Abstract
Cholangiocytes, the epithelial cells lining the bile ducts, represent the unique target of a group of progressive diseases known as cholangiopathies whose pathogenesis remain largely unknown. In normal conditions, cholangiocytes are quiescent and participate to the final bile volume and composition. Following exogenous or endogenous stimuli, cholangiocytes undergo extensive modifications of their phenotype. Reactive cholangiocytes actively proliferate and release a set of proinflammatory molecules, which act in autocrine/paracrine manner mediating the cross-talk with other liver cell types and innate and adaptive immune cells. Cholangiocytes themselves activate innate immune responses against gut-derived microorganisms or bacterial products that reach the liver via enterohepatic circulation. Gut microbiota has been implicated in the development and progression of the two most common cholangiopathies, i.e., primary sclerosing cholangitis (PSC) and primary biliary cholangitis (PBC), which have distinctive microbiota composition compared to healthy individuals. The impairment of intestinal barrier functions or gut dysbiosis expose cholangiocytes to an increasing amount of microorganisms and may exacerbate inflammatory responses thus leading to fibrotic remodeling of the organ. The present review focuses on the complex interactions between the activation of innate immune responses in reactive cholangiocytes, dysbiosis, and gut permeability to bacterial products in the pathogenesis of PSC and PBC.
1. Introduction
The biliary tree is composed of a tridimensional network of ducts that drain the bile into the lumen of the intestine and are lined by epithelial cells termed cholangiocytes [1,2]. Cholangiocytes are a heterogeneous cell population from both a functional and a morphological point of view. Cholangiocytes are divided into small and large cholangiocytes, which encircle small and large intrahepatic bile ducts, respectively. Large cholangiocytes actively participate to modification of bile composition and volume through secretory and absorptive mechanisms tightly regulated by the action of different molecules (e.g., hormones, peptides, and neurotransmitters) [3]. On the other hand, small cholangiocytes are able to modify their phenotype in response to exogenous/endogenous noxious stimuli (e.g., microorganisms, toxins/drugs, or hormones), thus participating to the inflammatory response during biliary tree damage [3,4], and serve as liver progenitor cells under certain conditions [5]. Cholangiocytes are the central target of a group of diseases known as cholangiopathies with different etiologies (e.g., genetic, autoimmune, infectious, drug-induced, idiopathic malignant, secondary sclerosing cholangitis). In response to injury, cholangiocytes that are in a quiescent state enhance proliferation and start to release mediators through which they interact with both resident and non-resident liver cells, maintaining tissue homeostasis and function, and contributing to immune cells and myofibroblasts chemotaxis at the damage site.
In health conditions, an enormous amount of gut-derived molecules reaches the liver via the portal blood circulation without eliciting any inflammatory response. In disease state, when the intestinal barrier function is defective or in case of modifications of gut bacterial homeostasis, an altered composition of gut-derived products reaches the liver where it may induce or exacerbate hepatic inflammation eliciting complex responses in hepatic cells including cholangiocytes.
An increasing body of clinical and experimental data has highlighted the prominent role of an altered-gut microbiota in the onset and perpetuation of a subgroup of chronic cholestatic liver diseases, particularly of primary sclerosing cholangitis (PSC) and primary biliary cholangitis (PBC). Both diseases are characterized by portal inflammation, bile duct damage, and subsequent fibrosis development, and progress to cirrhosis leading to end-stage liver failure. Despite these disorders share common features, PSC targets mainly medium to large extrahepatic and intrahepatic bile ducts while PBC is an autoimmune disease characterized by serum positivity to antimitochondrial antibody (AMA) and damage of small intrahepatic bile ducts [6,7]. Thus, awareness of biliary epithelium heterogeneity is function to understand the molecular mechanisms underlying the pathophysiology of cholangiopathies and possibly to devise new selective therapeutic targets. Ursodeoxycholic acid (UDCA) administration is currently the primary therapeutic approach for the treatment of PBC patients [3]. The 40% of patients who do not respond to UDCA, are subjected to obeticholic acid (OCA) administration. Therefore, the combination of the two existing therapies is not effective in whole PBC patient’s treatment. By contrast, up until now, no approved therapies for PSC patients exist. The only therapeutic strategy is liver transplantation. Several clinical trials based on the administration of drugs targeting specific cholangiocytes pathways, such as agonists of nuclear receptors and membrane transporters involved in bile acids homeostasis and metabolisms, are currently ongoing [8].
2. Cholangiocyte Adaptation to Injury
In response to exogenous/endogenous damage, cholangiocytes undergo noteworthy modifications of their phenotype and become “reactive”. In this setting, biliary epithelial cells release a wide range of proinflammatory and fibrogenic mediators orchestrating cholangiocyte proliferation, senescence, and apoptosis as well as immune and mesenchymal cells chemotaxis to repair the injured tissue and remodel the biliary tree [3,9]. These events determine cholangiocyte proliferation, which is intended to compensate for the anatomic loss of biliary cells and also to sustain secretory activities, tissue infiltration of immune cells, deposition of matrix proteins and activation of liver progenitor cells [10]. Unless reversed, these mechanisms lead to periportal fibrosis and ductopenia, when the balance between cell proliferation and cell death events is lost and, eventually, to biliary cirrhosis [11,12]. Cholangiocytes differentially proliferate in response to liver injury and toxins. Several stimuli, including bile acids, acetylcholine, estrogen, hepatocyte growth factor, and IL-6 may induce cholangiocyte proliferation by binding to specific receptors. Antiproliferative mediators such as somatostatin, gastrin, and interferon γ (INFγ) are also known [13,14].
Along with proliferation, cholangiocyte response to injury is characterized by the so-called “neuroendocrine-like transdifferentiation”, which plays an essential role in immune responses, hepatic inflammation and development of liver fibrosis in addition to sustaining biliary proliferation itself [4,15]. Indeed, activated cholangiocytes express and secrete various proinflammatory cytokines and chemokines (e.g., IL-6, IL-8, TNFα, and various growth factors) and respond to signaling cascades such as Notch and Hedgehog [16].
The signaling pathway of the gastrointestinal peptide hormone secretin (SCT), which binds the secretin receptor (SR), is one of the most studied in cholestatic liver injury. It has been supposed that the SCT/SR axis is associated with cholestatic liver injury because rats show elevated expression of SR after bile duct ligation (BDL) [17]. When the SCT binds to SR, which is expressed only in the basolateral membrane of large cholangiocytes, its activation elevates intracellular adenosine 3,5′-cyclic monophosphate (cAMP) levels leading to enhanced cell proliferation, exocytosis, and ductular secretion in cholangiocytes [18,19]. The increase in intracellular cAMP levels induces bicarbonate secretion from cholangiocytes into the bile [20], and the activation of ERK1/2 and Elk-1 signaling pathway [21,22]. Since small cholangiocytes do not express CFTR and SR, only large cholangiocytes are responsible for CFTR- or SR-dependent proliferation and bicarbonate secretion in the liver, and are involved in SCT-induced ductular secretion [23].
In small cholangiocytes, Ca2+ signaling is more important for proliferation. During histamine-induced small cholangiocyte proliferation, inositol 1,4,5-trisphosphate (IP3) levels are increased [24]. The binding of IP3 to IP3 receptor (IP3R), activates the release of Ca2+ into the cytosol and the subsequent activation of calcineurin (CN) and calmodulin (CaM). An elegant study showed that CN mediates the phosphorylation of the nuclear factor of activated T-cells (NFAT) proteins [25], while activation of CaM leads to the activation of CaM-dependent kinase (CaMK) [26]. In this way, small cholangiocytes proliferate through IP3/Ca2+/CaMK signaling [24,27]. Furthermore, the activation and nuclear translocation of NFAT2 increases in proliferating small cholangiocytes [28]. This pathway has been also identified in hepatocellular carcinoma contributing to the maintenance of cell proliferation [29]. In large cholangiocytes, Ca2+ signaling may not be critical for cell proliferation during cholestatic liver injury, since expression levels of IP3R are decreased in large cholangiocytes during BDL [30].
Although the cAMP pathway may be more important than the IP3/Ca2+ pathway for large cholangiocyte proliferation, Ca2+ signaling is essential for ductular secretion in large cholangiocytes [23], and a study has demonstrated that trigger-induced Cl− secretion is Ca2+ dependent [31]. An in vivo experiment carried out on rats showed that the expression of SR and CFTR as well as intracellular cAMP levels and cholangiocyte proliferation, were increased after SCT administration [32]. Moreover, SCT is expressed by cholangiocytes and S cells which are located primarily in the mucosa of the duodenum [33]. High levels of SCT expression were found in both large cholangiocytes and S cells during BDL [34]. In contrast, SCT knockout (SCT−/−) mice and SR−/− mice show an attenuated bile duct hyperplasia after BDL compared to wild-type mice [34,35].
Another recent study has highlighted a new possible mediator in cholangiocyte response to damage. In particular, it has been shown that cholangiocytes express gonadotropin-releasing hormone (GnRH) receptor 1 (GnRHR1) and its expression levels are elevated during BDL in rats [36]. GnRH is a tropic peptide hormone that modulates cell proliferation and act, depending on the cell type, by stimulating or inhibiting the proliferation [37]. Administration of GnRH increases bile duct mass and intracellular cAMP levels, expression of SR, CFTR, and AE2 in cholangiocytes in vivo. On the other hand, in vivo knockdown of GnRH decreased intrahepatic bile duct mass and fibrosis induced by BDL [36]. Moreover, enhanced GnRH serum levels as well as GnRHR1 expression in the liver, were observed in human PSC patients compared to healthy individuals [38].
A different known mediator of the cholangiocyte response to damage is the neuropeptide substance P (SP). The binding of SP to neurokinin-1 receptor (NK-1R) increases cytokine expression thus leading to the inflammatory response [39]. Patients with chronic liver disease show increased serum levels of SP compared to respective controls, also reproduced in cholestatic rat models [40]. Elevated SP secretion is also identified in cholangiocarcinoma (CCA) cell lines and treatment with an antagonist inhibits proliferation of CCA cells [41]. Moreover, the increase of SP serum levels and NK-1R expression in the liver were observed in human PSC patients compared to healthy group [42]. Elevated NK-1R expression was found in large cholangiocytes of BDL mice, and NK-1R−/− mice show attenuated large cholangiocyte proliferation and bile duct hyperplasia [43].
