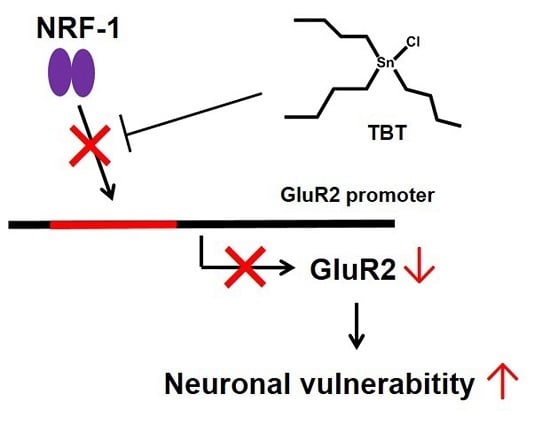
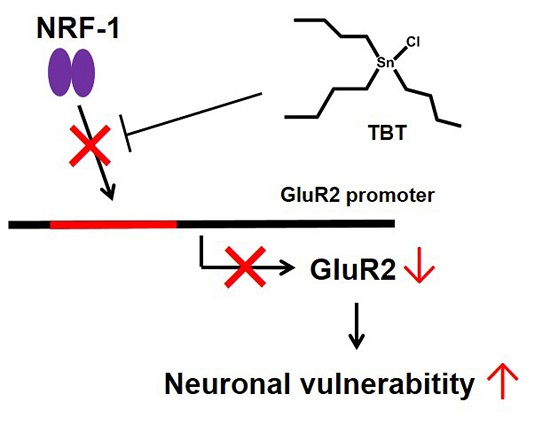
Low-Concentration Tributyltin Decreases GluR2 Expression via Nuclear Respiratory Factor-1 Inhibition

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
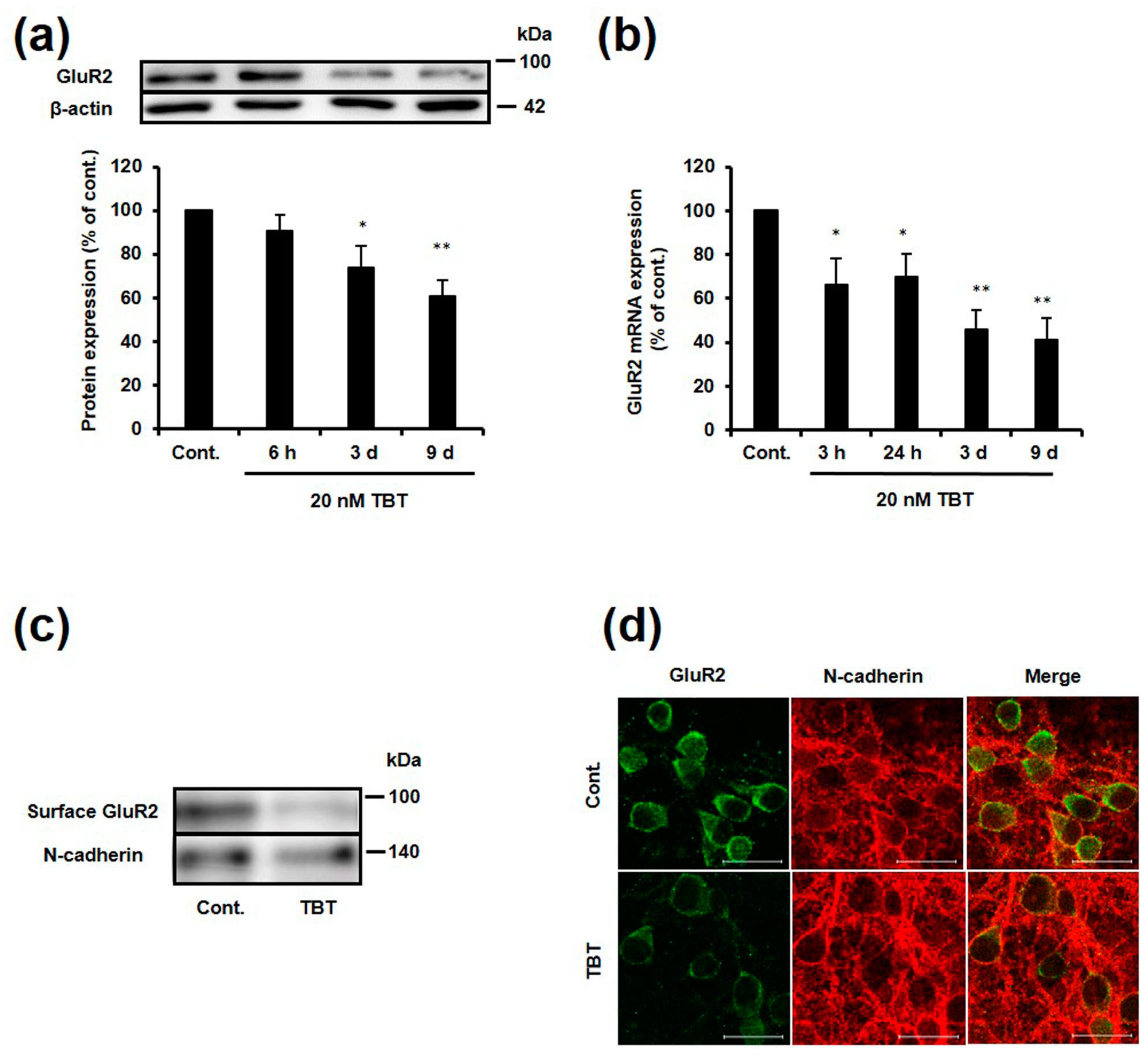
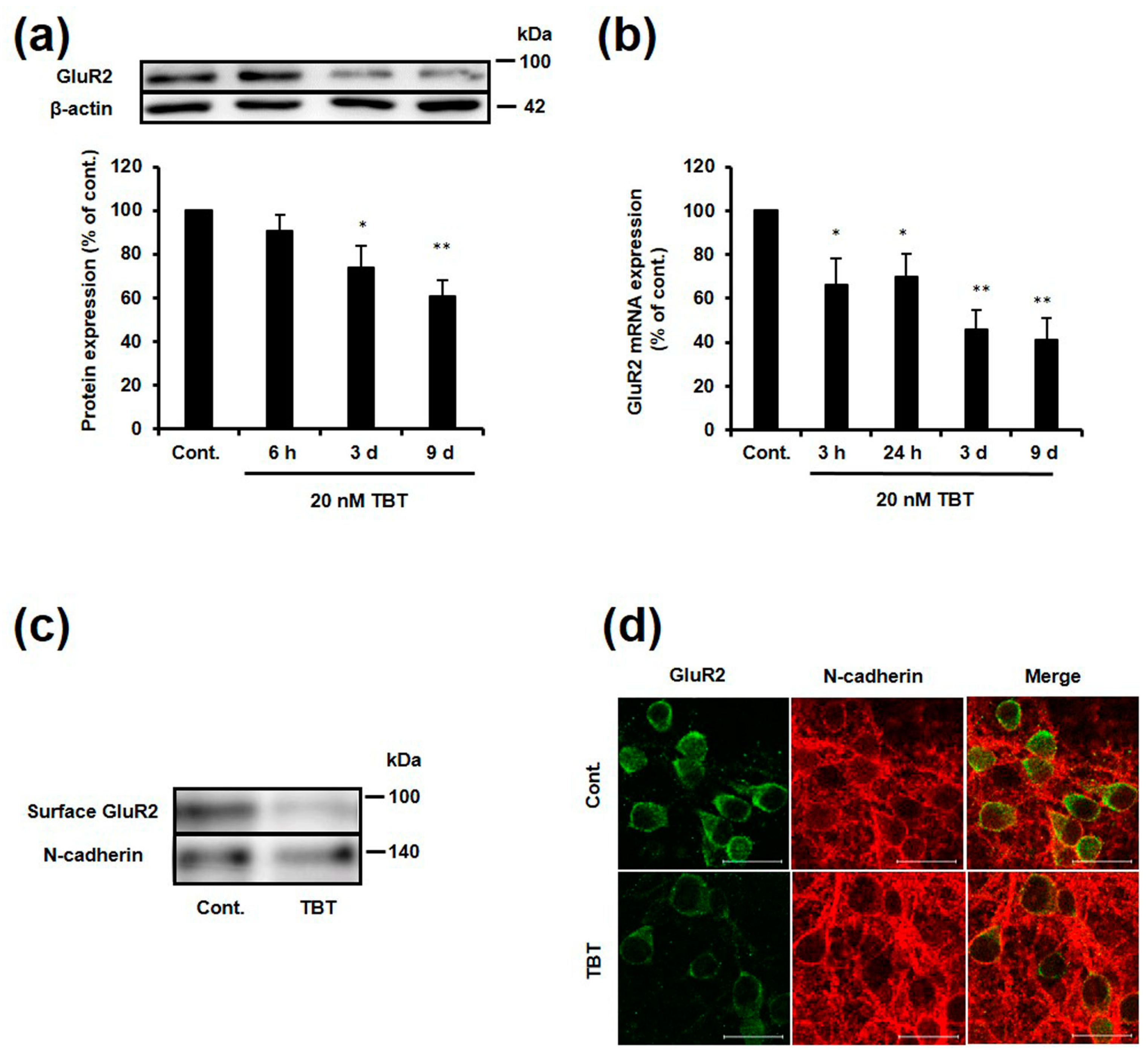
2.1. Effect of TBT Exposure on GluR2 Expression
2.2. Effect of TBT Exposure on GluR2 Expression
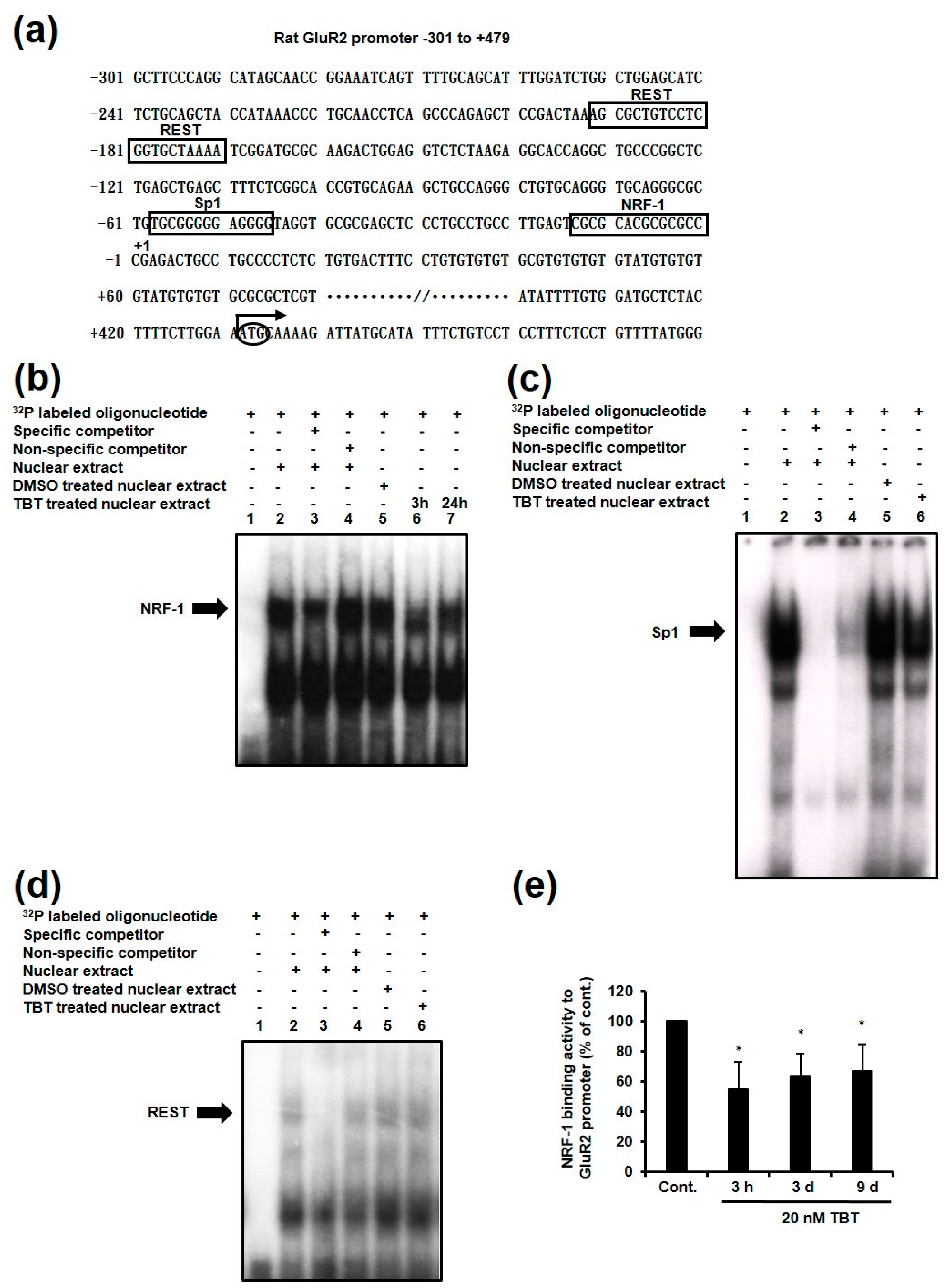
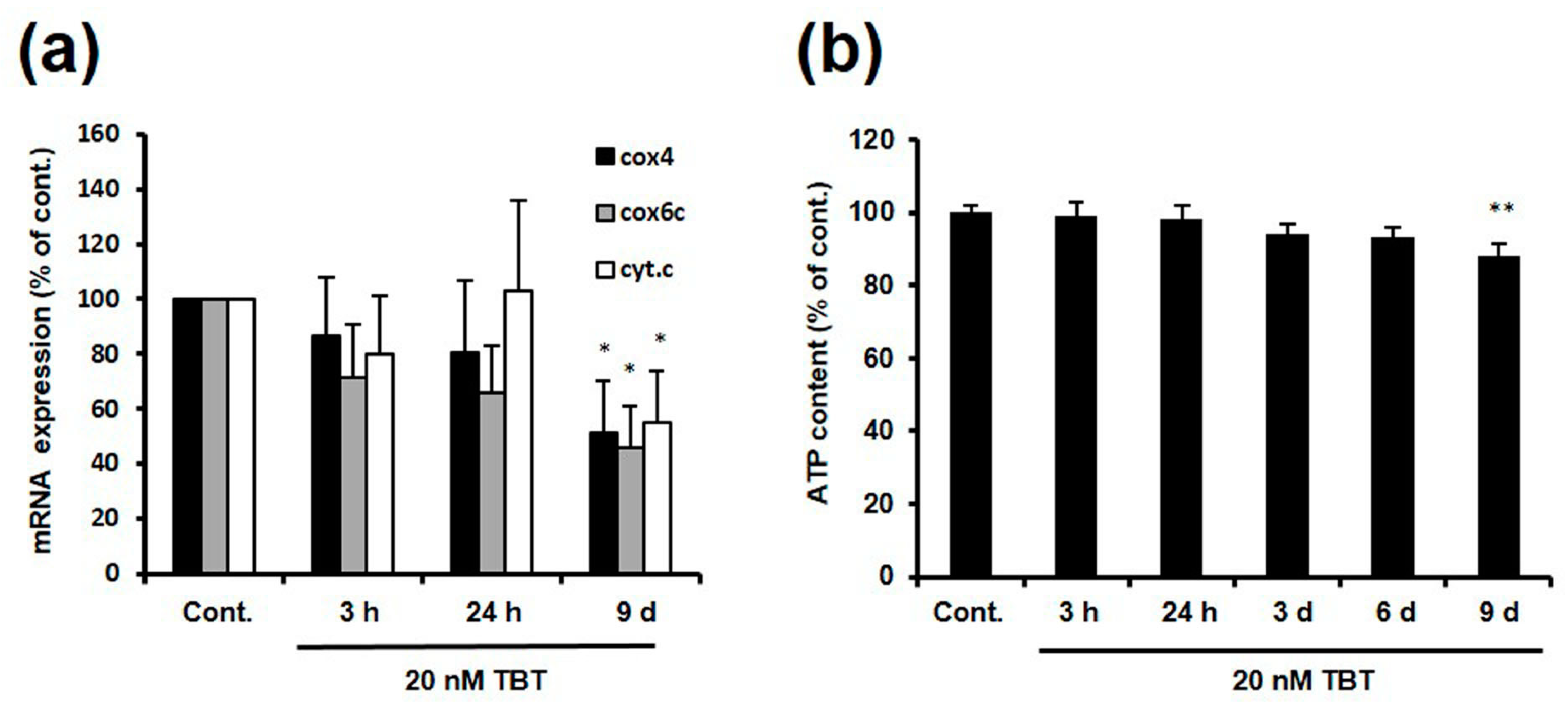
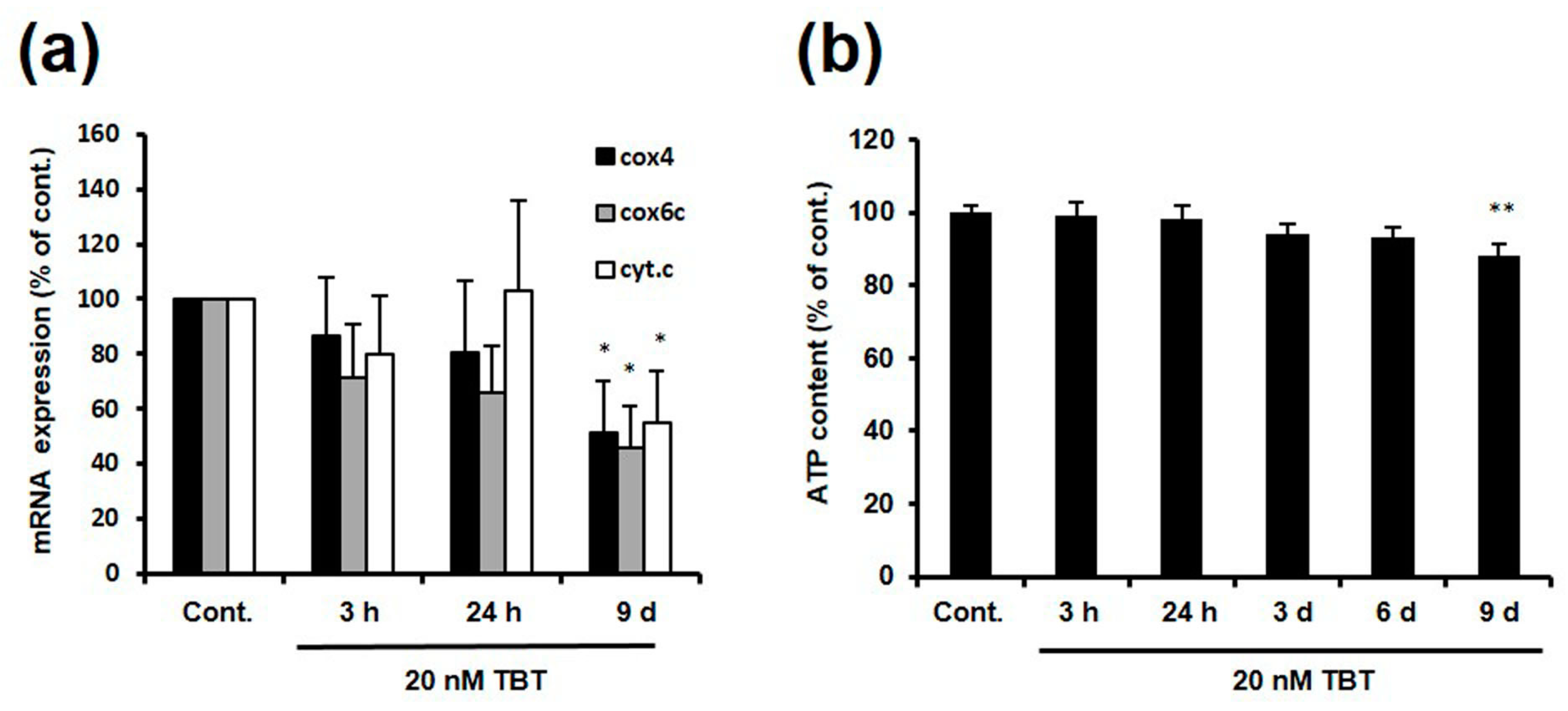
2.3. Effect of TBT on the Expression of NRF-1 Downstream Genes
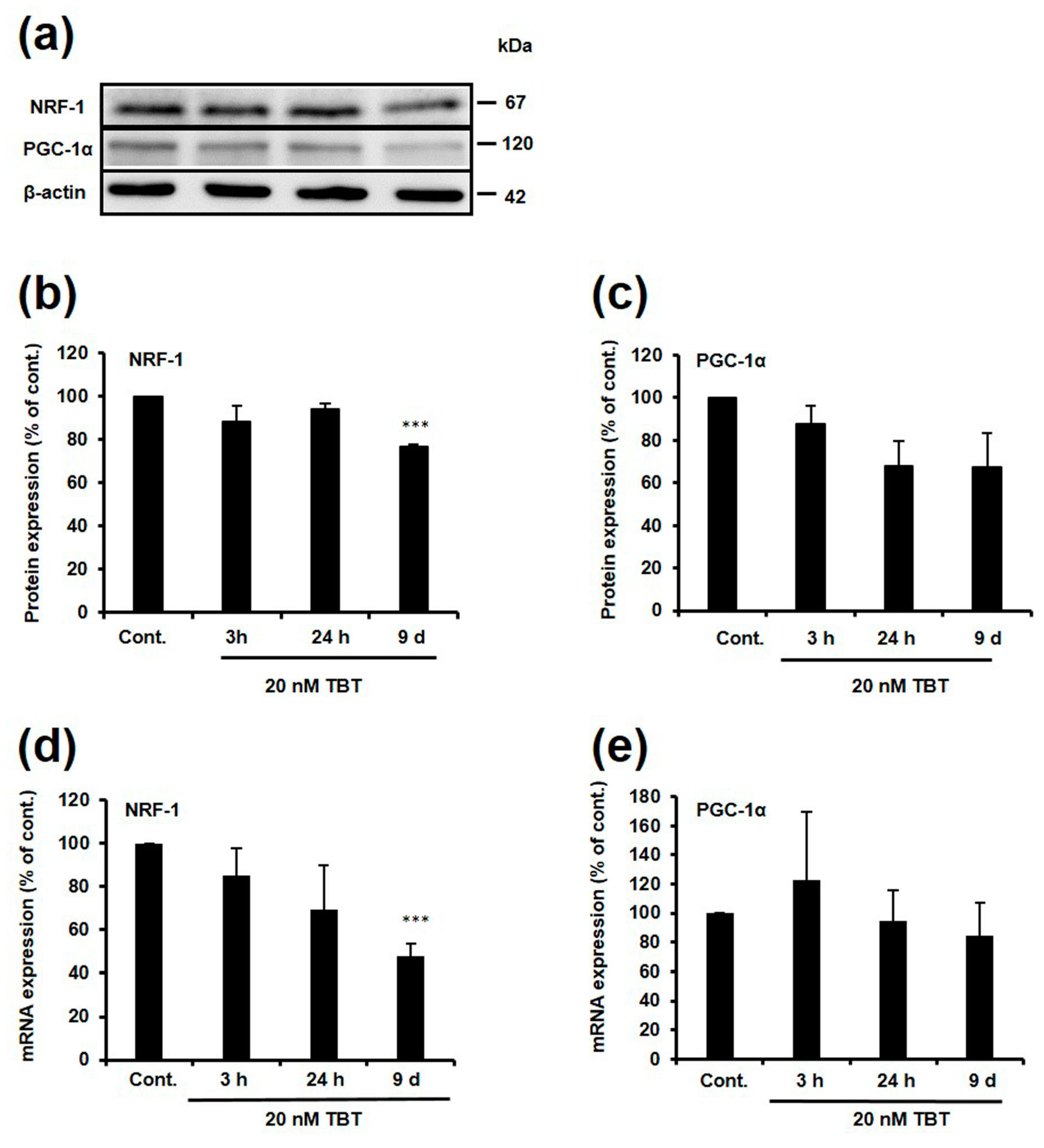
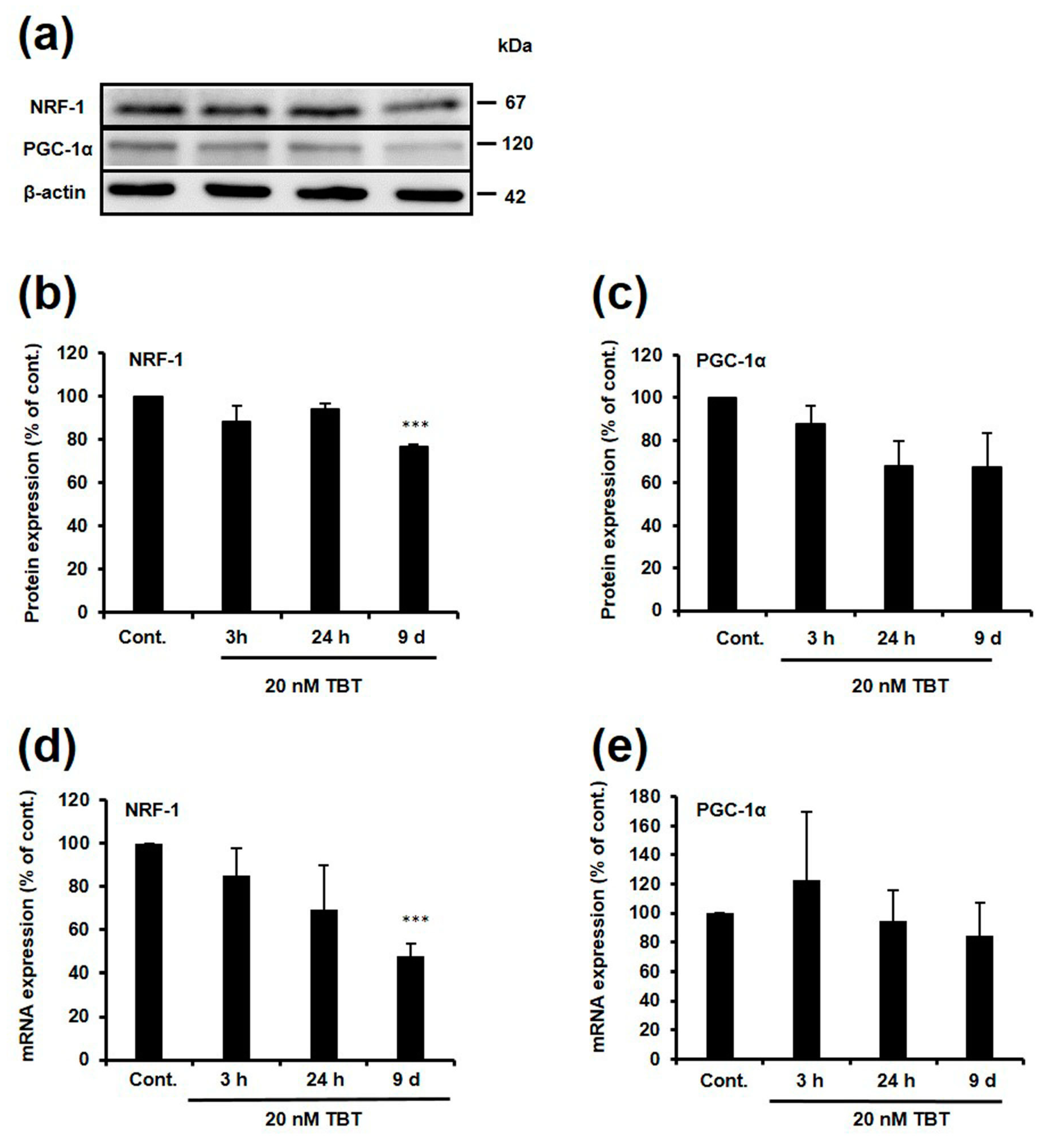
2.4. Effect of TBT on Expression of NRF-1 and PGC-1α
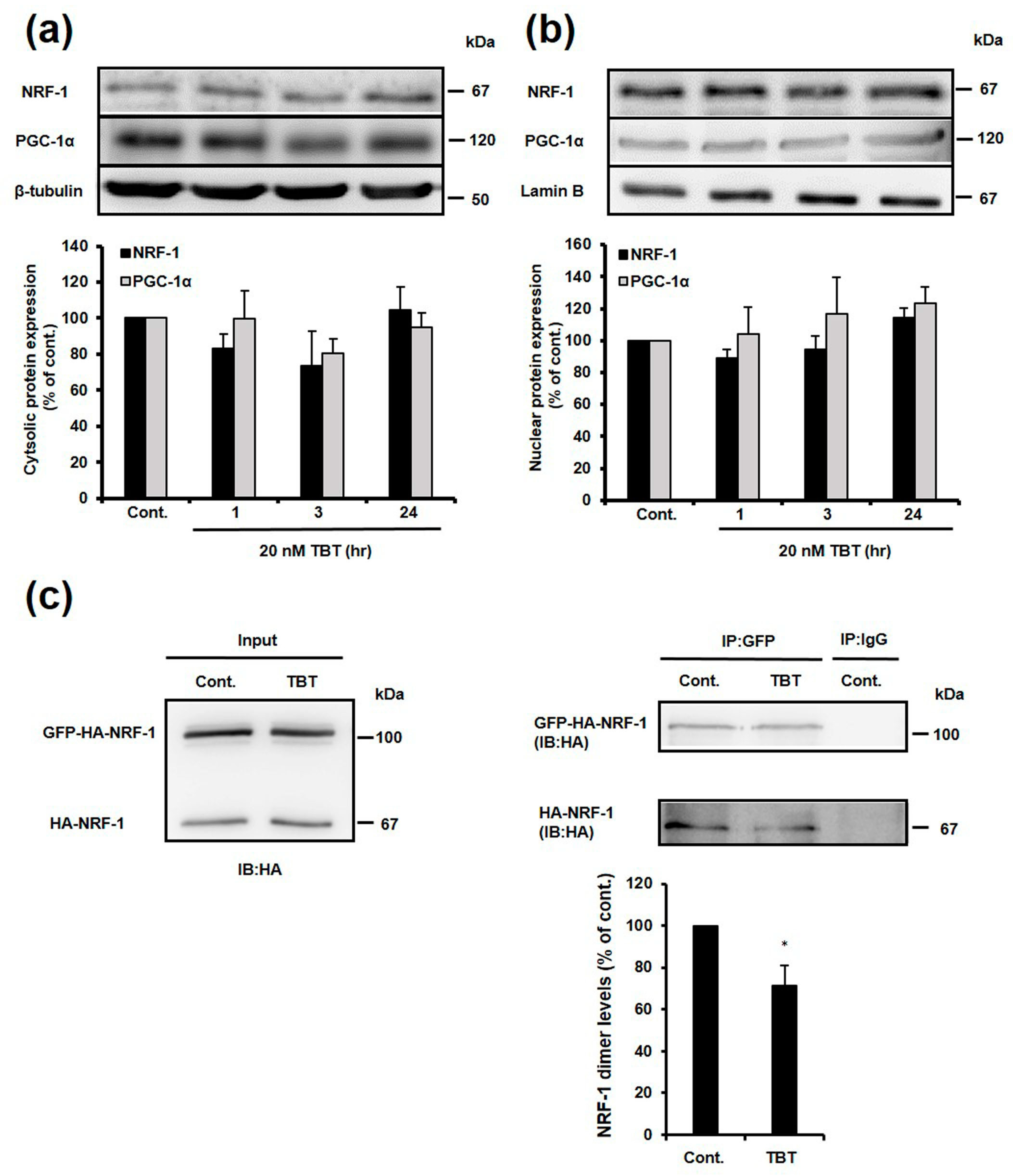
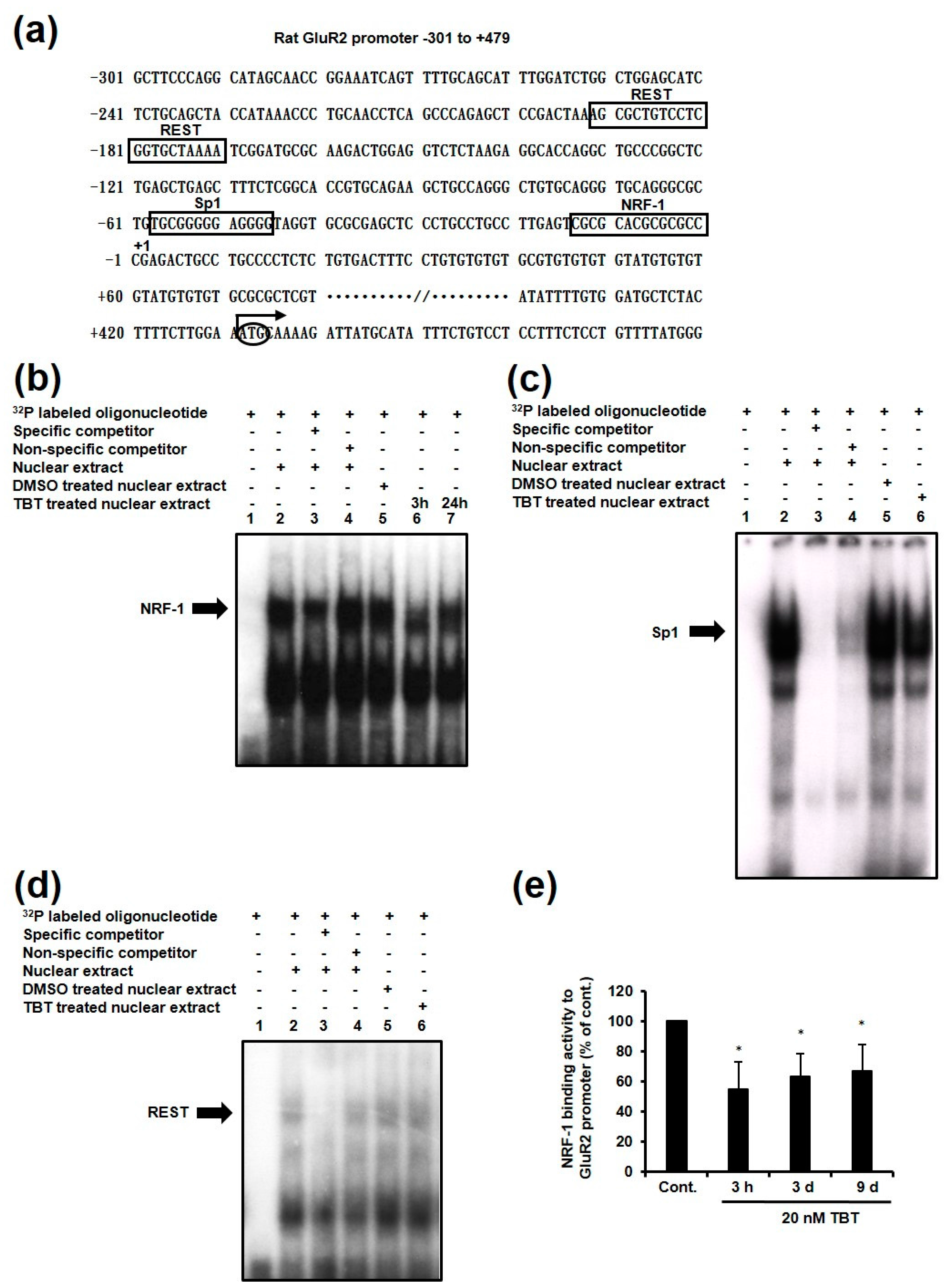
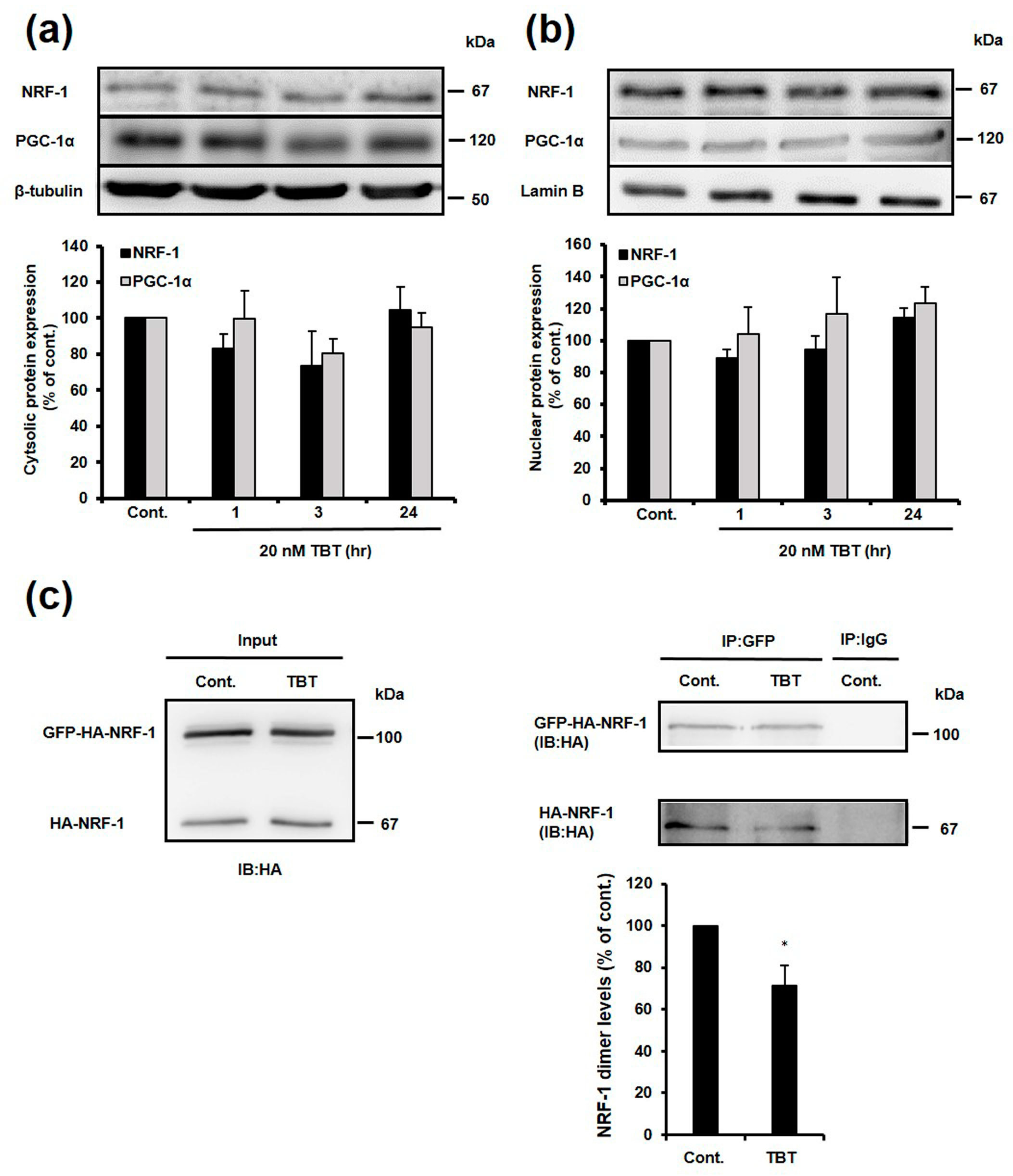
