The Retinoblastoma (RB) Tumor Suppressor: Pushing Back against Genome Instability on Multiple Fronts



Abstract
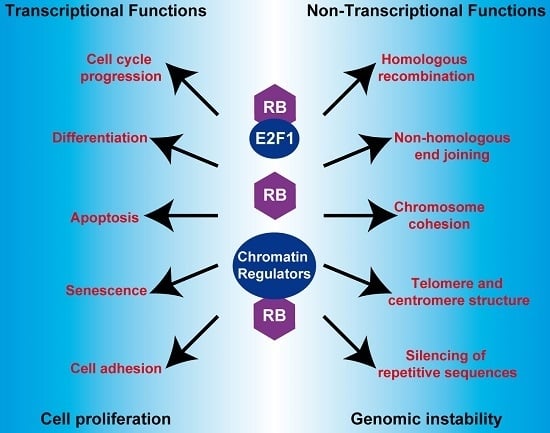
:
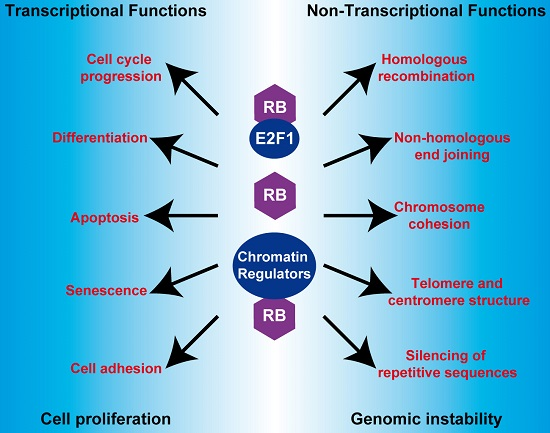{kind=link}
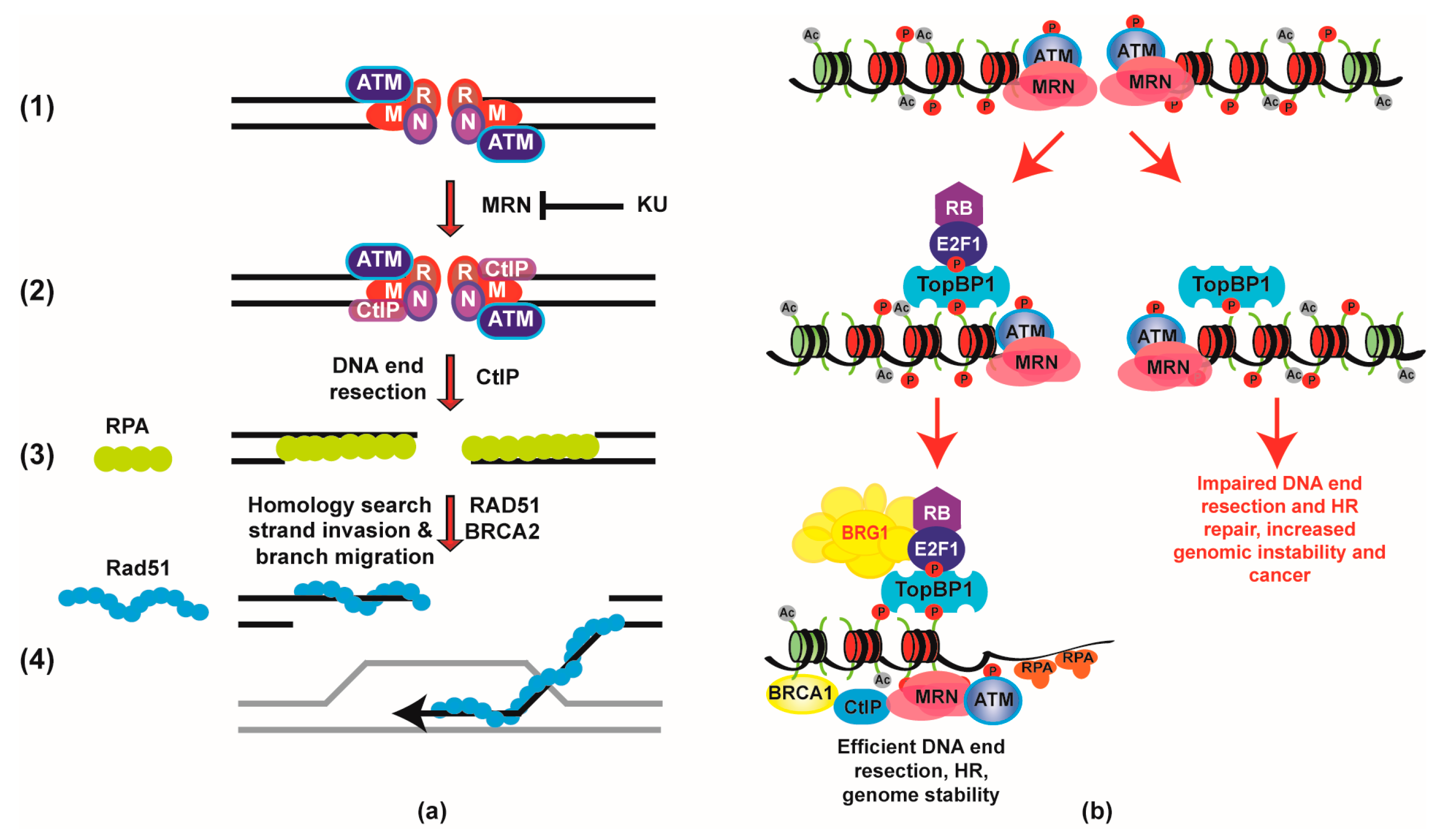{kind=link}
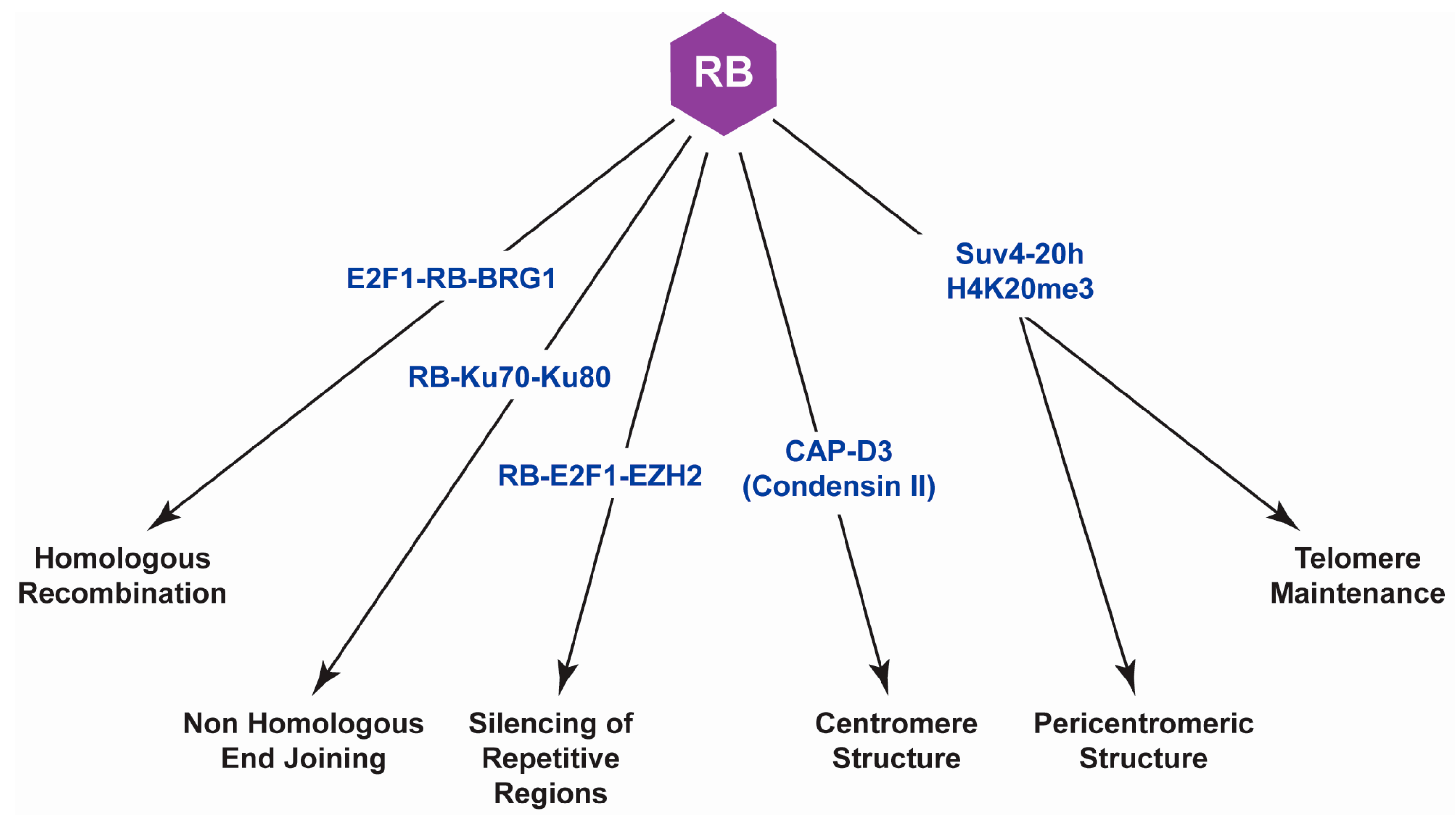{kind=link}
1. Introduction
2. Retinoblastoma (RB) Role in Double Strand Breaks (DSB) Repair
3. RB Role Silencing Repetitive Sequences
4. RB Role in Telomere Maintenance
5. RB Role in Centromere Structure and Chromosome Instability (CIN)
6. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| RB | Retinoblastoma |
| CDK | Cyclin-dependent kinases |
| PP1 | Protein phosphatase 1 |
| HAT | histone acetyltransferases |
| HDAC | Histone deacetylase |
| BRCA1 | Breast cancer susceptibility gene 1 |
| RPA | Replication protein A |
| ATM | Ataxia telangiectasia mutated kinase |
| ATR | Ataxia telangiectasia RAD3-related kinase |
| DSB | DNA double strand breaks |
| IR | Ionizing radiation |
| HR | Homologous recombination |
| NHEJ | Non-homologous end joining |
| PARPi | Poly (ADP-ribose) polymerase inhibitors |
| MRN | MRE11-RAD50-NBS1 complex |
| ssDNA | Single-stranded DNA |
| Chk1 | Checkpoint kinase 1 |
| BRCT | BRCA1 C terminal domain |
| MEF | Mouse embryonic fibroblasts |
| NER | Nucleotide excision repair |
| DNA-PK | DNA protein kinase |
| DKO | Double knock-out (p107−/−; p130−/−) |
| TKO | Triple knock-out (RB−/−;p107−/−; p130−/−) |
| CIN | Chromosome instability |
References
- Dick, F.A.; Rubin, S.M. Molecular mechanisms underlying RB protein function. Nat. Rev. Mol. Cell Biol. 2013, 14, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Chinnam, M.; Goodrich, D.W. RB1, development, and cancer. Curr. Top. Dev. Biol. 2011, 94, 129–169. [Google Scholar] [PubMed]
- Van Den Heuvel, S.; Dyson, N.J. Conserved functions of the pRB and E2F families. Nat. Rev. Mol. Cell Biol. 2008, 9, 713–724. [Google Scholar] [CrossRef] [PubMed]
- Burke, J.R.; Deshong, A.J.; Pelton, J.G.; Rubin, S.M. Phosphorylation-induced conformational changes in the Retinoblastoma Protein Inhibit E2F Transactivation Domain Binding. J. Biol. Chem. 2010, 285, 16286–16293. [Google Scholar] [CrossRef] [PubMed]
- Buchkovich, K.; Duffy, L.A.; Harlow, E. The retinoblastoma protein is phosphorylated during specific phases of the cell cycle. Cell 1989, 58, 1097–1105. [Google Scholar] [CrossRef]
- Narasimha, A.M.; Kaulich, M.; Shapiro, G.S.; Choi, Y.J.; Sicinski, P.; Dowdy, S.F. Cyclin D activates the RB tumor suppressor by mono-phosphorylation. Elife 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Kolupaeva, V.; Janssens, V. PP1 and PP2A phosphatases--cooperating partners in modulating retinoblastoma protein activation. FEBS J. 2013, 280, 627–643. [Google Scholar] [CrossRef] [PubMed]
- Morris, E.J.; Dyson, N.J. Retinoblastoma protein partners. Adv. Cancer Res. 2001, 82, 1–54. [Google Scholar] [PubMed]
- Dyson, N.J. RB1: A prototype tumor suppressor and an enigma. Genes Dev. 2016, 30, 1492–1502. [Google Scholar] [CrossRef] [PubMed]
