A Novel Naphthyridine Derivative, 3u, Induces Necroptosis at Low Concentrations and Apoptosis at High Concentrations in Human Melanoma A375 Cells


Abstract
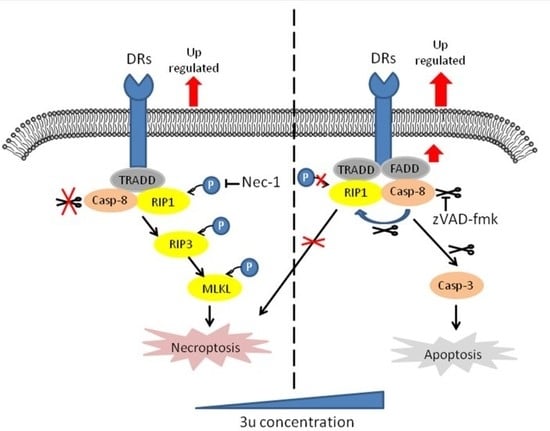
1. Introduction
2. Results
2.1. 3u Decreases Cell Viability in Human Cancer Cells
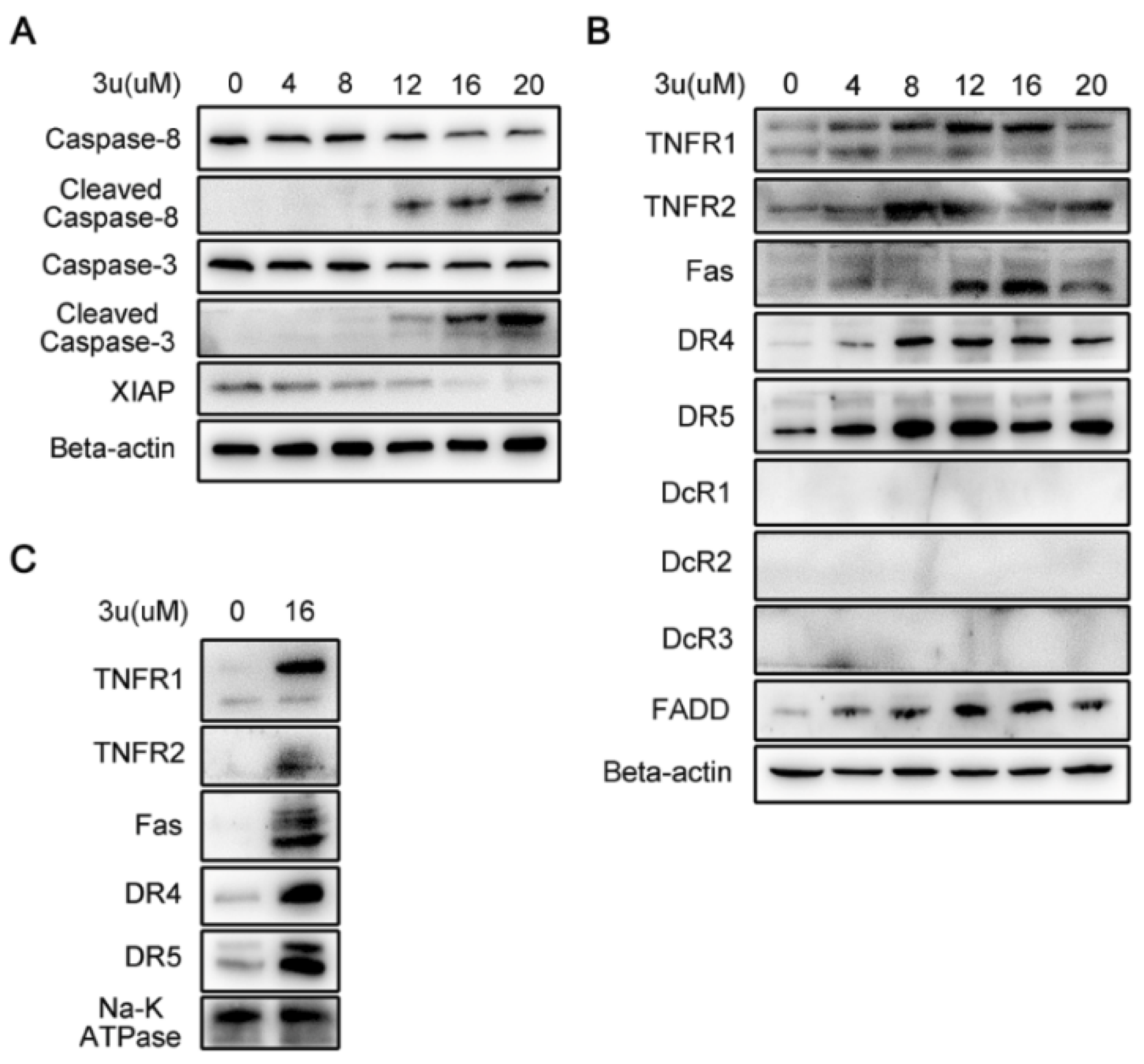
2.2. 3u-Induced Apoptosis at High Concentrations of 3u Depending on Caspase-3 Was Not Caused by ER Stress or Mitochondrial Pathway
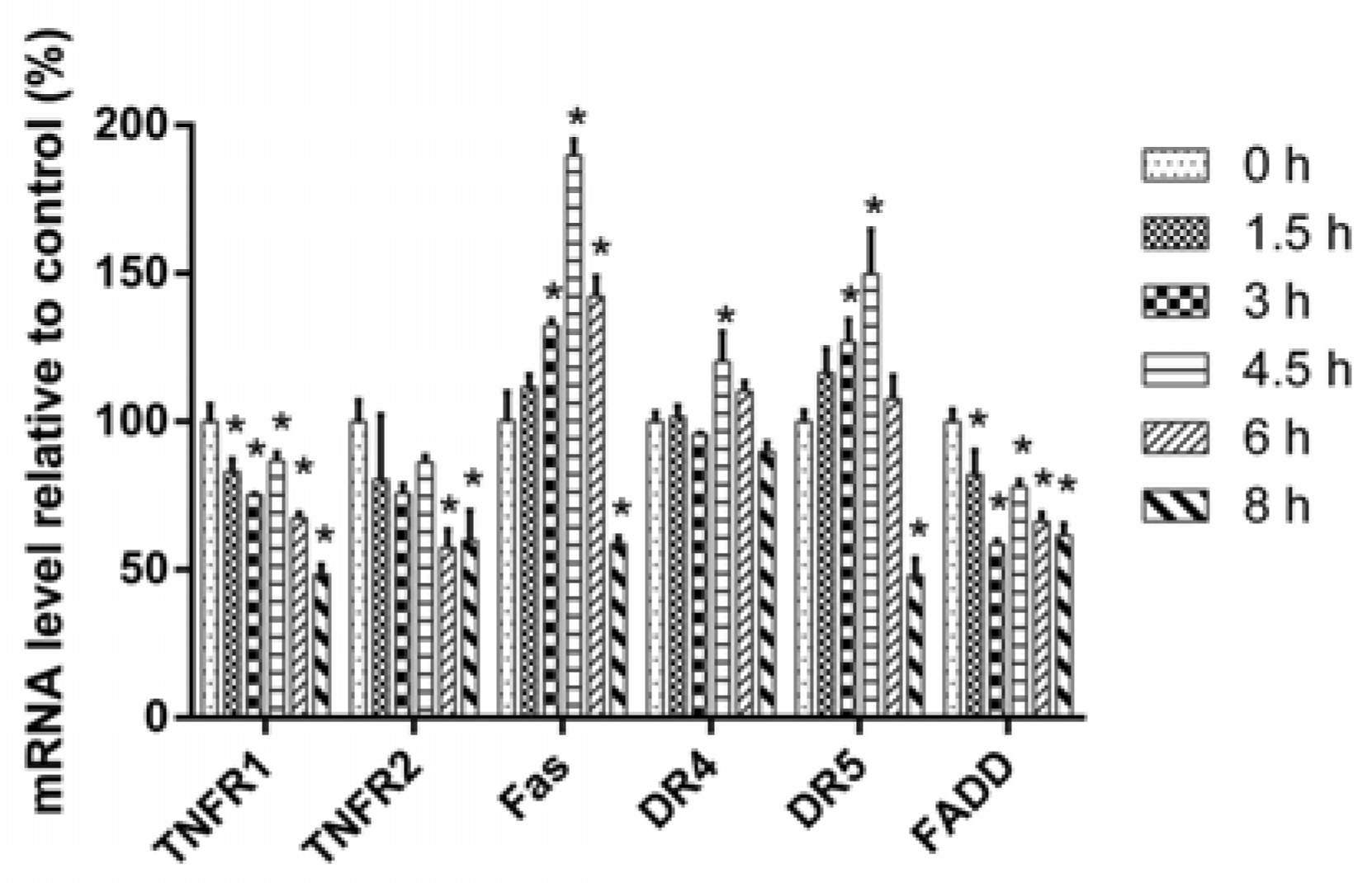
2.3. 3u-Induced Apoptosis at High Concentrations of 3u Was Caused by the Upregulation of DRs and FADD
2.4. The Mechanism of Cell Death Induced by Low Concentrations of 3u Was Not Apoptosis But Rather Necroptosis
2.5. Caspase Inhibitor zVAD-fmk Promoted Necroptosis at High Concentrations of 3u While Inhibiting Apoptosis
2.6. RIP1 Kinase Inhibitor Necrostatin-1 (Nec-1) Inhibited Necroptosis While Apoptosis Was Not Affected
3. Discussion
4. Materials and Methods
4.1. Chemicals and Antibodies
4.2. Cell Lines and Culture Conditions
4.3. Cell Viability Assay
4.4. Extraction of Mitochondrial and Membranous Proteins
4.5. SDS Electrophoresis and Immunoblotting
4.6. Quantitative Real-Time Polymerase Chain Reaction (qPCR)
4.7. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wahid, M.; Jawed, A.; Mandal, R.K.; Dar, S.A.; Akhter, N.; Somvanshi, P.; Khan, F.; Lohani, M.; Areeshi, M.Y.; Haque, S. Recent developments and obstacles in the treatment of melanoma with BRAF and MEK inhibitors. Crit. Rev. Oncol. Hematol. 2018, 125, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Gray-Schopfer, V.; Wellbrock, C.; Marais, R. Melanoma biology and new targeted therapy. Nature 2007, 445, 851–857. [Google Scholar] [CrossRef] [PubMed]
- Lens, M.B.; Dawes, M. Global perspectives of contemporary epidemiological trends of cutaneous malignant melanoma. Br. J. Dermatol. 2004, 150, 179–185. [Google Scholar] [CrossRef] [PubMed]
- MacKie, R.M.; Hauschild, A.; Eggermont, A.M. Epidemiology of invasive cutaneous melanoma. Ann. Oncol. 2009, 20 (Suppl. 6), vi1–vi7. [Google Scholar] [CrossRef]
- Geller, A.C.; Clapp, R.W.; Sober, A.J.; Gonsalves, L.; Mueller, L.; Christiansen, C.L.; Shaikh, W.; Miller, D.R. Melanoma epidemic: An analysis of six decades of data from the Connecticut Tumor Registry. J. Clin. Oncol. 2013, 31, 4172–4178. [Google Scholar] [CrossRef] [PubMed]
