A Comparison of Lysosomal Enzymes Expression Levels in Peripheral Blood of Mild- and Severe-Alzheimer’s Disease and MCI Patients: Implications for Regenerative Medicine Approaches

 , , , ,
, , , ,  ,
,  ,
, 

Abstract
:
1. Introduction
2. Results
2.1. Assessment of Lysosomal Enzymes in Peripheral Blood from Mild and Severe AD Patients and MCI Patients: Experimental Platform
2.1.1. Patients Enrolled in the Study
2.1.2. Lysosomal Enzymes Screening
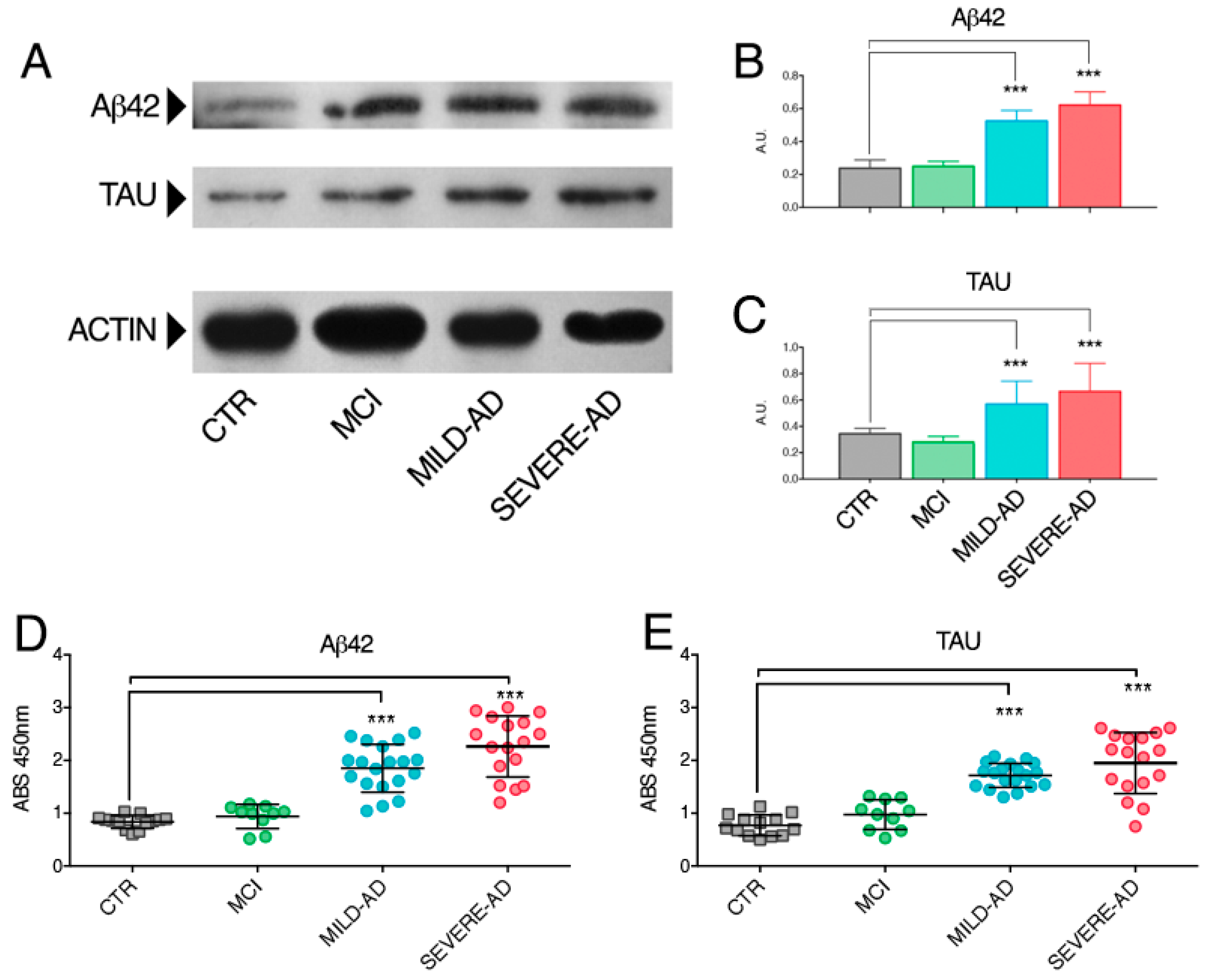
2.2. β-Amyloid and Tau Proteins Expression in PBMCs from Mild- and Severe AD and MCI Patients
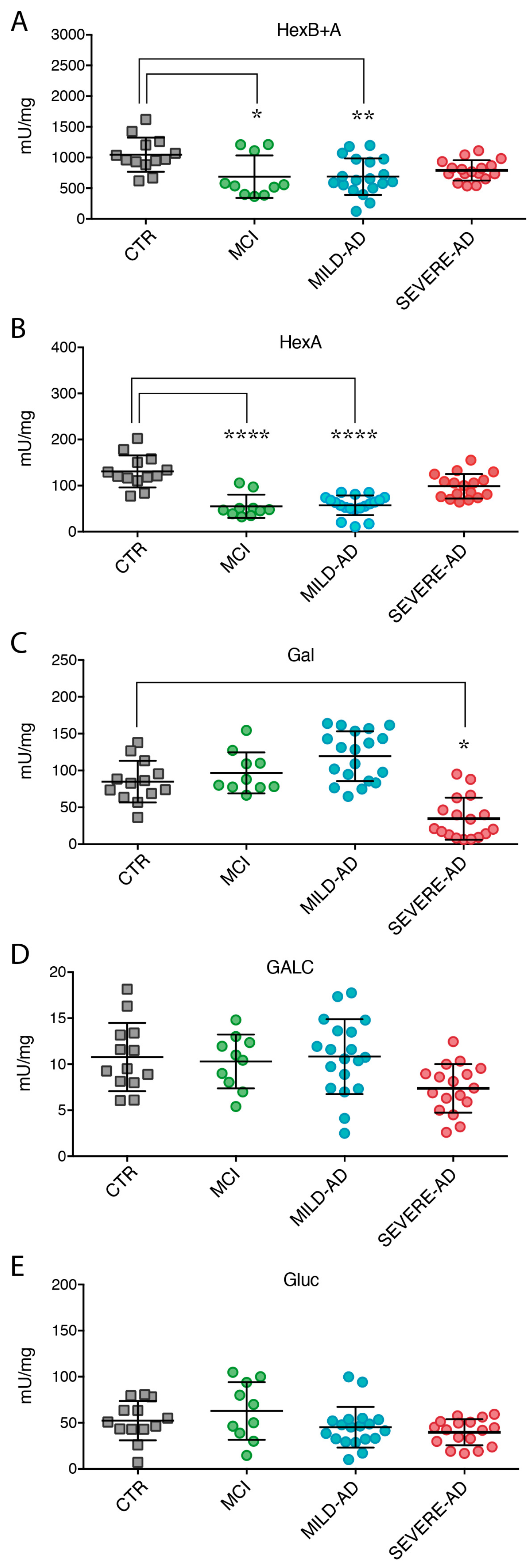
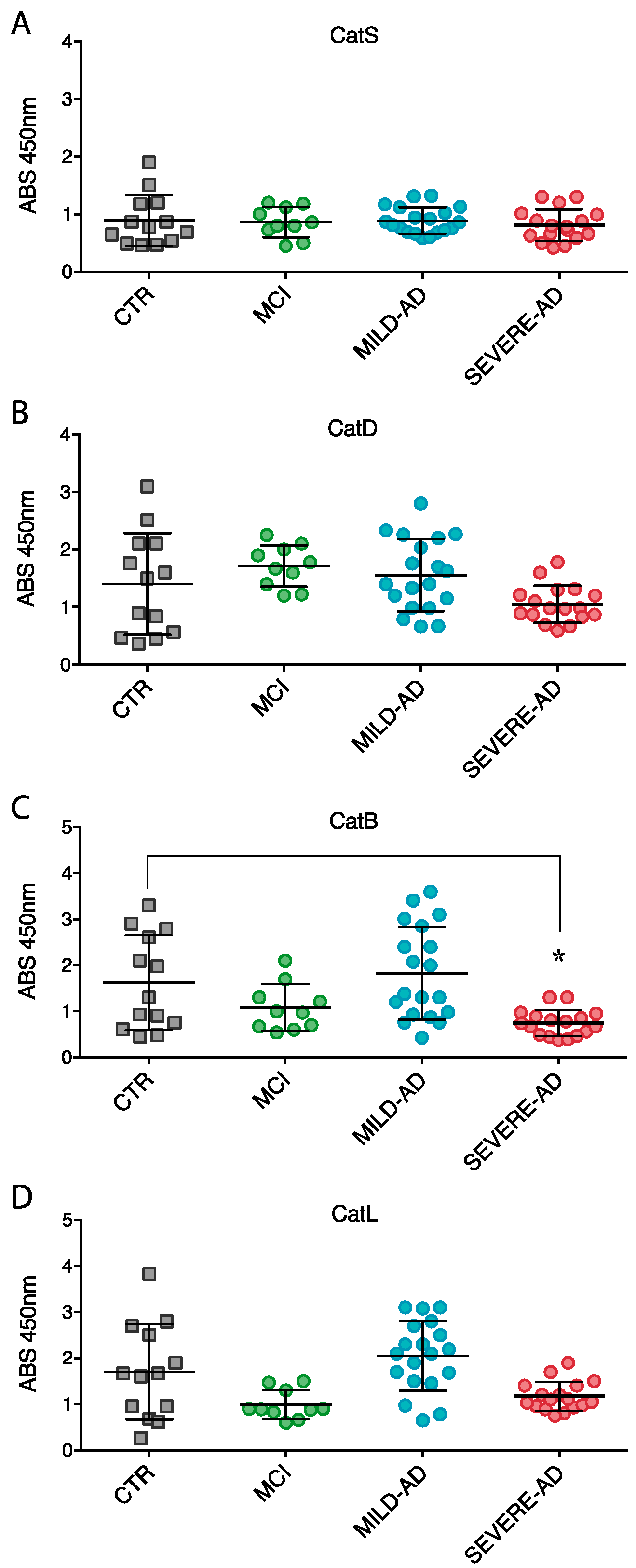
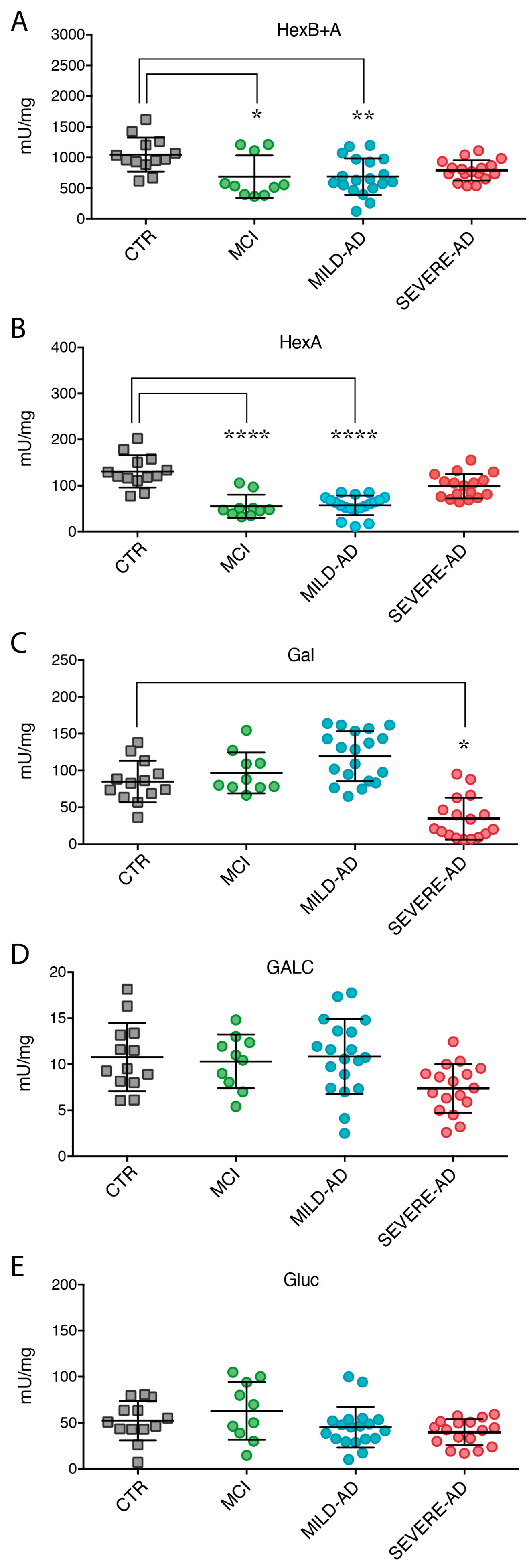
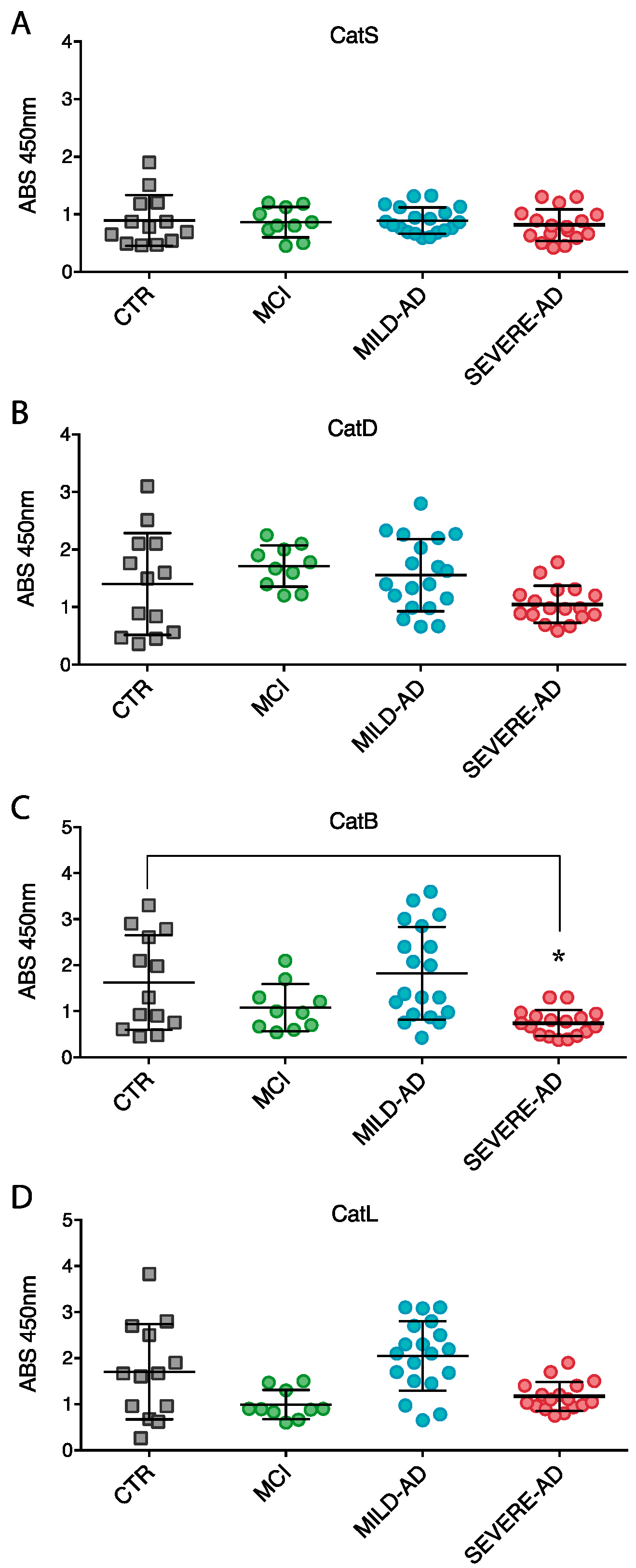
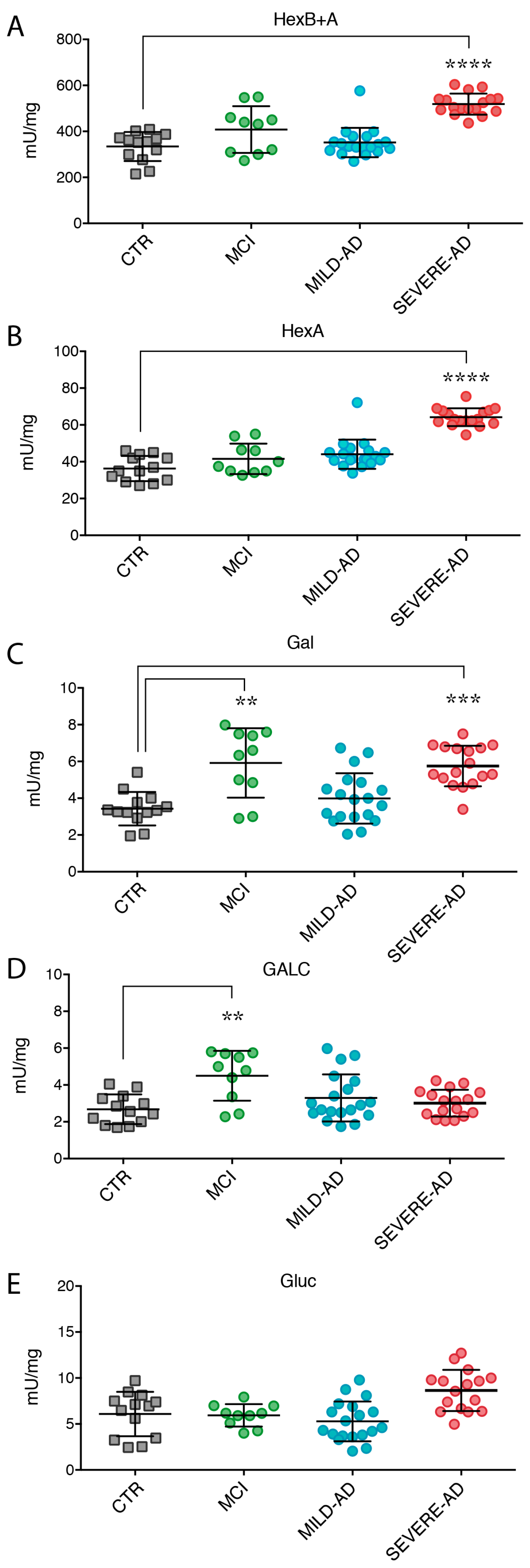
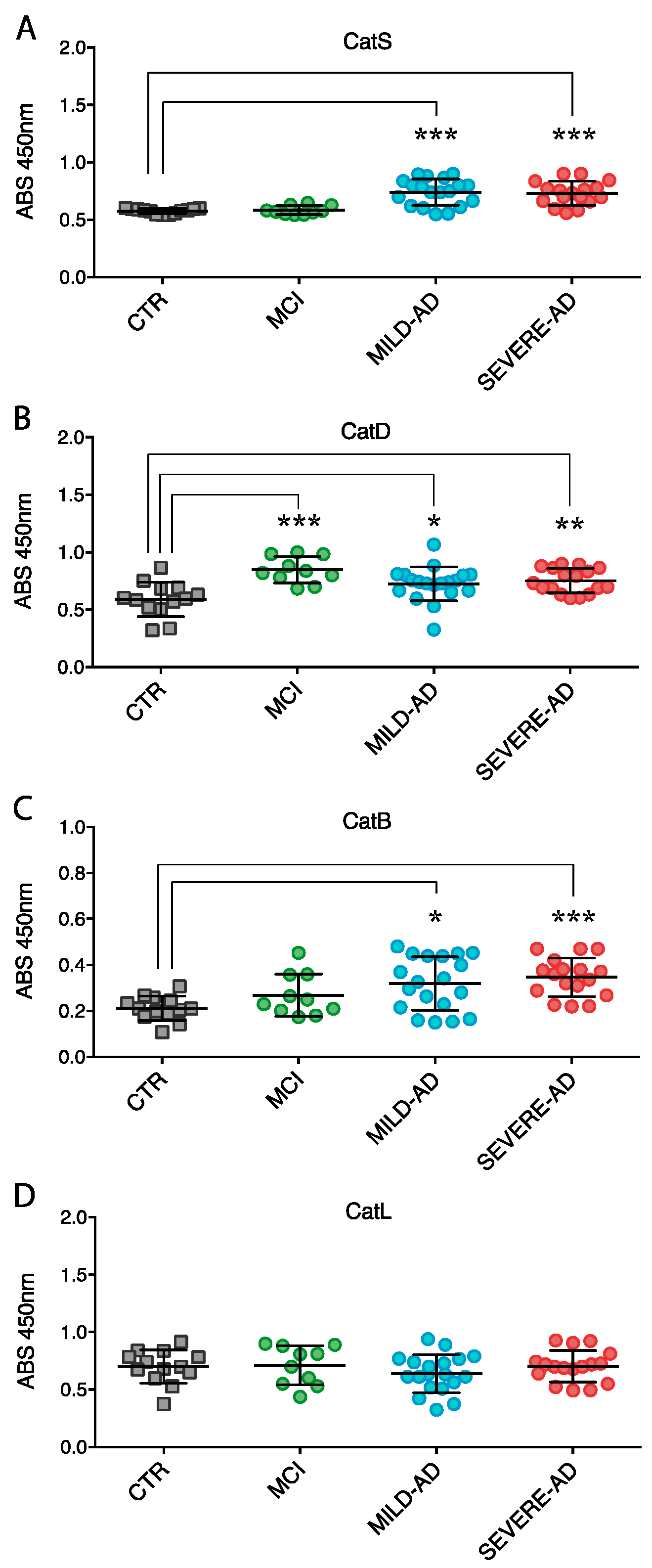
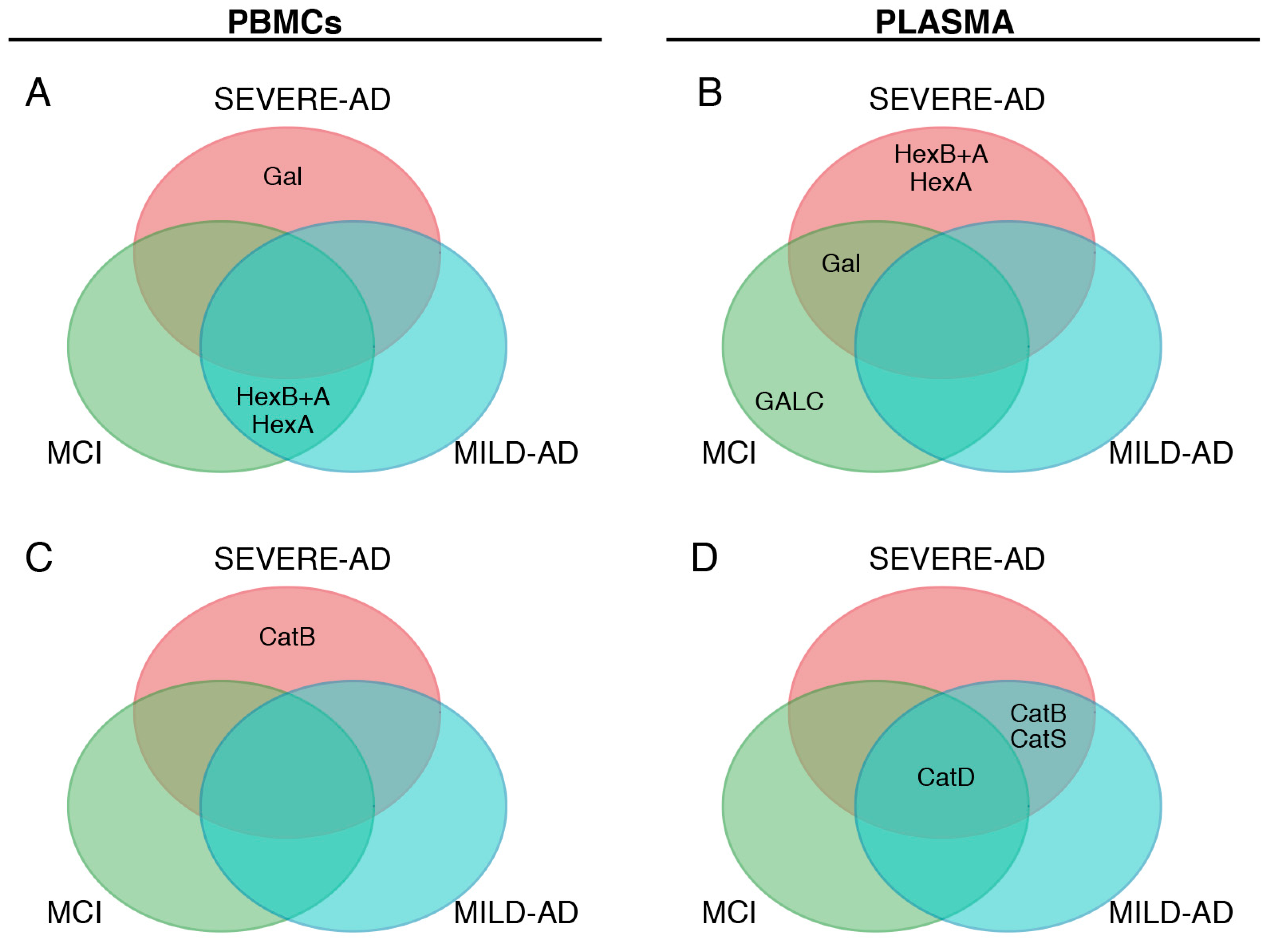
2.3. Lysosomal Enzymes Levels Are Altered in PBMCs from Patients with Mild and Severe AD and MCI
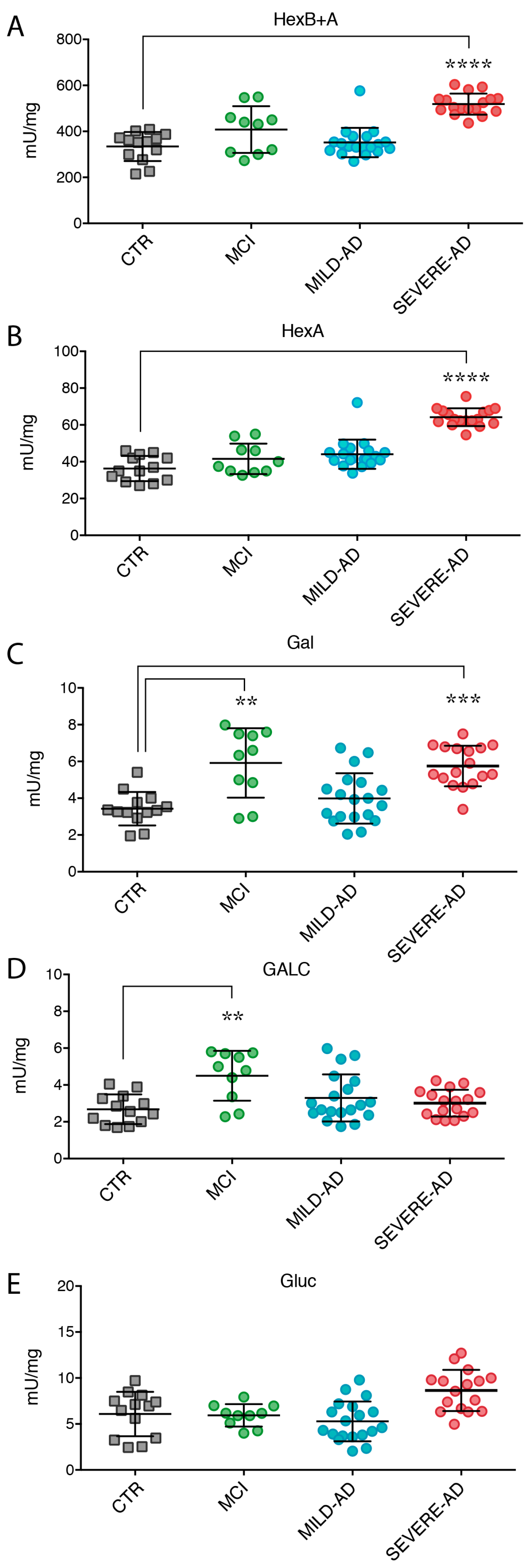
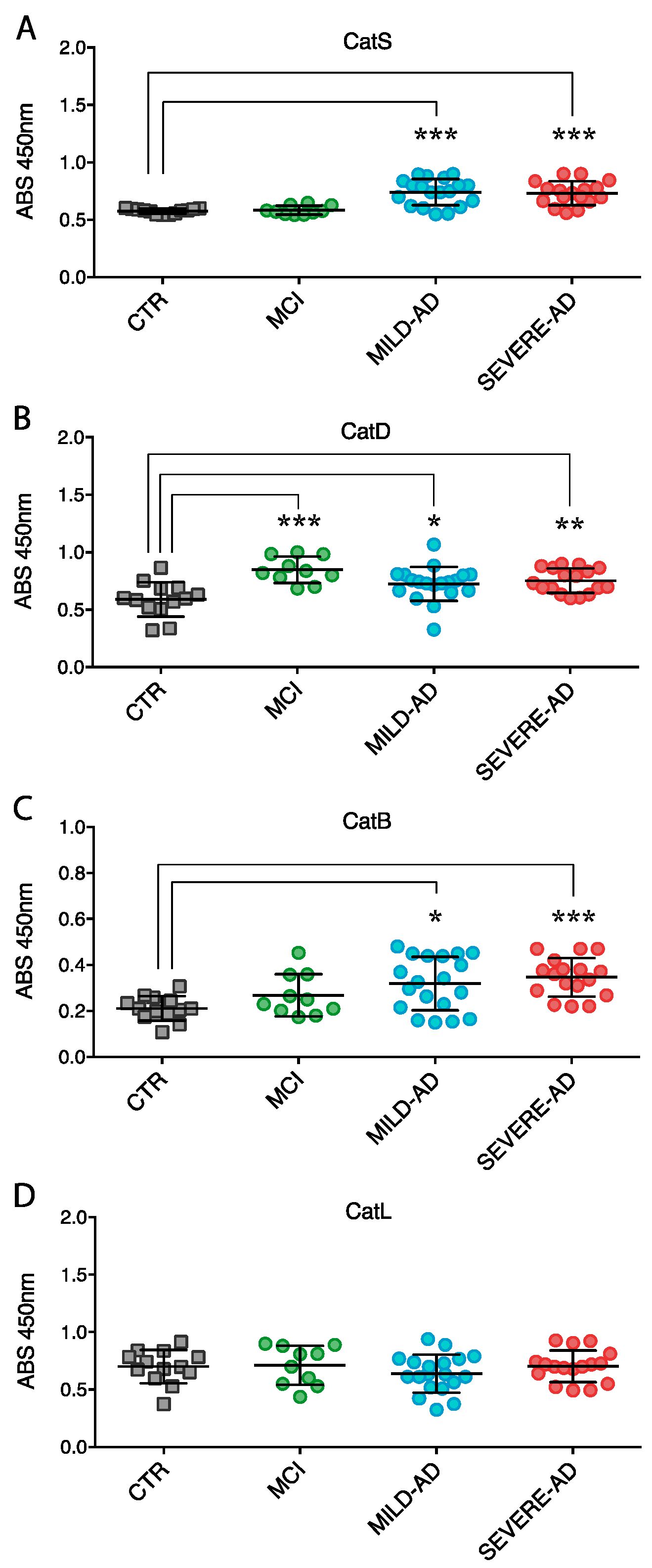
2.4. Secrete Lysosomal Enzymes Are Altered in Plasma from Patients with Mild and Severe AD and MCI
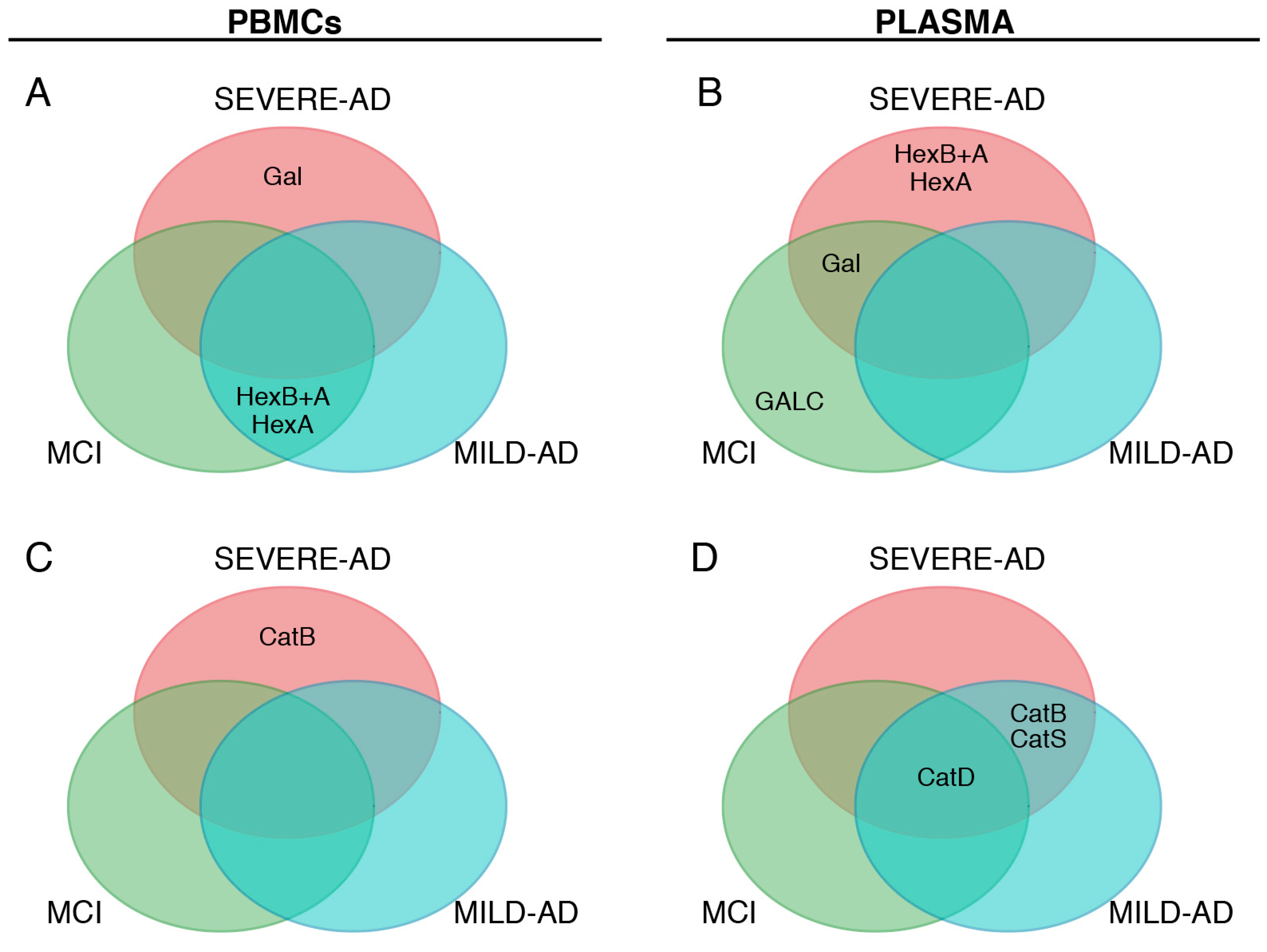
2.5. Comparison of Lysosomal Enzymes Expression Levels, Progression of AD and MCI
3. Discussion
4. Materials and Methods
4.1. Biological Samples Collection
4.2. Sample Preparation
4.3. Cell Extracts
4.4. Enzyme Assays
4.5. Western Blotting
4.6. ELISA
4.7. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Scheltens, P.; Blennow, K.; Breteler, M.M.; de Strooper, B.; Frisoni, G.B.; Salloway, S.; van der Flier, W.M. Alzheimer’s disease. Lancet 2016, 388, 505–517. [Google Scholar] [CrossRef]
- Scarano, S.; Lisi, S.; Ravelet, C.; Peyrin, E.; Minunni, M. Detecting Alzheimer’s disease biomarkers: From antibodies to new bio-mimetic receptors and their application to established and emerging bioanalytical platforms—A critical review. Anal. Chim. Acta 2016, 940, 21–37. [Google Scholar] [CrossRef] [PubMed]
- Rivero-Santana, A.; Ferreira, D.; Perestelo-Pérez, L.; Westman, E.; Wahlund, L.O.; Sarría, A.; Serrano-Aguilar, P. Cerebrospinal fluid biomarkers for the differential diagnosis between Alzheimer’s disease and frontotemporal lobar degeneration: Systematic review, HSROC analysis, and confounding factors. J. Alzheimers Dis. 2016, 55, 625–644. [Google Scholar] [CrossRef] [PubMed]
- Bicchi, I.; Emiliani, C.; Vescovi, A.; Martino, S. The big bluff of amyotrophic lateral sclerosis diagnosis: The role of neurodegenerative disease mimics. Neurodegener Dis. 2015, 15, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Crispoltoni, L.; Stabile, A.M.; Pistilli, A.; Venturelli, M.; Cerulli, G.; Fonte, C.; Smania, N.; Schena, F.; Rende, M. Changes in plasma β-NGF and its receptors expression on peripheral blood monocytes during Alzheimer’s disease progression. J. Alzheimers Dis. 2017, 55, 1005–1017. [Google Scholar] [CrossRef] [PubMed]