- Friend, S.H.; Bernards, R.; Rogelj, S.; Weinberg, R.A.; Rapaport, J.M.; Albert, D.M.; Dryja, T.P. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature 1986, 323, 643–646. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J.; McCormick, F. The RB and p53 pathways in cancer. Cancer Cell 2002, 2, 103–112. [Google Scholar] [CrossRef]
- Yamasaki, L.; Bronson, R.; Williams, B.O.; Dyson, N.J.; Harlow, E.; Jacks, T. Loss of E2F-1 reduces tumorigenesis and extends the lifespan of RB1(+/−)mice. Nat. Genet. 1998, 18, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, E.S.; Knudsen, K.E. Tailoring to RB: Tumour suppressor status and therapeutic response. Nat. Rev. Cancer 2008, 8, 714–724. [Google Scholar] [CrossRef] [PubMed]
- Jacks, T.; Fazeli, A.; Schmitt, E.M.; Bronson, R.T.; Goodell, M.A.; Weinberg, R.A. Effects of an RB mutation in the mouse. Nature 1992, 359, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.C.; Skapek, S.X.; Lee, E.Y. Genes in the RB pathway and their knockout in mice. Semin. Cancer Biol. 1996, 7, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.H.; Williams, B.O.; Mulligan, G.; Mukai, S.; Bronson, R.T.; Dyson, N.; Harlow, E.; Jacks, T. Targeted disruption of p107: Functional overlap between p107 and Rb. Genes Dev. 1996, 10, 1621–1632. [Google Scholar] [CrossRef] [PubMed]
- Sage, J.; Mulligan, G.J.; Attardi, L.D.; Miller, A.; Chen, S.; Williams, B.; Theodorou, E.; Jacks, T. Targeted disruption of the three RB-related genes leads to loss of G(1) control and immortalization. Genes Dev. 2000, 14, 3037–3050. [Google Scholar] [CrossRef] [PubMed]
- Ianari, A.; Natale, T.; Calo, E.; Ferretti, E.; Alesse, E.; Screpanti, I.; Haigis, K.; Gulino, A.; Lees, J.A. Proapoptotic function of the retinoblastoma tumor suppressor protein. Cancer Cell 2009, 15, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Sage, J. The retinoblastoma tumor suppressor and stem cell biology. Genes Dev. 2012, 26, 1409–1420. [Google Scholar] [CrossRef] [PubMed]
- Flowers, S.; Beck, G.R.; Moran, E. Transcriptional activation by pRB and its coordination with SWI/SNF recruitment. Cancer Res. 2010, 70, 8282–8287. [Google Scholar] [CrossRef] [PubMed]
- Engel, B.E.; Cress, W.D.; Santiago-Cardona, P.G. The retinoblastoma protein: A master tumor suppressor acts as a link between cell cycle and cell adhesion. Cell. Health Cytoskelet. 2015, 7, 1–10. [Google Scholar] [PubMed]
- Munro, S.; Carr, S.M.; La Thangue, N.B. Diversity within the pRb pathway: Is there a code of conduct? Oncogene 2012, 31, 4343–4352. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, J.I.; Dick, F.A. Posttranslational modifications of the retinoblastoma tumor suppressor protein as determinants of function. Genes Cancer 2013, 3, 619–633. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Lee, W.-H. Retinoblastoma tumor suppressor and genome stability. Adv. Cancer Res. 2002, 85, 13–50. [Google Scholar] [PubMed]