More and more new studies appear to deepen the role of some possible mediators of the cholangiocyte adaptation to injury. The role of histamine is already known in the induction of small and large cholangiocyte proliferation, where agonists for H1 histamine receptor induce small cholangiocyte proliferation, and agonists for H2 induce large cholangiocyte proliferation [24,27]. The inhibition of mast cell-derived histamine secretion has been shown to attenuate bile duct hyperplasia during BDL in vivo [44]. On the other hand, during cholestatic liver injury such as PBC, the number of mast cells in the liver is increased, probably due to their role in histamine secretion [45].
Another possible mediator is the melatonin, a hormone produced by the pineal gland, small intestine and liver [46]. An in vivo experiment carried out on BDL rats showed that the melatonin administration reduces liver fibrosis and serum cytokine levels for IL-1β and IL-6 [47], but it has recently been shown that melatonin administration inhibits GnRH secretion and reduces bile duct hyperplasia and liver fibrosis in BDL rats [48]. Other studies indicate that numerous other pathways, such as pancreatic duodenal homeobox-1 (PDX-1), vascular endothelial growth factor (VEGF) A/C and relative receptor VEGFR-2/3, and galanin and galanin receptor 1 (GalR1) are associated with cholangiocyte proliferation and liver fibrosis in cholestatic liver disease such as during BDL in rats [49,50].
A relevant role in shaping cholangiocyte response to injury is played by genetic variants, epigenetic mechanisms, and post-transcriptional phenomena (including the influence of microRNAs on protein expression [51]), which together determine whether reactive cholangiocytes regress to a normal phenotype or lead to chronic inflammation of the bile duct, with progression of the cholangiopathy. An elegant study of Lazaridis and LaRusso [11], summarized the recent findings concerning the individual genetic variants which may be implicated in cholangiopathies onset and disease progression. Data collected by four different genome wide-association studies (GWASs) have unveiled the presence of 30 susceptibility loci associated with PBC related to immune cells functions and processes. With regards to PSC, 31 risk loci have been identified by GWASs [52]. However, nine of these susceptibility loci do not reach the genome-wide significance [52]. As for PBC, PSC-associated risk loci are involved in innate and adaptive immune cells responses. In addition, genetic variants of fucosyltransferase 2 (FUT2), described in detail beyond, have been associated to gut microbiota modification in PSC patients [53].
Further investigations are needed to delineate the role of genetic factors in the pathophysiology of cholangiopathies; these studies prove to be complex also because of the difficulty in establishing the environmental contribution to the progression of the disease. With this regard, the recognition of the heterogeneity of cholangiocytes along the biliary tree is of fundamental importance, in order to better understand the cholangiopathies [54,55].
3. Innate Immune Response Activation
Biliary epithelial cells represent the first line of defense of the biliary system against microbial products and other potential noxious mediators that are continuously translocating from the gut to hepatic sinusoids [16]. Cholangiocytes are provided with pathogen recognition receptors (PRRs), adaptor proteins and related signaling pathways. PRRs recognize and bind to bacterial components known as pathogen-associated molecular patterns (PAMPs) such as lipopolysaccharide (LPS) or bacterial DNA fragments. Molecules released in the extracellular environment from damaged cells defined as damage-associated molecular patterns (DAMPs) can also activate PRRs of cholangiocytes [56]. The best characterized PRRs family is the Toll-like receptors (TLRs) consisting of a number of type 1 transmembrane glycoprotein receptors (10 members in human and 13 in mammalian cells) activated in response to specific conserved components shared among microorganisms [57]. Exposure of cholangiocytes to LPS leads to TLR4 activation, which in turn triggers intracellular signals culminating in the activation of nuclear factor-kappa B (Nf-κB) or activator protein-1 (AP-1) [58,59], genes involved in the biosynthesis of proinflammatory cytokines [60]. Beside TLRs family, the nucleotide-binding oligomerization domain (NOD)-like receptor (NLR) family consists of soluble PRRs, which sense intracellular pathogens and endogenous dangerous stimuli through the DAMPs [61]. Based on N-terminal structures, NLR family proteins are classified into different subfamilies [62]. Among these, the nucleotide-binding oligomerization domain (NOD)-like receptor (NLR) family, pyrin domain-containing protein 3 (Nlrp3) has been recently investigated and described as a member of innate immune system activated by both PAMPs and DAMPs. Nlrp3 is assembled into a multiprotein complex known as the “inflammasome” which, upon activation, triggers the proteolytic cleavage of cytokine precursors mediated by caspase-1 [61,63], leading to the secretion of IL-1β and IL-18 in the mature and biologically active form [56].
Our group of research has recently shown that the Nlrp3 inflammasome is upregulated in injured cholangiocytes, both in a murine model of sclerosing cholangitis and in biliary cells of PSC patients. In vitro, activation of Nlrp3 induced the expression of IL-18 and influenced the epithelial barrier function of cholangiocytes by blunting the increased epithelial permeability stimulated by LPS incubation [64]. Reactive cholangiocytes actively participate to host immune response by releasing a panel of cytokines and chemokines, which act in autocrine and paracrine fashion on resident liver cells and immune cells thus increasing the expression of surface adhesion molecules and producing antimicrobial peptides, which directly exert host defense activities (Figure 1).
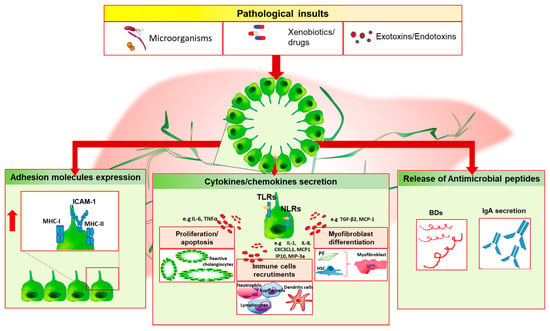
Figure 1.
Cholangiocyte immune response. Biliary epithelial cells are exposed to different dangerous stimuli such as microorganisms, drugs or toxins, and others exotoxins/endotoxins which trigger tissue damage. In this setting, cholangiocytes may modify their biology and phenotype by increasing the release of proinflammatory mediators (e.g., IL-6, IL-8, IL-1), which act in autocrine/paracrine fashion on both resident and non-resident cells, by upregulating the expression of surface proteins (i.e., MHC-I, MHC-II and ICAM-1) and by releasing antimicrobial molecules such as beta-defensins (BDs) or IgA following epithelial transcytosis. These events lead to ductules’ proliferation, immune cells chemotaxis and myofibroblast differentiation. In case of persistent biliary damage, these processes could lead to chronic inflammation and fibrosis establishment. PF, portal fibroblast; HSC, hepatic stellate cell.
Following an initial insult, cholangiocytes secrete IL-6, which determines cholangiocyte proliferation to compensate biliary mass loss and preserve biliary epithelial cells functions. LPS, TNFα, or IL-1 trigger the releasing of IL-8, the epithelial cell derived neutrophil activating protein (ENA-78) and growth-related oncogene (GRO), which mediate neutrophil recruitment at the inflammation site [65]. Differently, dendritic cells chemotaxis is driven by cholangiocyte secretion of macrophage inflammatory protein-3a (MIP-3a) following IL-1β, TNFα, IL-17 stimulus, or TLRs activation [66]. The chemoattraction ability of biliary epithelial cells comprises the releasing of the C-X-3-C motif chemokine ligand 1 (CX3CL1), also known as fractalkine. This chemokine act as chemoattractant molecule in its soluble form by promoting monocytes recruitment and lymphocytes T homing, and as adhesion molecule in its membrane-bound form, by mediating the adhesion of leukocyte-expressing the chemokine receptor (CXC3CR1) to endothelium [67]. IL-6 also exerts a pleiotropic effect by promoting the terminal differentiation of lymphocytes B cells, which secrete serum immunoglobulins [68]. A subtype of epithelial inflammatory cytokines modulate the expression of adhesion molecules on the surface of cholangiocytes, which serve for cell-mediated immune response. In vitro experiments have shown that TNFα, IL-1, and INFγ drive the upregulation of the adhesion molecules MHC-I, MHC-II, and ICAM-1 on cholangiocyte membrane [69,70]. Conversely, the negative regulation of this mechanism is mediated by TGFβ, which decreases the expression of these surface proteins [71]. Alternatively to MHC-I, CD8+ lymphocytes activation during biliary injury can be triggered by the upregulation of CD40 ligand on cholangiocytes surface upon INFγ or TNFα stimulus [71].
During chronic injury of the bile ducts, the array of proinflammtory mediators released undergoes extensive modifications. Following INFγ stimulation, cholangiocytes increase the secretion of monocyte chemoattractant protein-1 (MCP-1), monokine induced by INFγ (Mig), interferon inducible T cell alpha chemoattractant (ITAC) and interferon gamma inducible protein-10 (IP-10) [65]. Although MCP-1 is a well-known chemotactic factors for monocytes and lymphocytes, it also mediates fibrogenic effects by stimulating portal fibroblast differentiation and collagen-I secretion, and by inducing HSC homing [72,73]. With this regard, proliferating cholangiocytes upregulates MCP-1 expression whose levels correlate with a poor prognosis of disease [74]. One of the key features of chronic biliary injury is represented by fibrogenesis, which involves the participation of different cell subtypes. Biliary epithelial cells increase the releasing of a multifunctional cytokine, the TGFβ; in particular, high levels of β2 transcripts have been detected in reactive biliary cells of CCl4-treated fibrotic rat livers, while no expression has been evidenced in the other cell subtypes [75]. TGFβ induces the differentiation of portal fibroblast, the release of Endothelin-1 by cholangiocytes and the inhibition of biliary epithelial cells proliferation [76,77].
Bile duct loss is a consequence of chronic biliary inflammation. Co-incubation of TNFα and Actinomycin D determines cholangiocyte apoptosis and loss of secretion capabilities in BDL rat, suggesting that during cholestasis reactive cholangiocytes are more susceptible to TNFα cytotoxic effect [78]. Accordingly, the administration of taurocholate prevents TNFα-induced cholangiocyte damage through the PI3K molecular pathway activation [79]. In addition, in experimental biliary atresia it has been shown the ability of TNFα to promote cholangiocyte programmed cell-death through the activation of TNFα/TNFR2 molecular pathway [80].