- Mattia, G.; Puglisi, R.; Ascione, B.; Malorni, W.; Care, A.; Matarrese, P. Cell death-based treatments of melanoma: Conventional treatments and new therapeutic strategies. Cell Death Dis. 2018, 9, 112. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, S.M.; Dunagin, M.C.; Torborg, S.R.; Torre, E.A.; Emert, B.; Krepler, C.; Beqiri, M.; Sproesser, K.; Brafford, P.A.; Xiao, M.; et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 2017, 546, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Lockshin, R.A.; Zakeri, Z. Programmed cell death and apoptosis: Origins of the theory. Nat. Rev. Mol. Cell Biol. 2001, 2, 545–550. [Google Scholar] [CrossRef] [PubMed]
- Wallach, D.; Kang, T.B.; Kovalenko, A. Concepts of tissue injury and cell death in inflammation: A historical perspective. Nat. Rev. Immunol. 2014, 14, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Vanden Berghe, T.; Linkermann, A.; Jouan-Lanhouet, S.; Walczak, H.; Vandenabeele, P. Regulated necrosis: The expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 2014, 15, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Degterev, A.; Boyce, M.; Yuan, J. A decade of caspases. Oncogene 2003, 22, 8543–8567. [Google Scholar] [CrossRef] [PubMed]
- Walczak, H.; Krammer, P.H. The CD95 (APO-1/Fas) and the TRAIL (APO-2L) apoptosis systems. Exp. Cell Res. 2000, 256, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Lavrik, I.; Golks, A.; Krammer, P.H. Death receptor signaling. J. Cell Sci. 2005, 118, 265–267. [Google Scholar] [CrossRef] [PubMed]
- Hengartner, M.O. The biochemistry of apoptosis. Nature 2000, 407, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Saelens, X.; Festjens, N.; Vande Walle, L.; van Gurp, M.; van Loo, G.; Vandenabeele, P. Toxic proteins released from mitochondria in cell death. Oncogene 2004, 23, 2861–2874. [Google Scholar] [CrossRef] [PubMed]
- Vercammen, D.; Beyaert, R.; Denecker, G.; Goossens, V.; Van Loo, G.; Declercq, W.; Grooten, J.; Fiers, W.; Vandenabeele, P. Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J. Exp. Med. 1998, 187, 1477–1485. [Google Scholar] [CrossRef] [PubMed]
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Jagtap, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; Moskowitz, M.A.; Yuan, J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 2005, 1, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Devin, A.; Rodriguez, Y.; Liu, Z.G. Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev. 1999, 13, 2514–2526. [Google Scholar] [CrossRef] [PubMed]