- Trimarchi, J.M.; Lees, J.A. Sibling rivalry in the E2F family. Nat. Rev. Mol. Cell Biol. 2002, 3, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Degregori, J.; Johnson, D.G. Distinct and overlapping roles for E2F family members in transcription, proliferation and apoptosis. Curr. Mol. Med. 2006, 6, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Polager, S.; Ginsberg, D. E2F—At the crossroads of life and death. Trends Cell Biol. 2008, 18, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Biswas, A.K.; Johnson, D.G. Transcriptional and nontranscriptional functions of E2F1 in response to DNA damage. Cancer Res. 2012, 72, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.C.; Lin, F.T.; Nevins, J.R. Selective induction of E2F1 in response to DNA damage, mediated by ATM-dependent phosphorylation. Genes Dev. 2001, 15, 1833–1844. [Google Scholar] [PubMed]
- Wang, B.; Liu, K.; Lin, F.-T.; Lin, W.-C. A role for 14-3-3 tau in E2F1 stabilization and DNA damage-induced apoptosis. J. Biol. Chem. 2004, 279, 54140–54152. [Google Scholar] [CrossRef] [PubMed]
- Velez-Cruz, R.; Johnson, D.G. E2F1 and p53 Transcription factors as accessory factors for nucleotide excision repair. Int. J. Mol. Sci. 2012, 13, 13554–13568. [Google Scholar] [CrossRef] [PubMed]
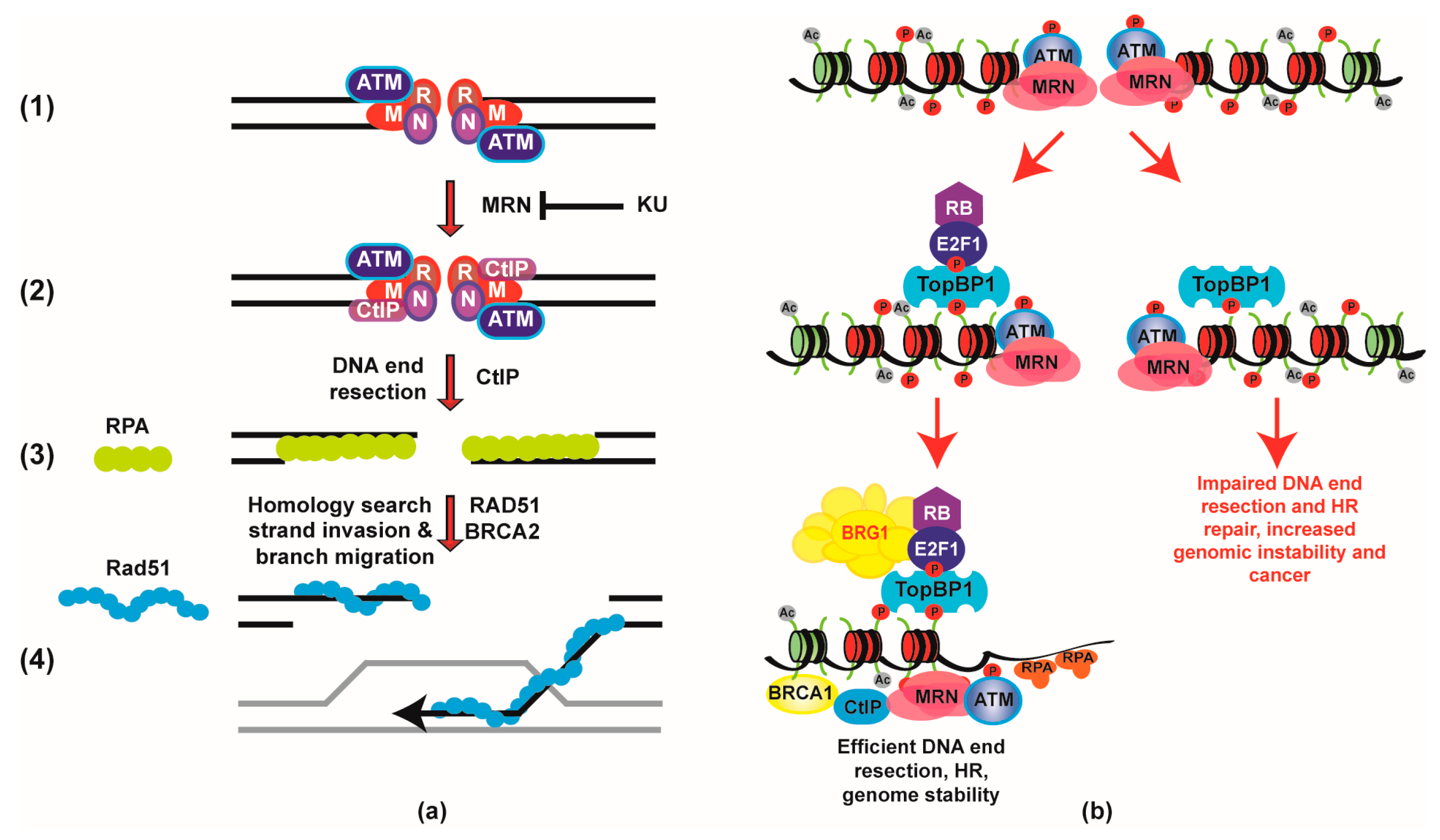
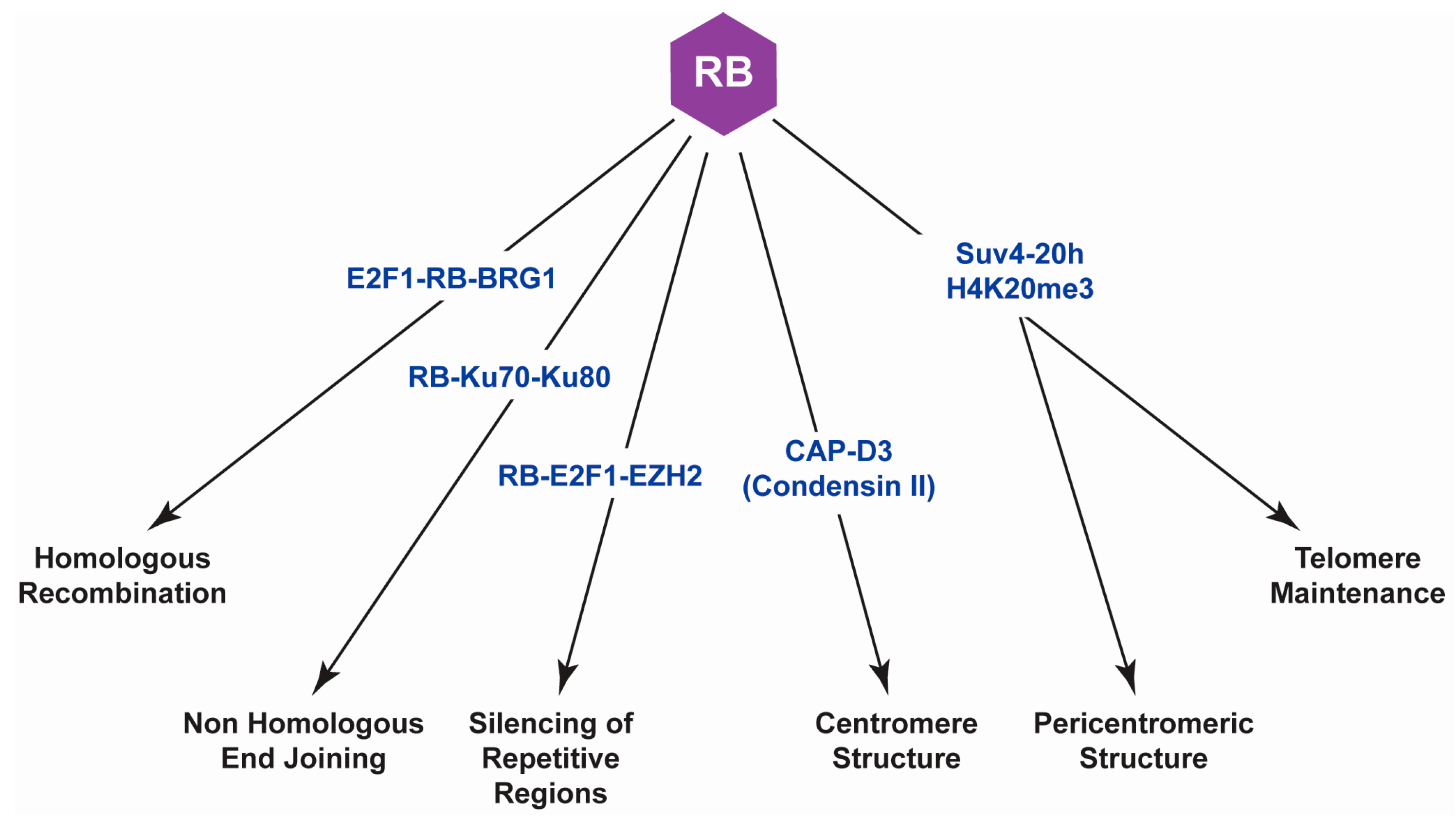
- Velez-Cruz, R.; Manickavinayaham, S.; Biswas, A.K.; Clary, R.W.; Premkumar, T.; Cole, F.; Johnson, D.G. RB localizes to DNA double-strand breaks and promotes DNA end resection and homologous recombination through the recruitment of BRG1. Genes Dev. 2016, 30, 2500–2512. [Google Scholar] [CrossRef] [PubMed]
- Biswas, A.K.; Mitchell, D.L.; Johnson, D.G. E2F1 responds to ultraviolet radiation by directly stimulating DNA repair and suppressing carcinogenesis. Cancer Res. 2014, 74, 3369–3377. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Andor, N.; Maley, C.C.; Ji, H.P. Genomic instability in cancer: Teetering on the limit of tolerance. Cancer Res. 2017, 77, 2179–2185. [Google Scholar] [CrossRef] [PubMed]
- Tubbs, A.; Nussenzweig, A. Endogenous DNA damage as a source of genomic instability in cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, M.J. Targeting the DNA damage response in cancer. Mol. Cell 2015, 60, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Jasin, M.; Rothstein, R. Repair of strand breaks by homologous recombination. Cold Spring Harb. Perspect. Biol. 2013, 5, a012740. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.J.; Chen, D.J. DNA double strand break repair via non-homologous end-joining. Transl. Cancer Res. 2013, 2, 130–143. [Google Scholar] [PubMed]
- Bunting, S.F.; Nussenzweig, A. End-joining, translocations and cancer. Nat. Rev. Cancer 2013, 13, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Yu, V.P.; Koehler, M.; Steinlein, C.; Schmid, M.; Hanakahi, L.A.; van Gool, A.J.; West, S.C.; Venkitaraman, A.R. Gross chromosomal rearrangements and genetic exchange between nonhomologous chromosomes following BRCA2 inactivation. Genes Dev. 2000, 14, 1400–1406. [Google Scholar] [PubMed]
- Talens, F.; Jalving, M.; Gietema, J.A.; Van Vugt, M.A. Therapeutic targeting and patient selection for cancers with homologous recombination defects. Exp. Opin. Drug Discov. 2017, 12, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Carden, C.P.; Yap, T.A.; Kaye, S.B. PARP inhibition: Targeting the Achilles’ heel of DNA repair to treat germline and sporadic ovarian cancers. Curr. Opin. Oncol. 2010, 22, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Paull, T.T. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science 2005, 308, 551–554. [Google Scholar] [CrossRef] [PubMed]