In vitro studies have shown that murine and rat cholangiocytes treated with conditional medium collected by myofibroblast hepatic stellate cells, increase the production of soluble mediators involved in immune cell chemotaxis, antigen presentation and processing via activation of Hedgehog (Hh) signaling, as confirmed by Hh-neutralizing antibody challenge [81]. The upregulation of these immune mediators has been confirmed in vivo in patched (ptc) haplo-insufficient (ptc+/−) mice (with impaired capacity to deactivate Hh signal) subjected to BDL, a model of biliary fibrosis associated with liver Hh pathway activation [81,82]. Previous studies demonstrated the ability of biliary epithelial cells and hepatic stellate cells to express and respond to Hh ligands [83,84]. The perpetuation of Hedgehog pathway activation has been related to biliary mass loss and fibrosis due in part to the induction of epithelial-mesenchimal transition of hepatic progenitors [85] and in part to myofibroblastic phenotype retention through an autocrine mechanism [84,86].
Induction of biliary senescence has been recently related to cholangiocyte immune response deregulation during cholangiopathies. PSC cholangiocytes show an increased expression of cellular senescence markers and of the senescence-associated secretory phenotype (SASP) [87]. Cholangiocytes from multi-drug resistance 2 knockout (mdr2−/−) mice, a model of PSC, undergo senescence [87]. Mdr2 is a canalicular phospholipid flippase encoded by the abcb4 gene, involved in the transport of phospholipids into bile. At this level, phospholipids assemble together with bile acids in mixed micelles that prevent the damage of biliary epithelium induced by toxic bile acids [88]. In the mouse model, the absence of mdr2 causes bile duct injury mediated by free bile acids which results in sclerosing cholangitis development. Lesions are characterized by portal inflammation, onion-skin lesions and obliterative cholangitis resembling PSC hallmarks [88]. As mentioned above, cholangiocytes are equipped of TLRs family receptors through which recognize PAMPs. The activation of TLR4 by transient LPS stimulus rapidly determines N-Ras activation and subsequently induces downstream events culminating in cholangiocyte IL-6 secretion [89]. Conversely, human cultured cholangiocytes subjected to persistent LPS treatment acquire a senescent phenotype characterized by the upregulation of proteins involved in cell cycle arrest (p16INK4a and p21WAF1/Cip) and the expression of SASP components [87]. Despite the precisely mechanism of cholangiocyte senescence is poorly understood, these fascinating results suggest an important role of non-replicative cholangiocytes in the modulation of biliary microenvironment and organ homeostasis [87,90]. Senescent cholangiocytes contribute to progressive ductopenia due to the loss of proliferative and regenerative capabilities that make non-replicative cells prone to subsequent injuries with worsening inflammation [87].
A novel attracting mechanism through which cholangiocytes mediate intercellular communication in the setting of microbial infection has been recently reported. The extracellular vesicles (EVs)-mediated cell–cell communication has been demonstrated in cultured immortalized cholangiocyte H69 cell lines following LPS challenge [91]. Endotoxin treatment determines an increase of LPS-derived EVs secretion in immortalized human cultured cholangiocyte. Cultured cholangiocytes stimulated with LPS-derived EVs enhance the inflammatory response by upregulating mRNA levels of proinflammatory cytokines. Knock-down of either SCT and SR determines a reduction of EVs released by large cholangiocyte following LPS treatment. Moreover, EVs collected by STC and SR knocked-down large cholangiocytes do not elicit an inflammatory response in control large cholangiocytes. By taking into account these data, it is possible to hypothesize that inflammatory mediators secreted in response to LPS-derived EVs and influenced by the STC/SR axis could act on immune cells to contain biliary damage [91].
Cholangiocyte immune capability relies also on secretory immunoglobulin A (sIgA) transcytosis into the bile to preserve mucosal integrity during pathogens infection. The opsonization of circulating bacterial antigen mediated by sIgA prevents microbial attachment on cholangiocyte surface [92]. The secretory capacity of cholangiocyte includes the synthesis of antimicrobial peptides. Defensins are small proteins rich in cysteine that bind to the microbial surface inducing membrane disruption, intracellular ion release and ultimately cell death. Of the two existing subfamilies, α and β-defensin, the latter serves as mucosal defense against local infections. In humans six isoforms (Hbd-1 to Hbd-6) have been identified. In vitro, the Hbd-1 is constitutively expressed in the cytoplasm of cultured biliary epithelial cells lining the intrahepatic bile ducts; instead, the expression of Hbd-2 occurs upon LPS challenge, Escherichia coli infection [93] or stimulation by TNFα and IL-1β whose levels are reported to be increased in cholangiopathies in the portal tract [94].
4. Leaky-Gut Hypothesis
The term “gut microbiota” refers to commensal and pathogen bacteria, and other microorganisms (i.e., Fungii, viruses), including metabolites, bacterial products or genomes, that colonize the intestinal tract [95]. The gut-colonizing microorganisms are in symbiosis with the host and possess functional roles in human metabolism, (e.g., by production of vitamins or secondary biliary acids) [96]. The lack of this beneficial association is known as dysbiosis and it depends on qualitative changes of gut microbiota, due to the increased extent of pathogen bacteria with respect to commensal flora, or to quantitative changes caused by bacterial overgrowth [97]. In health conditions, the gastrointestinal tract exhibits physical (intestinal barrier and mucus layer) and chemical barriers (IgA or antimicrobial peptides and proteins) that prevent the translocation of bacteria towards the liver. A portion of gut microbial antigens enters in the portal blood circulation and is recognized by liver immune cells without eliciting an immunological response because of the existing mechanism of “immune tolerance” [98,99]. The impairment of intestinal barrier functions observed during gut inflammation exposes the liver to an increasing number of bacteria, bacterial components or metabolites, which may trigger hepatic inflammation and fibrotic remodeling (Figure 2).
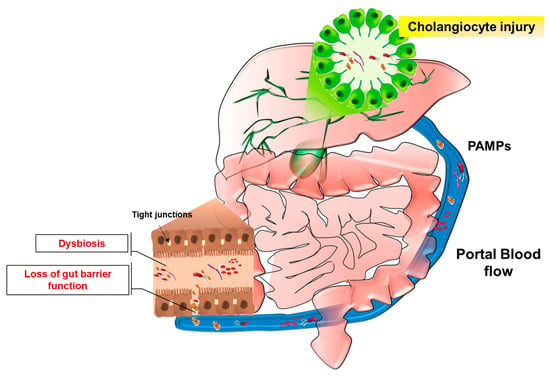
Figure 2.
The leaky-gut hypothesis. Portal blood flow ensures the metabolic connection between intestine and liver. During gut inflammation, which leads to impairment of intestinal barrier functions (e.g., downregulation of tight junctions), or as a consequence of dysbiosis establishment, a considerable amount of microorganisms and related molecules (PAMPs) reach the liver and may determine hepatobiliary inflammation. In this context, deregulation of cholangiocyte response to injury is thought to contribute to disease onset and progression.
These notions establish the “leaky-gut” hypothesis which may contribute to the pathogenesis of hepatobiliary diseases.
A clear association between intestinal inflammation, gut microbiota modifications and the development of cholangiopathies is exemplified in PSC. Indeed, 75% of PSC patients are also affected with inflammatory bowel disease (IBD), more often ulcerative colitis (UC), whose pathogenesis is thought to be linked to gut dysbiosis [16,100,101]. Clinical observations have shown that PSC patients display blood and bile bacteria colonization [102,103] and an increase of 16 S ribosomal RNA (rRNA) in the bile [104]. Intriguingly, colectomy after or during liver transplantation, reduced PSC recurrence (which occurs in up to 37% of PSC-transplanted patients), reflecting the role of gut microbiota in PSC relapse [105,106]. Oral antibiotics have been shown to be effective in PSC patients’ treatment [87,107].
A possible pathogenic role of bacterial translocation has also been proposed for PBC. Lipoteichoic acid, a component of bacterial wall, has been detected in damaged bile ducts of PBC patients [108]. Moreover, long-term exposure of BALB/c mice to bacterial antigens induces autoantibody production which determines histological features resembling PBC [109]. Different risk loci involved in immune regulation and immune response have been identified and genetic susceptibility seems to contribute to chronic biliary diseases establishment [110,111,112].
In particular, GWASs have evidenced a correlation between PSC and IBD that share, among others, the genetic risk locus FUT2. The gene FUT2 encodes for galactoside 2-alpha-l-fucosyltransferase 2 enzyme involved in the synthesis of soluble A and B blood antigens. Inactivating variants of FUT2, which define patients as “non-secretors”, influence biliary bacterial diversity with a reduction of Proteobacteria and Actinobacteria, the enrichment of dominant microbial flora (e.g., Firmicutes) and the increase of biliary tree infection rate and incidence of dominant stenosis [113,114]. Knock-out of Fut2 expression in mice has been shown to cause hepatobiliary abnormalities related to the development of porto-systemic shunts [115].
The impact of gut microbiota in chronic biliary inflammation has been examined in distinct animal models displaying gut dysbiosis or subjected to bacterial-derived antigens inoculation. In both cases, hepatic changes recapitulating different hallmarks of cholangiopathies, have been reported. Germ-free (GF) mdr2−/− mice model showed a worse biliary injury and higher rate of senescent cholangiocytes, as compared to control mdr2−/− mice conventionally raised [116]. The potential protective role of commensal flora metabolites in the setting of biliary inflammation has been confirmed in vitro. Treatment of senescent cholangiocytes with UDCA, a microbial metabolite absent in GF mice, induced a partial reversal of the senescent phenotype [116]. In line with these findings, decreased levels of secondary bile acids, which are commensal flora metabolites exerting anti-inflammatory functions in vitro, have been found in PSC and IBD patients compared to healthy controls [116]. This is not the case of PBC [116]. Contrasting results have been reported in GF-NOD.c3c4 mice, a mouse model that develops spontaneously intra and extra-hepatic biliary tree inflammation. GF-NOD.c3c4 mice are protected from disease development when compared to conventionally raised (CONV-R) NOD.c3c4. Likewise, biliary disease improvement could be achieved by antibiotic-treatment of CONV-R NOD.c3c4. [117]. The potential role of intestinal luminal bacteria in hepatobiliary inflammation has been explored in genetically susceptible rats, which develop small-intestine bacterial overgrowth (SIBO) after the surgical jejunal self-filling of blind loops. Portal inflammation and bile duct proliferations and loss have been reported in this animal model [118]. The pathological phenotype could be prevented by treatment with Mutanolysin, a muralytic enzyme which breaks the β1-4 linkage of N-acetylmuramyl acid and N-acetylglucosamine of peptidoglycan [118].