2.5. Effect of TBT on Nuclear and Cytosolic Localization of NRF-1 and PGC-1α, and Dimerization of NRF-1
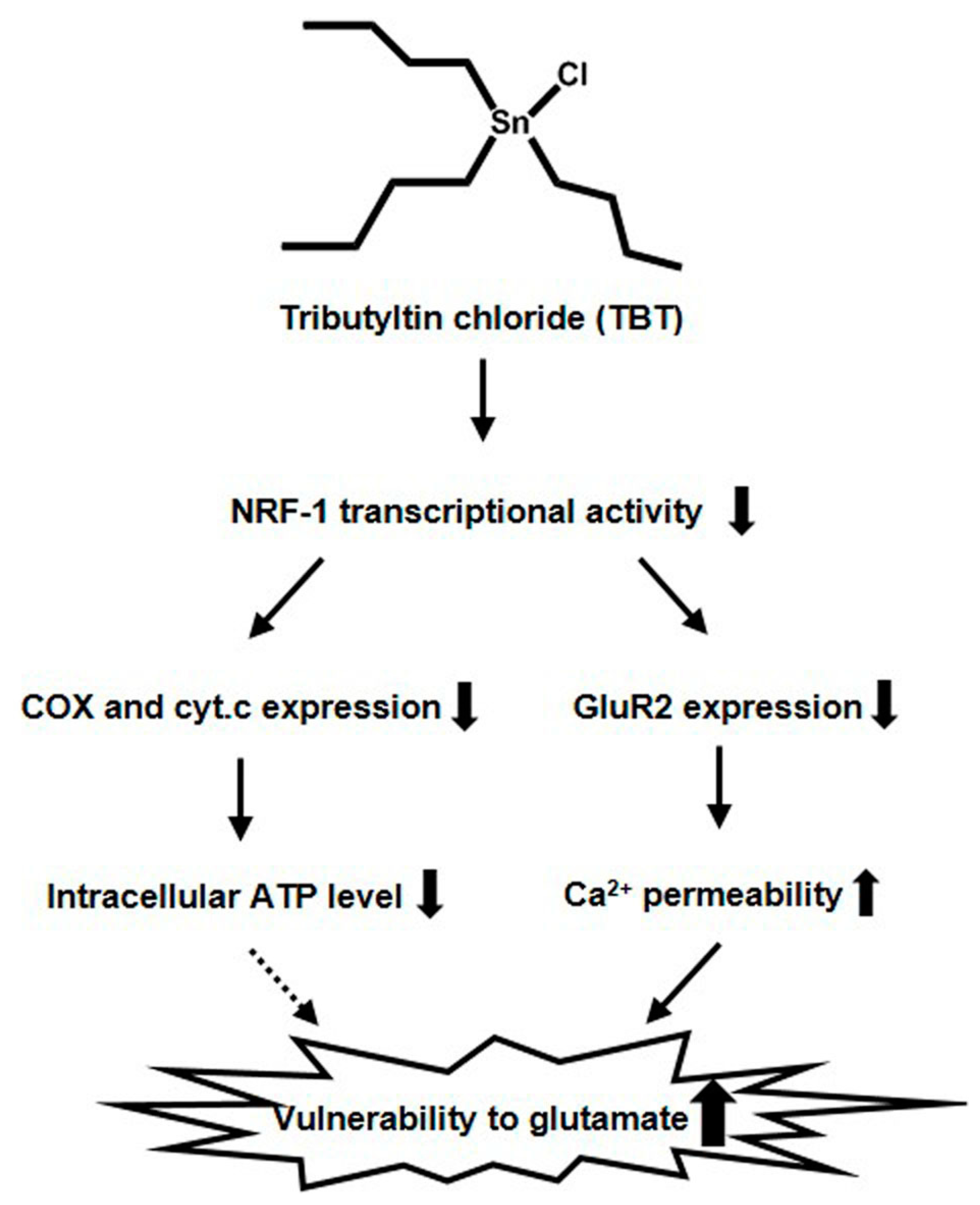
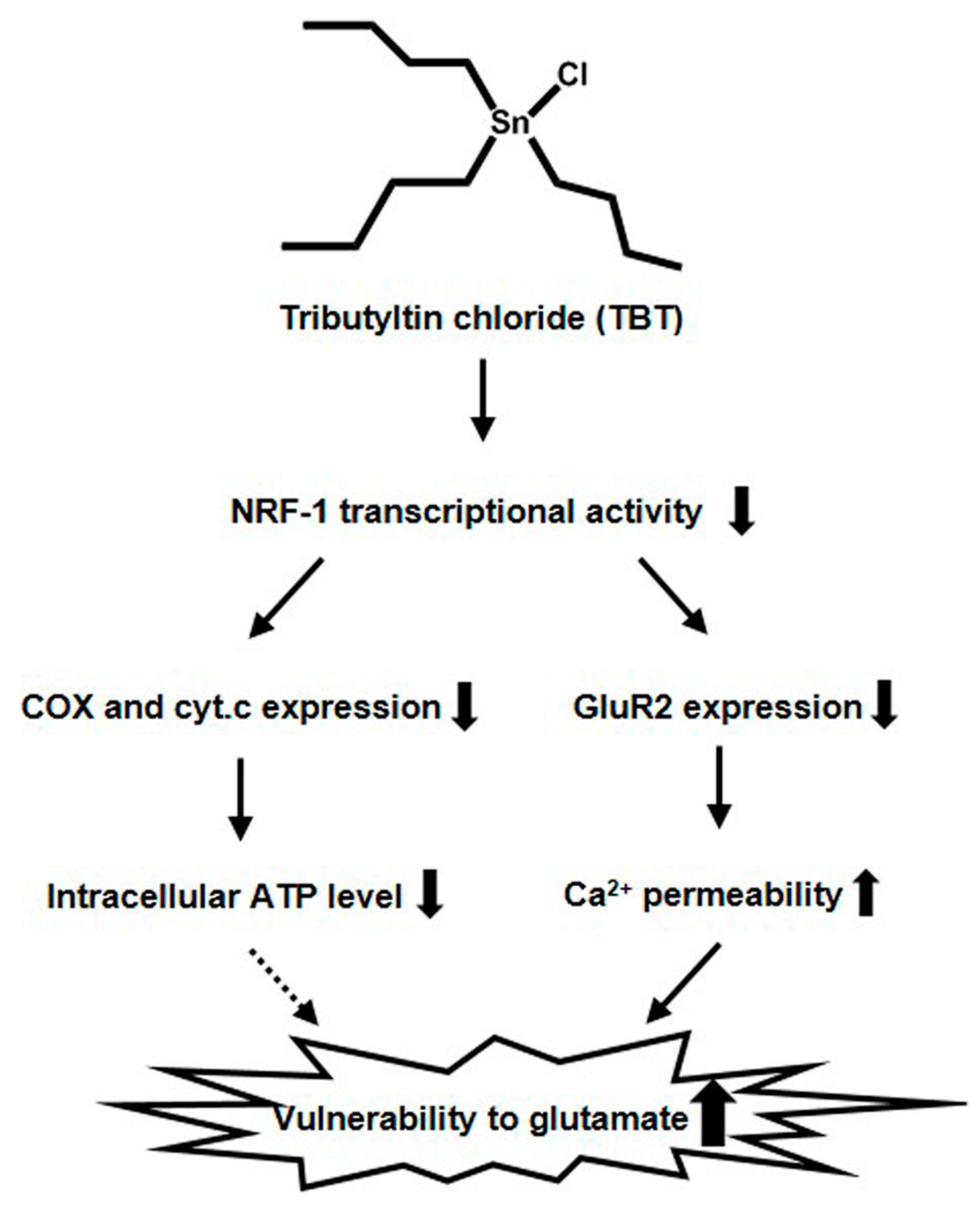
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cortical Neuron Culture
4.3. Protein Isolation
4.3.1. Whole-Cell Extracts
4.3.2. Nuclear Extracts
4.3.3. Membrane Extract
4.4. Western Blotting
4.5. Quantitative Real-Time PCR
4.6. Immunocytochemistry
4.7. ChIP Assay
4.8. Electrophoresis Mobility Shift Assay (EMSA)
4.9. Plasmid Constructions
4.10. Human Embryonic Kidney (HEK) 293T Cell Culture and Transfection
4.11. Immunoprecipitation
4.12. ATP Measurement
4.13. Statistics Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Viglino, L.; Pelletier, E.; St-Louis, R. Highly persistent butyltins in northern marine sediments: A long-term threat for the Saguenay Fjord (Canada). Environ. Toxicol. Chem. 2004, 23, 2673–2681. [Google Scholar] [CrossRef] [PubMed]