- Sartori, A.A.; Lukas, C.; Coates, J.; Mistrik, M.; Fu, S.; Bartek, J.; Baer, R.; Lukas, J.; Jackson, S.P. Human CtIP promotes DNA end resection. Nature 2007, 450, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Elledge, S.J. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003, 300, 1542–1548. [Google Scholar] [CrossRef] [PubMed]
- Mimitou, E.P.; Symington, L.S. DNA end resection--unraveling the tail. DNA Repair 2011, 10, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Price, B.D.; D’andrea, A.D. Chromatin remodeling at DNA double-strand breaks. Cell 2013, 152, 1344–1354. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhu, F.; Weaks, R.L.; Biswas, A.K.; Guo, R.; Li, Y.; Johnson, D.G. E2F1 promotes the recruitment of DNA repair factors to sites of DNA double-strand breaks. Cell Cycle 2011, 10, 1287–1294. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Lin, F.-T.; Ruppert, J.M.; Lin, W.-C. Regulation of E2F1 by BRCT domain-containing protein TopBP1. Mol. Cell. Biol. 2003, 23, 3287–3304. [Google Scholar] [CrossRef] [PubMed]
- Wardlaw, C.P.; Carr, A.M.; Oliver, A.W. TopBP1: A BRCT-scaffold protein functioning in multiple cellular pathways. DNA Repair 2014, 22, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Luo, Y.; Lin, F.-T.; Lin, W.-C. TopBP1 recruits Brg1/Brm to repress E2F1-induced apoptosis, a novel pRb-independent and E2F1-specific control for cell survival. Genes Dev. 2004, 18, 673–686. [Google Scholar] [CrossRef] [PubMed]
- Campanero, M.R.; Flemington, E.K. Regulation of E2F through ubiquitin-proteasome-dependent degradation: Stabilization by the pRB tumor suppressor protein. Proc. Natl. Acad. Sci. USA 1997, 94, 2221–2226. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, F.; Martelli, F.; Livingston, D.M.; Wang, Z. The retinoblastoma gene product protects E2F-1 from degradation by the ubiquitin-proteasome pathway. Genes Dev. 1996, 10, 2949–2959. [Google Scholar] [CrossRef] [PubMed]
- Hateboer, G.; Kerkhoven, R.M.; Shvarts, A. Degradation of E2F by the ubiquitin-proteasome pathway: Regulation by retinoblastoma family proteins and adenovirus transforming proteins. Genes Dev. 1996, 10, 2960–2970. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Chen, J.; Zhu, F.; Biswas, A.K.; Berton, T.R.; Mitchell, D.L.; Johnson, D.G. E2F1 localizes to sites of UV-induced DNA damage to enhance nucleotide excision repair. J. Biol. Chem. 2010, 285, 19308–19315. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Chen, J.; Mitchell, D.L.; Johnson, D.G. GCN5 and E2F1 stimulate nucleotide excision repair by promoting H3K9 acetylation at sites of damage. Nucleic Acids Res. 2011, 39, 1390–1397. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.S.; Mcpherson, L.A.; Chen, A.Y.; Sage, J.; Ford, J.M. The role of the retinoblastoma/E2F1 tumor suppressor pathway in the lesion recognition step of nucleotide excision repair. DNA Repair 2009, 8, 795–802. [Google Scholar] [CrossRef] [PubMed]
- Hardy, T.M.; Kumar, M.S.; Smith, M.L. RB stabilizes XPC and promotes cellular NER. Anticancer Res. 2010, 30, 2483–2488. [Google Scholar] [PubMed]