Others in vivo studies rely on bacteria or microbial-derived components animal inoculation to trigger hepatobiliary inflammation. These models resemble various characteristic of diseases of the biliary tree. A clear example derives from an experimental study carried out on BALB/c mice subjected to long-term inoculation of Streptococcus intermedius. The prolonged bacterial exposure determines a non-suppurative cholangitis and the production of autoantibodies, resembling histological features of PBC [109]. Moreover, intraperitoneal injection of peptidoglycan in rats causes cholangiographic irregularities of both small and large ductules, focal areas of sacculations and loss of bile mass [119]. Rectal inoculation of N-formyl l-methionine l-leucine l-tyrosine (fMLT), an E. coli-chemotactic peptide that reaches the liver via portal blood circulation, resulted in small duct cholangitis with leukocytes infiltration, inducing bile duct abnormalities similar to those observed in the earlier stages of PSC [120]. The link between enteric microbes and inflammatory cholangiopathies has been observed in mice lacking the expression of functional CFTR (CFTR−/−) and subjected to dextran sodium sulfate (DSS) colitis model [121,122]. DSS treatment increases the intestinal permeability and induces portal endotoxemia [123]. DSS-treated CFTR−/− mice exhibit signs of hepatic injury including the activation of ductular reaction, the establishment of portal inflammation and the infiltration of immune cells, as compared to wild-type (WT) counterpart [124]. In vitro, LPS challenge induces the increase of inflammatory cytokines releasing in CFTR−/− isolated cholangiocytes with respect to WT cultured cells, via upregulation of TLR4 and NF-kB pathway [124]. Taken together these experimental findings support the involvement of microorganisms, PAMPs or bacterial products in the development and progression of cholangiopathies. Nonetheless, it is not yet clear the precise mechanism through which gut-derived products determine biliary tree damage and the degree of injury. Intestinal dysbiosis, loss of intestinal epithelial barrier functions or a deregulated and exacerbated host immune responses are all mechanisms proposed as causal links in the development and progression of chronic biliary inflammatory diseases.
5. Microbiota Modification in Cholangiopathies
Several attempts have been made in recent years to define the gut microbiota signature of a group of cholangiopathies. Advances in high-throughput technologies allowed a deeper understanding of gut microbiota composition and modification in the setting of biliary inflammatory diseases. The composition of the microbiota harboring the intestine is the result of different variables such as patient genotype, eating habits, age, health conditions and antibiotic assumption [95]. Typically, the microbiota observed in fecal samples of healthy individuals is mainly enriched of two phyla, Firmicutes and Bacterioides. Proteobacteria accounts for up to 2–3% of gut milieu [125]. Commensal flora possess functional role in nutritive metabolism, by synthetizing nutrients in absorbable substrates for the host, in drugs or xenobiotics metabolism, in intestinal mucosal barrier integrity maintenance, in immune system development and in preventing pathogen bacteria colonization [126].
Multiple sequencing studies have been carried out to precisely characterize the microbiota composition of patients with chronic cholestatic liver diseases, both at fecal and mucosal level.
A recent study highlighted the existing differences in fecal microbiota composition between PSC-patients and both IBD and healthy patients [127]. At the genus level, PSC patients, with or without concomitant IBD, had increased abundance of Enterococcus, Lactobacillus, Streptococcus, and Fusobacterium genera compared to the healthy group. Oral antibiotic treatment has been shown to be effective in Streptococcus abundance abolishment [127]. The remaining three genera, independently from oral assumption of UDCA and after removal of confounding factors such as the correlation with IBD, set a “PSC-microbiota signature”, which allowed disease diagnosis with a 71% of accuracy [127]. The overrepresentation of Enterococcus, Lactobacillus, and Fusobacterium genera has been already reported in different diseases such as liver cirrhosis and IBD [128,129,130]. In particular Enterococcus faecalis, which along Enterococcus faecium is the most represented species revealed in PSC-patients bile with dominant strictures, has been linked to intestinal barrier perturbation due to its ability to produce enzymes belonging to matrix metalloproteases (MMPs) such as gelatinase (GelE) [131]. A positive correlation between Enterococcus abundance and increased serum levels of the alkaline phosphatase (ALP), a cholestatic marker associated to a poor prognosis in cholestatic liver diseases, has been reported. However, the results were not confirmed after eliminating confounding factors [127].
Additional experimental evidences confirmed a distinct microbiota composition of PSC independently to IBD co-presentation, compared to control group. Sequencing data obtained from stool samples revealed the reduction of alpha diversity and the modification of different bacterial taxa richness in PSC patients with respect to controls. The abundance of Veillonella, which belongs to the phylum Firmicutes, resulted highly enriched in PSC patients compared to both hepatocellular carcinoma (HCC) and UC patients [132]. Interestingly, the reduced alpha diversity observed in PSC patients as compared to HCC patients is not influenced by oral antibiotics administration in the last 12 months [132]. Thus, the lowering of intra-individual microbiota diversity seems to be a unique characteristic of PSC and not of other liver diseases [129]. Veillonella, which has been found to be the most abundant genus enriched in PSC patients, has been previously linked to others inflammatory and fibrotic diseases (pulmonary cystic fibrosis or idiopathic pulmonary fibrosis) and to Crohn’s Disease (CD) relapse after ileo-caecal resection [132]. Accordingly, PSC gut microbiota profile is not influenced by IBD type, suggesting the concept that the dysbiosis is determined by liver disease [132].
With regard to pediatric PSC-patients dysbiosis, an increased abundance of Streptococcus parasanguinis, Veillonella spp., Enterococcus faecium and Enterococcus spp. has been evidenced compared to HCCs [133]. The overall findings suggest the association of three major genera, Enteroccocus, Streptococcus, and Veillonella to pediatric PSC-development. The alteration of gut microbiota has been also investigated in fecal samples of PBC patients [134]. The sequencing of 16S rRNA of microbiota performed in stool samples of PBC patients prior to UDCA treatment has shown a reduction in microbiota diversity with respect to the healthy group [134]. Contrasting results have been reported in a separate study, probably due to UDCA treatment of PBC patients selected for the study [135]. As in PSC, a microbial signature consisting of 12 genera has been identified that could be hypothetically used for PBC diagnosis [134]. Microbiota profile analysis highlighted the enrichment of the Enterobacteriaceae family, followed by Pseudomonas, Veillonella, and Clostridium genera in PBC patients. Conversely, Oscillospira and Sutterella resulted as less represented [136]. Six months of UDCA treatment reverted gut microbiota alteration in six of the 12 genera associated to PBC with respect to healthy group [136].
The interaction between gut microbiota and host takes play at intestinal mucosa level suggesting that the suitable way to explore dysbiosis, proposed as the basis of “leaky-gut hypothesis”, is represented by mucosal microbiota profiling. In a recent study, 16S rRNA microarray analysis carried out in ileocecal biopsies collected by 12 PSC patients, evidenced differences in mucosa-adherent microbiota composition with respect to UC (11 individuals) or non-inflammatory patients (nine individuals) [137]. Microbiota profiling showed lower abundance of uncultured Clostridiales II, members of Firmicutes, in PSC patients compared to both UC and control patients, and a significant reduction of microbiota diversity and abundance in PSC patients with respect to healthy group [137]. A correlation between Firmicutes and the health state has been previously reported [138]. The mucosa-adherent microbiota profiling has been performed from ileocecal biopsies collected by 20 PSC patients with (19 individuals) or without IBD (one patient only), 15 IBD-affected individuals and nine healthy controls [139]. Despite the results showed no differences in microbiota composition throughout the ileum or cecum mucosa, an increased abundance of Blautia and Barnesiellaceae genera and a shift under Clostridiales order have been reported in PSC patients [139]. These data are in contrast with a previous study that showed a reduction of uncultured Clostridiales in PSC patients [137].
6. Conclusions
Intense research over recent years has unveiled many aspects of the pathophysiology of chronic biliary diseases. Cholangiocytes have shifted from passive bystander of cholangiopathies to the central cells orchestrating the complex cellular and molecular interactions occurring in response to damage. Gut-derived bacterial products elicit the activation of strong immune responses in cholangiocytes, which, in genetically susceptible individuals, may contribute to the development or sustain biliary injury. Moreover, disturbances of the intestinal barrier function, with or without intestinal inflammation, may favor the uncontrolled permeation of bacterial products form the gut to the portal circulation, thus perpetuating biliary inflammation. In this context, a specific microbiota composition in patients affected by cholangiopathies is emerging in recent studies and could in part be responsible for cholangiocyte activation. A deeper understanding of the complex relations between biliary inflammation, cholangiocytes response to injury and the role of gut-derived antigens in the pathogenesis of cholangiopathies may prove essential to devise novel effective therapeutic strategies.
