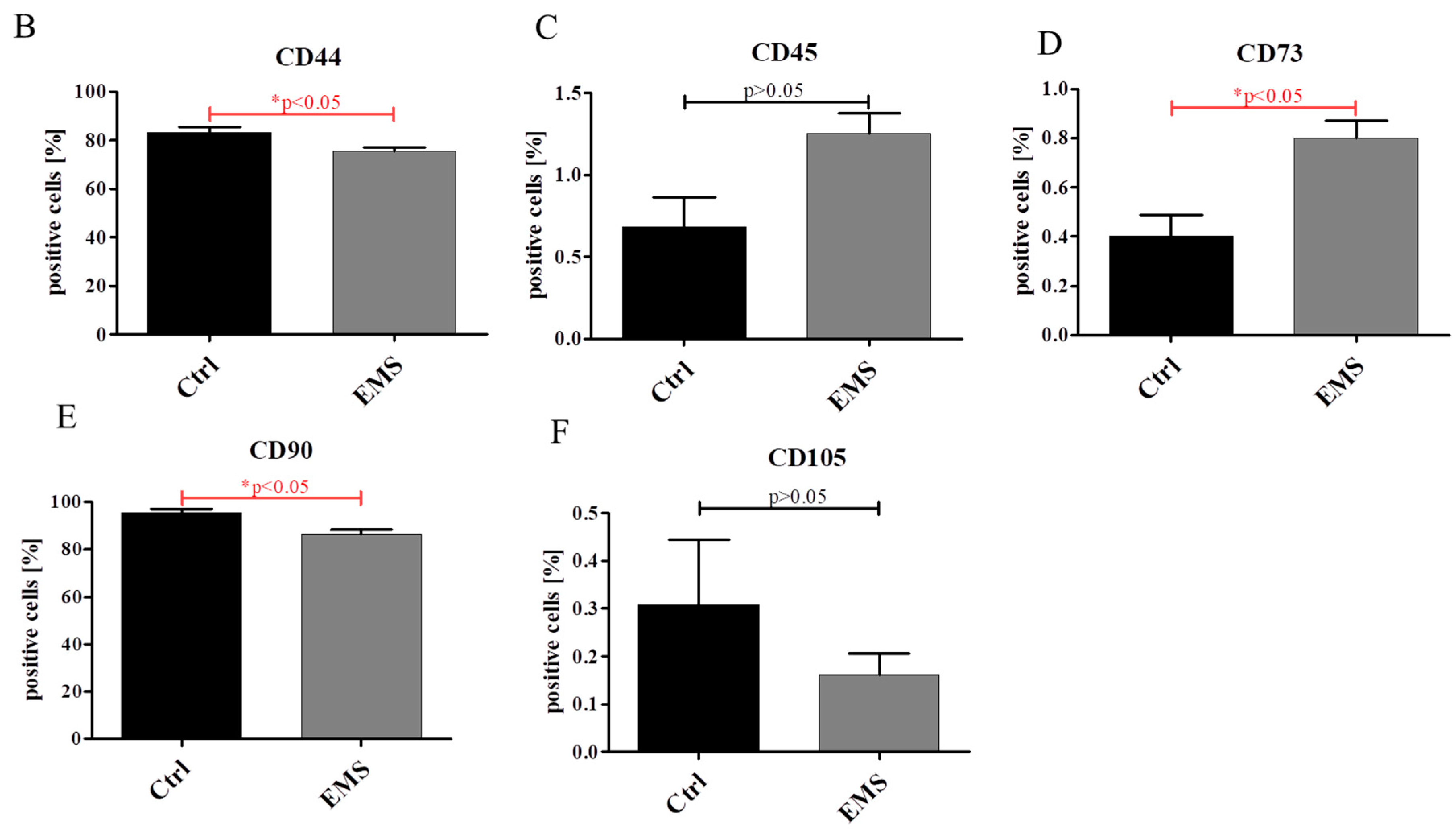
2.1. Expression of Surface Antigens in IECs
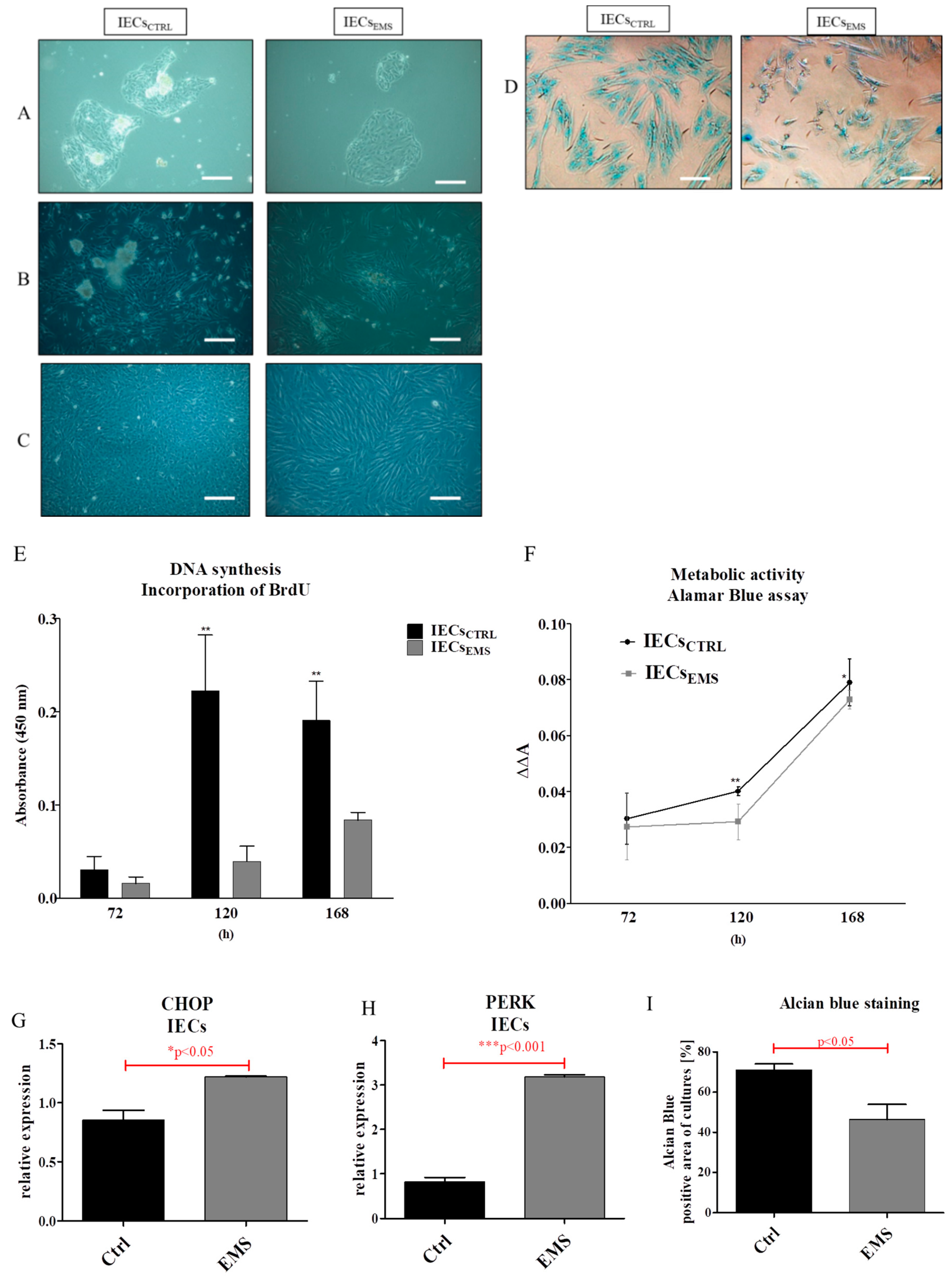
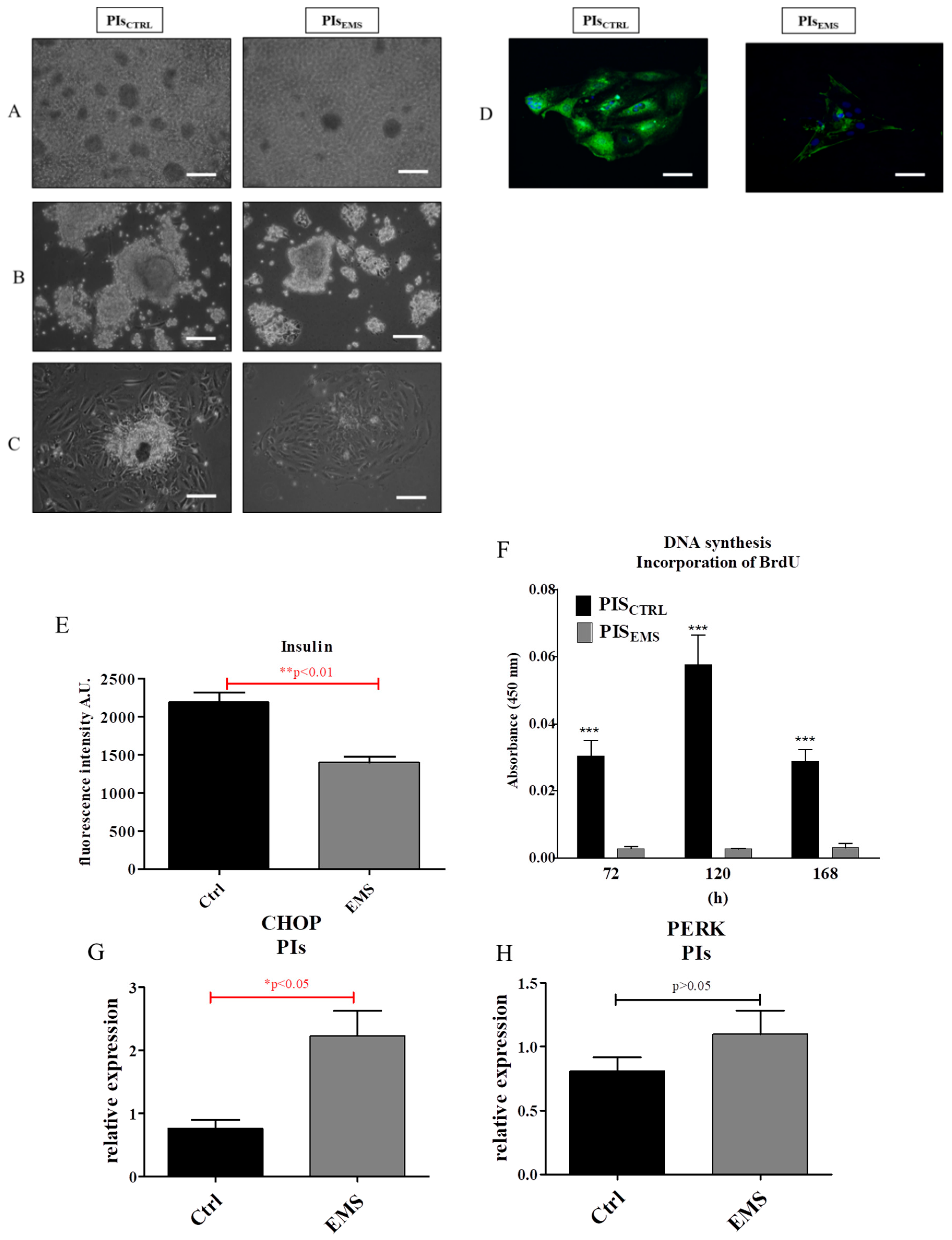
2.2. Morphology and Proliferation of IECs and PIs in Culture
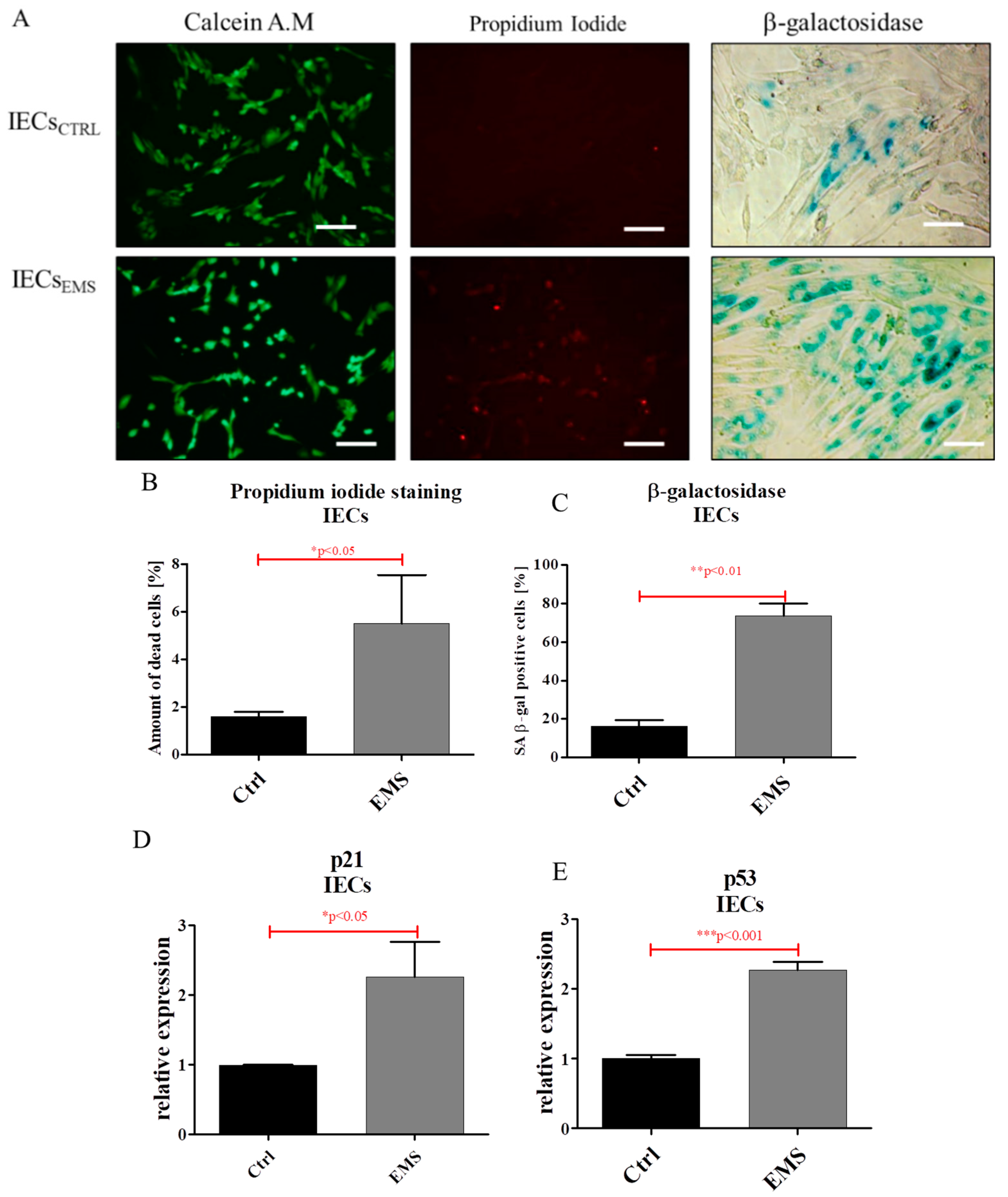
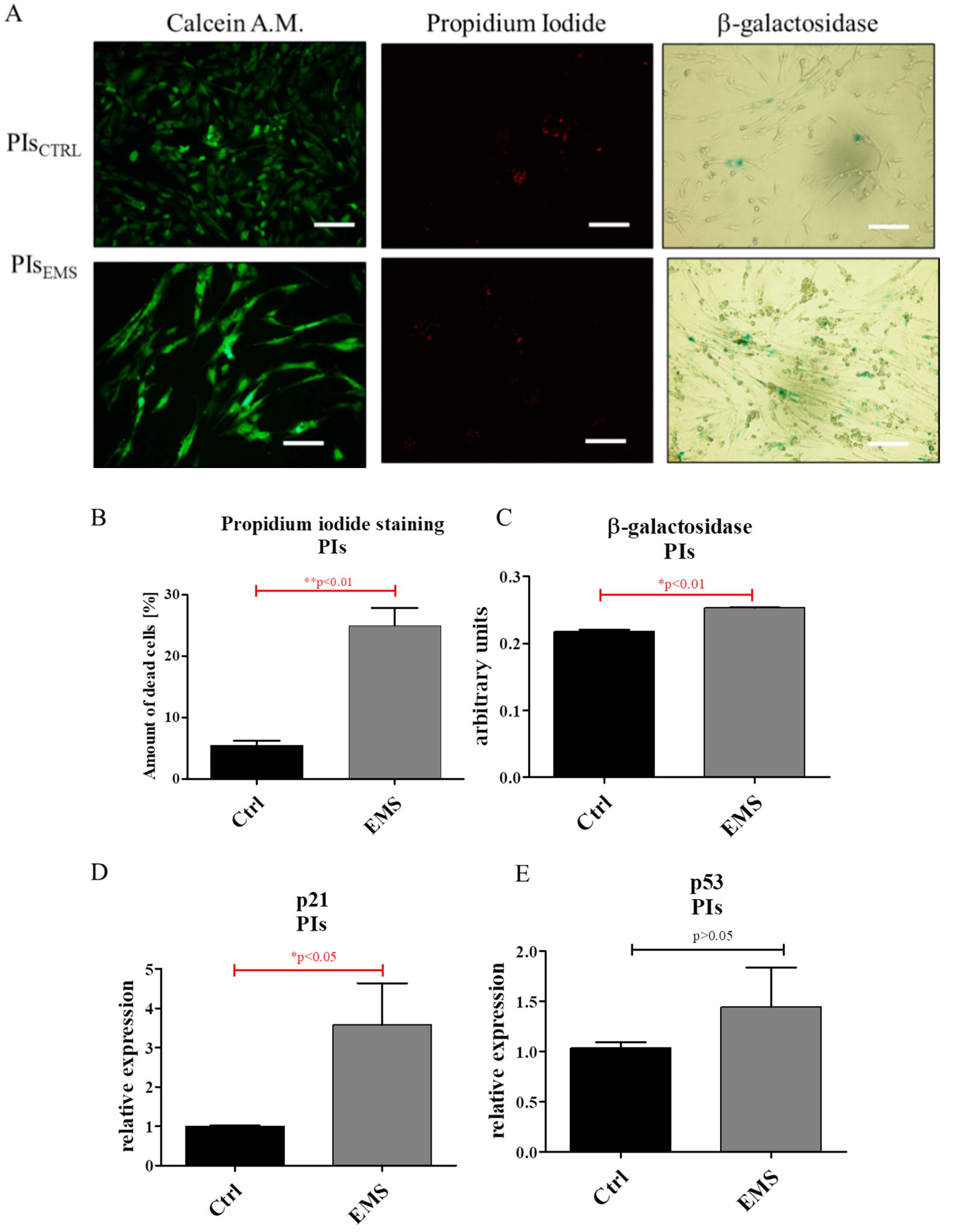
2.3. Assessment of Senescence and Apoptosis in IECs and PIs Cultures
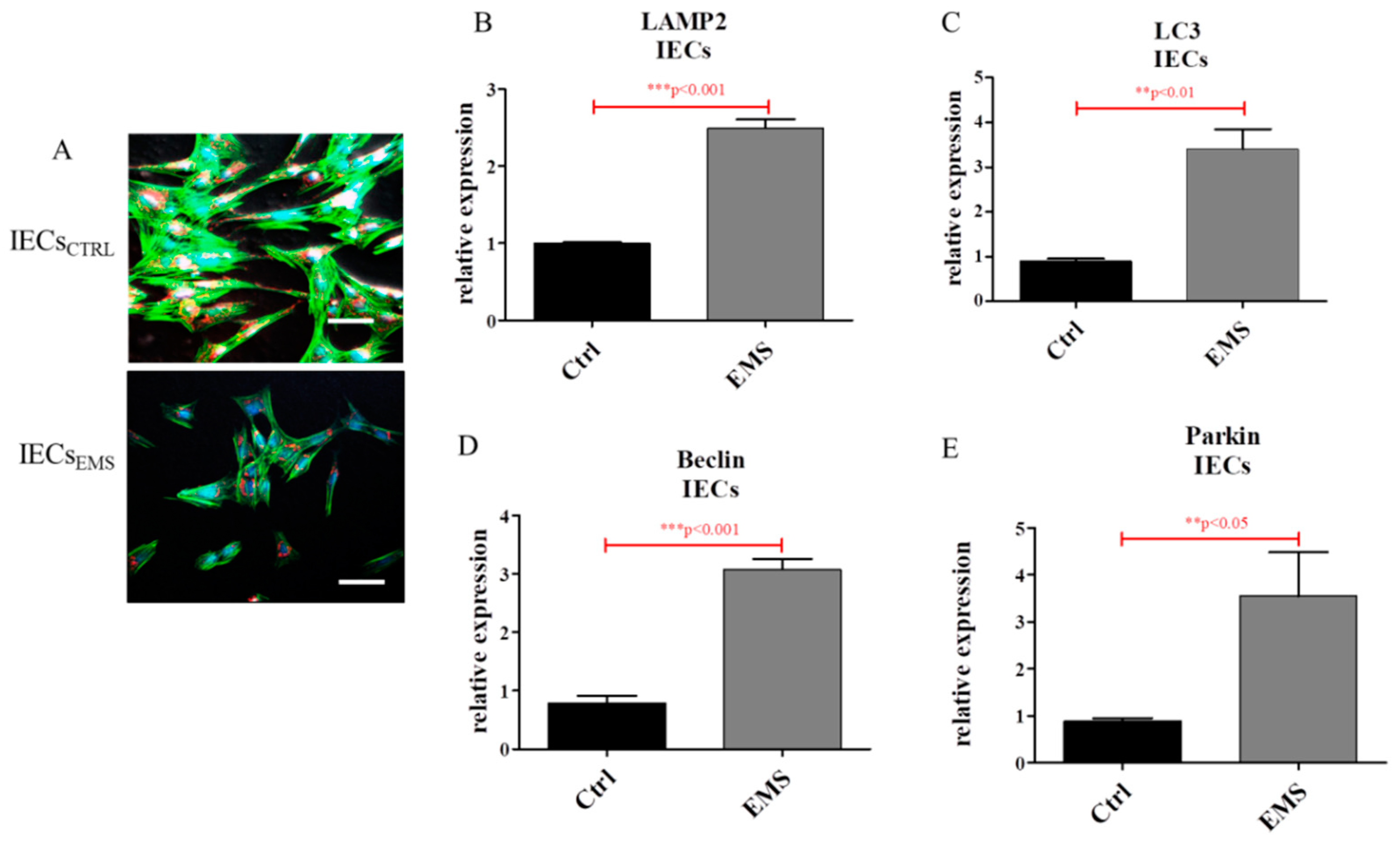
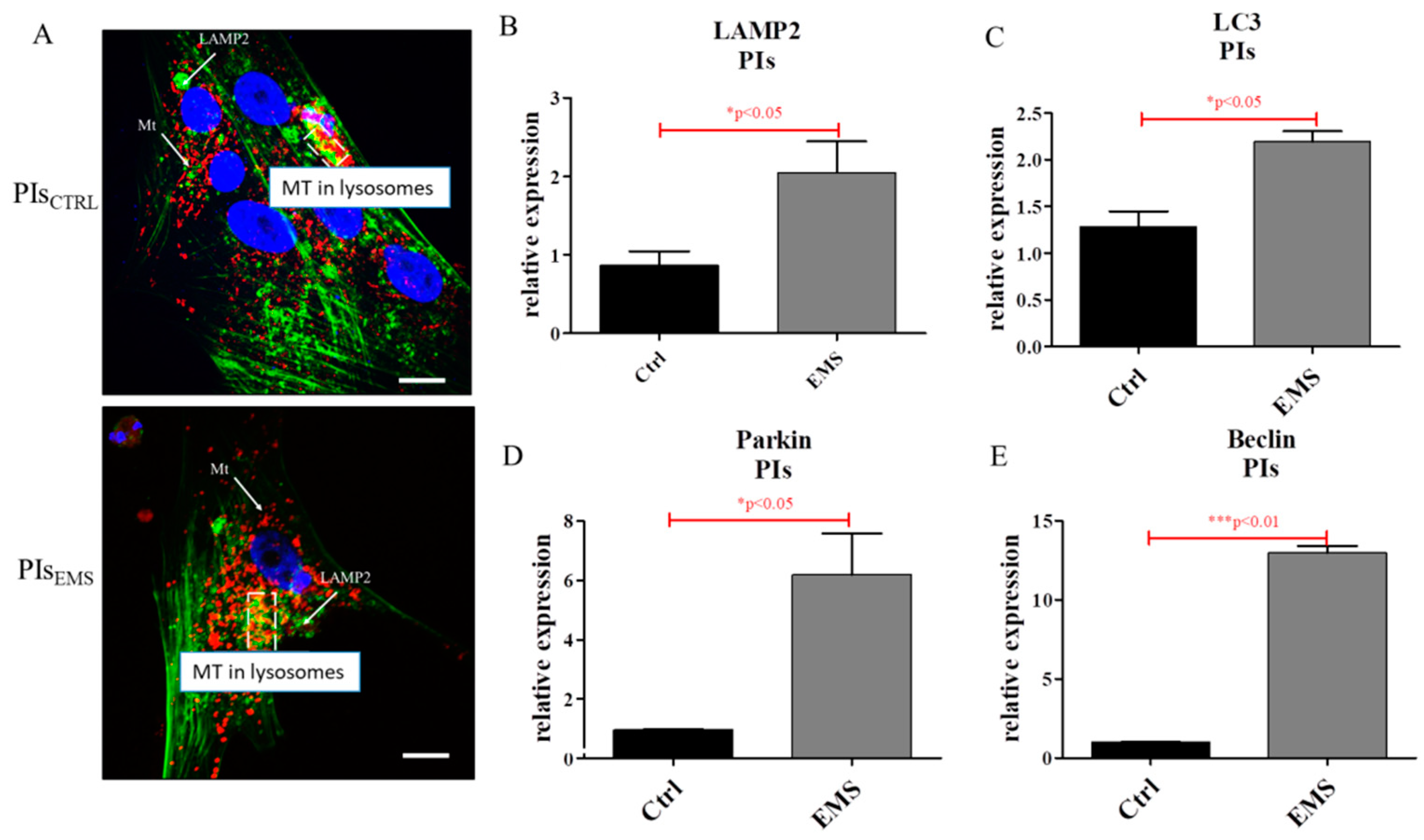
2.4. Autophagy in IECs and PIs
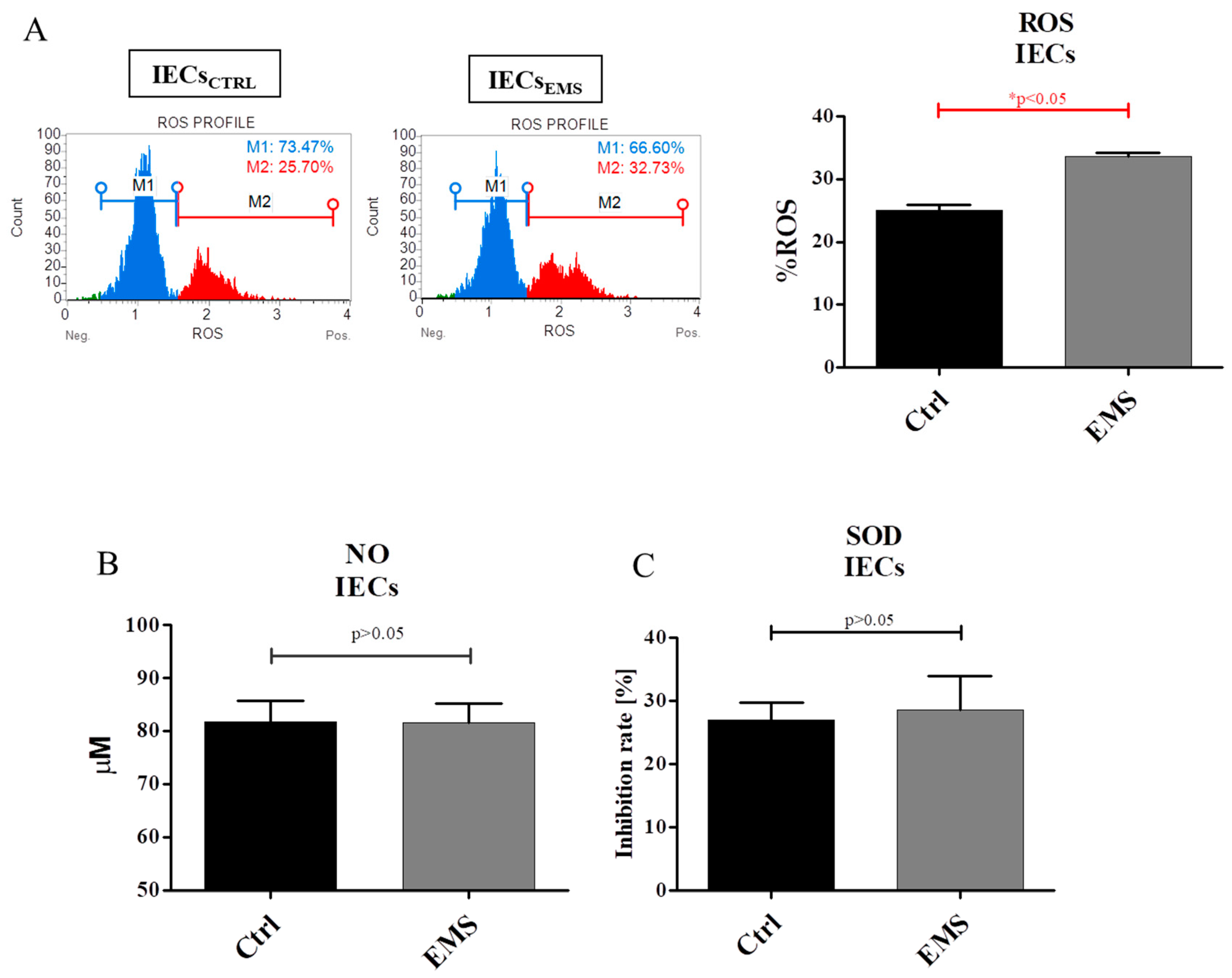
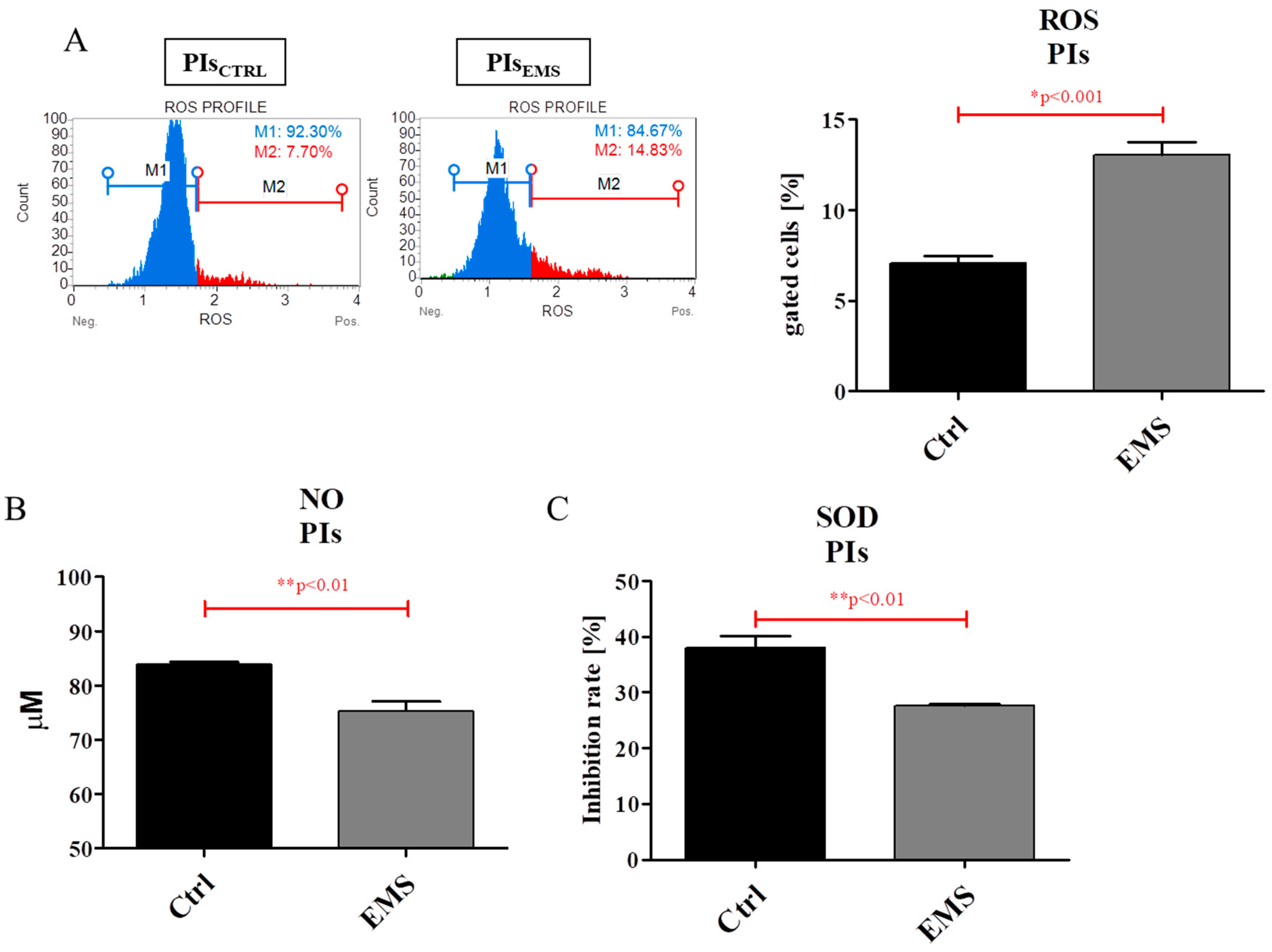
2.5. Oxidative Stress Factors in IECs and PIs
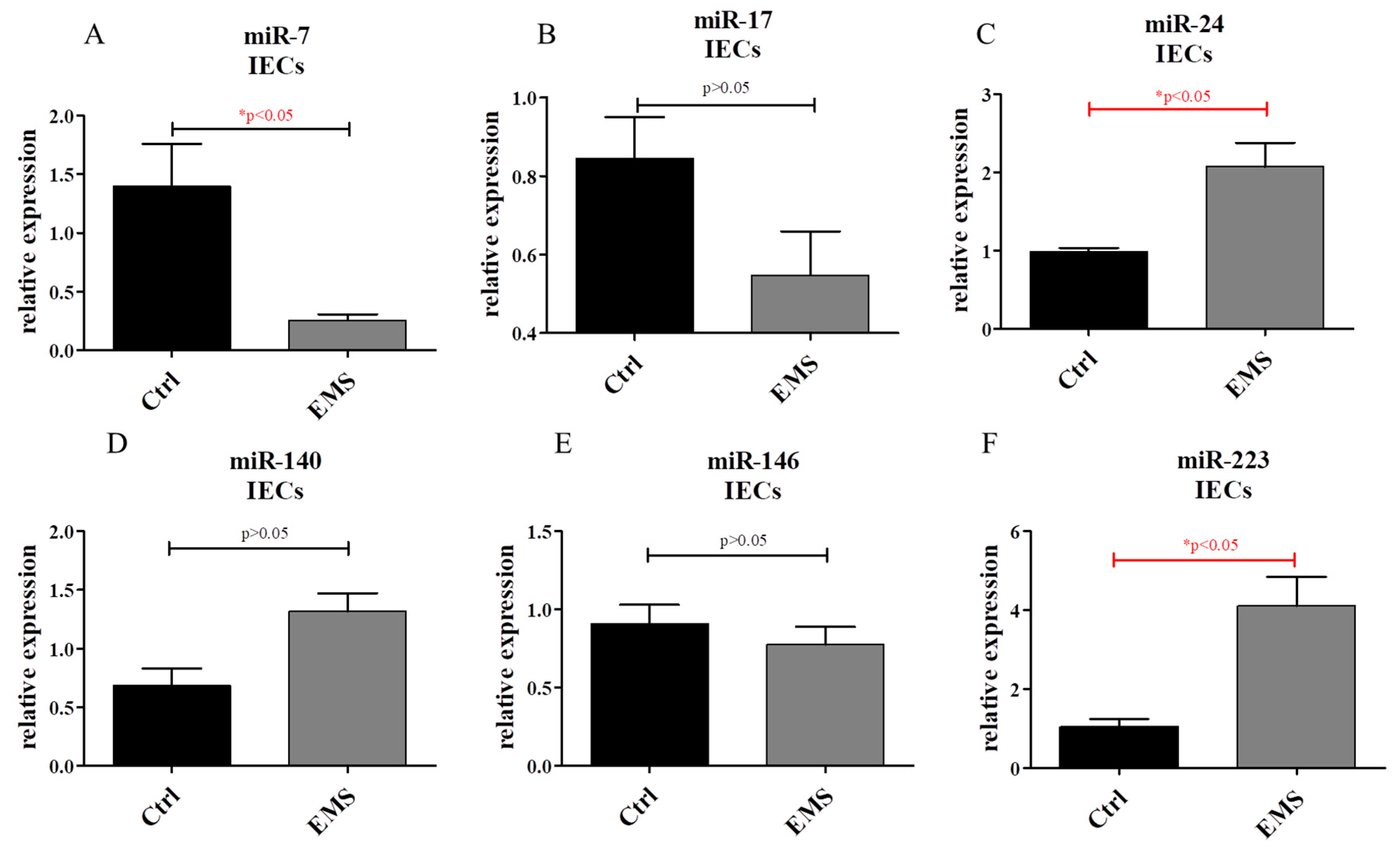
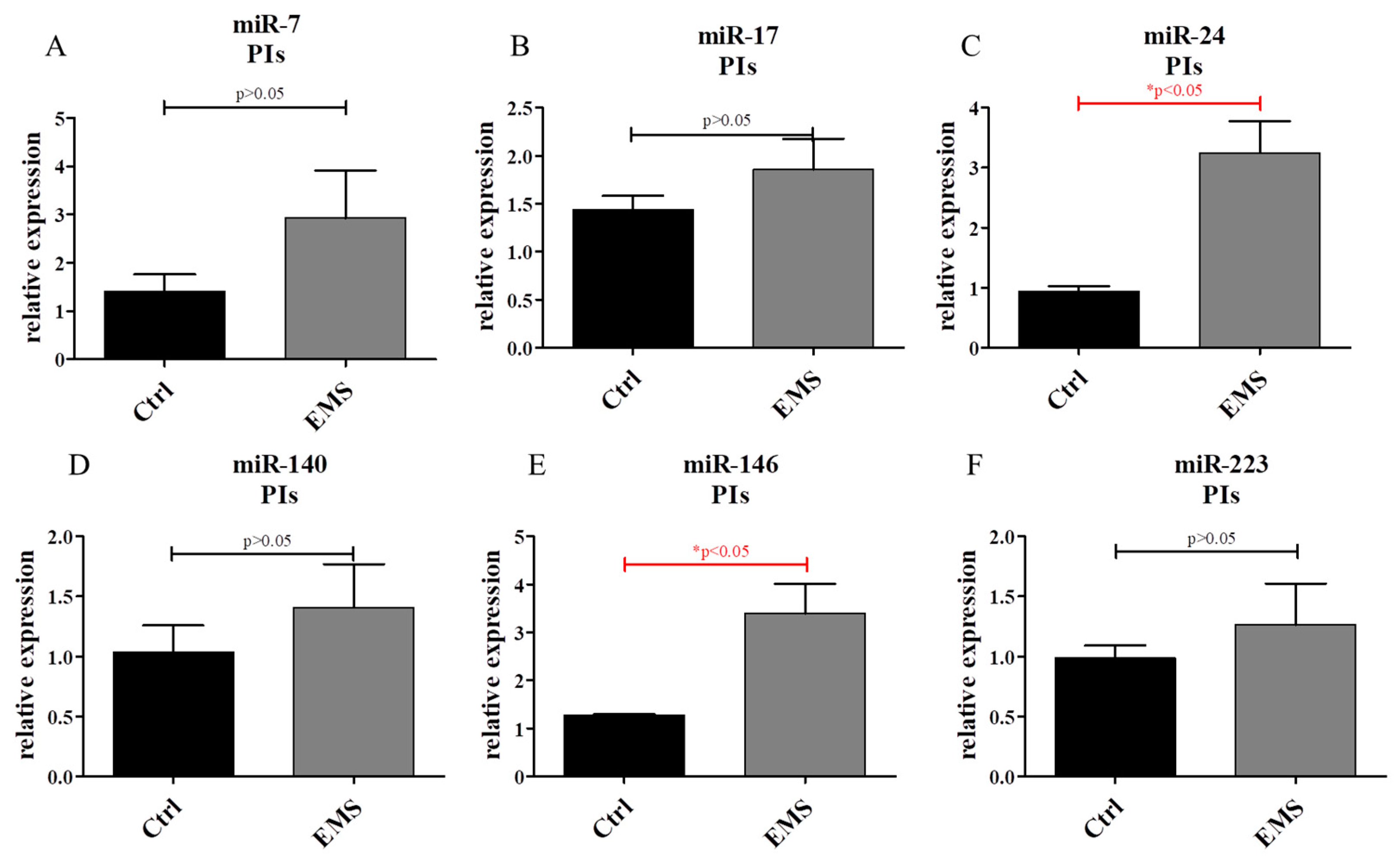
2.6. miR Expression in IECs and PIs
3. Discussion
4. Materials and Methods
4.1. All Reagents Were Purchased in Sigma Aldrich (Saint Louis, Missouri, USA), Unless Otherwise Indicated
4.1.1. IEC Isolation and Culture