- Hodges, J.R. Is pathology-specific diagnosis of the dementia possible in life? J. Neurol. Neurosurg. Psychiatry 2017, 2017, 316016. [Google Scholar] [CrossRef] [PubMed]
- Cha, M.Y.; Kwon, Y.W.; Ahn, H.S.; Jeong, H.; Lee, Y.Y.; Moon, M.; Baik, S.H.; Kim, D.K.; Song, H.; Yi, E.C.; et al. Protein-induced pluripotent stem cells ameliorate cognitive dysfunction and reduce Aβ deposition in a mouse model of Alzheimer’s disease. Stem Cells Transl. Med. 2017, 6, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.; Solanki, A.; Lee, K.B. Nanotechnology-based approaches for guiding neural regeneration. Acc. Chem. Res. 2016, 49, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Laske, C.; Sohrabi, H.R.; Frost, S.M.; López-de-Ipiña, K.; Garrard, P.; Buscema, M.; Dauwels, J.; Soekadar, S.R.; Mueller, S.; Linnemann, C.; et al. Innovative diagnostic tools for early detection of Alzheimer’s disease. Alzheimers Dement. 2015, 11, 561–578. [Google Scholar] [CrossRef] [PubMed]
- O’Bryant, S.E.; Mielke, M.M.; Rissman, R.A.; Lista, S.; Vanderstichele, H.; Zetterberg, H.; Lewczuk, P.; Posner, H.; Hall, J.; Johnson, L.; et al. Blood-based biomarkers in Alzheimer disease: Current state of the science and a novel collaborative paradigm for advancing from discovery to clinic. Alzheimers Dement. 2017, 13, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Lista, S.; O’Bryant, S.E.; Blennow, K.; Dubois, B.; Hugon, J.; Zetterberg, H.; Hampel, H. Biomarkers in sporadic and familial Alzheimer’s disease. J. Alzheimers Dis. 2015, 47, 291–317. [Google Scholar] [CrossRef] [PubMed]
- Deng, B.; Cai, L.; Li, S.; Wang, R.; Yu, H.; Chen, Y.; Wang, J. Multivariate multi-scaleweighted permutation entropy analysis of EEG complexity for Alzheimer’s disease. Cogn. Neurodyn. 2017, 11, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Pansieri, J.; Plissonneau, M.; Stransky-Heilkron, N.; Dumoulin, M.; Heinrich-Balard, L.; Rivory, P.; Morfin, J.F.; Toth, E.; Saraiva, M.J.; Allémann, E.; et al. Multimodal imaging Gd-nanoparticles functionalized with Pittsburgh compound B or a nanobody for amyloid plaques targeting. Nanomedicine 2017, 12, 1675–1687. [Google Scholar] [CrossRef] [PubMed]
- Martino, S.; Di Girolamo, I.; Orlacchio, A.; Datti, A.; Orlacchio, A. MicroRNA implications across neurodevelopment and neuropathology. J. Biomed. Biotechnol. 2009. [Google Scholar] [CrossRef] [PubMed]
- Khan, T.K.; Alkon, D.L. Peripheral biomarkers of Alzheimer’s disease. J. Alzheimers Dis. 2015, 44, 729–744. [Google Scholar] [PubMed]
- Bicchi, I.; Morena, F.; Montesano, S.; Polidoro, M.; Martino, S. MicroRNAs and molecular mechanisms of neurodegeneration. Genes 2013, 4, 244–263. [Google Scholar] [CrossRef] [PubMed]
