Determination of Sphingosine-1-Phosphate in Human Plasma Using Liquid Chromatography Coupled with Q-Tof Mass Spectrometry

Abstract
:1. Introduction
2. Results
2.1. Optimising Chromatography
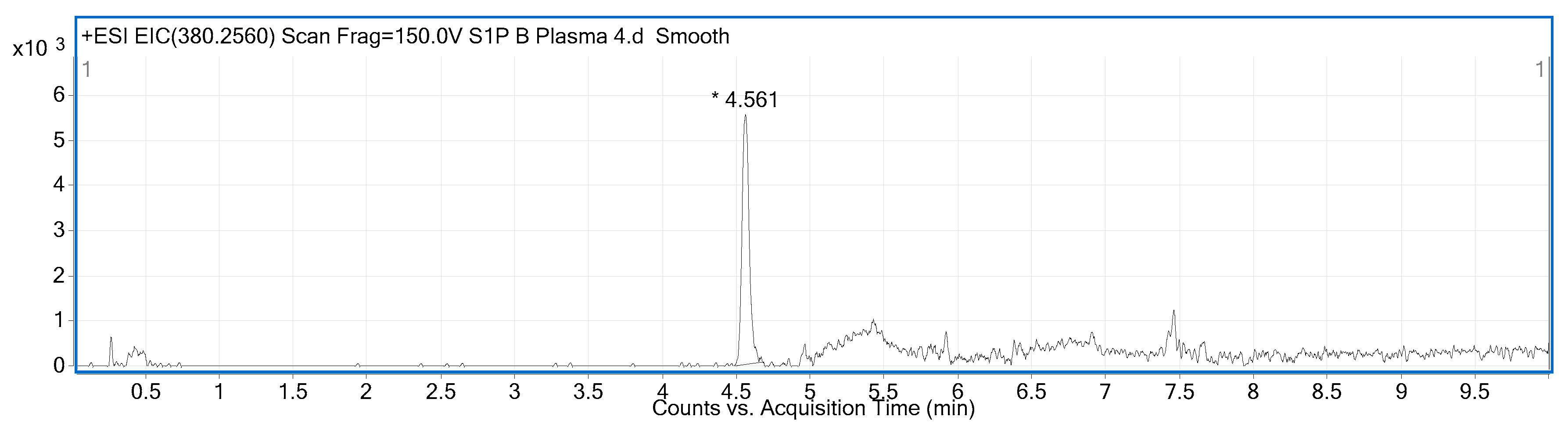
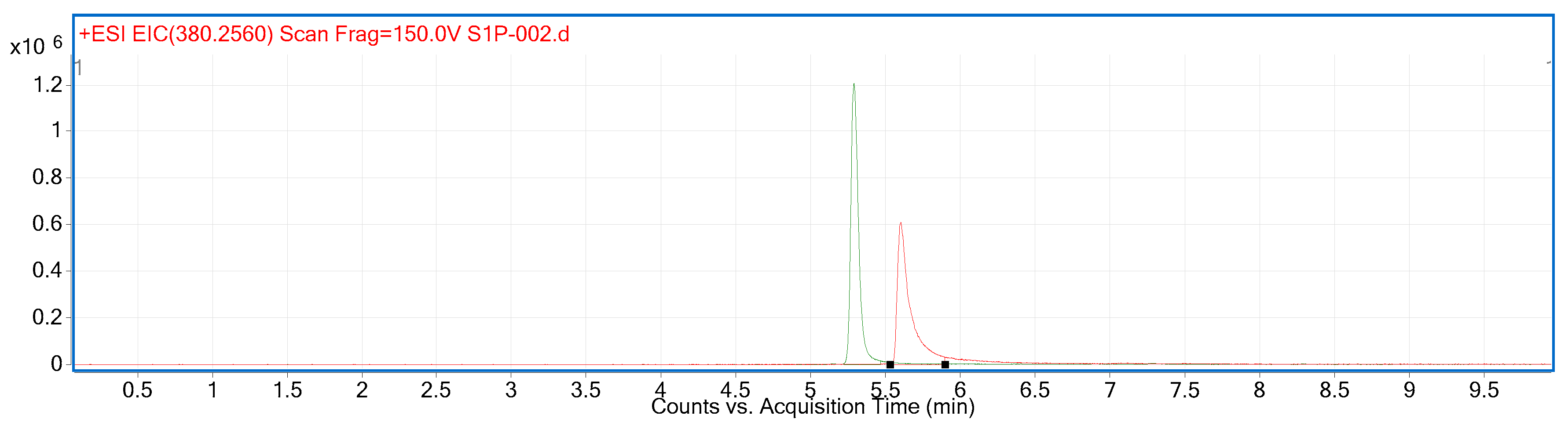
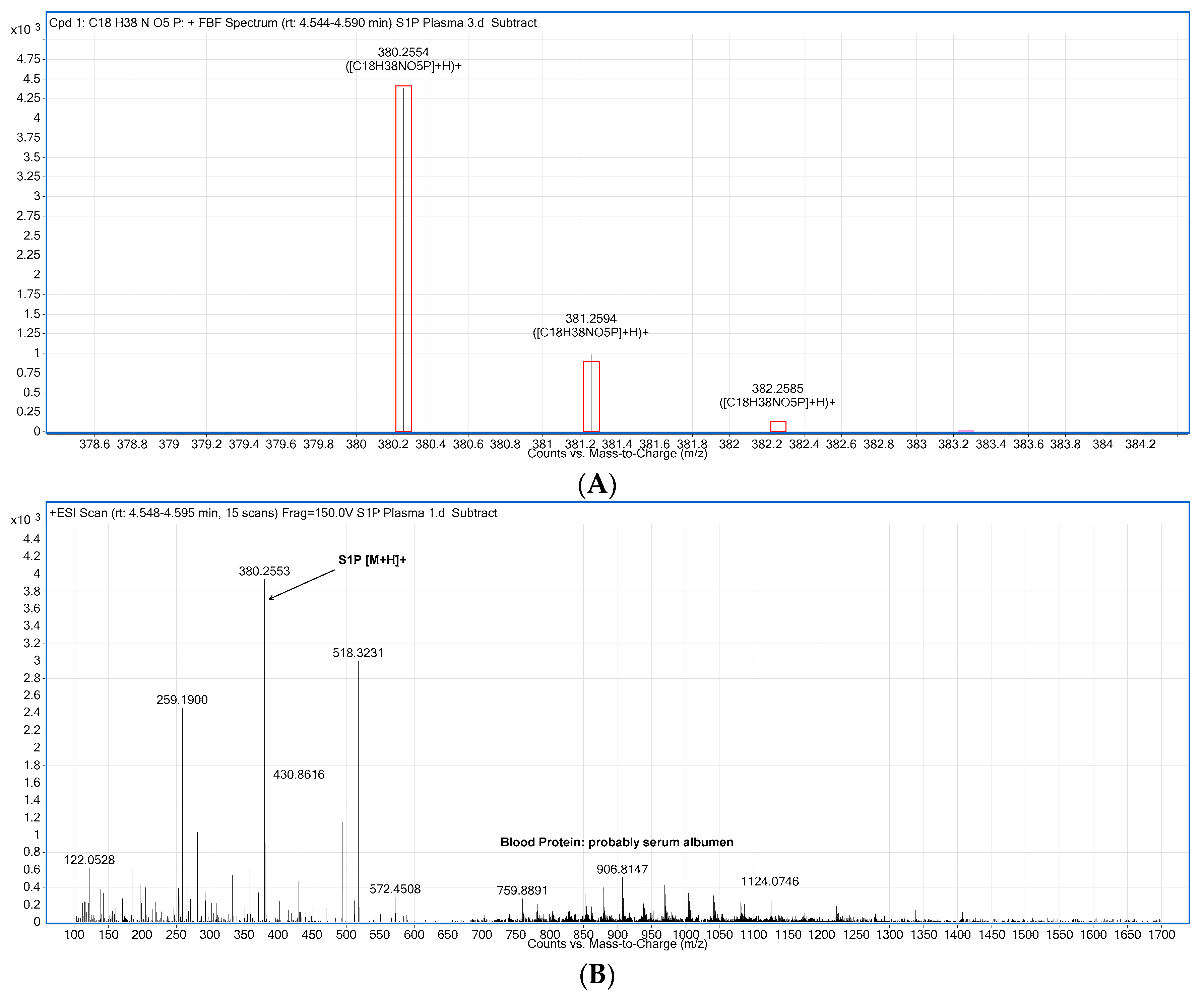
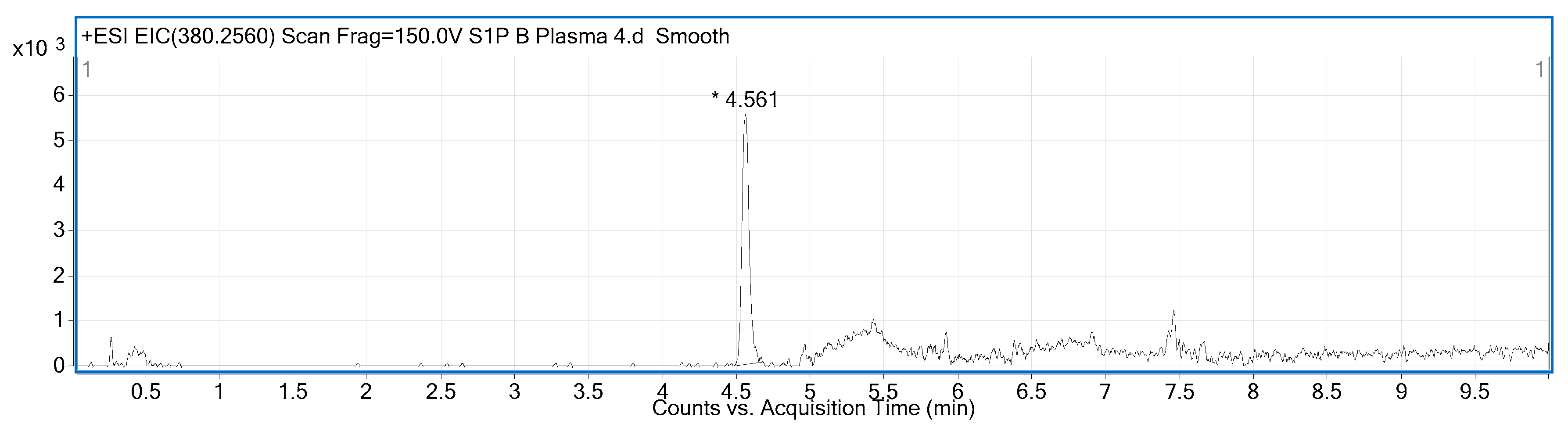