4.1.2. Pancreatic Islets (PIs) Isolation and Culture
4.2. Phenotyping of IECs
4.3. Detection of Mucin Complexes
4.4. Measurement of DNA Synthesis: BrdU Assay
4.5. Alamar Blue Viability Assay
4.6. Visualization of PIs Morphology and Senescence Markers
4.7. Immunofluorescence Staining
4.8. Superoxide Dismutase (SOD) Activity and Nitric Oxide (NO) Levels
4.9. ROS Accumulation
4.10. Quantitative Real-Time Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
4.11. The Analysis of miRNA Expression
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Frank, N. Equine Metabolic Syndrome. J. Equine Vet. Sci. 2009, 29, 259–267. [Google Scholar] [CrossRef]
- Frank, N. Equine metabolic syndrome. Vet. Clin. N. Am. Equine Pract. 2011, 27, 73–92. [Google Scholar] [CrossRef] [PubMed]
- Marycz, K.; Michalak, I.; Kornicka, K. Advanced nutritional and stem cells approaches to prevent equine metabolic syndrome. Res. Vet. Sci. 2018, 118, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Nawrocka, D.; Kornicka, K.; Śmieszek, A.; Marycz, K. Spirulina platensis Improves Mitochondrial Function Impaired by Elevated Oxidative Stress in Adipose-Derived Mesenchymal Stromal Cells (ASCs) and Intestinal Epithelial Cells (IECs), and Enhances Insulin Sensitivity in Equine Metabolic Syndrome (EMS) Horses. Mar. Drugs 2017, 15. [Google Scholar] [CrossRef] [PubMed]
- Basinska, K.; Marycz, K.; Śmieszek, A.; Nicpoń, J. The production and distribution of IL-6 and TNF-α in subcutaneous adipose tissue and their correlation with serum concentrations in Welsh ponies with equine metabolic syndrome. J. Vet. Sci. 2015, 16, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Marycz, K.; Kornicka, K.; Basinska, K.; Czyrek, A. Equine Metabolic Syndrome Affects Viability, Senescence, and Stress Factors of Equine Adipose-Derived Mesenchymal Stromal Stem Cells: New Insight into EqASCs Isolated from EMS Horses in the Context of Their Aging. Oxid. Med. Cell. Longev. 