- Kaur, J.; Debnath, J. Autophagy at the crossroads of catabolism and anabolism. Nat. Rev. Mol. Cell Biol. 2015, 16, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Arikkath, J.; Yang, L.; Guo, M.L.; Periyasamy, P.; Buch, S. Interplay of endoplasmic reticulum stress and autophagy in neurodegenerative disorders. Autophagy 2016, 12, 225–244. [Google Scholar] [CrossRef] [PubMed]
- McBrayer, M.; Nixon, R.A. Lysosome and calcium dysregulation in Alzheimer’s disease: Partners in crime. Biochem. Soc. Trans. 2013, 41, 1495–1502. [Google Scholar] [CrossRef] [PubMed]
- Gowrishankar, S.; Yuan, P.; Wu, Y.; Schrag, M.; Paradise, S.; Grutzendler, J.; de Camilli, P.; Ferguson, S.M. Massive accumulation of luminal protease-deficient axonal lysosomes at Alzheimer’s disease amyloid plaques. Proc. Natl. Acad. Sci. USA 2015, 112, 3699–3708. [Google Scholar] [CrossRef] [PubMed]
- Magini, A.; Polchi, A.; Tozzi, A.; Tancini, B.; Tantucci, M.; Urbanelli, L.; Borsello, T.; Calabresi, P.; Emiliani, C. Abnormal cortical lysosomal β-Hexosaminidase and β-Galactosidase activity at post-synaptic sites during Alzheimer’s disease progression. Int. J. Biochem. Cell. Biol. 2015, 58, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, A.; Mattsson, N.; Appelqvist, H.; Janefjord, C.; Sandin, L.; Agholme, L.; Olsson, B.; Svensson, S.; Blennow, K.; Zetterberg, H.; et al. Lysosomal network proteins as potential novel CSF biomarkers for Alzheimer’s disease. Neuromol. Med. 2014, 16, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Mueller-Steiner, S.; Zhou, Y.; Arai, H.; Roberson, E.D.; Sun, B.; Chen, J.; Wang, X.; Yu, G.; Esposito, L.; Mucke, L.; et al. Antiamyloidogenic and neuroprotective functions of Cathepsin B: Implications for Alzheimer’s disease. Neuron 2006, 51, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Ladror, U.S.; Snyder, S.W.; Wang, G.T.; Holzman, T.F.; Krafft, G.A. Cleavage at the amino and carboxyl termini of Alzheimer’s amyloid-β by Cathepsin D. J. Biol. Chem. 1994, 269, 18422–18428. [Google Scholar] [PubMed]
- Tiribuzi, R.; Orlacchio, A.; Crispoltoni, L.; Maiotti, M.; Zampolini, M.; de Angelis, M.; Mecocci, P.; Cecchetti, R.; Bernardi, G.; Datti, A.; et al. Lysosomal β-Galactosidase and β-Hexosaminidase activities correlate with clinical stages of dementia associated with Alzheimer’s disease and type 2 diabetes mellitus. J. Alzheimers Dis. 2011, 24, 785–797. [Google Scholar] [PubMed]