Author Contributions
D.M.G., writing–original draft preparation and preparing figures, C.P., writing–original draft preparation, L.M., writing–original draft preparation, A.B. and M.M., writing–review and editing, and supervision.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Cheung, A.C.; Lorenzo Pisarello, M.J.; LaRusso, N.F. Pathobiology of biliary epithelia. Biochim. Biophys. Acta 2018, 1864, 1220–1231. [Google Scholar] [CrossRef] [PubMed]
- Yoo, K.S.; Lim, W.T.; Choi, H.S. Biology of cholangiocytes: From bench to bedside. Gut Liver 2016, 10, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Maroni, L.; Haibo, B.; Ray, D.; Zhou, T.; Wan, Y.; Meng, F.; Marzioni, M.; Alpini, G. Functional and Structural Features of Cholangiocytes in Health and Disease. Cell Mol. Gastroenterol. Hepatol. 2015, 1, 368–380. [Google Scholar] [CrossRef] [PubMed]
- Franchitto, A.; Onori, P.; Renzi, A.; Carpino, G.; Mancinelli, R.; Alvaro, D.; Gaudio, E. Recent advances on the mechanisms regulating cholangiocyte proliferation and the significance of the neuroendocrine regulation of cholangiocyte pathophysiology. Ann. Transl. Med. 2013, 1, 27. [Google Scholar] [CrossRef] [PubMed]
- Sell, S. Heterogeneity and plasticity of hepatocyte lineage cells. Hepatology 2001, 33, 738–750. [Google Scholar] [CrossRef] [PubMed]
- Sclair, S.N.; Little, E.; Levy, C. Current Concepts in Primary Biliary Cirrhosis and Primary Sclerosing Cholangitis. Clin. Transl. Gastroenterol. 2015, 6, e109. [Google Scholar] [CrossRef] [PubMed]
- Marchioni Beery, R.M.; Vaziri, H.; Forouhar, F. Primary Biliary Cirrhosis and Primary Sclerosing Cholangitis: A Review Featuring a Women’s Health Perspective. J. Clin. Transl. Hepatol. 2014, 24, 266–284. [Google Scholar] [CrossRef]
- Goldstein, J.; Levy, C. Novel and emerging therapies for cholestatic liver diseases. Liver Int. 2018, 38, 1520–1535. [Google Scholar] [CrossRef] [PubMed]
- Fukata, M.; Vamadevan, A.S.; Abreu, M.T. Toll-like receptors (TLRs) and Nod-like receptors (NLRs) in inflammatory disorders. Semin. Immunol. 2009, 21, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Lesage, G.; Glaser, S.S.; Gubba, S.; Robertson, W.E.; Phinizy, J.L.; Lasater, J.; Rodgers, R.E.; Alpini, G. Regrowth of the rat biliary tree after 70% partial hepatectomy is coupled to increased secretin-induced ductal secretion. Gastroenterology 1996, 111, 1633–1644. [Google Scholar] [CrossRef]
- Lazaridis, K.N.; LaRusso, N.F. The cholangiopathies. Mayo. Clin. Proc. 2015, 90, 791–800. [Google Scholar] [CrossRef] [PubMed]
- Desmet, V.J. Ductal plates in hepatic ductular reactions. Hypothesis and implications. I. Types of ductular reaction reconsidered. Virchows. Archiv. 2011, 458, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Carpino, G.; Cardinale, V.; Onori, P.; Franchitto, A.; Berloco, P.B.; Rossi, M.; Wang, Y.; Semeraro, R.; Anceschi, M.; Brunelli, R.; et al. Biliary tree stem/progenitor cells in glands of extrahepatic and intraheptic bile ducts: An anatomical in situ study yielding evidence of maturational lineages. J. Anat. 2012, 220, 186–199. [Google Scholar] [CrossRef] [PubMed]
- Alvaro, D.; Alpini, G.; Onori, P.; Perego, L.; Baroni, G.S.; Franchitto, A.; Baiocchi, L.; Glaser, S.S.; Le Sage, G.; Folli, F.; et al. Estrogens stimulate proliferation of intrahepatic biliary epithelium in rats. Gastroenterology 2000, 119, 1681–1691. [Google Scholar] [CrossRef] [PubMed]
- Alvaro, D.; Mancino, M.G.; Glaser, S.; Gaudio, E.; Marzioni, M.; Francis, H.; Alpini, G. Proliferating Cholangiocytes: A Neuroendocrine Compartment in the Diseased Liver. Gastroenterology 2007, 132, 415–431. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, S.P.; Tabibian, J.H.; Splinter, P.L.; LaRusso, N.F. The dynamic biliary epithelia: Molecules, pathways, and disease. J. Hepatol. 2013, 58, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Alpini, G.; Ulrich, C.D.; Phillips, J.O.; Pham, L.D.; Miller, L.J.; LaRusso, N.F. Upregulation of secretin receptor gene expression in rat cholangiocytes after bile duct ligation. Am. J. Physiol. 1994, 266, G922–G928. [Google Scholar] [CrossRef] [PubMed]
- Lenzen, R.; Alpini, G.; Tavoloni, N. Secretin stimulates bile ductular secretory activity through the cAMP system. Am. J. Physiol. 1992, 263, G527–G532. [Google Scholar] [CrossRef] [PubMed]
- Kato, A.; Gores, G.J.; LaRusso, N.F. Secretin stimulates exocytosis in isolated bile duct epithelial cells by a cyclic AMP-mediated mechanism. J. Biol. Chem. 1992, 267, 15523–15529. [Google Scholar] [PubMed]
- Minagawa, N.; Nagata, J.; Shibao, K.; Masyuk, A.I.; Gomes, D.A.; Rodrigues, M.A.; Lesage, G.; Akiba, Y.; Kaunitz, J.D.; Ehrlich, B.E.; et al. Cyclic AMP Regulates Bicarbonate Secretion in Cholangiocytes Through Release of ATP Into Bile. Gastroenterology 2007, 133, 1592–1602. [Google Scholar] [CrossRef] [PubMed]
- Francis, H.; Glaser, S.; Ueno, Y.; Lesage, G.; Marucci, L.; Benedetti, A.; Taffetani, S.; Marzioni, M.; Alvaro, D.; Venter, J.; et al. CAMP stimulates the secretory and proliferative capacity of the rat intrahepatic biliary epithelium through changes in the PKA/Src/MEK/ERK1/2 pathway. J. Hepatol. 2004, 41, 528–537. [Google Scholar] [CrossRef] [PubMed]
- Mancinelli, R.; Onori, P.; Gaudio, E.; DeMorrow, S.; Franchitto, A.; Francis, H.; Glaser, S.; Carpino, G.; Venter, J.; Alvaro, D.; et al. Follicle-stimulating hormone increases cholangiocyte proliferation by an autocrine mechanism via cAMP-dependent phosphorylation of ERK1/2 and Elk-1. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 297, G11–G26. [Google Scholar] [CrossRef] [PubMed]
- Alpini, G.; Glaser, S.; Robertson, W.; Rodgers, R.E.; Phinizy, J.L.; Lasater, J.; LeSage, G.D. Large but not small intrahepatic bile ducts are involved in secretin- regulated ductal bile secretion. Am. J. Physiol. 1997, 272, G1064–G1074. [Google Scholar] [CrossRef] [PubMed]
- Francis, H.L.; Demorrow, S.; Franchitto, A.; Venter, J.K.; Mancinelli, R.A.; White, M.A.; Meng, F.; Ueno, Y.; Carpino, G.; Renzi, A.; et al. Histamine stimulates the proliferation of small and large cholangiocytes by activation of both IP3/Ca2+and cAMP-dependent signaling mechanisms. Lab Investig 2012, 92, 282–294. [Google Scholar] [CrossRef] [PubMed]
- Hogan, P.G.; Chen, L.; Nardone, J.; Rao, A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 2003, 17, 2205–2232. [Google Scholar] [CrossRef] [PubMed]
- Swulius, M.T.; Waxham, M.N. Ca2+/Calmodulin-dependent Protein Kinases. Cell Mol. Life Sci. 2013, 65, 2637–2657. [Google Scholar] [CrossRef] [PubMed]
- Francis, H.; Glaser, S.; DeMorrow, S.; Gaudio, E.; Ueno, Y.; Venter, J.; Dostal, D.; Onori, P.; Franchitto, A.; Marzioni, M.; et al. Small mouse cholangiocytes proliferate in response to H1 histamine receptor stimulation by activation of the IP3/CaMK I/CREB pathway. AJP Cell Physiol. 2008, 295, C499–G513. [Google Scholar] [CrossRef] [PubMed]
- Alpini, G.; Franchitto, A.; Demorrow, S.; Onori, P.; Gaudio, E.; Wise, C.; Francis, H.; Venter, J.; Kopriva, S.; Mancinelli, R.; et al. Activation of alpha(1)-adrenergic receptors stimulate the growth of small mouse cholangiocytes via calcium-dependent activation of nuclear factor of activated T cells 2 and specificity protein 1. Hepatology 2011, 53, 628–639. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Kang, X.; Cao, S.; Cheng, H.; Wang, D.; Geng, J. Calcineurin/NFATc1 pathway contributes to cell proliferation in hepatocellular carcinoma. Dig. Dis. Sci. 2012, 57, 3184–3188. [Google Scholar] [CrossRef] [PubMed]
- Shibao, K.; Hirata, K.; Robert, M.E.; Nathanson, M.H. Loss of inositol 1,4,5-trisphosphate receptors from bile duct epithelia is a common event in cholestasis. Gastroenterology 2003, 125, 1175–1187. [Google Scholar] [CrossRef]
- Feranchak, A.P.; Doctor, R.B.; Troetsch, M.; Brookman, K.; Johnson, S.M.; Fitz, J.G. Calcium-dependent regulation of secretion in biliary epithelial cells: The role of apamin-sensitive SK channels. Gastroenterology 2004, 127, 903–913. [Google Scholar] [CrossRef] [PubMed]
- Guerrier, M.; Attili, F.; Alpini, G.; Glaser, S. Prolonged administration of secretin to normal rats increases biliary proliferation and secretin-induced ductal secretory activity. Hepatobiliary Surg. Nutr. 2014, 3, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Polak, J.M.; Coulling, I.; Bloom, S.; Pearse, A.G. Immunofluorescent localization of secretin and enteroglucagon in human intestinal mucosa. Scand. J. Gastroenterol. 1971, 6, 739–744. [Google Scholar] [CrossRef] [PubMed]
- Glaser, S.; Meng, F.; Han, Y.; Onori, P.; Chow, B.K.; Francis, H.; Venter, J.; McDaniel, K.; Marzioni, M.; Invernizzi, P.; et al. Secretin stimulates biliary cell proliferation by regulating expression of microRNA 125b and MicroRNA let7a in mice. Gastroenterology 2014, 146, 1795–1808. [Google Scholar] [CrossRef] [PubMed]