- Miki, S.; Ikeda, K.; Oba, Y.; Satone, H.; Honda, M.; Shimasaki, Y.; Onikura, N.; Arakawa, O.; Oshima, Y. Tributyltin in blood of marine fish collected from a coastal area of northern kyushu, Japan. Mar. Pollut. Bull. 2011, 62, 2533–2536. [Google Scholar] [CrossRef] [PubMed]
- Oshima, Y.; Nirmala, K.; Go, J.; Yokota, Y.; Koyama, J. High accumulation of tributyltin in blood among the tissues of fish and applicability to environmental monitoring. Environ. Toxicol. Chem. 1997, 16, 1515–1517. [Google Scholar] [CrossRef]
- Kannan, K.; Senthilkumar, K.; Giesy, J. Occurrence of butyltin compounds in human blood. Environ. Sci. Technol. 1999, 33, 1776–1779. [Google Scholar] [CrossRef]
- Whalen, M.M.; Loganathan, B.G.; Kannan, K. Immunotoxicity of environmentally relevant concentrations of butyltins on human natural killer cells in vitro. Environ. Res. 1999, 81, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Horiguchi, T.; Shiraishi, H.; Shimizu, M.; Morita, M. Effects of triphenyltin chloride and five other organotin compounds on the development of imposex in the rock shell, thais clavigera. Environ. Pollut. 1997, 95, 85–91. [Google Scholar] [CrossRef]
- Bettin, C.; Oehlmann, J.; Stroben, E. TBT-induced imposex in marine neogastropods is mediated by an increasing androgen level. Helgolander Wiss. Meeresunters. 1996, 50, 299–317. [Google Scholar] [CrossRef]
- Heidrich, D.D.; Steckelbroeck, S.; Klingmuller, D. Inhibition of human cytochrome p450 aromatase activity by butyltins. Steroids 2001, 66, 763–769. [Google Scholar] [CrossRef]