- Tiribuzi, R.; Crispoltoni, L.; Porcellati, S.; Di Lullo, M.; Florenzano, F.; Pirro, M.; Bagaglia, F.; Kawarai, T.; Zampolini, M.; Orlacchio, A.; et al. MiR128 up-regulation correlates with impaired amyloid β(1–42) degradation in monocytes from patients with sporadic Alzheimer’s disease. Neurobiol. Aging 2014, 35, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Sato, Y.; Nixon, R.A. Primary lysosomal dysfunction causes cargo-specific deficits of axonal transport leading to Alzheimer-like neuritic dystrophy. Autophagy 2011, 7, 1562–1563. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Yu, W.H.; Kumar, A.; Lee, S.; Mohan, P.S.; Peterhoff, C.M.; Wolfe, D.M.; Martinez-Vicente, M.; Massey, A.C.; Sovak, G.; et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 2010, 141, 1146–1158. [Google Scholar] [CrossRef] [PubMed]
- Emiliani, C.; Urbanelli, L.; Racanicchi, L.; Orlacchio, A.; Pelicci, G.; Sorbi, S.; Bernardi, G.; Orlacchio, A. Up-regulation of glycohydrolases in Alzheimer’s Disease fibroblasts correlates with Ras activation. J. Biol. Chem. 2003, 278, 38453–38460. [Google Scholar] [CrossRef] [PubMed]
- Urbanelli, L.; Emiliani, C.; Massini, C.; Persichetti, E.; Orlacchio, A.; Pelicci, G.; Sorbi, S.; Hasilik, A.; Bernardi, G.; Orlacchio, A. Cathepsin D expression is decreased in Alzheimer’s disease fibroblasts. Neurobiol. Aging 2008, 29, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Martino, S.; Montesano, S.; di Girolamo, I.; Tiribuzi, R.; di Gregorio, M.; Orlacchio, A.; Datti, A.; Calabresi, P; Sarchielli, P.; Orlacchio, A. Expression of cathepsins S and D signals a distinctive biochemical trait in CD34+ hematopoietic stem cells of relapsing–remitting multiple sclerosis patients. Mult. Scler. 2013. [Google Scholar] [CrossRef] [PubMed]
- Martino, S.; Tiribuzi, R.; Ciraci, E.; Makrypidi, G.; D’Angelo, F.; di Girolamo, I.; Gritti, A.; de Angelis, G.M.; Papaccio, G.; Sampaolesi, M.; et al. Coordinated involvement of cathepsins S, D and cystatin C in the commitment of hematopoietic stem cells to dendritic cells. Int. J. Biochem. Cell Biol. 2011, 43, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Tiribuzi, R.; D’Angelo, F.; Berardi, A.C.; Martino, S.; Orlacchio, A. Knock-down of HEXA and HEXB genes correlate with the absence of the immunostimulatory function of HSC-derived dendritic cells. Cell. Biochem. Funct. 2012, 30, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Morena, F.; di Girolamo, I.; Emiliani, C.; Gritti, A.; Biffi, A.; Martino, S. A new analytical bench assay for the determination of arylsulfatase a activity toward galactosyl-3-sulfate ceramide: Implication for metachromatic leukodystrophy diagnosis. Anal. Chem. 2013, 86, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Martino, S.; Emiliani, C.; Tancini, B.; Severini, G.M.; Chigorno, V.; Bordignon, C.; Sonnino, S.; Orlacchio, A. Absence of metabolic cross-correction in Tay-Sachs cells: Implications for gene therapy. J. Biol. Chem. 2002, 277, 20177–20184. [Google Scholar] [CrossRef] [PubMed]