- Oberst, A.; Dillon, C.P.; Weinlich, R.; McCormick, L.L.; Fitzgerald, P.; Pop, C.; Hakem, R.; Salvesen, G.S.; Green, D.R. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 2011, 471, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Holler, N.; Zaru, R.; Micheau, O.; Thome, M.; Attinger, A.; Valitutti, S.; Bodmer, J.L.; Schneider, P.; Seed, B.; Tschopp, J. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 2000, 1, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.S.; Challa, S.; Moquin, D.; Genga, R.; Ray, T.D.; Guildford, M.; Chan, F.K. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009, 137, 1112–1123. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Jin, Y.; Yang, W.; Zhang, L.; Jin, X.; Liu, X.; He, Y.; Li, X. Necroptosis in cancer: An angel or a demon? Tumour Biol. 2017, 39. [Google Scholar] [CrossRef] [PubMed]
- Surivet, J.P.; Lange, R.; Hubschwerlen, C.; Keck, W.; Specklin, J.L.; Ritz, D.; Bur, D.; Locher, H.; Seiler, P.; Strasser, D.S.; et al. Structure-guided design, synthesis and biological evaluation of novel DNA ligase inhibitors with in vitro and in vivo anti-staphylococcal activity. Bioorg. Med. Chem. Lett. 2012, 22, 6705–6711. [Google Scholar] [CrossRef] [PubMed]
- Bhuniya, D.; Umrani, D.; Dave, B.; Salunke, D.; Kukreja, G.; Gundu, J.; Naykodi, M.; Shaikh, N.S.; Shitole, P.; Kurhade, S.; et al. Discovery of a potent and selective small molecule hGPR91 antagonist. Bioorg. Med. Chem. Lett. 2011, 21, 3596–3602. [Google Scholar] [CrossRef] [PubMed]
- Manera, C.; Saccomanni, G.; Malfitano, A.M.; Bertini, S.; Castelli, F.; Laezza, C.; Ligresti, A.; Lucchesi, V.; Tuccinardi, T.; Rizzolio, F.; et al. Rational design, synthesis and anti-proliferative properties of new CB2 selective cannabinoid receptor ligands: An investigation of the 1,8-naphthyridin-2(1H)-one scaffold. Eur. J. Med. Chem. 2012, 52, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Zhang, A.; Chen, D.; Jia, Z.; Li, X. 4-Substituted 4-(1H-1,2,3-triazol-1-yl)piperidine: Novel C7 moieties of fluoroquinolones as antibacterial agents. Bioorg. Med. Chem. Lett. 2010, 20, 2859–2863. [Google Scholar] [CrossRef] [PubMed]
- Roma, G.; Di Braccio, M.; Grossi, G.; Piras, D.; Ballabeni, V.; Tognolini, M.; Bertoni, S.; Barocelli, E. 1,8-Naphthyridines VIII. Novel 5-aminoimidazo[1,2-a] [1,8]naphthyridine-6-carboxamide and 5-amino[1,2,4]triazolo[4,3-a] [1,8]naphthyridine-6-carboxamide derivatives showing potent analgesic or anti-inflammatory activity, respectively, and completely devoid of acute gastrolesivity. Eur. J. Med. Chem. 2010, 45, 352–366. [Google Scholar] [CrossRef] [PubMed]