- Glaser, S.; Lam, I.P.; Franchitto, A.; Gaudio, E.; Onori, P.; Chow, B.K.; Wise, C.; Kopriva, S.; Venter, J.; White, M.; et al. Knockout of secretin receptor reduces large cholangiocyte hyperplasia in mice with extrahepatic cholestasis induced by bile duct ligation. Hepatology 2010, 52, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Ray, D.; Han, Y.; Franchitto, A.; Demorrow, S.; Meng, F.; Venter, J.; McMillin, M.; Kennedy, L.; Francis, H.; Onori, P.; et al. Gonadotropin-releasing hormone stimulates biliary proliferation by paracrine/autocrine mechanisms. Am. J. Pathol. 2015, 185, 1061–1072. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, M.; Park, M.K. GnRH as a cell proliferation regulator: mechanism of action and evolutionary implications. Zoolog. Sci. 2004, 21, 1005–1013. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, D.P.; Dangott, L.J.; Timothy Lightfoot, J. Lessons learned from vivo-morpholinos: How to avoid vivo-morpholino toxicity. Biotechniques 2014, 56, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Rameshwar, P.; Gascon, P.; Ganea, D. Immunoregulatory effects of neuropeptides. Stimulation of interleukin-2 production by substance P. J. Neuroimmunol. 1992, 37, 65–74. [Google Scholar] [CrossRef]
- Trivedi, M.; Bergasa, N.V. Serum concentrations of substance P in cholestasis. Ann. Hepatol. 2010, 9, 177–180. [Google Scholar] [PubMed]
- Meng, F.; Demorrow, S.; Venter, J.; Frampton, G.; Han, Y.; Francis, H.; Standeford, H.; Avila, S.; McDaniel, K.; McMillin, M.; et al. Overexpression of membrane metalloendopeptidase inhibits substance P stimulation of cholangiocarcinoma growth. Am. J. Physiol. Gastrointest Liver Physiol. 2014, 306, G759–G768. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Meng, F.; Wu, N.; Zhou, T.; Venter, J.; Francis, H.; Kennedy, L.; Glaser, T.; Bernuzzi, F.; Invernizzi, P.; et al. Substance P increases liver fibrosis by differential changes in senescence of cholangiocytes and hepatic stellate cells. Hepatology 2017, 66, 528–541. [Google Scholar] [CrossRef] [PubMed]
- Glaser, S.; Gaudio, E.; Renzi, A.; Mancinelli, R.; Ueno, Y.; Venter, J.; White, M.; Kopriva, S.; Chiasson, V.; DeMorrow, S.; et al. Knockout of the neurokinin-1 receptor reduces cholangiocyte proliferation in bile duct-ligated mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G297–G305. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, L.L.; Hargrove, L.A.; Graf, A.B.; Francis, T.C.; Hodges, K.M.; Nguyen, Q.P.; Ueno, Y.; Greene, J.F.; Meng, F.; Huynh, V.D.; et al. Inhibition of mast cell-derived histamine secretion by cromolyn sodium treatment decreases biliary hyperplasia in cholestatic rodents. Lab Investig. 2014, 94, 1406–1418. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Yamazaki, K.; Suzuki, K.; Sato, S. Increased portal tract infiltration of mast cells and eosinophils in primary biliary cirrhosis. Am. J. Gastroenterol 1997, 92, 2245–2249. [Google Scholar] [PubMed]
- Bubenik, G.A. Gastrointestinal melatonin: Localization, function, and clinical relevance. Dig. Dis. Sci. 2002, 47, 2336–2348. [Google Scholar] [CrossRef] [PubMed]
- Tahan, G.; Akin, H.; Aydogan, F.; Ramadan, S.S.; Yapicier, O.; Tarcin, O.; Uzun, H.; Tahan, V.; Zengin, K. Melatonin ameliorates liver fibrosis induced by bile-duct ligation in rats. Can J. Surg. 2010, 53, 313–318. [Google Scholar] [PubMed]
- McMillin, M.; Frampton, G.; Grant, S.; DeMorrow, S. The Neuropeptide Galanin Is Up-Regulated during Cholestasis and Contributes to Cholangiocyte Proliferation. Am. J. Pathol. 2017, 187, 819–830. [Google Scholar] [CrossRef] [PubMed]
- Gaudio, E.; Barbaro, B.; Alvaro, D.; Glaser, S.; Francis, H.; Ueno, Y.; Meininger, C.J.; Franchitto, A.; Onori, P.; Marzioni, M.; et al. Vascular Endothelial Growth Factor Stimulates Rat Cholangiocyte Proliferation Via an Autocrine Mechanism. Gastroenterology 2006, 130, 1270–1282. [Google Scholar] [CrossRef] [PubMed]
- Marzioni, M.; Saccomanno, S.; Candelaresi, C.; Rychlicki, C.; Agostinelli, L.; Shanmukhappa, K.; Trozzi, L.; Pierantonelli, I.; De Minicis, S.; Benedetti, A. Pancreatic Duodenal Homeobox-1 de novo expression drives cholangiocyte neuroendocrine-like transdifferentiation. J Hepatol. 2010, 53, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Garrido, P.; Marzioni, M.; Hijona, E.; Bujanda, L.; Banales, J.M. MicroRNAs in Liver Diseases. In MicroRNAs in Medicine, 1st ed.; Charles, H.L., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; pp. 509–522. ISBN 9781118300398. [Google Scholar]
- Jiang, X.; Karlsen, T.H. Genetics of primary sclerosing cholangitis and pathophysiological implications. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 279–295. [Google Scholar] [CrossRef] [PubMed]
- Rausch, P.; Rehman, A.; Künzel, S.; Häsler, R.; Ott, S.J.; Schreiber, S.; Rosenstiel, P.; Franke, A.; Baines, F.B. Colonic mucosa-associated microbiota is influenced by an interaction of Crohn disease and FUT2 (Secretor) genotype. Proc. Natl. Acad. Sci. USA 2011, 108, 19030–19035. [Google Scholar] [CrossRef] [PubMed]
- Marzioni, M.; Glaser, S.S.; Francis, H.; Phinizy, J.L.; LeSage, G.; Alpini, G. Functional heterogeneity of cholangiocytes. Semin. Liver Dis. 2002, 22, 227–240. [Google Scholar] [CrossRef] [PubMed]
- Kanno, N.; LeSage, G.; Glaser, S.; Alvaro, D.; Alpini, G. Functional Heterogeneity of the Intrahepatic Biliary Epithelium. Hepatology 2016, 31, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.K.; Wen, H.; Ting, J.P. The Inflammasome NLRs in Immunity, Inflammation, and Associated Diseases. Annu. Rev. Immunol. 2011, 29, 707–735. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Kaisho, T.; Akira, S. Toll-like receptors. Annu. Rev. Immunol. 2003, 21, 335–376. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, T.; Komori, A.; Nakamura, M.; Takii, Y.; Kamihira, T.; Shimoda, S.; Mori, T.; Fujiwara, S.; Koyabu, M.; Taniguchi, K.; et al. Human intrahepatic biliary epithelial cells function in innate immunity by producing IL-6 and IL-8 via the TLR4-NF-κB and -MAPK signaling pathways. Liver Int. 2006, 26, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.M.; O’Hara, S.P.; LaRusso, N.F. The immunobiology of cholangiocytes. Immunol. Cell Biol. 2008, 86, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Takeda, K. Toll-like receptor signaling. Nature 2004, 4, 88. [Google Scholar] [CrossRef] [PubMed]
- Tschopp, J.; Schroder, K. NLRP3 inflammasome activation: The convergence of multiple signaling pathways on ROS production? Nat. Rev. Immunol. 2010, 10, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Tschopp, J. The Inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Monack, D.M. Inflammasome adaptors and sensors: Intracellular regulators of infection and inflammation. Nat. Rev. Immunol. 2007, 7, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Maroni, L.; Agostinelli, L.; Saccomanno, S.; Pinto, C.; Giordano, D.M.; Rychlicki, C.; De Minicis, S.; Trozzi, L.; Banales, J.M.; Melum, E.; et al. Nlrp3 Activation Induces Il-18 Synthesis and Affects the Epithelial Barrier Function in Reactive Cholangiocytes. Am. J. Pathol. 2017, 187, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.H.; Afford, S.C. The role of cholangiocytes in the development of chronic inflammatory liver disease. Front. Biosci. 2002, 7, 276–285. [Google Scholar]
- Harada, K.; Shimoda, S.; Ikeda, H.; Chiba, M.; Hsu, M.; Sato, Y.; Kobayashi, M.; Ren, X.S.; Ohta, H.; Kasashima, S.; et al. Significance of periductal Langerhans cells and biliary epithelial cell-derived macrophage inflammatory protein-3α in the pathogenesis of primary biliary cirrhosis. Liver Int. 2011, 31, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Imai, T.; Hieshima, K.; Haskell, C.; Baba, M.; Nagira, M.; Nishimura, M.; Kakizaki, M.; Takagi, S.; Nomiyama, H.; Schall, T.J.; et al. Identification and molecular characterization of fractalkine receptor CX3CR1, which mediates both leukocyte migration and adhesion. Cell 1997, 91, 521–530. [Google Scholar] [CrossRef]
- Ezure, T.; Sakamoto, T.; Tsuji, H.; Lunz, J.G.; Murase, N.; Fung, J.J.; Demetris, A.J. The development and compensation of biliary cirrhosis in interleukin-6-deficient mice. Am. J. Pathol. 2000, 156, 1627–1639. [Google Scholar] [CrossRef]
- Wu, C.T.; Davis, P.A.; Luketic, V.A.; Gershwin, M.E. A review of the physiological and immunological functions of biliary epithelial cells: Targets for primary biliary cirrhosis, primary sclerosing cholangitis and drug-induced ductopenias. Clin. Dev. Immunol. 2004, 11, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Ayres, R.C.; Neuberger, J.M.; Shaw, J.; Joplin, R.; Adams, D.H. Intercellular adhesion molecule-1 and MHC antigens on human intrahepatic bile duct cells: effect of pro-inflammatory cytokines. Gut 1993, 34, 1245–1249. [Google Scholar] [CrossRef] [PubMed]
- Cruickshank, S.M.; Southgate, J.; Selby, P.J.; Trejdosiewicz, L.K. Expression and cytokine regulation of immune recognition elements by normal human biliary epithelial and established liver cell lines in vitro. J. Hepatol. 1998, 29, 550–558. [Google Scholar] [CrossRef]