- Wiest, N.E.; Houghtaling, S.; Sanchez, J.C.; Tomkinson, A.E.; Osley, M.A. The SWI/SNF ATP-dependent nucleosome remodeler promotes resection initiation at a DNA double-strand break in yeast. Nucleic Acids Res. 2017, 45, 5887–5900. [Google Scholar] [CrossRef] [PubMed]
- Mimitou, E.P.; Yamada, S.; Keeney, S. A global view of meiotic double-strand break end resection. Science 2017, 355, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Lang, S.E.; McMahon, S.B.; Cole, M.D.; Hearing, P. E2F transcriptional activation requires TRRAP and GCN5 cofactors. J. Biol. Chem. 2001, 276, 32627–32634. [Google Scholar] [CrossRef] [PubMed]
- Dunaief, J.L.; Strober, B.E.; Guha, S.; Khavari, P.A.; Alin, K.; Luban, J.; Begemann, M.; Crabtree, G.R.; Goff, S.P. The retinoblastoma protein and BRG1 form a complex and cooperate to induce cell cycle arrest. Cell 1994, 79, 119–130. [Google Scholar] [CrossRef]
- Malewicz, M.; Perlmann, T. Function of transcription factors at DNA lesions in DNA repair. Exp. Cell Res. 2014, 329, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.J.; Chen, B.P.C.; Chen, D.J. DNA-PK: A dynamic enzyme in a versatile DSB repair pathway. DNA Repair 2014, 17, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Cook, R.; Zoumpoulidou, G.; Luczynski, M.T.; Rieger, S.; Moquet, J.; Spanswick, V.J.; Hartley, J.A.; Rothkamm, K.; Huang, P.H.; Mittnacht, S. Direct involvement of retinoblastoma family proteins in DNA repair by non-homologous end-joining. Cell Rep. 2015, 10, 2006–2018. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.M.; Tjeertes, J.V.; Coates, J.; Legube, G.; Polo, S.E.; Britton, S.; Jackson, S.P. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat. Struct. Mol. Biol. 2010, 17, 1144–1151. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.M.; Jackson, S.P. Histone marks: Repairing DNA breaks within the context of chromatin. Biochem. Soc. Trans. 2012, 40, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Tyler, J.K. Nucleosome disassembly during human non-homologous end joining followed by concerted HIRA- and CAF-1-dependent reassembly. Elife 2016, 5, e15129. [Google Scholar] [CrossRef] [PubMed]
- Granata, M.; Panigada, D.; Galati, E.; Lazzaro, F.; Pellicioli, A.; Plevani, P.; Muzi-Falconi, M. To trim or not to trim: Progression and control of DSB end resection. Cell Cycle 2013, 12, 1848–1860. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Chen, L.; Hou, X.; Li, Z.; Kabra, N.; Ma, Y.; Nemoto, S.; Finkel, T.; Gu, W.; Cress, W.D.; et al. Interactions between E2F1 and SirT1 regulate apoptotic response to DNA damage. Nat. Cell Biol. 2006, 8, 1025–1031. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.M.; Krstic-Demonacos, M.; Smith, L.; Demonacos, C.; La Thangue, N.B. Acetylation control of the retinoblastoma tumour-suppressor protein. Nat. Cell Biol. 2001, 3, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Moehlenbrink, J.; Lu, Y.-C.; Zalmas, L.-P.; Sagum, C.A.; Carr, S.; McGouran, J.F.; Alexander, L.; Fedorov, O.; Munro, S.; et al. Arginine methylation-dependent reader-writer interplay governs growth control by E2F-1. Mol. Cell 2013, 52, 37–51. [Google Scholar] [CrossRef] [PubMed]