- Martino, S.; di Girolamo, I.; Cavazzin, C.; Tiribuzi, R.; Galli, R.; Rivaroli, A.; Valsecchi, M.; Sandhoff, K.; Sonnino, S.; Vescovi, A.; et al. Neural precursor cell cultures from GM2 gangliosidosis animal models recapitulate the biochemical and molecular hallmarks of the brain pathology. J. Neurochem. 2009, 109, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Martino, S.; Tiribuzi, R.; Tortori, A.; Conti, D.; Visigalli, I.; Lattanzi, A.; Biffi, A.; Gritti, A.; Orlacchio, A. Specific determination of beta-galactocerebrosidase activity via competitive inhibition of beta-galactosidase. Clin. Chem. 2009, 55, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Sessa, M.; Lorioli, L.; Fumagalli, F.; Acquati, S.; Redaelli, D.; Baldoli, C.; Canale, S.; Lopez, I.D.; Morena, F.; Calabria, A.; et al. Lentiviral haemopoietic stem-cell gene therapy in early-onset metachromatic leukodystrophy: An ad-hoc analysis of a non-randomised, open-label, phase 1/2 trial. Lancet 2016, 388, 476–487. [Google Scholar] [CrossRef]
- Orlacchio, A.; Martino, S.; Sarchielli, P.; Gallai, V.; Emiliani, C. β-N-Acethyl-Hexosaminidase in pheripheral blood lymphocytes and monocytes in the different forms and stages of multiple sclerosis. J. Neurochem. 1998, 71, 1168–1176. [Google Scholar] [CrossRef] [PubMed]
- Martino, S.; Consiglio, A.; Cavalieri, C.; Tiribuzi, R.; Costanzi, E.; Severini, G.M.; Emiliani, C.; Bordignon, C.; Orlacchio, A. Expression and purification of a human, soluble Arylsulfatase A for Metachromatic Leukodystrophy enzyme replacement therapy. J. Biotechnol. 2005, 117, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Ricca, A.; Rufo, N.; Ungari, S.; Morena, F.; Martino, S.; Kulik, W.; Alberizzi, V.; Bolino, A.; Bianchi, F.; Del Carro, U.; et al. Combined gene/cell therapies provide long-term and pervasive rescue of multiple pathological symptoms in a murine model of globoid cell leukodystrophy. Hum. Mol. Genet. 2015, 24, 3372–3389. [Google Scholar] [CrossRef] [PubMed]
- Lorioli, L.; Cesani, M.; Regis, S.; Morena, F.; Grossi, S.; Fumagalli, F.; Acquati, S.; Redaelli, D.; Pini, A.; Sessa, M.; et al. Critical issues for the proper diagnosis of Methachromatic Leukodystrophy. Gene 2013, 537, 348–351. [Google Scholar] [CrossRef] [PubMed]
- Meneghini, V.; Frati, G.; Sala, D.; de Cicco, S.; Luciani, M.; Cavazzin, C.; Paulis, M.; Mentzen, W.; Morena, F.; Giannelli, S.; et al. Generation of human induced pluripotent stem cell-derived bona fide neural stem cells for ex vivo gene therapy of metachromatic leukodystrophy. Stem Cells Transl. Med. 2017, 6, 352–368. [Google Scholar] [CrossRef] [PubMed]
- Biffi, A.; Montini, E.; Lorioli, L.; Cesani, M.; Fumagalli, F.; Plati, T.; Baldoli, C.; Martino, S.; Calabria, A.; Canale, S.; et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science 2013, 341, 1233158. [Google Scholar] [CrossRef] [PubMed]