- Kanayama, T.; Kobayashi, N.; Mamiya, S.; Nakanishi, T.; Nishikawa, J. Organotin compounds promote adipocyte differentiation as agonists of the peroxisome proliferator-activated receptor gamma/retinoid X receptor pathway. Mol. Pharmacol. 2005, 67, 766–774. [Google Scholar] [CrossRef] [PubMed]
- Castro, L.F.; Lima, D.; Machado, A.; Melo, C.; Hiromori, Y.; Nishikawa, J.; Nakanishi, T.; Reis-Henriques, M.A.; Santos, M.M. Imposex induction is mediated through the retinoid x receptor signalling pathway in the neogastropod nucella lapillus. Aquat. Toxicol. 2007, 85, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, T. Endocrine disruption induced by organotin compounds; organotins function as a powerful agonist for nuclear receptors rather than an aromatase inhibitor. J. Toxicol. Sci. 2008, 33, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Hara, K.; Yoshizuka, M.; Fujimoto, S. Toxic effects of bis (tributyltin) oxide on the synthesis and secretion of zymogen granules in the rat exocrine pancreas. Arch. Histol. Cytol. 1994, 57, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Tsunoda, M.; Konno, N.; Nakano, K.; Liu, Y. Altered metabolism of dopamine in the midbrain of mice treated with tributyltin chloride via subacute oral exposure. Environ. Sci. 2004, 11, 209–219. [Google Scholar] [PubMed]
- Ema, M.; Itami, T.; Kawasaki, H. Behavioral effects of acute exposure to tributyltin chloride in rats. Neurotoxicol. Teratol. 1991, 13, 489–493. [Google Scholar] [CrossRef]
- Konno, N.; Tsunoda, M.; Sugita-Konishi, Y. Effect of tributyltin compound on N-methyl-d-aspartate (NMDA) receptors in brain of preweanling mice. Environ. Health Prev. Med. 2005, 10, 335–337. [Google Scholar] [CrossRef] [PubMed]