- Markham, D.; Munro, S.; Soloway, J.; O’Connor, D.P.; La Thangue, N.B. DNA-damage-responsive acetylation of pRb regulates binding to E2F-1. EMBO Rep. 2006, 7, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Pediconi, N.; Guerrieri, F.; Vossio, S.; Bruno, T.; Belloni, L.; Schinzari, V.; Scisciani, C.; Fanciulli, M.; Levrero, M. hSirT1-dependent regulation of the PCAF-E2F1-p73 apoptotic pathway in response to DNA damage. Mol. Cell. Biol. 2009, 29, 1989–1998. [Google Scholar] [CrossRef] [PubMed]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. International human genome sequencing consortium initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [PubMed]
- Jachowicz, J.W.; Torres-Padilla, M.-E. LINEs in mice: Features, families, and potential roles in early development. Chromosoma 2016, 125, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Nishibuchi, G.; Déjardin, J. The molecular basis of the organization of repetitive DNA-containing constitutive heterochromatin in mammals. Chromosome Res. 2017, 25, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Slotkin, R.K.; Martienssen, R. Transposable elements and the epigenetic regulation of the genome. Nat. Publ. Group 2007, 8, 272–285. [Google Scholar] [CrossRef] [PubMed]
- Ishak, C.A.; Marshall, A.E.; Passos, D.T.; White, C.R.; Kim, S.J.; Cecchini, M.J.; Ferwati, S.; MacDonald, W.A.; Howlett, C.J.; Welch, I.D.; et al. An RB-EZH2 complex mediates silencing of repetitive DNA sequences. Mol. Cell 2016, 64, 1074–1087. [Google Scholar] [CrossRef] [PubMed]
- Dick, F.A.; Dyson, N. pRB contains an E2F1-specific binding domain that allows E2F1-induced apoptosis to be regulated separately from other E2F activities. Mol. Cell 2003, 12, 639–649. [Google Scholar] [CrossRef]
- Cecchini, M.J.; Dick, F.A. The biochemical basis of CDK phosphorylation-independent regulation of E2F1 by the retinoblastoma protein. Biochem. J. 2011, 434, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, L.; Jacks, T.; Bronson, R.; Goillot, E.; Harlow, E.; Dyson, N.J. Tumor induction and tissue atrophy in mice lacking E2F-1. Cell 1996, 85, 537–548. [Google Scholar] [CrossRef]
- Maciejowski, J.; de Lange, T. Telomeres in cancer: Tumour suppression and genome instability. Nat. Rev. Mol. Cell. Biol. 2017, 18, 175–186. [Google Scholar] [CrossRef] [PubMed]
- García-Cao, M.; Gonzalo, S.; Dean, D.; Blasco, M.A. A role for the Rb family of proteins in controlling telomere length. Nat. Genet. 2002, 32, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Gonzalo, S.; García-Cao, M.; Fraga, M.F.; Schotta, G.; Peters, A.H.F.M.; Cotter, S.E.; Eguía, R.; Dean, D.C.; Esteller, M.; Jenuwein, T.; et al. Role of the RB1 family in stabilizing histone methylation at constitutive heterochromatin. Nat. Cell Biol. 2005, 7, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Vasconcellos, I.; Schneider, R.; Anastasov, N.; Alonso-Rodriguez, S.; Sanli-Bonazzi, B.; Fernández, J.L.; Atkinson, M.J. The Rb1 tumour suppressor gene modifies telomeric chromatin architecture by regulating TERRA expression. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Gonzalo, S.; Blasco, M.A. Role of Rb family in the epigenetic definition of chromatin. Cell Cycle 2005, 4, 752–755. [Google Scholar] [CrossRef] [PubMed]