- Appelqvist, H.; Wäster, P.; Kågedal, K.; Öllinger, K. The lysosome: From waste bag to potential therapeutic target. J. Mol. Cell. Biol. 2013, 5, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Cataldo, A.M.; Nixon, R.A. Enzymatically active lysosomal proteases are associated with amyloid deposits in Alzheimer brain. Proc. Natl. Acad. Sci. USA 1990, 87, 3861–3865. [Google Scholar] [CrossRef] [PubMed]
- Peall, K.; Robertson, N.P. Parkinsonism, dementia and glucocerebrosidase mutations. J. Neurol. 2013, 260, 1441–1444. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A.; Yang, D.S. Autophagy failure in Alzheimer’s disease locating the primary defect. Neurobiol. Dis. 2011, 43, 38–45. [Google Scholar] [CrossRef] [PubMed]
- DePaolo, J.; Goker-Alpan, O.; Samaddar, T.; Lopez, G.; Sidransky, E. The association between mutations in the lysosomal protein glucocerebrosidase and parkinsonism. Mov. Disord. 2009, 24, 1571–1578. [Google Scholar] [CrossRef] [PubMed]
- Migdalska, R.A.; Schapira, A.H. The relationship between glucocerebrosidase mutations and Parkinson disease. J. Neurochem. 2016, 139, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef] [PubMed]
- Erie, C.; Sacino, M.; Houle, L.; Lu, M.L.; Wei, J. Altered lysosomal positioning affects lysosomal functions in a cellular model of Huntington’s disease. Eur. J. Neurosci. 2015, 42, 1941–1951. [Google Scholar] [CrossRef] [PubMed]
- Yung, C.; Sha, D.; Li, L.; Chin, L.S. Parkin Protects Against Misfolded SOD1 Toxicity by Promoting Its Aggresome Formation and Autophagic Clearance. Mol. Neurobiol. 2016, 53, 6270–6287. [Google Scholar] [CrossRef] [PubMed]
- Coen, K.; Flannagan, R.S.; Baron, S.; Carraro-Lacroix, L.R.; Wang, D.; Vermeire, W.; Michiels, C.; Munck, S.; Baert, V.; Sugita, S.; et al. Lysosomal calcium homeostasis defects, not proton pump defects, cause endo-lysosomal dysfunction in PSEN-deficient cells. J. Cell. Biol. 2012, 198, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Meneghini, V.; Lattanzi, A.; Tiradani, L.; Bravo, G.; Morena, F.; Sanvito, F.; Calabria, A.; Bringas, J.; Fisher-Perkins, J.M.; Dufour, J.P.; et al. Pervasive supply of therapeutic lysosomal enzymes in the CNS of normal and Krabbe-affected non-human primates by intracerebral lentiviral gene therapy. EMBO Mol. Med. 2016, 8, 489–510. [Google Scholar] [CrossRef] [PubMed]
- Mielke, M.M.; Haughey, N.J.; Bandaru, V.V.; Schech, S.; Carrick, R.; Carlson, M.C.; Mori, S.; Miller, M.I.; Ceritoglu, C.; Brown, T.; et al. Plasma ceramides are altered in mild cognitive impairment and predict cognitive decline and hippocampal volume loss. Alzheimers Dement. 2010, 6, 378–385. [Google Scholar] [CrossRef] [PubMed]
- De la Monte, S.M.; Tong, M.; Nguyen, V.; Setshedi, M.; Longato, L.; Wands, J.R. Ceramide-mediated insulin resistance and impairment of cognitive-motor functions. J. Alzheimers Dis. 2010, 21, 967–984. [Google Scholar] [CrossRef] [PubMed]