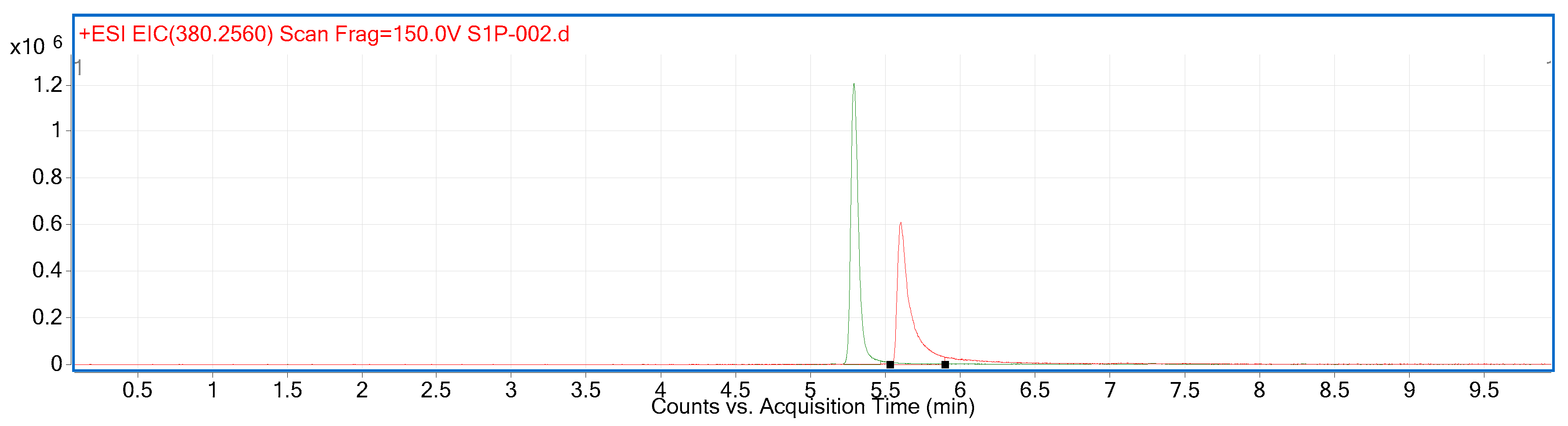
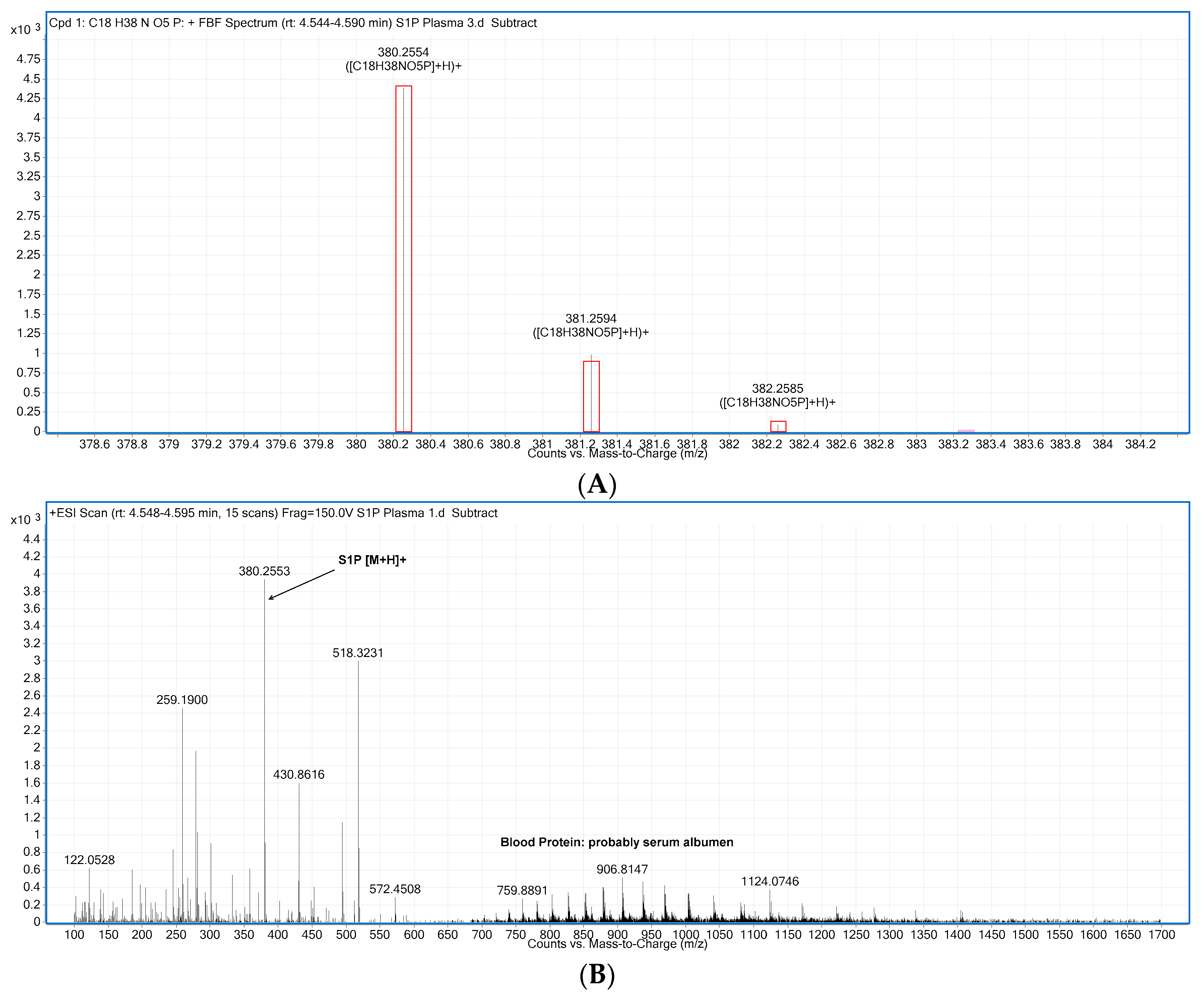
2.2. Compound Identification and Selectivity
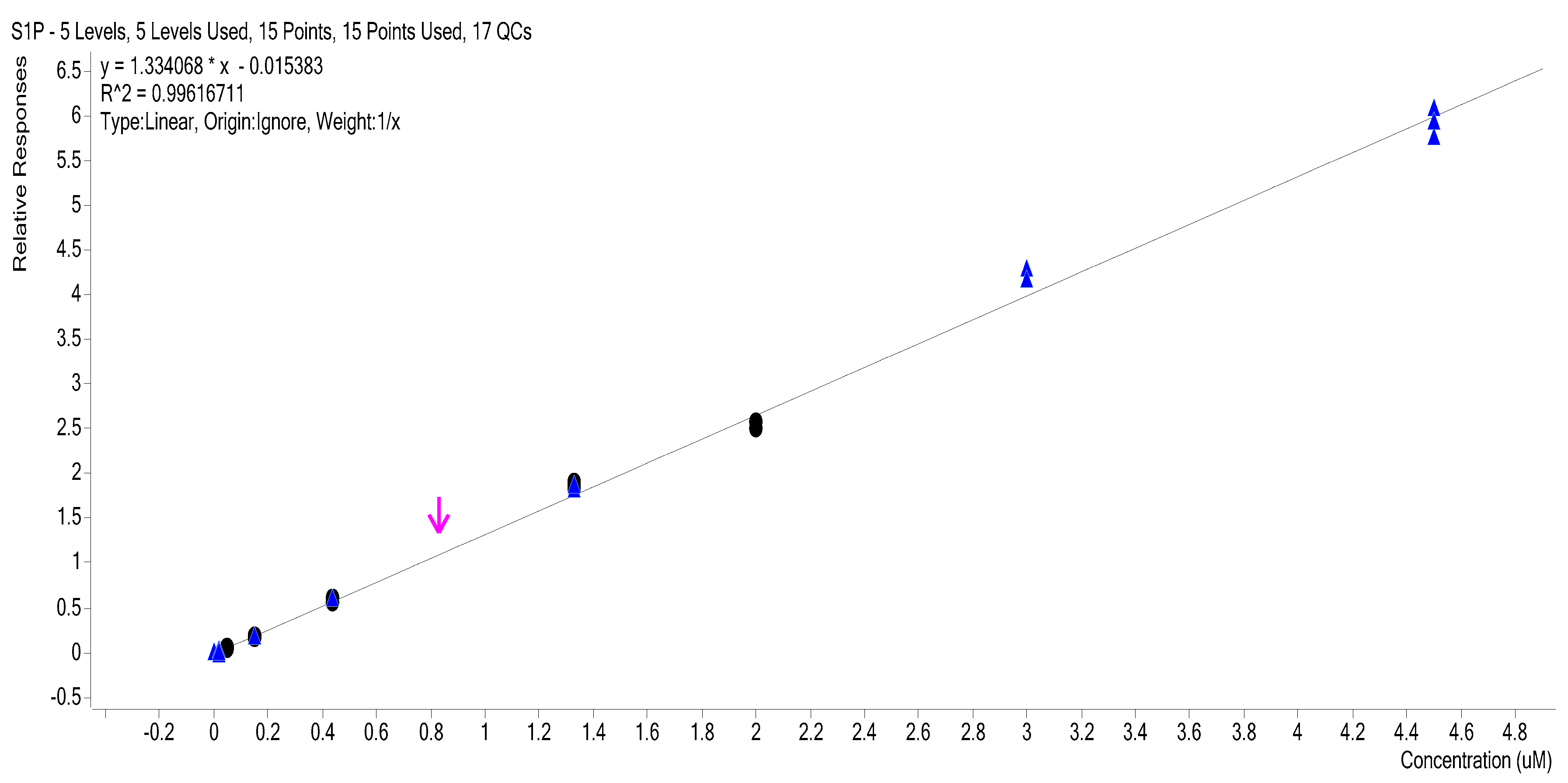
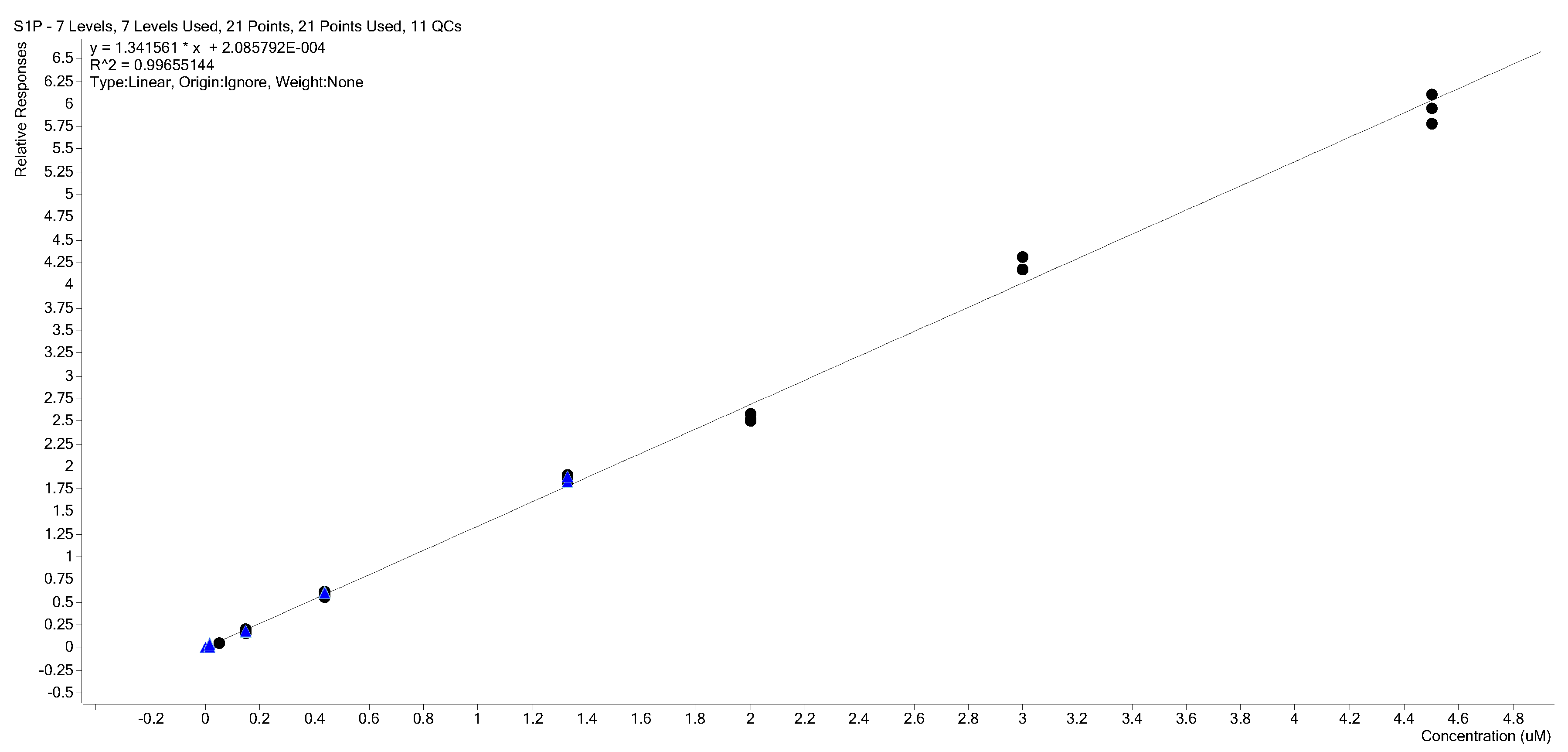
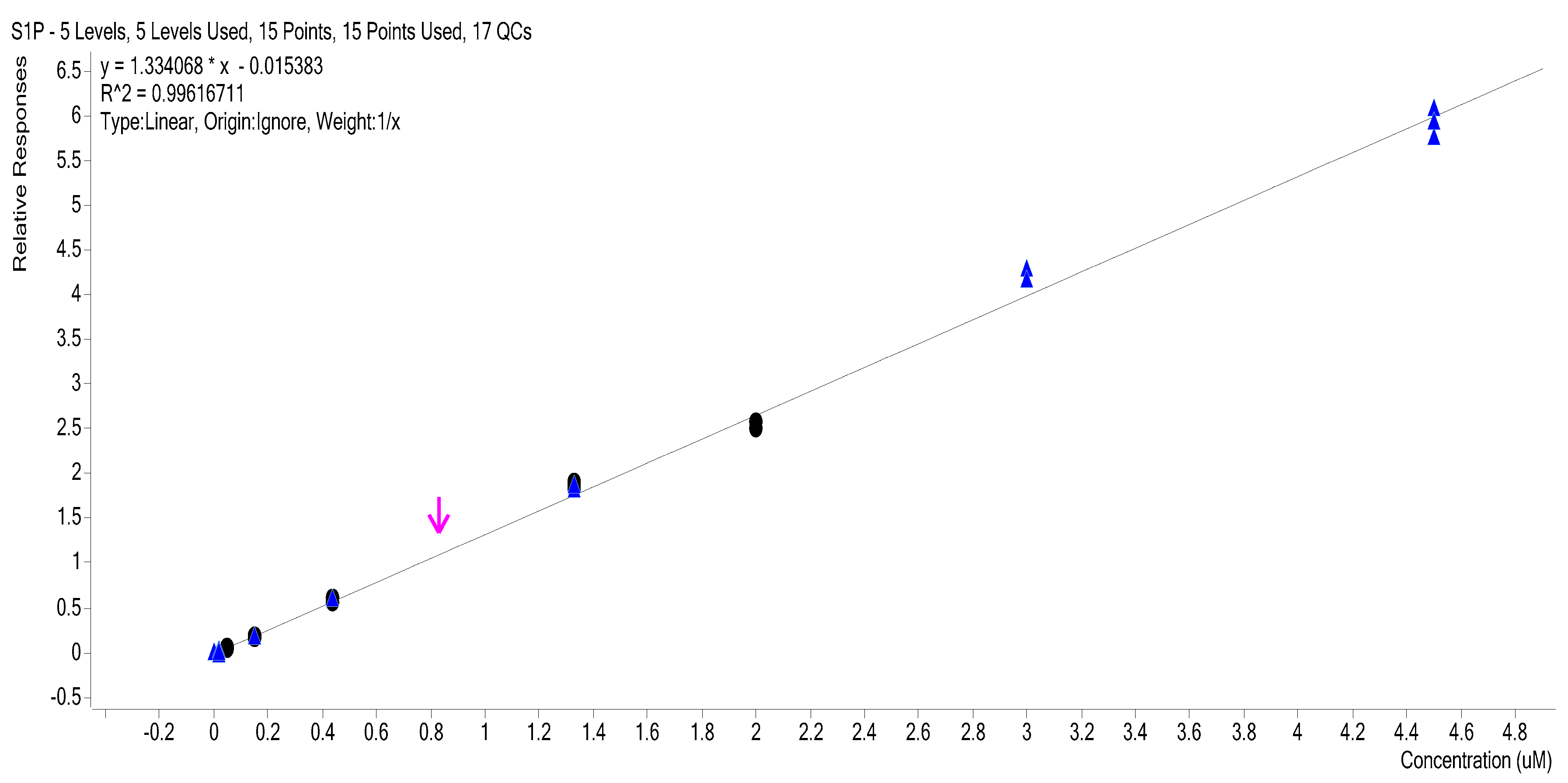
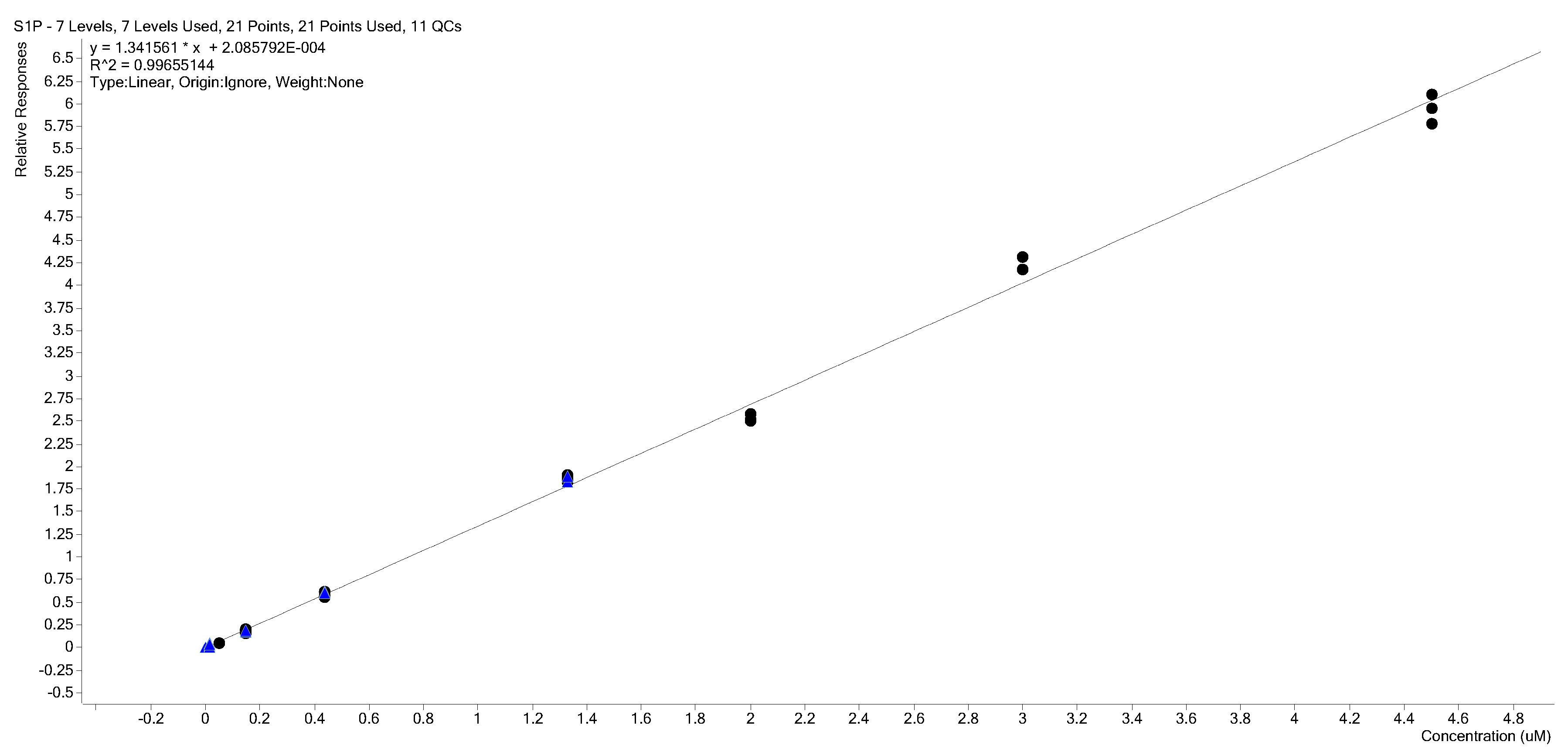
2.3. Calibration Curves
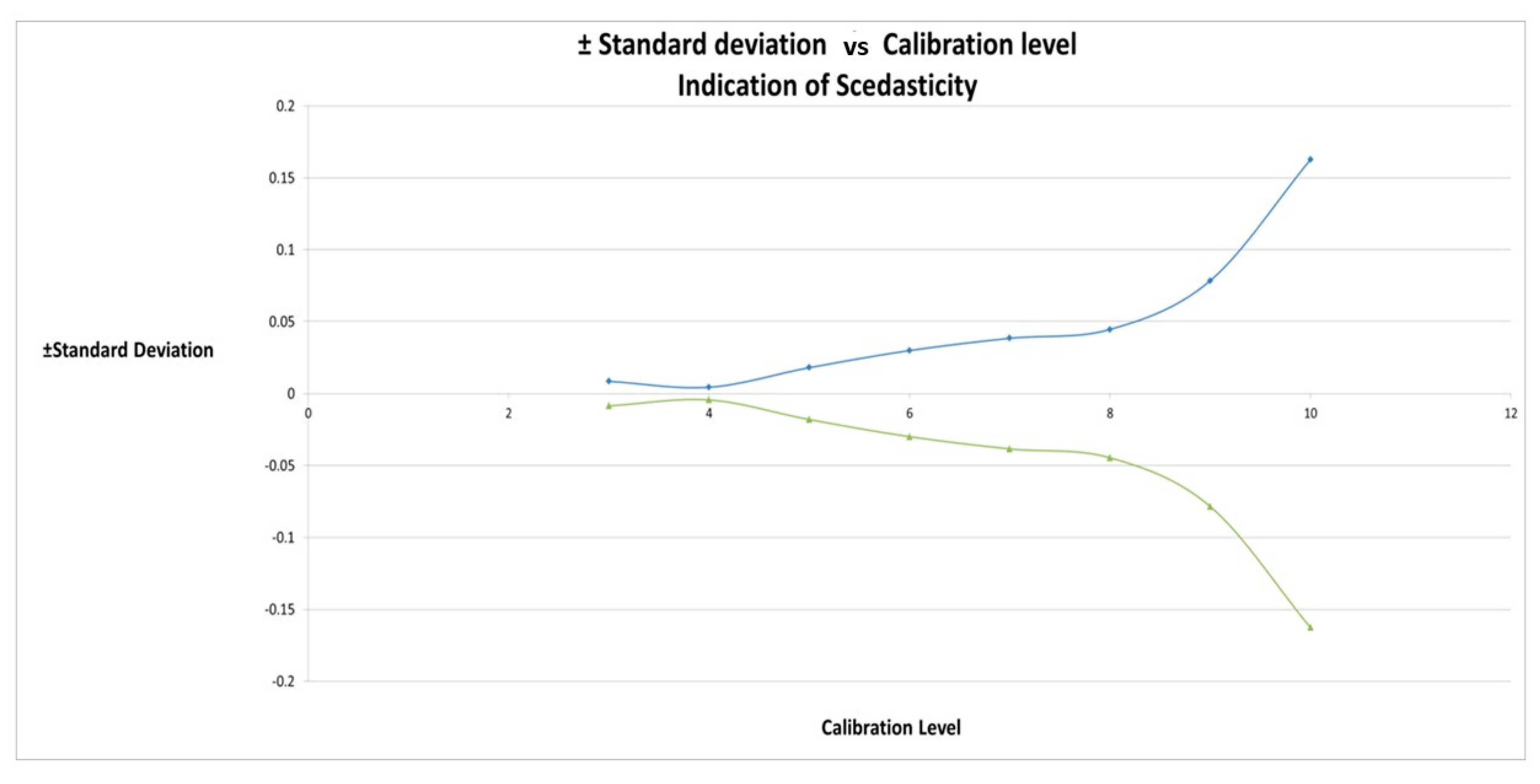