- O’sullivan, R.J.; Karlseder, J. Telomeres: Protecting chromosomes against genome instability. Nat. Rev. Mol. Cell Biol. 2010, 11, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Müller, S.; Almouzni, G. Chromatin dynamics during the cell cycle at centromeres. Nat. Rev. Genet. 2017, 18, 192–208. [Google Scholar] [CrossRef] [PubMed]
- Coschi, C.H.; Dick, F.A. Chromosome instability and deregulated proliferation: An unavoidable duo. Cell. Mol. Life Sci. 2012, 69, 2009–2024. [Google Scholar] [CrossRef] [PubMed]
- Sage, J.; Straight, A.F. RB’s original CIN? Genes Dev. 2010, 24, 1329–1333. [Google Scholar] [CrossRef] [PubMed]
- Manning, A.L.; Dyson, N.J. RB: Mitotic implications of a tumour suppressor. Nat. Rev. Cancer 2012, 12, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Manning, A.L.; Longworth, M.S.; Dyson, N.J. Loss of pRB causes centromere dysfunction and chromosomal instability. Genes Dev. 2010, 24, 1364–1376. [Google Scholar] [CrossRef] [PubMed]
- Longworth, M.S.; Herr, A.; Ji, J.-Y.; Dyson, N.J. RBF1 promotes chromatin condensation through a conserved interaction with the Condensin II protein dCAP-D3. Genes Dev. 2008, 22, 1011–1024. [Google Scholar] [CrossRef] [PubMed]
- Manning, A.L.; Yazinski, S.A.; Nicolay, B.; Bryll, A.; Zou, L.; Dyson, N.J. Suppression of Genome Instability in pRB-Deficient Cells by Enhancement of Chromosome Cohesion. Mol. Cell 2014, 53, 993–1004. [Google Scholar] [CrossRef] [PubMed]
- Van Harn, T.; Foijer, F.; van Vugt, M.; Banerjee, R.; Yang, F.; Oostra, A.; Joenje, H.; Riele, H. Loss of Rb proteins causes genomic instability in the absence of mitogenic signaling. Genes Dev. 2010, 24, 1377–1388. [Google Scholar] [CrossRef] [PubMed]
- Coschi, C.H.; Martens, A.L.; Ritchie, K.; Francis, S.M.; Chakrabarti, S.; Berube, N.G.; Dick, F.A. Mitotic chromosome condensation mediated by the retinoblastoma protein is tumor-suppressive. Genes Dev. 2010, 24, 1351–1363. [Google Scholar] [CrossRef] [PubMed]
- Isaac, C.E.; Francis, S.M.; Martens, A.L.; Julian, L.M.; Seifried, L.A.; Erdmann, N.; Binné, U.K.; Harrington, L.; Sicinski, P.; Berube, N.G.; et al. The retinoblastoma protein regulates pericentric heterochromatin. Mol. Cell. Biol. 2006, 26, 3659–3671. [Google Scholar] [CrossRef] [PubMed]
- Talluri, S.; Isaac, C.E.; Ahmad, M.; Henley, S.A.; Francis, S.M.; Martens, A.L.; Bremner, R.; Dick, F.A. A G1 checkpoint mediated by the retinoblastoma protein that is dispensable in terminal differentiation but essential for senescence. Mol. Cell. Biol. 2010, 30, 948–960. [Google Scholar] [CrossRef] [PubMed]
- Brady, C.A.; Jiang, D.; Mello, S.S.; Johnson, T.M.; Jarvis, L.A.; Kozak, M.M.; Broz, D.K.; Basak, S.; Park, E.J.; McLaughlin, M.E.; et al. Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression. Cell 2011, 145, 571–583. [Google Scholar] [CrossRef] [PubMed]
- Attardi, L.D.; Sage, J. RB goes mitochondrial. Genes Dev. 2013, 27, 975–979. [Google Scholar] [CrossRef] [PubMed]
- Abbas, T.; Keaton, M.A.; Dutta, A. Genomic Instability in Cancer. Cold Spring Harb. Perspect. Biol. 2013, 5, a012914. [Google Scholar] [CrossRef] [PubMed]