- Parnetti, L.; Chiasserini, D.; Persichetti, E.; Eusebi, P.; Varghese, S.; Qureshi, M.M. Cerebrospinal fluid lysosomal enzymes and α-synuclein in Parkinson’s disease. Mov. Disord. 2014, 29, 1019–1027. [Google Scholar] [CrossRef] [PubMed]
- Holt, O.J.; Gallo, F.; Griffiths, G.M. Regulating secretory lysosomes. J. Biochem. 2006, 140, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Motta, M.; Imbesi, R.; di Rosa, M.; Stivala, F.; Malaguarnera, L. Altered plasma cytokine levels in Alzheimer’s disease: Correlation with the disease progression. Immunol. Lett. 2007, 30, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Pickford, F.; Masliah, E.; Britschgi, M.; Lucin, K.; Narasimhan, R.; Jaeger, P.A.; Small, S.; Spencer, B.; Rockenstein, E.; Levine, B.; et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J. Clin. Investig. 2008, 118, 2190–2199. [Google Scholar] [PubMed]
- Caccamo, A.; Majumder, S.; Richardson, A.; Strong, R.; Oddo, S. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and Tau: Effects on cognitive impairments. J. Biol. Chem. 2010, 285, 13107–13120. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.S.; Stavrides, P.; Mohan, P.S.; Kaushik, S.; Kumar, A.; Ohno, M.; Schmidt, S.D.; Wesson, D.; Bandyopadhyay, U.; Jiang, Y.; et al. Reversal of autophagy dysfunction in the TgCRND8 mouse model of Alzheimer’s disease ameliorates amyloid pathologies and memory deficits. Brain 2011, 134, 258–277. [Google Scholar] [CrossRef] [PubMed]
- Pedrinolla, A.; Schena, F.; Venturelli, M. Resilience to Alzheimer’s disease: The role of physical activity. Curr. Alzheimer Res. 2017, 14, 1–8. [Google Scholar] [CrossRef]
- McKhann, G.; Drachman, D.; Folstein, M.; Katzman, R.; Price, D.; Stadlan, E.M. Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984, 34, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Folstein, M.F.; Folstein, S.E.; McHugh, P.R. Mini-Mental State. A practical method for grading the cognitive state of patients for the clinicians. J. Psychiatr. Res. 1975, 12, 189–198. [Google Scholar] [CrossRef]
- Morris, J.C. Clinical Dementia rating: A reliable and valid diagnostic and staging measure for dementia of the Alzheimer type. Int. Psychogeriatr. 1997, 9, 173–176. [Google Scholar] [CrossRef] [PubMed]
- Chaves, M.L.; Camozzato, A.L.; Godinho, C.; Kochhann, R.; Schuh, A.; de Almeida, V.L.; Kaye, J. Validity of the clinical dementia rating scale for the detection and staging of dementia in Brazilian patients. Alzheimer Dis. Assoc. Disord. 2007, 21, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.L.; Mega, M.; Gray, K.; Rosenberg-Thompson, S.; Carusi, D.A.; Gornbein, J. The Neuropsychiatric Inventory: Comprehensive assessment of psychopathology in dementia. Neurology 1994, 44, 2308–2314. [Google Scholar] [CrossRef] [PubMed]