- Ramm, G.A.; Shepherd, R.W.; Hoskins, A.C.; Greco, S.A.; Ney, A.D.; Pereira, T.N.; Bridle, K.R.; Doecke, J.D.; Meikle, P.J.; Turlin, B.; et al. Fibrogenesis in pediatric cholestatic liver disease: Role of taurocholate and hepatocyte-derived monocyte chemotaxis protein-1 in hepatic stellate cell recruitment. Hepatology 2009, 49, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Harada, K.; Chiba, M.; Okamura, A.; Hsu, M.; Sato, Y.; Igarashi, S.; Ren, X.S.; Ikeda, H.; Ohta, H.; Kasashima, S.; et al. Monocyte chemoattractant protein-1 derived from biliary innate immunity contributes to hepatic fibrogenesis. J. Clin. Pathol. 2011, 64, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Marra, F.; DeFranco, R.; Grappone, C.; Milani, S.; Pastacaldi, S.; Pinzani, M.; Romanelli, R.G.; Laffi, G.; Gentilini, P. Increased expression of monocyte chemotactic protein-1 during active hepatic fibrogenesis: correlation with monocyte infiltration. Am. J. Pathol. 1998, 152, 423–430. [Google Scholar] [PubMed]
- Milani, S.; Herbst, H.; Schuppan, D.; Stein, H.; Surrenti, C. Transforming growth factors beta 1 and beta 2 are differentially expressed in fibrotic liver disease. Am. J. Pathol. 1991, 139, 1221–1229. [Google Scholar] [PubMed]
- Li, Z.; Dranoff, J.A.; Chan, E.P.; Uemura, M.; Sévigny, J.; Wells, R.G. Transforming growth factor-beta and substrate stiffness regulate portal fibroblast activation in culture. Hepatology 2007, 46, 1246–1256. [Google Scholar] [CrossRef] [PubMed]
- Luo, B.; Tang, L.; Wang, Z.; Zhang, J.; Ling, Y.; Feng, W.; Sun, J.Z.; Stockard, C.R.; Frost, A.R.; Chen, Y.F.; et al. Cholangiocyte endothelin 1 and transforming growth factor β1 production in rat experimental hepatopulmonary syndrome. Gastroenterology 2005, 129, 682–695. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Gaudio, E.; Onori, P.; Wise, C.; Alpini, G.; Glaser, S.S. Mechanisms of Biliary Damage. J. Cell Death 2010, 3, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Ueno, Y.; Francis, H.; Glaser, S.; Demorrow, S.; Venter, J.; Benedetti, A.; Fava, G.; Marzioni, M.; Alpini, G. Taurocholic acid feeding prevents tumor necrosis factor-alpha-induced damage of cholangiocytes by a PI3K-mediated pathway. Exp. Biol. Med. (Maywood) 2007, 232, 942–949. [Google Scholar] [PubMed]
- Shivakumar, P.; Mizuochi, T.; Mourya, R.; Gutta, S.; Yang, L.; Luo, Z.; Bezerra, J.A. Preferential TNF α signaling via TNFR2 regulates epithelial injury and duct obstruction in experimental biliary atresia. JCI Insight. 2017, 2, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Omenetti, A.; Syn, W.K.; Jung, Y.; Francis, H.; Porrello, A.; Witek, R.P.; Choi, S.S.; Yang, L.; Mayo, M.J.; Gershwin, M.E.; et al. Repair-related activation of hedgehog signaling promotes cholangiocyte chemokine production. Hepatology 2009, 50, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Omenetti, A.; Popov, Y.; Jung, Y.; Choi, S.S.; Witek, R.P.; Yang, L.; Brown, K.D.; Schuppan, D.; Diehl, A.M. The hedgehog pathway regulates remodelling responses to biliary obstruction in rats. Gut 2008, 57, 1275–1282. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.; McCall, S.J.; Li, Y.X.; Diehl, A.M. Bile ductules and stromal cells express hedgehog ligands and/or hedgehog target genes in primary biliary cirrhosis. Hepatology 2007, 45, 1091–1096. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wang, Y.; Mao, H.; Fleig, S.; Omenetti, A.; Brown, K.D.; Sicklick, J.K.; Li, Y.X.; Diehl, A.M. Sonic hedgehog is an autocrine viability factor for myofibroblastic hepatic stellate cells. J. Hepatol. 2008, 48, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Omenetti, A.; Porrello, A.; Jung, Y.; Yang, L.; Popov, Y.; Choi, S.S.; Witek, R.P.; Alpini, G.; Venter, J.; Vandongen, H.M.; et al. Hedgehog signaling regulates epithelial-mesenchymal transition during biliary fibrosis in rodents and humans. J. Clin. Investig. 2008, 118, 3331–3342. [Google Scholar] [CrossRef] [PubMed]
- Omenetti, A.; Yang, L.; Li, Y.X.; McCall, S.J.; Jung, Y.; Sicklick, J.K.; Huang, J.; Choi, S.; Suzuki, A.; Diehl, A.M. Hedgehog-mediated mesenchymal-epithelial interactions modulate hepatic response to bile duct ligation. Lab Investig. 2007, 87, 499–514. [Google Scholar] [CrossRef] [PubMed]
- Tabibian, J.H.; O’Hara, S.P.; Splinter, P.L.; Trussoni, C.E.; Larusso, N.F. Cholangiocyte senescence by way of N-Ras activation is a characteristic of primary sclerosing cholangitis. Hepatology 2014, 59, 2263–2275. [Google Scholar] [CrossRef] [PubMed]
- Fickert, P.; Fuchsbichler, A.; Wagner, M.; Zollner, G.; Kaser, A.; Tilg, H.; Krause, R.; Lammert, F.; Langner, C.; Zatloukal, K.; et al. Regurgitation of bile acids from leaky bile ducts causes sclerosing cholangitis in Mdr2 (Abcb4) knockout mice. Gastroenterology 2004, 127, 261–274. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, S.P.; Splinter, P.L.; Trussoni, C.E.; Gajdos, G.B.; Lineswala, P.N.; LaRusso, N.F. Cholangiocyte N-Ras protein mediates lipopolysaccharide-induced interleukin 6 secretion and proliferation. J. Biol. Chem. 2011, 286, 30352–30360. [Google Scholar] [CrossRef] [PubMed]
- Aravinthan, A.D.; Alexander, G.J.M. Senescence in chronic liver disease: Is the future in aging? J. Hepatol. 2016, 65, 825–834. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Meng, F.; Venter, J.; Giang, T.; Glaser, S.; Alpini, G. The role of the secretin/secretin receptor axis in inflammatory cholangiocyte communication via extracellular vesicles. Sci. Rep. 2018, 8, 11238. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.Y.; Costerton, J.W.; Shaffer, E.A. Defense system in the biliary tract against bacterial infection. Dig. Dis. Sci. 1992, 37, 689–696. [Google Scholar] [CrossRef] [PubMed]
- Harada, K.; Nakanuma, Y. Biliary innate immunity: Function and modulation. Mediat. Inflamm. 2010, 2010, 373878. [Google Scholar] [CrossRef] [PubMed]
- Harada, K.; Ohba, K.; Ozaki, S.; Isse, K.; Hirayama, T.; Wada, A.; Nakanuma, Y. Peptide antibiotic human beta-defensin-1 and -2 contribute to antimicrobial defense of the intrahepatic biliary tree. Hepatology 2004, 40, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Tabibian, J.H.; Varghese, C.; LaRusso, N.F.; O’Hara, S.P. The enteric microbiome in hepatobiliary health and disease. Liver Int. 2016, 36, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Kummen, M.; Vesterhus, M.; Trøseid, M.; Moum, B.; Svardal, A.; Boberg, K.M.; Aukrust, P.; Karlsen, T.H.; Berge, R.K.; Hov, J.R. Elevated trimethylamine-N-oxide (TMAO) is associated with poor prognosis in primary sclerosing cholangitis patients with normal liver function. United Eur. Gastroenterol. J. 2017, 5, 532–541. [Google Scholar] [CrossRef] [PubMed]
- Llorente, C.; Schnabl, B. The Gut Microbiota and Liver Disease. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Cani, P.D.; Mayer, E.A. Gut microbiome and liver diseases. Gut 2016, 65, 2035–2344. [Google Scholar] [CrossRef] [PubMed]
- Harada, K.; Isse, K.; Sato, Y.; Ozaki, S.; Nakanuma, Y. Endotoxin tolerance in human intrahepatic biliary epithelial cells is induced by upregulation of IRAK-M. Liver Int. 2006, 26, 935–942. [Google Scholar] [CrossRef] [PubMed]
- Ngu, J.H.; Gearry, R.B.; Wright, A.J.; Stedman, C.A. Inflammatory bowel disease is associated with poor outcomes of patients with primary sclerosing cholangitis. Clin. Gastroenterol. Hepatol. 2011, 9, 1092–1097. [Google Scholar] [CrossRef] [PubMed]
- Tabibian, J.H.; Talwalkar, J.A.; Lindor, K.D. Role of the microbiota and antibiotics in primary sclerosing cholangitis. Biomed. Res. Int. 2013, 2013, 389537. [Google Scholar] [CrossRef] [PubMed]
- Pohl, J.; Ring, A.; Stremmel, W.; Stiehl, A. The role of dominant stenoses in bacterial infections of bile ducts in primary sclerosing cholangitis. Eur. J. Gastroenterol. Hepatol. 2006, 18, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, G.; Gotthardt, D.; Klöters-Plachky, P.; Kulaksiz, H.; Rost, D.; Stiehl, A. Influence of dominant bile duct stenoses and biliary infections on outcome in primary sclerosing cholangitis. J. Hepatol. 2009, 51, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Hiramatsu, K.; Harada, K.; Tsuneyama, K.; Sasaki, M.; Fujita, S.; Hashimoto, T.; Kaneko, S.; Kobayashi, K.; Nakanuma, Y. Amplification and sequence analysis of partial bacterial 16S ribosomal RNA gene in gallbladder bile from patients with primary biliary cirrhosis. J. Hepatol. 2000, 33, 9–18. [Google Scholar] [CrossRef]
- Alabraba, E.; Nightingale, P.; Gunson, B.; Hubscher, S.; Olliff, S.; Mirza, D.; Neuberger, J. A re-evaluation of the risk factors for the recurrence of primary sclerosing cholangitis in liver allografts. Liver Transpl. 2009, 15, 330–340. [Google Scholar] [CrossRef] [PubMed]
- Vera, A.; Moledina, S.; Gunson, B.; Hubscher, S.; Mirza, D.; Olliff, S.; Neuberger, J. Risk factors for recurrence of primary sclerosing cholangitis of liver allograft. Lancet 2002, 360, 1943–1944. [Google Scholar] [CrossRef]
- Tabibian, J.H.; Weeding, E.; Jorgensen, R.A.; Petz, J.L.; Keach, J.C.; Talwalkar, J.A.; Lindor, K.D. Randomised clinical trial: vancomycin or metronidazole in patients with primary sclerosing cholangitis—A pilot study. Aliment. Pharmacol. Ther. 2013, 37, 604–612. [Google Scholar] [CrossRef] [PubMed]