- Nam, T.G.; Rector, C.L.; Kim, H.Y.; Sonnen, A.F.; Meyer, R.; Nau, W.M.; Atkinson, J.; Rintoul, J.; Pratt, D.A.; Porter, N.A. Tetrahydro-1,8-naphthyridinol analogues of alpha-tocopherol as antioxidants in lipid membranes and low-density lipoproteins. J. Am. Chem. Soc. 2007, 129, 10211–10219. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.C.; Liu, Z.C.; Yang, R.; Zhang, J.H.; Yan, S.J.; Lin, J. Regioselective construction of 1,3-diazaheterocycle fused [1,2-a][1,8]naphthyridine derivatives via cascade reaction of quinolines with heterocyclic ketene aminals: A joint experimental-computational approach. Org. Biomol. Chem. 2013, 11, 7276–7288. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Zhu, H.; Morishima, N.; Li, E.; Xu, J.; Yankner, B.A.; Yuan, J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature 2000, 403, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, D.N.; Reddy, E.P. JNK signaling in apoptosis. Oncogene 2008, 27, 6245–6251. [Google Scholar] [CrossRef] [PubMed]
- Nakano, K.; Vousden, K.H. PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell 2001, 7, 683–694. [Google Scholar] [CrossRef]
- Deveraux, Q.L.; Takahashi, R.; Salvesen, G.S.; Reed, J.C. X-linked IAP is a direct inhibitor of cell-death proteases. Nature 1997, 388, 300–304. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, R.; Deveraux, Q.; Tamm, I.; Welsh, K.; Assa-Munt, N.; Salvesen, G.S.; Reed, J.C. A single BIR domain of XIAP sufficient for inhibiting caspases. J. Biol. Chem. 1998, 273, 7787–7790. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.W.; Zhang, C.M.; Huang, X.J.; Zhang, X.X.; Zhang, L.K.; Li, J.H.; Hua, Z.C. Tumor-targeted delivery of a C-terminally truncated FADD (N-FADD) significantly suppresses the B16F10 melanoma via enhancing apoptosis. Sci. Rep. 2016, 6, 34178. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.; Xiong, J.; Goeddel, D.V. The TNF receptor 1-associated protein TRADD signals cell death and NF-kappa B activation. Cell 1995, 81, 495–504. [Google Scholar] [CrossRef]
- Boldin, M.P.; Varfolomeev, E.E.; Pancer, Z.; Mett, I.L.; Camonis, J.H.; Wallach, D. A novel protein that interacts with the death domain of Fas/APO1 contains a sequence motif related to the death domain. J. Biol. Chem. 1995, 270, 7795–7798. [Google Scholar] [CrossRef] [PubMed]
- Chinnaiyan, A.M.; O’Rourke, K.; Tewari, M.; Dixit, V.M. FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell 1995, 81, 505–512. [Google Scholar] [CrossRef]