- Lawton, M.P.; Brody, E.M. Assessment of older people: Self-maintaining and instrumental activities of daily living. Gerontologist 1969, 9, 179–186. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PARAMETERS | CTR | MCI | MILD AD | SEVERE-AD |
|---|---|---|---|---|
| Weight (kg) | 73 ± 12 | 75 ± 19 | 73 ± 12 | 73 ± 13 |
| Height (m) | 1.67 ± 0.1 | 1.65 ± 0.1 | 1.58 ± 0.1 | 1.62 ± 0.1 |
| SBP (mm Hg) | 129 ± 33 | 136 ± 37 | 132 ± 12 | 130 ± 9 |
| DBP (mm Hg) | 90 ± 24 | 92 ± 12 | 86 ± 10 | 90 ± 5 |
| Glucose (mg∙dL−1) | 95 ± 32 | 107 ± 21 † | 91 ± 8 | 95 ± 47 |
| HDL (mg∙dL−1) | 50 ± 17 | 58 ± 19 | 57 ± 21 | 57 ± 11 |
| LDL (mg∙dL−1) | 100 ± 23 | 102 ± 15 | 122 ± 32 † | 110 ± 12 |
| Pharmacological treatment n. (%) | ||||
| Cholinesterase Inhibitors | 0 | 2 (10) † | 10 (42) † | 5 (25) † |
| Antipsychotics | 0 | 0 | 1 (4) | 1 (5) |
| Antidepressants | 0 | 0 | 2 (8) | 4 (20) † |
| Benzodiazepines | 0 | 0 | 0 | 1 (5) |
| Comorbidity n. (%) | ||||
| CVD | 0 | 1 (5) | 4 (16) † | 2 (10) † |
| Diabetes | 0 | 1 (5) | 0 | 1 (5) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morena, F.; Argentati, C.; Trotta, R.; Crispoltoni, L.; Stabile, A.; Pistilli, A.; Di Baldassarre, A.; Calafiore, R.; Montanucci, P.; Basta, G.; et al. A Comparison of Lysosomal Enzymes Expression Levels in Peripheral Blood of Mild- and Severe-Alzheimer’s Disease and MCI Patients: Implications for Regenerative Medicine Approaches. Int. J. Mol. Sci. 2017, 18, 1806. https://doi.org/10.3390/ijms18081806
Morena F, Argentati C, Trotta R, Crispoltoni L, Stabile A, Pistilli A, Di Baldassarre A, Calafiore R, Montanucci P, Basta G, et al. A Comparison of Lysosomal Enzymes Expression Levels in Peripheral Blood of Mild- and Severe-Alzheimer’s Disease and MCI Patients: Implications for Regenerative Medicine Approaches. International Journal of Molecular Sciences. 2017; 18(8):1806. https://doi.org/10.3390/ijms18081806
Chicago/Turabian StyleMorena, Francesco, Chiara Argentati, Rosa Trotta, Lucia Crispoltoni, Anna Stabile, Alessandra Pistilli, Angela Di Baldassarre, Riccardo Calafiore, Pia Montanucci, Giuseppe Basta, and et al. 2017. "A Comparison of Lysosomal Enzymes Expression Levels in Peripheral Blood of Mild- and Severe-Alzheimer’s Disease and MCI Patients: Implications for Regenerative Medicine Approaches" International Journal of Molecular Sciences 18, no. 8: 1806. https://doi.org/10.3390/ijms18081806
APA StyleMorena, F., Argentati, C., Trotta, R., Crispoltoni, L., Stabile, A., Pistilli, A., Di Baldassarre, A., Calafiore, R., Montanucci, P., Basta, G., Pedrinolla, A., Smania, N., Venturelli, M., Schena, F., Naro, F., Emiliani, C., Rende, M., & Martino, S. (2017). A Comparison of Lysosomal Enzymes Expression Levels in Peripheral Blood of Mild- and Severe-Alzheimer’s Disease and MCI Patients: Implications for Regenerative Medicine Approaches. International Journal of Molecular Sciences, 18(8), 1806. https://doi.org/10.3390/ijms18081806