2016, 2016, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Kornicka, K.; Houston, J.; Marycz, K. Dysfunction of Mesenchymal Stem Cells Isolated from Metabolic Syndrome and Type 2 Diabetic Patients as Result of Oxidative Stress and Autophagy may Limit Their Potential Therapeutic Use. Stem Cell Rev. Rep. 2018, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kaniuk, N.A.; Kiraly, M.; Bates, H.; Vranic, M.; Volchuk, A.; Brumell, J.H. Ubiquitinated-protein aggregates form in pancreatic beta-cells during diabetes-induced oxidative stress and are regulated by autophagy. Diabetes 2007, 56, 930–939. [Google Scholar] [CrossRef] [PubMed]
- Matveyenko, A.V.; Butler, P.C. Relationship between beta-cell mass and diabetes onset. Diabetes Obes. Metab. 2008, 10, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Kasuga, M. Insulin resistance and pancreatic β cell failure. J. Clin. Investig. 2006, 116, 1756–1760. [Google Scholar] [CrossRef] [PubMed]
- Augstein, P.; Elefanty, A.G.; Allison, J.; Harrison, L.C. Apoptosis and beta-cell destruction in pancreatic islets of NOD mice with spontaneous and cyclophosphamide-accelerated diabetes. Diabetologia 1998, 41, 1381–1388. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.P.; Köhler, M.; Graff, C.; Oldfors, A.; Magnuson, M.A.; Berggren, P.O.; Larsson, N.G. Impaired insulin secretion and beta-cell loss in tissue-specific knockout mice with mitochondrial diabetes. Nat. Genet. 2000, 26, 336–340. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Klionsky, D.J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Li, Y.; Han, J.; Wang, J.; Yin, J.; Li, J.; Tian, H. The double-edged effect of autophagy in pancreatic beta cells and diabetes. Autophagy 2011, 7, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Kiyono, K.; Suzuki, H.I.; Matsuyama, H.; Morishita, Y.; Komuro, A.; Kano, M.R.; Sugimoto, K.; Miyazono, K. Autophagy is activated by TGF-beta and potentiates TGF-beta-mediated growth inhibition in human hepatocellular carcinoma cells. Cancer Res. 2009, 69, 8844–8852. [Google Scholar] [CrossRef] [PubMed]
- Fujitani, Y.; Kawamori, R.; Watada, H. The role of autophagy in pancreatic beta-cell and diabetes. Autophagy 2009, 5, 280–282. [Google Scholar] [CrossRef] [PubMed]
- Marycz, K.; Kornicka, K.; Grzesiak, J.; Mieszek, A.; Apka, J. Macroautophagy and Selective Mitophagy Ameliorate Chondrogenic Differentiation Potential in Adipose Stem Cells of Equine Metabolic Syndrome: New Findings in the Field of Progenitor Cells Differentiation. Available online: https://www.hindawi.com/journals/omcl/2016/3718468/ (accessed on 27 July 2017).