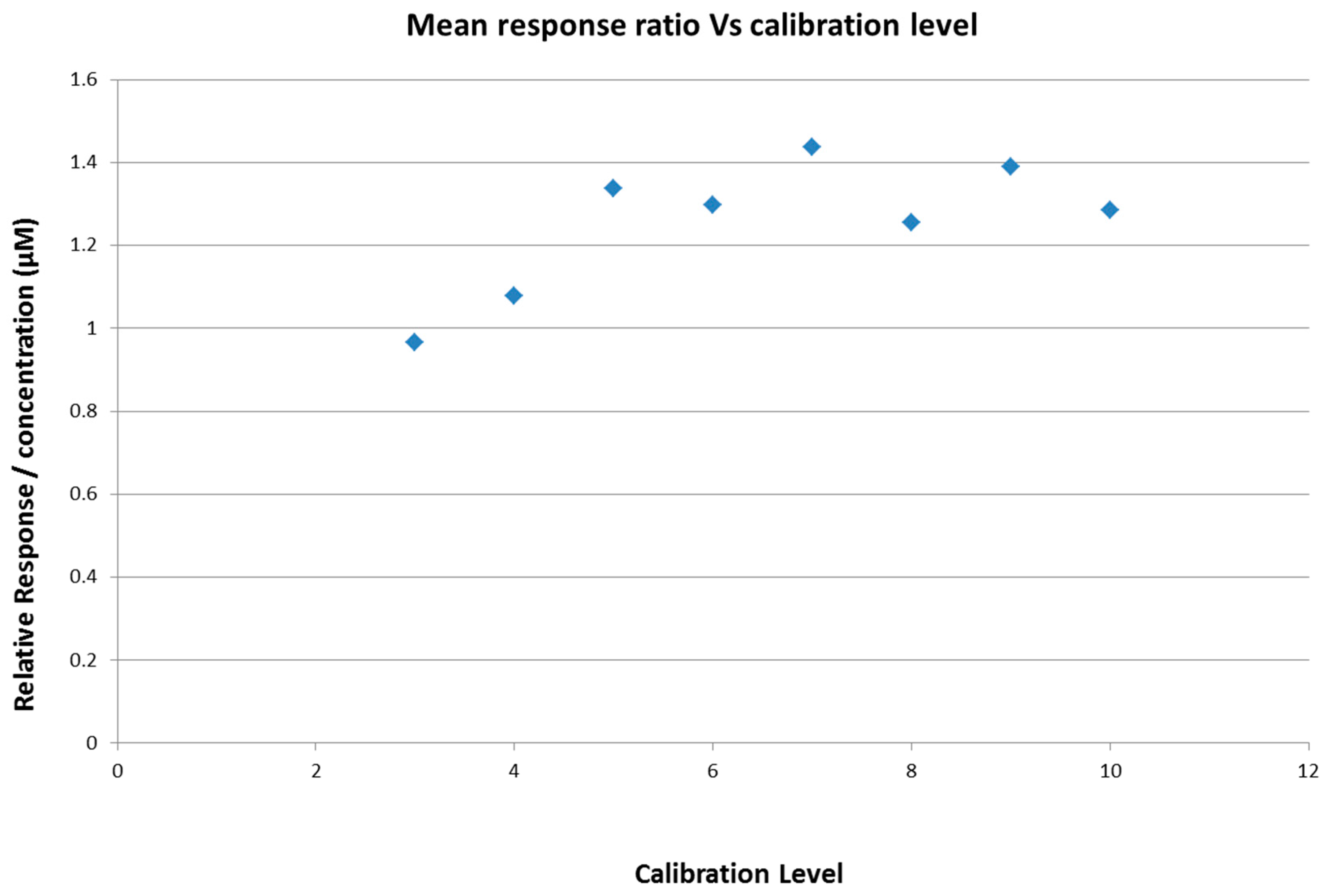
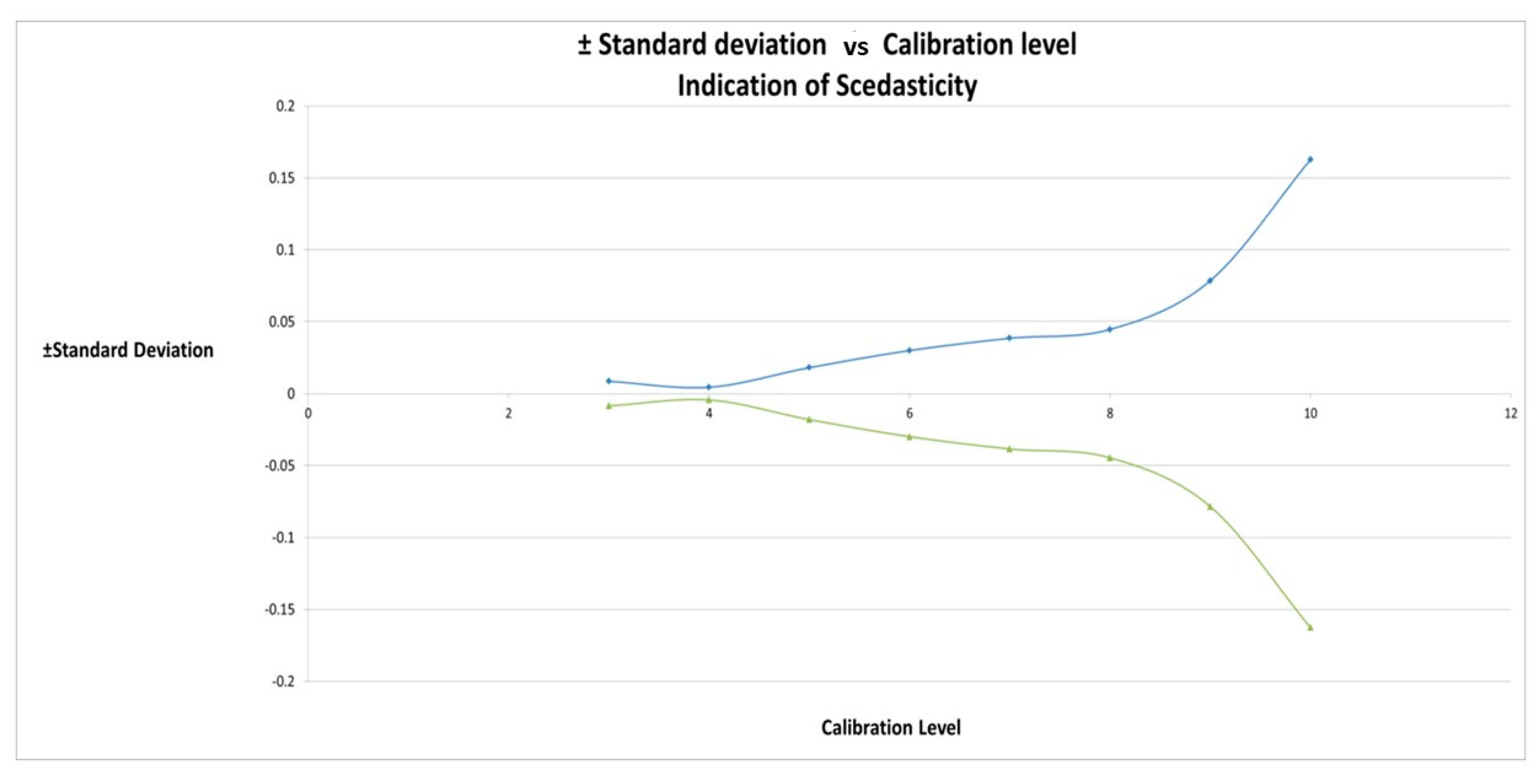
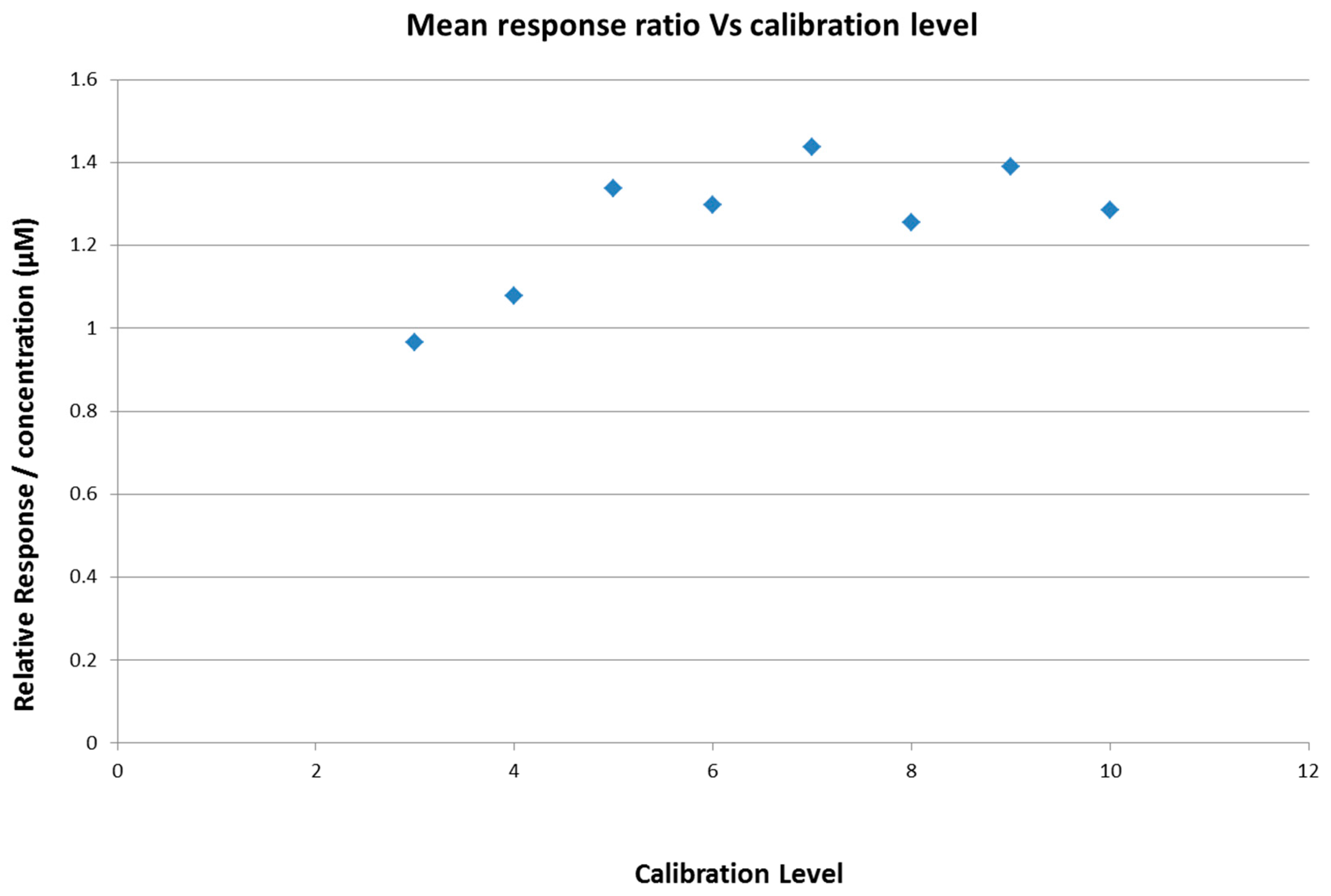
2.4. Calibration Model Selection
2.5. Range
2.6. Accuracy and Precision
2.7. Estimated Lower Level of Detection (LLOD) and LLOQ
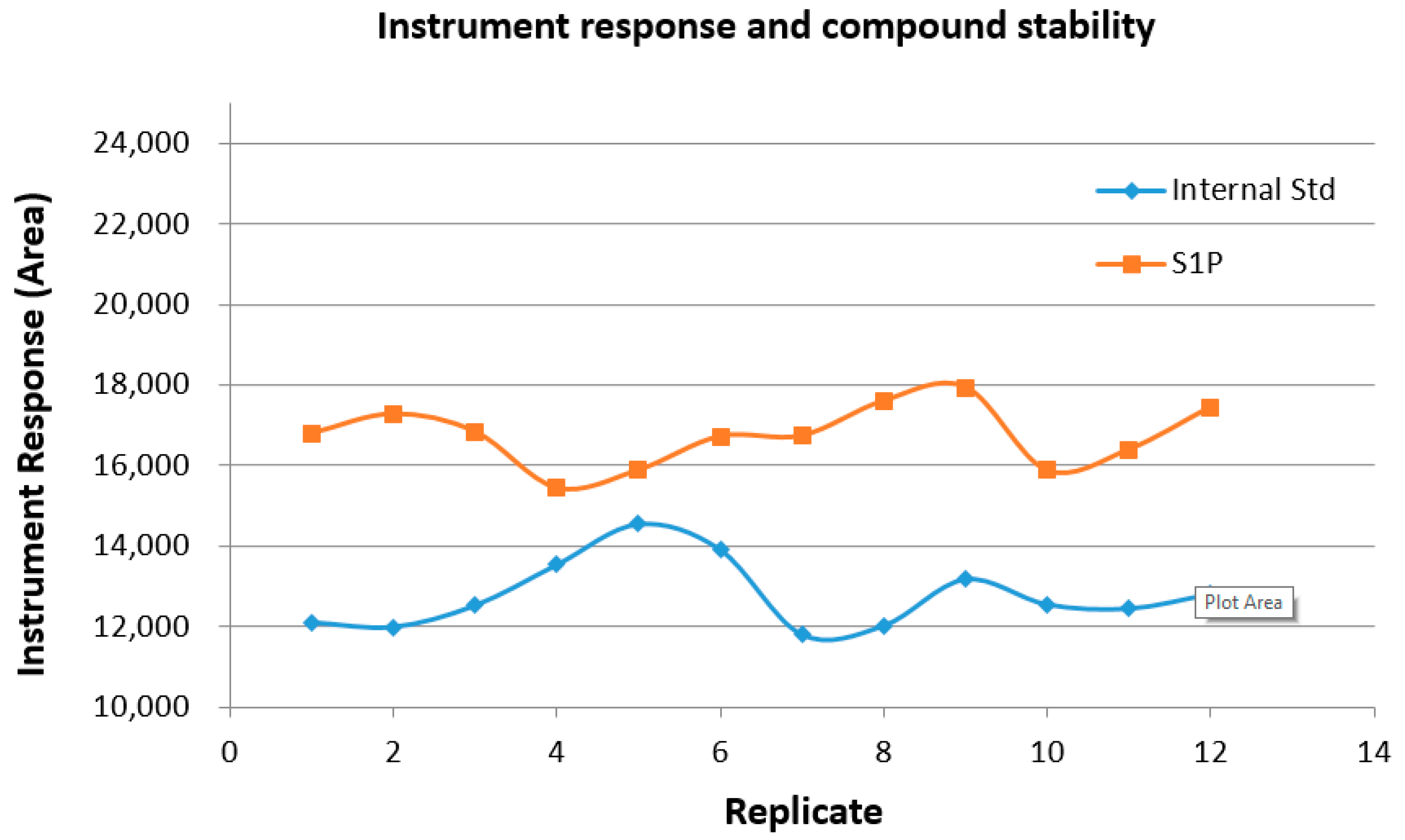
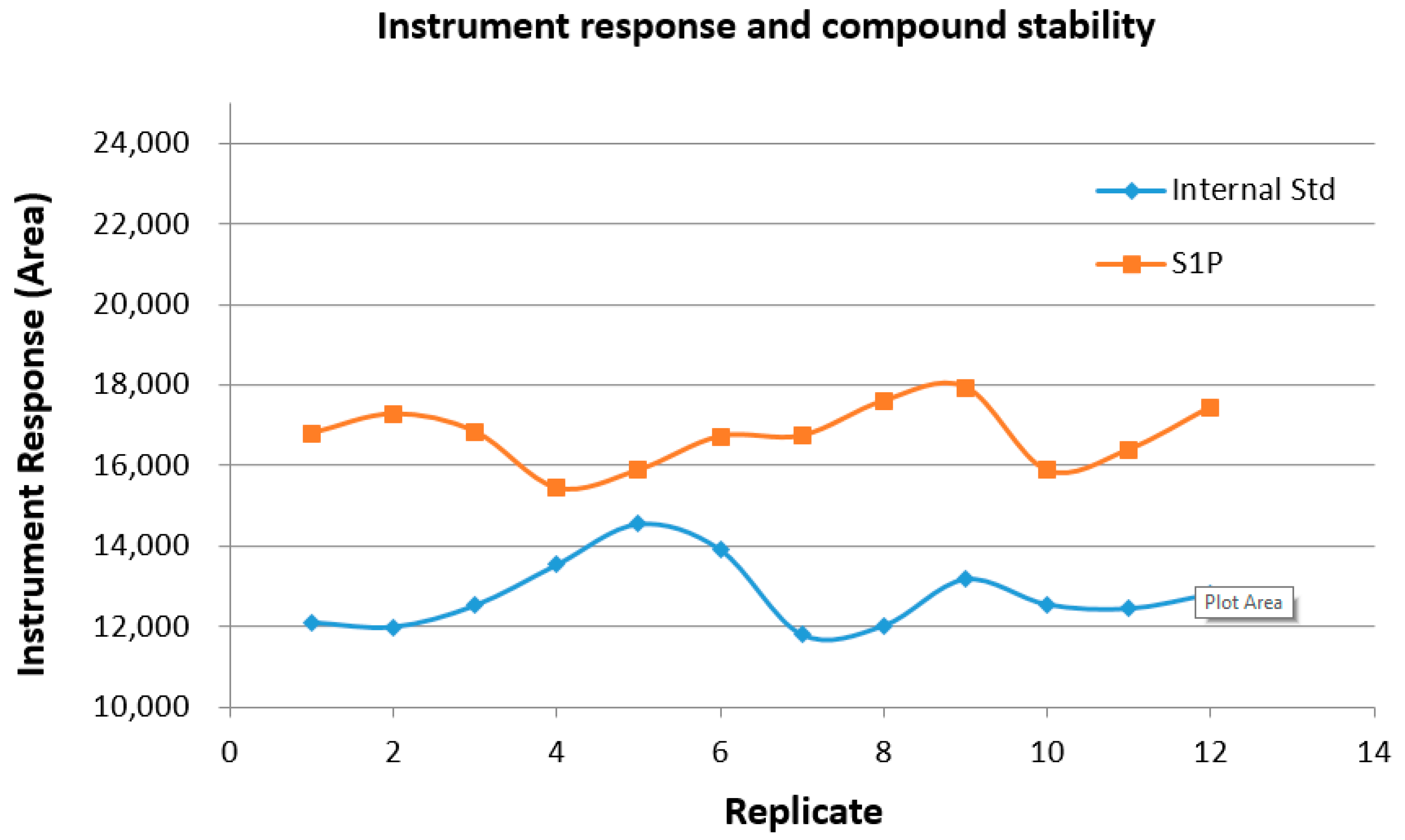
2.8. Sample and Instrument Stability over Time
2.9. Injection Carryover
3. Discussion
4. Materials and Methods
4.1. Study Population and Samples Preparation
4.2. Chemical, Reagents, and Instrumentation
4.3. Method Validation
4.3.1. Stability and Selectivity
4.3.2. Carryover
4.3.3. Linearity, Precision, and Accuracy
Acknowledgments
Author Contributions
Conflicts of Interest
References
- De Backer, G.; Ambrosionie, E.; Borch-Johnsen, K.; Brotons, C.; Cifkova, R.; Dallongeville, J.; Ebrahim, S.; Faergeman, O.; Graham, I.; Mancia, G. European guidelines on cardiovascular disease prevention in clinical practice: Third joint task force of european and other societies on cardiovascular disease prevention in clinical practice (constituted by representatives of eight societies and by invited experts). Eur. J. Cardiovasc. Prev. Rehabil. 2003, 10, S1–S10. [Google Scholar] [PubMed]
- World Health Organization. Cardiovascular Diseases (cvds). Fact Sheet n 317. September 2011. Available online: http://www.whoint/mediacentre/factsheets/fs317/en/index html (accessed on 5 November 2012).