- Dasgupta, A.; Nomura, M.; Shuck, R.; Yustein, J. Cancer’s Achilles’ Heel: Apoptosis and Necroptosis to the Rescue. Int. J. Mol. Sci. 2016, 18, 23. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Michels, J.; Brenner, C.; Szabadkai, G.; Harel-Bellan, A.; Castedo, M.; Kroemer, G. Systems biology of cisplatin resistance: Past, present and future. Cell Death Dis. 2014, 5, e1257. [Google Scholar] [CrossRef] [PubMed]
- Janicke, R.U.; Sprengart, M.L.; Wati, M.R.; Porter, A.G. Caspase-3 is required for DNA fragmentation and morphological changes associated with apoptosis. J. Biol. Chem. 1998, 273, 9357–9360. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yang, X.; Xu, T.; Kong, Q.; Zhang, Y.; Shen, Y.; Wei, Y.; Wang, G.; Chang, K.J. Overcoming resistance to TRAIL-induced apoptosis in solid tumor cells by simultaneously targeting death receptors, c-FLIP and IAPs. Int. J. Oncol. 2016, 49, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Owen-Schaub, L.B.; Zhang, W.; Cusack, J.C.; Angelo, L.S.; Santee, S.M.; Fujiwara, T.; Roth, J.A.; Deisseroth, A.B.; Zhang, W.W.; Kruzel, E.; et al. Wild-type human p53 and a temperature-sensitive mutant induce Fas/APO-1 expression. Mol. Cell. Biol. 1995, 15, 3032–3040. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.S.; Burns, T.F.; McDonald, E.R., 3rd; Jiang, W.; Meng, R.; Krantz, I.D.; Kao, G.; Gan, D.D.; Zhou, J.Y.; Muschel, R.; et al. KILLER/DR5 is a DNA damage-inducible p53-regulated death receptor gene. Nat. Genet. 1997, 17, 141–143. [Google Scholar] [CrossRef] [PubMed]
- Takimoto, R.; El-Deiry, W.S. Wild-type p53 transactivates the KILLER/DR5 gene through an intronic sequence-specific DNA-binding site. Oncogene 2000, 19, 1735–1743. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yue, P.; Khuri, F.R.; Sun, S.Y. p53 upregulates death receptor 4 expression through an intronic p53 binding site. Cancer Res. 2004, 64, 5078–5083. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhou, X.; McQuade, T.; Li, J.; Chan, F.K.; Zhang, J. Functional complementation between FADD and RIP1 in embryos and lymphocytes. Nature 2011, 471, 373–376. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: A multicentre, double-blind, phase 3 randomised controlled trial. Lancet 2015, 386, 444–451. [Google Scholar] [CrossRef]
- Little, A.S.; Smith, P.D.; Cook, S.J. Mechanisms of acquired resistance to ERK1/2 pathway inhibitors. Oncogene 2013, 32, 1207–1215. [Google Scholar] [CrossRef] [PubMed]
- Eisen, T.; Ahmad, T.; Flaherty, K.T.; Gore, M.; Kaye, S.; Marais, R.; Gibbens, I.; Hackett, S.; James, M.; Schuchter, L.M.; et al. Sorafenib in advanced melanoma: A Phase II randomised discontinuation trial analysis. Br. J. Cancer 2006, 95, 581–586. [Google Scholar] [CrossRef] [PubMed]