- Hilgendorf, K.I.; Leshchiner, E.S.; Nedelcu, S.; Maynard, M.A.; Calo, E.; Ianari, A.; Walensky, L.D.; Lees, J.A. The retinoblastoma protein induces apoptosis directly at the mitochondria. Genes Dev. 2013, 27, 1003–1015. [Google Scholar] [CrossRef] [PubMed]
- Burkhart, D.L.; Sage, J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat. Rev. Cancer 2008, 8, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Viatour, P.; Sage, J. Newly identified aspects of tumor suppression by RB. Dis. Model. Mech. 2011, 4, 581–585. [Google Scholar] [CrossRef] [PubMed]
- Burke, J.R.; Hura, G.L.; Rubin, S.M. Structures of inactive retinoblastoma protein reveal multiple mechanisms for cell cycle control. Genes Dev. 2012, 26, 1156–1166. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J. Principles of tumor suppression. Cell 2004, 116, 235–246. [Google Scholar] [CrossRef]
- Pelka, P.; Miller, M.S.; Cecchini, M.; Yousef, A.F.; Bowdish, D.M.; Dick, F.; Whyte, P.; Mymryk, J.S. Adenovirus E1A Directly Targets the E2F/DP-1 Complex. J. Virol. 2011, 85, 8841–8851. [Google Scholar] [CrossRef] [PubMed]
- Carr, S.M.; Munro, S.; Zalmas, L.P.; Fedorov, O.; Johansson, C.; Krojer, T.; Sagum, C.A.; Bedford, M.T.; Oppermann, U.; La Thangue, N.B. Lysine methylation-dependent binding of 53BP1 to the pRb tumor suppressor. Proc. Natl. Acad. Sci. USA 2014, 111, 11341–11346. [Google Scholar] [CrossRef] [PubMed]
- Heilmann, A.M.F.; Dyson, N.J. Phosphorylation puts the pRb tumor suppressor into shape. Genes Dev. 2012, 26, 1128–1130. [Google Scholar] [CrossRef] [PubMed]
- Carnevale, J.; Palander, O.; Seifried, L.A.; Dick, F.A. DNA Damage signals through differentially modified E2F1 molecules to induce apoptosis. Mol. Cell. Biol. 2012, 32, 900–912. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Peng, Y.; Wei, L.; Zhang, W.; Yang, L.; Lan, L.; Kapoor, P.; Ju, Z.; Mo, Q.; Shih, I.M.; et al. ARID1A deficiency impairs the DNA damage checkpoint and sensitizes cells to PARP inhibitors. Cancer Discov. 2015, 5, 752–767. [Google Scholar] [CrossRef] [PubMed]
- Bennett, G.; Peterson, C.L. SWI/SNF recruitment to a DNA double-strand break by the NuA4 and Gcn5 histone acetyltransferases. DNA Repair 2015, 30, 38–45. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vélez-Cruz, R.; Johnson, D.G. The Retinoblastoma (RB) Tumor Suppressor: Pushing Back against Genome Instability on Multiple Fronts. Int. J. Mol. Sci. 2017, 18, 1776. https://doi.org/10.3390/ijms18081776
Vélez-Cruz R, Johnson DG. The Retinoblastoma (RB) Tumor Suppressor: Pushing Back against Genome Instability on Multiple Fronts. International Journal of Molecular Sciences. 2017; 18(8):1776. https://doi.org/10.3390/ijms18081776
Chicago/Turabian StyleVélez-Cruz, Renier, and David G. Johnson. 2017. "The Retinoblastoma (RB) Tumor Suppressor: Pushing Back against Genome Instability on Multiple Fronts" International Journal of Molecular Sciences 18, no. 8: 1776. https://doi.org/10.3390/ijms18081776
APA StyleVélez-Cruz, R., & Johnson, D. G. (2017). The Retinoblastoma (RB) Tumor Suppressor: Pushing Back against Genome Instability on Multiple Fronts. International Journal of Molecular Sciences, 18(8), 1776. https://doi.org/10.3390/ijms18081776