- Reiner, Å.E.; Catapano, A.L.; De Backer, G.; Graham, I.; Taskinen, M.-R.; Wiklund, O.; Agewall, S.; Alegria, E.; Chapman, M.J.; Durrington, P. Esc/Eas guidelines for the management of dyslipidaemias. Eur. Heart J. 2011, 32, 1769–1818. [Google Scholar] [CrossRef] [PubMed]
- Gordon, T.; Castelli, W.P.; Hjortland, M.C.; Kannel, W.B.; Dawber, T.R. High density lipoprotein as a protective factor against coronary heart disease. The Framingham study. Am. J. Med. 1977, 62, 707–714. [Google Scholar] [CrossRef]
- Di Angelantonio, E.; Sarwar, N.; Perry, P.; Kaptoge, S.; Ray, K.K.; Thompson, A.; Wood, A.M.; Lewington, S.; Sattar, N.; Packard, C.J.; et al. Major lipids, apolipoproteins, and risk of vascular disease. JAMA 2009, 302, 1993–2000. [Google Scholar] [PubMed]
- Rosenson, R.S. Low HDL-C: A secondary target of dyslipidemia therapy. Am. J. Med. 2005, 118, 1067–1077. [Google Scholar] [CrossRef] [PubMed]
- Castelli, W.P. Cardiovascular disease and multifactorial risk: Challenge of the 1980s. Am. Heart J. 1983, 106, 1191–1200. [Google Scholar] [CrossRef]
- Nofer, J.R. Hyperlipidaemia and cardiovascular disease: The quantity does not turn into quality! Curr. Opin. Lipidol. 2013, 24, 366–368. [Google Scholar] [CrossRef] [PubMed]
- Rosenson, R.S.; Brewer, H.B., Jr.; Ansell, B.; Barter, P.; Chapman, M.J.; Heinecke, J.W.; Kontush, A.; Tall, A.R.; Webb, N.R. Translation of high-density lipoprotein function into clinical practice: Current prospects and future challenges. Circulation 2013, 128, 1256–1267. [Google Scholar] [PubMed]
- Egom, E.E.; Mamas, M.A.; Soran, H. Hdl quality or cholesterol cargo: What really matters—Spotlight on sphingosine-1-phosphate-rich hdl. Curr. Opin. Lipidol. 2013, 24, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Egom, E.E. Letter from egom regarding article, “high-density lipoprotein cholesterol, size, particle number, and residual vascular risk after potent statin therapy. Circulation 2014, 129, e480. [Google Scholar] [CrossRef] [PubMed]
- Egom, E.E. HDL-C/HDL-P ratio: A measure of reverse cholesterol transport rather than HDL functionality. J. Am. Coll. Cardiol. 2015, 65, 2576. [Google Scholar] [CrossRef] [PubMed]
- Rye, K.A.; Barter, P.J. Regulation of high-density lipoprotein metabolism. Circ. Res. 2014, 114, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Silva, R.A.; Jerome, W.G.; Kontush, A.; Chapman, M.J.; Curtiss, L.K.; Hodges, T.J.; Davidson, W.S. Apolipoprotein AI structural organization in high-density lipoproteins isolated from human plasma. Nat. Struct. Mol. Biol. 2011, 18, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.S.; Tan, L.; Long, J.L.; Davidson, W.S. Proteomic diversity of high density lipoproteins: Our emerging understanding of its importance in lipid transport and beyond. J. Lipid Res. 2013, 54, 2575–2585. [Google Scholar] [CrossRef] [PubMed]
- Vickers, K.C.; Palmisano, B.T.; Shoucri, B.M.; Shamburek, R.D.; Remaley, A.T. Micrornas are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat. Cell Biol. 2011, 13, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Poti, F.; Simoni, M.; Nofer, J.R. Atheroprotective role of high-density lipoprotein (HDL)-associated sphingosine-1-phosphate (S1P). Cardiovasc. Res. 2014, 103, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Sachinidis, A.; Kettenhofen, R.; Seewald, S.; Gouni-Berthold, I.; Schmitz, U.; Seul, C.; Ko, Y.; Vetter, H. Evidence that lipoproteins are carriers of bioactive factors. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 2412–2421. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Sato, K.; Kuwabara, A.; Tomura, H.; Ishiwara, M.; Kobayashi, I.; Ui, M.; Okajima, F. Sphingosine 1-phosphate may be a major component of plasma lipoproteins responsible for the cytoprotective actions in human umbilical vein endothelial cells. J. Biol. Chem. 2001, 276, 31780–31785. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Tomura, H.; Kuwabara, A.; Kimura, T.; Miura, S.; Noda, K.; Okajima, F.; Saku, K. Correlation of high density lipoprotein (HDL)-associated sphingosine 1-phosphate with serum levels of HDL-cholesterol and apolipoproteins. Atherosclerosis 2005, 178, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Blaho, V.A.; Hla, T. An update on the biology of sphingosine 1-phosphate receptors. J. Lipid Res. 2014, 55, 1596–1608. [Google Scholar] [CrossRef] [PubMed]
- Maceyka, M.; Harikumar, K.B.; Milstien, S.; Spiegel, S. Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol. 2012, 22, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Egom, E.E.; Mamas, M.A.; Clark, A.L. The potential role of sphingolipid-mediated cell signaling in the interaction between hyperglycemia, acute myocardial infarction and heart failure. Exp. Opin. Ther. Targets 2012, 16, 791–800. [Google Scholar] [CrossRef] [PubMed]
- Egom, E.E. Sphingosine-1-phosphate signalling as a therapeutic target for patients with abnormal glucose metabolism and ischaemic heart disease. J. Cardiovasc. Med. 2014, 15, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Egom, E.E.; Ke, Y.; Solaro, R.J.; Lei, M. Cardioprotection in ischemia/reperfusion injury: Spotlight on sphingosine-1-phosphate and bradykinin signalling. Prog. Biophys. Mol. Biol. 2010, 103, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Bode, C.; Graler, M.H. Quantification of sphingosine-1-phosphate and related sphingolipids by liquid chromatography coupled to tandem mass spectrometry. Methods Mol. Biol. 2012, 874, 33–44. [Google Scholar] [PubMed]
- Ceglarek, U.; Dittrich, J.; Helmschrodt, C.; Wagner, K.; Nofer, J.R.; Thiery, J.; Becker, S. Preanalytical standardization of sphingosine-1-phosphate, sphinganine-1-phosphate and sphingosine analysis in human plasma by liquid chromatography-tandem mass spectrometry. Clin. Chim. Acta 2014, 435, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Lan, T.; Bi, H.; Liu, W.; Xie, X.; Xu, S.; Huang, H. Simultaneous determination of sphingosine and sphingosine 1-phosphate in biological samples by liquid chromatography-tandem mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2011, 879, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Liebisch, G.; Scherer, M. Quantification of bioactive sphingo- and glycerophospholipid species by electrospray ionization tandem mass spectrometry in blood. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2012, 883–884, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Luth, A.; Neuber, C.; Kleuser, B. Novel methods for the quantification of (2e)-hexadecenal by liquid chromatography with detection by either ESI QTOF tandem mass spectrometry or fluorescence measurement. Anal. Chim. Acta 2012, 722, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Saigusa, D.; Shiba, K.; Inoue, A.; Hama, K.; Okutani, M.; Iida, N.; Saito, M.; Suzuki, K.; Kaneko, T.; Suzuki, N.; et al. Simultaneous quantitation of sphingoid bases and their phosphates in biological samples by liquid chromatography/electrospray ionization tandem mass spectrometry. Anal. Bioanal. Chem. 2012, 403, 1897–1905. [Google Scholar] [CrossRef] [PubMed]
- Frej, C.; Andersson, A.; Larsson, B.; Guo, L.J.; Norstrom, E.; Happonen, K.E.; Dahlback, B. Quantification of sphingosine 1-phosphate by validated LC-MS/MS method revealing strong correlation with apolipoprotein m in plasma but not in serum due to platelet activation during blood coagulation. Anal. Bioanal. Chem. 2015, 407, 8533–8542. [Google Scholar] [CrossRef] [PubMed]