- Marycz, K.; Kornicka, K.; Marędziak, M.; Golonka, P.; Nicpoń, J. Equine metabolic syndrome impairs adipose stem cells osteogenic differentiation by predominance of autophagy over selective mitophagy. J. Cell. Mol. Med. 2016, 20, 2384–2404. [Google Scholar] [CrossRef] [PubMed]
- Marycz, K.; Kornicka, K.; Irwin-Houston, J.M.; Weiss, C. Combination of resveratrol and 5-azacytydine improves osteogenesis of metabolic syndrome mesenchymal stem cells. J. Cell. Mol. Med. 2018, 22, 4771–4793. [Google Scholar] [CrossRef] [PubMed]
- Marycz, K.; Weiss, C.; Śmieszek, A.; Kornicka, K. Evaluation of Oxidative Stress and Mitophagy during Adipogenic Differentiation of Adipose-Derived Stem Cells Isolated from Equine Metabolic Syndrome (EMS) Horses. Stem Cells Int. 2018, 2018. [Google Scholar] [CrossRef] [PubMed]
- Marycz, K.; Kornicka, K.; Szlapka-Kosarzewska, J.; Weiss, C. Excessive Endoplasmic Reticulum Stress Correlates with Impaired Mitochondrial Dynamics, Mitophagy and Apoptosis, in Liver and Adipose Tissue, but Not in Muscles in EMS Horses. Int. J. Mol. Sci. 2018, 19, 165. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, U.; Cao, Q.; Yilmaz, E.; Lee, A.-H.; Iwakoshi, N.N.; Ozdelen, E.; Tuncman, G.; Görgün, C.; Glimcher, L.H.; Hotamisligil, G.S. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004, 306, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Oyadomari, S.; Koizumi, A.; Takeda, K.; Gotoh, T.; Akira, S.; Araki, E.; Mori, M. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J. Clin. Investig. 2002, 109, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Lenzen, S.; Drinkgern, J.; Tiedge, M. Low antioxidant enzyme gene expression in pancreatic islets compared with various other mouse tissues. Free Radic. Biol. Med. 1996, 20, 463–466. [Google Scholar] [CrossRef]
- Baynes, J.W.; Thorpe, S.R. Role of oxidative stress in diabetic complications: A new perspective on an old paradigm. Diabetes 1999, 48, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Robertson, R.P. Chronic Oxidative Stress as a Central Mechanism for Glucose Toxicity in Pancreatic Islet Beta Cells in Diabetes. J. Biol. Chem. 2004, 279, 42351–42354. [Google Scholar] [CrossRef] [PubMed]
- Peterson, L.W.; Artis, D. Intestinal epithelial cells: Regulators of barrier function and immune homeostasis. Nat. Rev. Immunol. 2014, 14, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Wyse, C.A.; McNie, K.A.; Tannahill, V.J.; Tannahil, V.J.; Murray, J.K.; Love, S. Prevalence of obesity in riding horses in Scotland. Vet. Rec. 2008, 162, 590–591. [Google Scholar] [CrossRef] [PubMed]
- Leahy, J.L.; Bonner-Weir, S.; Weir, G.C. Beta-cell dysfunction induced by chronic hyperglycemia. Current ideas on mechanism of impaired glucose-induced insulin secretion. Diabetes Care 1992, 15, 442–455. [Google Scholar] [CrossRef] [PubMed]
- Ferrannini, E. Insulin resistance versus insulin deficiency in non-insulin-dependent diabetes mellitus: Problems and prospects. Endocr. Rev. 1998, 19, 477–490. [Google Scholar] [CrossRef] [PubMed]
- Eizirik, D.L. Beta-cell defence and repair mechanisms in human pancreatic islets. Horm. Metab. Res. 1996, 28, 302–305. [Google Scholar] [CrossRef] [PubMed]
- Donath, M.Y.; Gross, D.J.; Cerasi, E.; Kaiser, N. Hyperglycemia-induced beta-cell apoptosis in pancreatic islets of Psammomys obesus during development of diabetes. Diabetes 1999, 48, 738–744. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Moley, K.H.; Chi, M.M.; Knudson, C.M.; Korsmeyer, S.J.; Mueckler, M.M. Hyperglycemia induces apoptosis in pre-implantation embryos through cell death effector pathways. Nat. Med. 1998, 4, 1421–1424. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Lv, Y.; Zhao, N.; Guan, G.; Wang, J. Protein kinase R-like ER kinase and its role in endoplasmic reticulum stress-decided cell fate. Cell Death Dis. 2015, 6, e1822. [Google Scholar] [CrossRef] [PubMed]
- Karaskov, E.; Scott, C.; Zhang, L.; Teodoro, T.; Ravazzola, M.; Volchuk, A. Chronic palmitate but not oleate exposure induces endoplasmic reticulum stress, which may contribute to INS-1 pancreatic beta-cell apoptosis. Endocrinology 2006, 147, 3398–3407. [Google Scholar] [CrossRef] [PubMed]
- Behrends, C.; Sowa, M.E.; Gygi, S.P.; Harper, J.W. Network organization of the human autophagy system. Nature 2010, 466, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Back, S.H.; Scheuner, D.; Han, J.; Song, B.; Ribick, M.; Wang, J.; Gildersleeve, R.D.; Pennathur, S.; Kaufman, R.J. Translation attenuation through eIF2alpha phosphorylation prevents oxidative stress and maintains the differentiated state in beta cells. Cell Metab. 2009, 10, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Kaneto, H.; Katakami, N.; Matsuhisa, M.; Matsuoka, T.; Kaneto, H.; Katakami, N.; Matsuhisa, M.; Matsuoka, T. Role of Reactive Oxygen Species in the Progression of Type 2 Diabetes and Atherosclerosis, Role of Reactive Oxygen Species in the Progression of Type 2 Diabetes and Atherosclerosis. Mediat. Inflamm. Mediat. Inflamm. 2010, 2010, e453892. [Google Scholar] [CrossRef]
- Frei, B. Reactive oxygen species and antioxidant vitamins: Mechanisms of action. Am. J. Med. 1994, 97, S5–S13, discussion S22–S28. [Google Scholar] [CrossRef]
- Busik, J.V.; Mohr, S.; Grant, M.B. Hyperglycemia-Induced Reactive Oxygen Species Toxicity to Endothelial Cells Is Dependent on Paracrine Mediators. Diabetes 2008, 57, 1952–1965. [Google Scholar] [CrossRef] [PubMed]
- Sakuraba, H.; Mizukami, H.; Yagihashi, N.; Wada, R.; Hanyu, C.; Yagihashi, S. Reduced beta-cell mass and expression of oxidative stress-related DNA damage in the islet of Japanese Type II diabetic patients. Diabetologia 2002, 45, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Kaneto, H.; Kajimoto, Y.; Fujitani, Y.; Matsuoka, T.; Sakamoto, K.; Matsuhisa, M.; Yamasaki, Y.; Hori, M. Oxidative stress induces p21 expression in pancreatic islet cells: Possible implication in beta-cell dysfunction. Diabetologia 1999, 42, 1093–1097. [Google Scholar] [CrossRef] [PubMed]