- Jilaveanu, L.; Zito, C.; Lee, S.J.; Nathanson, K.L.; Camp, R.L.; Rimm, D.L.; Flaherty, K.T.; Kluger, H.M. Expression of sorafenib targets in melanoma patients treated with carboplatin, paclitaxel and sorafenib. Clin. Cancer Res. 2009, 15, 1076–1085. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.T.; Infante, J.R.; Daud, A.; Gonzalez, R.; Kefford, R.F.; Sosman, J.; Hamid, O.; Schuchter, L.; Cebon, J.; Ibrahim, N.; et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N. Engl. J. Med. 2012, 367, 1694–1703. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.T.; Robert, C.; Hersey, P.; Nathan, P.; Garbe, C.; Milhem, M.; Demidov, L.V.; Hassel, J.C.; Rutkowski, P.; Mohr, P.; et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N. Engl. J. Med. 2012, 367, 107–114. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Cell Lines IC50 (µM) | ||||
|---|---|---|---|---|---|
| 3u | A375 | A549 | HCT116 | HeLa | HT29 |
| 1.52 ± 0.10 | 3.69 ± 0.63 | 2.54 ± 0.20 | 4.31 ± 0.71 | 2.45 ± 0.13 | |
| LOVO | MCF7 | U2OS | SY5Y | HSF | |
| 5.72 ± 0.37 | 10.24 ± 1.03 | 4.70 ± 0.76 | 4.60 ± 0.64 | 7.27 ± 0.03 | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kong, Q.; Lv, J.; Yan, S.; Chang, K.-J.; Wang, G. A Novel Naphthyridine Derivative, 3u, Induces Necroptosis at Low Concentrations and Apoptosis at High Concentrations in Human Melanoma A375 Cells. Int. J. Mol. Sci. 2018, 19, 2975. https://doi.org/10.3390/ijms19102975
Kong Q, Lv J, Yan S, Chang K-J, Wang G. A Novel Naphthyridine Derivative, 3u, Induces Necroptosis at Low Concentrations and Apoptosis at High Concentrations in Human Melanoma A375 Cells. International Journal of Molecular Sciences. 2018; 19(10):2975. https://doi.org/10.3390/ijms19102975
Chicago/Turabian StyleKong, Qinghong, Jianxin Lv, Shengjiao Yan, Kwen-Jen Chang, and Guanlin Wang. 2018. "A Novel Naphthyridine Derivative, 3u, Induces Necroptosis at Low Concentrations and Apoptosis at High Concentrations in Human Melanoma A375 Cells" International Journal of Molecular Sciences 19, no. 10: 2975. https://doi.org/10.3390/ijms19102975
APA StyleKong, Q., Lv, J., Yan, S., Chang, K.-J., & Wang, G. (2018). A Novel Naphthyridine Derivative, 3u, Induces Necroptosis at Low Concentrations and Apoptosis at High Concentrations in Human Melanoma A375 Cells. International Journal of Molecular Sciences, 19(10), 2975. https://doi.org/10.3390/ijms19102975