- Van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Bourquin, F.; Capitani, G.; Grütter, M.G. PLP-dependent enzymes as entry and exit gates of sphingolipid metabolism. Protein Sci. 2011, 20, 1492–1508. [Google Scholar] [CrossRef] [PubMed]
- Helsel, D.R.; Hirsch, R.M. Statistical Methods in Water Resources; Elsevier: Amsterdam, The Netherlands, 1992; p. 49. [Google Scholar]
- Bernal, E. Limit of detection and limit of quantification determination in gas chromatography. Adv. Gas Chromatogr. 2014, 3, 57–63. [Google Scholar]
- Saigusa, D.; Okudaira, M.; Wang, J.; Kano, K.; Kurano, M.; Uranbileg, B.; Ikeda, H.; Yatomi, Y.; Motohashi, H.; Aoki, J. Simultaneous quantification of sphingolipids in small quantities of liver by LC-MS/MS. Mass Spectrom 2014, 3, S0046. [Google Scholar] [CrossRef] [PubMed]
- Scherer, M.; Schmitz, G.; Liebisch, G. High-throughput analysis of sphingosine 1-phosphate, sphinganine 1-phosphate, and lysophosphatidic acid in plasma samples by liquid chromatography-tandem mass spectrometry. Clin. Chem. 2009, 55, 1218–1222. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.; Schmidt, R.; Geisslinger, G. LC-MS/MS-analysis of sphingosine-1-phosphate and related compounds in plasma samples. Prostaglandins Other Lipid Mediat. 2006, 81, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Hu, J.; Li, Y. Simultaneous determination of syl-1119 and syl-1119-p in rat plasma using HPLC coupled with tandem mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2014, 945–946, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Samiei, N.; Foroutan, S.M.; Shafaati, A.; Zarghi, A. Development and validation of an HPLC method for determination of amifostine and/or its metabolite (WR-1065) in human plasma using OPA derivatization and UV detection. Iran. J. Pharm. Res. IJPR 2015, 14, 1051. [Google Scholar] [PubMed]
- Majors, R.E. The cleaning and regeneration of reversed-phase HPLC columns. LC GC N. Am. 2003, 21, 19–27. [Google Scholar]
- Li, P.; Bartlett, M.G. A review of sample preparation methods for quantitation of small-molecule analytes in brain tissue by liquid chromatography tandem mass spectrometry (LC-MS/MS). Anal. Methods 2014, 6, 6183–6207. [Google Scholar] [CrossRef]
- Viswanathan, C.; Bansal, S.; Booth, B.; DeStefano, A.J.; Rose, M.J.; Sailstad, J.; Shah, V.P.; Skelly, J.P.; Swann, P.G.; Weiner, R. Quantitative bioanalytical methods validation and implementation: Best practices for chromatographic and ligand binding assays. Pharm. Res. 2007, 24, 1962–1973. [Google Scholar] [CrossRef] [PubMed]
- Uney, K.; Altan, F.; Elmas, M. Development and validation of a high-performance liquid chromatography method for determination of cefquinome concentrations in sheep plasma and its application to pharmacokinetic studies. Antimicrob. Agents Chemother. 2011, 55, 854–859. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Level | 1/x Weighted Curve Levels 4–8 3 Replicates (R2: 0.9962) | |||||||
|---|---|---|---|---|---|---|---|---|
| Known Conc | Calculated Conc | Accuracy | Conc Mean | Mean Accuracy | Conc SD | Precision | ||
| (μM) | (μM) | % | (μM) | % | (μM) | RSD % | ||
| 1 | 0.0016 | 0.0141 | 883.8 | |||||
| 2 | 0.005 | |||||||
| 3 | 0.016 | 0.014 | 87.7 | |||||
| 3 | 0.016 | 0.036 | 225.2 | |||||
| 3 | 0.016 | 0.05 | 312.5 | |||||
| 3 | 0.016 | 0.0231 | 144.4 | 0.036 | 157.43 | 0.011 | 30.21 | |
| 4 | 0.05 | Cal | 0.0453 | 90.7 | ||||
| 4 | 0.05 | Cal | 0.0482 | 96.4 | ||||
| 4 | 0.05 | Cal | 0.0519 | 103.8 | 0.048 | 93.38 | 0.003 | 5.57 |
| 5 | 0.15 | Cal | 0.1352 | 90.1 | ||||
| 5 | 0.15 | Cal | 0.1523 | 101.5 | ||||
| 5 | 0.15 | Cal | 0.1619 | 107.9 | 0.150 | 92.53 | 0.011 | 7.37 |
| 6 | 0.44 | Cal | 0.47 | 106.8 | ||||
| 6 | 0.44 | Cal | 0.4262 | 96.9 | ||||
| 6 | 0.44 | Cal | 0.4393 | 99.8 | 0.445 | 101.34 | 0.018 | 4.12 |
| 7 | 1.33 | Cal | 1.3863 | 104.2 | ||||
| 7 | 1.33 | Cal | 1.4165 | 106.5 | ||||
| 7 | 1.33 | Cal | 1.4439 | 108.6 | 1.416 | 98.04 | 0.024 | 1.66 |
| 8 | 2.00 | Cal | 1.8894 | 94.5 | ||||
| 8 | 2.00 | Cal | 1.9494 | 97.5 | ||||
| 8 | 2.00 | Cal | 1.894 | 94.7 | 1.911 | 100.89 | 0.027 | 1.43 |
| 9 | 3.00 | 3.1447 | 104.8 | |||||
| 9 | 3.00 | 3.2439 | 108.1 | |||||
| 9 | 3.00 | 3.1393 | 104.6 | 3.176 | 101.17 | 0.048 | 1.51 | |
| 10 | 4.50 | 4.4759 | 99.5 | |||||
| 10 | 4.50 | 4.5893 | 102 | |||||
| 10 | 4.50 | 4.3456 | 96.6 | 4.470 | 102.87 | 0.100 | 2.23 | |
| Level | Known Conc | Calculated Conc | Accuracy | Conc Mean | Mean Accuracy | Conc SD | Precision |
|---|---|---|---|---|---|---|---|
| (µM) | (µM) | % | (µM) | % | (µM) | RSD % | |
| 5 | 0.15 | 0.1561 | 104.1 | ||||
| 5 | 0.15 | 0.1454 | 96.9 | 0.151 | 103.7 | 0.008 | 5.02 |
| 6 | 0.44 | 0.4672 | 106.2 | ||||
| 6 | 0.44 | 0.4771 | 108.4 | 0.472 | 99.0 | 0.007 | 1.48 |
| 7 | 1.33 | 1.4309 | 107.6 | ||||
| 7 | 1.33 | 1.3808 | 103.8 | 1.406 | 101.8 | 0.035 | 2.52 |
| Mean Accuracy | 104.5 | ||||||
| ± | 4.2 |
| Sample | RT (min) | Response (area) | Calculated | |||||
|---|---|---|---|---|---|---|---|---|
| S1P | IS | Conc (μM) | Mean (μM) | SD | CV (%) | |||
| 1 | S1P Plasma 1-1.d | 4.502 | 16,799 | 12,098 | 1.05 | |||
| 2 | S1P Plasma 1-2.d | 4.499 | 17,279 | 11,983 | 1.09 | |||
| 3 | S1P Plasma 1-3.d | 4.498 | 16,837 | 12,520 | 1.02 | 1.05 | 0.04 | 3.46 |
| 4 | S1P Plasma 2-1.d | 4.502 | 15,446 | 13,537 | 0.87 | |||
| 5 | S1P Plasma 2-2.d | 4.498 | 15,889 | 14,551 | 0.83 | |||
| 6 | S1P Plasma 2-3.d | 4.496 | 16,723 | 13,915 | 0.91 | 0.87 | 0.04 | 4.75 |
| 7 | S1P Plasma 3-1.d | 4.499 | 16,739 | 11,792 | 1.08 | |||
| 8 | S1P Plasma 3-2.d | 4.501 | 17,601 | 12,009 | 1.11 | |||
| 9 | S1P Plasma 3-3.d | 4.493 | 17,945 | 13,172 | 1.03 | 1.07 | 0.04 | 3.61 |
| 10 | S1P Plasma 4-1.d | 4.504 | 15,883 | 12,545 | 0.96 | |||
| 11 | S1P Plasma 4-2.d | 4.503 | 16,393 | 12,439 | 1.00 | |||
| 12 | S1P Plasma 4-3.d | 4.494 | 17,445 | 12,800 | 1.03 | 1.00 | 0.04 | 3.64 |
| Mean (μM) | 16,748.25 | 12,780.08 | 1.00 | |||||
| SD | 753.58 | 856.09 | 0.09 | |||||
| RSD (%) | 4.50 | 6.70 | 8.93 | |||||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Egom, E.E.; Fitzgerald, R.; Canning, R.; Pharithi, R.B.; Murphy, C.; Maher, V. Determination of Sphingosine-1-Phosphate in Human Plasma Using Liquid Chromatography Coupled with Q-Tof Mass Spectrometry. Int. J. Mol. Sci. 2017, 18, 1800. https://doi.org/10.3390/ijms18081800
Egom EE, Fitzgerald R, Canning R, Pharithi RB, Murphy C, Maher V. Determination of Sphingosine-1-Phosphate in Human Plasma Using Liquid Chromatography Coupled with Q-Tof Mass Spectrometry. International Journal of Molecular Sciences. 2017; 18(8):1800. https://doi.org/10.3390/ijms18081800
Chicago/Turabian StyleEgom, Emmanuel E., Ross Fitzgerald, Rebecca Canning, Rebabonye B. Pharithi, Colin Murphy, and Vincent Maher. 2017. "Determination of Sphingosine-1-Phosphate in Human Plasma Using Liquid Chromatography Coupled with Q-Tof Mass Spectrometry" International Journal of Molecular Sciences 18, no. 8: 1800. https://doi.org/10.3390/ijms18081800
APA StyleEgom, E. E., Fitzgerald, R., Canning, R., Pharithi, R. B., Murphy, C., & Maher, V. (2017). Determination of Sphingosine-1-Phosphate in Human Plasma Using Liquid Chromatography Coupled with Q-Tof Mass Spectrometry. International Journal of Molecular Sciences, 18(8), 1800. https://doi.org/10.3390/ijms18081800