- Piro, S.; Anello, M.; Di Pietro, C.; Lizzio, M.N.; Patanè, G.; Rabuazzo, A.M.; Vigneri, R.; Purrello, M.; Purrello, F. Chronic exposure to free fatty acids or high glucose induces apoptosis in rat pancreatic islets: Possible role of oxidative stress. Metab. Clin. Exp. 2002, 51, 1340–1347. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-E.; Kim, J.-W.; Lee, E.-M.; Ahn, Y.-B.; Song, K.-H.; Yoon, K.-H.; Kim, H.-W.; Park, C.-W.; Li, G.; Liu, Z.; et al. Chronic resveratrol treatment protects pancreatic islets against oxidative stress in db/db mice. PLoS ONE 2012, 7, e50412. [Google Scholar] [CrossRef] [PubMed]
- Coskun, O.; Kanter, M.; Korkmaz, A.; Oter, S. Quercetin, a flavonoid antioxidant, prevents and protects streptozotocin-induced oxidative stress and β-cell damage in rat pancreas. Pharmacol. Res. 2005, 51, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wang, Q.; Evers, B.M.; Chung, D.H. Oxidative Stress-Induced Intestinal Epithelial Cell Apoptosis is Mediated By p38 Mapk. Biochem. Biophys. Res. Commun. 2006, 350, 860–865. [Google Scholar] [CrossRef] [PubMed]
- Lal, A.; Navarro, F.; Maher, C.; Maliszewski, L.E.; Yan, N.; O’Day, E.; Chowdhury, D.; Dykxhoorn, D.M.; Tsai, P.; Hofman, O.; et al. miR-24 inhibits cell proliferation by suppressing expression of E2F2, MYC and other cell cycle regulatory genes by binding to “seedless” 3′UTR microRNA recognition elements. Mol. Cell 2009, 35, 610–625. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, C.; Doss, C.G.P.; Bandyopadhyay, S.; Agoramoorthy, G. Influence of miRNA in insulin signaling pathway and insulin resistance: Micro-molecules with a major role in type-2 diabetes. Wiley Interdiscip. Rev. RNA 2014, 5, 697–712. [Google Scholar] [CrossRef] [PubMed]
- Chuang, T.-Y.; Wu, H.-L.; Chen, C.-C.; Gamboa, G.M.; Layman, L.C.; Diamond, M.P.; Azziz, R.; Chen, Y.-H. MicroRNA-223 Expression Is Upregulated in Insulin Resistant Human Adipose Tissue. J. Diabetes Res. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Buchan, R.J.; Cook, S.A. MicroRNA-223 regulates Glut4 expression and cardiomyocyte glucose metabolism. Cardiovasc. Res. 2010, 86, 410–420. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Park, E.J.; Yuki, Y.; Ahmad, S.; Mizuguchi, K.; Ishii, K.J.; Shimaoka, M.; Kiyono, H. Profiles of microRNA networks in intestinal epithelial cells in a mouse model of colitis. Sci. Rep. 2015, 5, 18174. [Google Scholar] [CrossRef] [PubMed]
- Lovis, P.; Roggli, E.; Laybutt, D.R.; Gattesco, S.; Yang, J.-Y.; Widmann, C.; Abderrahmani, A.; Regazzi, R. Alterations in microRNA expression contribute to fatty acid-induced pancreatic beta-cell dysfunction. Diabetes 2008, 57, 2728–2736. [Google Scholar] [CrossRef] [PubMed]
- Kornicka, K.; Marycz, K.; Tomaszewski, K.A.; Marędziak, M.; Śmieszek, A. The Effect of Age on Osteogenic and Adipogenic Differentiation Potential of Human Adipose Derived Stromal Stem Cells (hASCs) and the Impact of Stress Factors in the Course of the Differentiation Process. Oxid. Med. Cell. Longev. 2015, 2015, 309169. [Google Scholar] [CrossRef] [PubMed]
- Kornicka, K.; Marycz, K.; Marędziak, M.; Tomaszewski, K.A.; Nicpoń, J. The effects of the DNA methyltranfserases inhibitor 5-Azacitidine on ageing, oxidative stress and DNA methylation of adipose derived stem cells. J. Cell. Mol. Med. 2017, 21, 387–401. [Google Scholar] [CrossRef] [PubMed]
- Chomczynski, P.; Sacchi, N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987, 162, 156–159. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer | Sequence 5′-3′ | Amplicon Length (bp) | Accesion No. |
|---|---|---|---|---|
| LC3 | F: | TTACTGCTTTGCTCTGCCAC | 213 | XM_005608485.2 |
| R: | AGCTGCTTCTCCCCCTTGT | |||
| Beclin | F: | GATGCGTTATGCCCAGATGC | 147 | XM_014729146.1 |
| R: | ATCCAGCGAACACTCTTGGG | |||
| LAMP2 | F: | GCACCCCTGGGAAGTTCTTA | 139 | XM_014733098.1 |
| R: | TTCGAGGATCTGTGCCAATCA | |||
| GAPDH | F: | GATGCCCCAATGTTTGTGA | 250 | NM_001163856.1 |
| R: | AAGCAGGGATGATGTTCTGG | |||
| CHOP | F: | AGCCAAAATCAGAGCCGGAA | 272 | XM_014844003.1 |
| R: | GGGGTCAAGAGTGGTGAAGG | |||
| PERK | F: | GTGACTGCAATGGACCAGGA | 283 | XM_014852775.1 |
| R: | TCACGTGCTCACGAGGATATT | |||
| PARKIN | F: | TCCCAGTGGAGGTCGATTCT | 218 | XM_014858374.1 |
| R: | CCCTCCAGGTGTGTTCGTTT | |||
| p53 | F: | TACTCCCCTGCCCTCAACAA | 252 | U37120.1 |
| R: | AGGAATCAGGGCCTTGAGGA | |||
| p21 | F: | GAAGAGAAACCCCCAGCTCC | 241 | XM_003365840.2 |
| R: | TGACTGCATCAAACCCCACA |
| miRNA | Sequence of Specific Primer | Accesion Number of Sequence |
|---|---|---|
| miR-140-3p | TACCACAGGGTAGAACCACGGA | MIMAT0012926 |
| h/m/r-miR-146a-5p | TGAGAACTGAATTCCATGGGTT | MI0012809 |
| h/m/r-miR-223-3p | TGTCAGTTTGTCAAATACCCCA | MI0012953 |
| h/e/m/r-miR-17-5p | CAAAGTGCTTACAGTGCAGGTAG | MI0012831 |
| h/e/m/r-miR-24-3p | TGGCTCAGTTCAGCAGGAACAG | MI0012738 |
| h/e/m/r-miR-7a-5p | TGGAAGACTAGTGATTTTGTTGT | MIMAT001290 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kornicka, K.; Śmieszek, A.; Szłapka-Kosarzewska, J.; Irwin Houston, J.M.; Roecken, M.; Marycz, K. Characterization of Apoptosis, Autophagy and Oxidative Stress in Pancreatic Islets Cells and Intestinal Epithelial Cells Isolated from Equine Metabolic Syndrome (EMS) Horses. Int. J. Mol. Sci. 2018, 19, 3068. https://doi.org/10.3390/ijms19103068
Kornicka K, Śmieszek A, Szłapka-Kosarzewska J, Irwin Houston JM, Roecken M, Marycz K. Characterization of Apoptosis, Autophagy and Oxidative Stress in Pancreatic Islets Cells and Intestinal Epithelial Cells Isolated from Equine Metabolic Syndrome (EMS) Horses. International Journal of Molecular Sciences. 2018; 19(10):3068. https://doi.org/10.3390/ijms19103068
Chicago/Turabian StyleKornicka, Katarzyna, Agnieszka Śmieszek, Jolanta Szłapka-Kosarzewska, Jennifer M. Irwin Houston, Michael Roecken, and Krzysztof Marycz. 2018. "Characterization of Apoptosis, Autophagy and Oxidative Stress in Pancreatic Islets Cells and Intestinal Epithelial Cells Isolated from Equine Metabolic Syndrome (EMS) Horses" International Journal of Molecular Sciences 19, no. 10: 3068. https://doi.org/10.3390/ijms19103068
APA StyleKornicka, K., Śmieszek, A., Szłapka-Kosarzewska, J., Irwin Houston, J. M., Roecken, M., & Marycz, K. (2018). Characterization of Apoptosis, Autophagy and Oxidative Stress in Pancreatic Islets Cells and Intestinal Epithelial Cells Isolated from Equine Metabolic Syndrome (EMS) Horses. International Journal of Molecular Sciences, 19(10), 3068. https://doi.org/10.3390/ijms19103068