- Haruta, I.; Hashimoto, E.; Kato, Y.; Kikuchi, K.; Kato, H.; Yagi, J.; Uchiyama, T.; Kobayash, M.; Shiratori, K. Lipoteichoic acid may affect the pathogenesis of bile duct damage in primary biliary cirrhosis. Autoimmunity 2006, 39, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Haruta, I.; Kikuchi, K.; Hashimoto, E.; Nakamura, M.; Miyakawa, H.; Hirota, K.; Shibata, N.; Kato, H.; Arimura, Y.; Kato, Y.; et al. Long-term bacterial exposure can trigger nonsuppurative destructive cholangitis associated with multifocal epithelial inflammation. Lab Investig. 2010, 90, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Karlsen, T.H.; Franke, A.; Melum, E.; Kaser, A.; Hov, J.R.; Balschun, T.; Lie, B.A.; Bergquist, A.; Schramm, C.; Weismüller, T.J.; et al. Genome-Wide Association Analysis in Primary Sclerosing Cholangitis. Gastroenterology 2010, 138, 1102–1111. [Google Scholar] [CrossRef] [PubMed]
- Melum, E.; Franke, A.; Schramm, C.; Weismuller, T.J.; Gotthardt, D.N.; Offner, F.A.; Juran, B.D.; Laerdahl, J.K.; Labi, V.; Björnsson, E.; et al. Genome-wide association analysis in primary sclerosing cholangitis identifies two non-HLA susceptibility loci. Nat. Genet. 2011, 43, 17–19. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Z.; Hov, J.R.; Folseraas, T.; Ellinghaus, E.; Rushbrook, S.M.; Doncheva, N.T.; Andreassen, O.A.; Weersma, R.K.; Weismüller, T.J.; Eksteen, B.; et al. Dense genotyping of immune-related disease regions identifies nine new risk loci for primary sclerosing cholangitis. Nat. Genet. 2013, 45, 670–675. [Google Scholar] [CrossRef] [PubMed]
- Maroni, L.; van de Graaf, S.F.; Hohenester, S.D.; Oude Elferink, R.P.; Beuers, U. Fucosyltransferase 2: A Genetic Risk Factor for Primary Sclerosing Cholangitis and Crohn’s Disease—A Comprehensive Review. Clin. Rev. Allergy Immunol. 2015, 48, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Wannhoff, A.; Rupp, C.; Friedrich, K.; Brune, M.; Knierim, J.; Flechtenmacher, C.; Sauer, P.; Stremmel, W.; Hov, J.R.; Schirmacher, P.; et al. Inflammation but Not Biliary Obstruction Is Associated with Carbohydrate Antigen 19-9 Levels in Patients With Primary Sclerosing Cholangitis. Clin. Gastroenterol. Hepatol 2015, 13, 2372–2379. [Google Scholar] [CrossRef] [PubMed]
- Maroni, L.; Hohenester, S.D.; van de Graaf, S.F.; Tolenaars, D.; van Lienden, K.; Verheij, J.; Marzioni, M.; Karlsen, T.H.; Oude Elferink, R.P.J.; Beuers, U. Knockout of the primary sclerosing cholangitis-risk gene Fut2 causes liver disease in mice. Hepatology 2017, 66, 542–554. [Google Scholar] [CrossRef] [PubMed]
- Tabibian, J.H.; O’Hara, S.P.; Trussoni, C.E.; Tietz, P.S.; Splinter, P.L.; Mounajjed, T.; Hagey, L.R.; LaRusso, N.F. Absence of the intestinal microbiota exacerbates hepatobiliary disease in a murine model of primary sclerosing cholangitis. Hepatology 2016, 63, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Schrumpf, E.; Kummen, M.; Valestrand, L.; Greiner, T.U.; Holm, K.; Arulampalam, V.; Reims, H.M.; Baines, J.; Bäckhed, F.; Karlsen, T.H.; et al. The gut microbiota contributes to a mouse model of spontaneous bile duct inflammation. J. Hepatol. 2017, 66, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Lichtman, S.N.; Okoruwa, E.E.; Keku, J.; Schwab, J.H.; Sartor, R.B. Degradation of endogenous bacterial cell wall polymers by the muralytic enzyme mutanolysin prevents hepatobiliary injury in genetically susceptible rats with experimental intestinal bacterial overgrowth. J. Clin. Investig. 1992, 90, 1313–1322. [Google Scholar] [CrossRef] [PubMed]
- Lichtman, S.N.; Wang, J.; Clark, R.L. A microcholangiographic study of liver disease models in rats. Acad. Radiol. 1995, 2, 515–521. [Google Scholar] [CrossRef]
- Yamada, S.; Ishii, M.; Liang, L.S.; Yamamoto, T.; Toyota, T. Small duct cholangitis induced by N-formyl l-methionine l-leucine l-tyrosine in rats. J. Gastroenterol. 1994, 29, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Chassaing, B.; Aitken, J.D.; Malleshappa, M.; Vijay-Kumar, M. Dextran sulfate sodium (DSS)-induced colitis in mice. Curr. Protoc. Immunol. 2014, 104, 15.25.1–15.25.14. [Google Scholar] [CrossRef] [PubMed]
- Eichele, D.D.; Kharbanda, K.K. Dextran sodium sulfate colitis murine model: An indispensable tool for advancing our understanding of inflammatory bowel diseases pathogenesis. World J. Gastroenterol. 2017, 23, 6016–6029. [Google Scholar] [CrossRef] [PubMed]
- Masubuchi, Y.; Horie, T. Endotoxin-mediated disturbance of hepatic cytochrome P450 function and development of endotoxin tolerance in the rat model of dextran sulfate sodium-induced experimental colitis. Drug Metab. Dispos. 2004, 32, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Fiorotto, R.; Scirpo, R.; Trauner, M.; Fabris, L.; Hoque, R.; Spirli, C.; Strazzabosco, M. Loss of CFTR affects biliary epithelium innate immunity and causes TLR4NF-κB-mediated inflammatory response in mice. Gastroenterology 2011, 141, 1498–1508.e5. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, J.S. The relationship between the gut microbiota and liver disease. Gastroenterol. Hepatol. 2015, 11, 626–628. [Google Scholar]
- Thursby, E.; Juge, N. Introduction to the human gut microbiota. Biochem. J. 2017, 474, 1823–1836. [Google Scholar] [CrossRef] [PubMed]
- Sabino, J.; Vieira-Silva, S.; Machiels, K.; Joossens, M.; Falony, G.; Ballet, V.; Ferrante, M.; Van Assche, G.; Van der Merwe, S.; Vermeire, S.; et al. Primary sclerosing cholangitis is characterised by intestinal dysbiosis independent from IBD. Gut 2016, 65, 1681–1689. [Google Scholar] [CrossRef] [PubMed]
- Qin, N.; Yang, F.; Li, A.; Prifti, E.; Chen, Y.; Shao, L.; Guo, J.; Le Chatelier, E.; Yao, J.; Wu, L.; et al. Alterations of the human gut microbiome in liver cirrhosis. Nature 2014, 513, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yang, F.; Lu, H.; Wang, B.; Chen, Y.; Lei, D.; Wang, Y.; Zhu, B.; Li, L. Characterization of fecal microbial communities in patients with liver cirrhosis. Hepatology 2011, 54, 562–572. [Google Scholar] [CrossRef] [PubMed]
- Gevers, D.; Kugathasan, S.; Denson, L.A.; Vázquez-Baeza, Y.; Van Treuren, W.; Ren, B.; Schwager, E.; Knights, D.; Song, S.J.; Yassour, M.; et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe 2014, 15, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Steck, N.; Hoffmann, M.; Sava, I.G.; Kim, S.C.; Hahne, H.; Tonkonogy, S.L.; Mair, K.; Krueger, D.; Pruteanu, M.; Shanahan, F.; et al. Enterococcus faecalis metalloprotease compromises epithelial barrier and contributes to intestinal inflammation. Gastroenterology 2011, 141, 959–971. [Google Scholar] [CrossRef] [PubMed]
- Kummen, M.; Holm, K.; Anmarkrud, J.A.; Nygård, S.; Vesterhus, M.; Høivik, M.L.; Trøseid, M.; Marschall, H.U.; Schrumpf, E.; Moum, B.; et al. The gut microbial profile in patients with primary sclerosing cholangitis is distinct from patients with ulcerative colitis without biliary disease and healthy controls. Gut 2017, 66, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Iwasawa, K.; Suda, W.; Tsunoda, T.; Oikawa-Kawamoto, M.; Umetsu, S.; Inui, A.; Fujisawa, T.; Morita, H.; Sogo, T.; Hattori, M. Characterisation of the fecal microbiota in Japanese patients with paediatric-onset primary sclerosing cholangitis. Gut 2017, 66, 1344–1346. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Selmi, C.; Tang, R.; Gershwin, M.E.; Ma, X. The microbiome and autoimmunity: a paradigm from the gut–liver axis. Cell Mol. Immunol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Lv, L.X.; Fang, D.Q.; Shi, D.; Chen, D.Y.; Yan, R.; Zhu, Y.X.; Chen, Y.F.; Shao, L.; Guo, F.F.; Wu, W.R.; et al. Alterations and correlations of the gut microbiome, metabolism and immunity in patients with primary biliary cirrhosis. Environ. Microbiol. 2016, 18, 2272–2286. [Google Scholar] [CrossRef] [PubMed]
- Tang, R.; Wei, Y.; Li, Y.; Chen, W.; Chen, H.; Wang, Q.; Yang, F.; Miao, Q.; Xiao, X.; Zhang, H.; et al. Gut microbial profile is altered in primary biliary cholangitis and partially restored after UDCA therapy. Gut 2018, 67, 534–571. [Google Scholar] [CrossRef] [PubMed]
- Rossen, N.G.; Fuentes, S.; Boonstra, K.; D’Haens, G.R.; Heilig, H.G.; Zoetendal, E.G.; de Vos, W.M.; Ponsioen, C.Y. The mucosa-associated microbiota of PSC patients is characterized by low diversity and low abundance of uncultured Clostridiales II. J. Crohns Colitis 2015, 9, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Lahti, L.; Salojärvi, J.; Salonen, A.; Scheffer, M.; De Vos, W.M. Tipping elements in the human intestinal ecosystem. Nat. Commun. 2014, 5, 4344. [Google Scholar] [CrossRef] [PubMed]
- Torres, J.; Bao, X.; Goel, A.; Colombel, J.F.; Pekow, J.; Jabri, B.; Williams, K.M.; Castillo, A.; Odin, J.A.; Meckel, K.; et al. The features of mucosa-associated microbiota in primary sclerosing cholangitis. Aliment. Pharmacol. Ther. 2016, 43, 790–801. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).