- Nakatsu, Y.; Kotake, Y.; Takishita, T.; Ohta, S. Long-term exposure to endogenous levels of tributyltin decreases GluR2 expression and increases neuronal vulnerability to glutamate. Toxicol. Appl. Pharmacol. 2009, 240, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Kotake, Y.; Demizu, Y.; Kurihara, M.; Sekino, Y.; Kanda, Y. NAD-dependent isocitrate dehydrogenase as a novel target of tributyltin in human embryonic carcinoma cells. Sci. Rep. 2014, 4, 5952. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Kotake, Y.; Nakano, M.; Sekino, Y.; Kanda, Y. Tributyltin induces mitochondrial fission through NAD-IDH dependent mitofusin degradation in human embryonic carcinoma cells. Metallomics 2015, 7, 1240–1246. [Google Scholar] [CrossRef] [PubMed]
- Ishida, K.; Saiki, T.; Umeda, K.; Miyara, M.; Sanoh, S.; Ohta, S.; Kotake, Y. Prenatal. Exposure to tributyltin decreases GluR2 expression in the mouse brain. Biol. Pharm Bull. 2017, 40, 1121–1124. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.J.; Zukin, R.S. Ca2+-permeable AMPA receptors in synaptic plasticity and neuronal death. Trends Neurosci. 2007, 30, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Kotake, Y. Molecular mechanisms of environmental organotin toxicity in mammals. Biol. Pharm Bull. 2012, 35, 1876–1880. [Google Scholar] [CrossRef] [PubMed]
- Ishida, K.; Kotake, Y.; Miyara, M.; Aoki, K.; Sanoh, S.; Kanda, Y.; Ohta, S. Involvement of decreased glutamate receptor subunit GluR2 expression in lead-induced neuronal cell death. J. Toxicol. Sci. 2013, 38, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, C.; Kotake, Y.; Yamaguchi, M.; Umeda, K.; Tsuyama, Y.; Sanoh, S.; Okuda, K.; Ohta, S. Development of a simple measurement method for GluR2 protein expression as an index of neuronal vulnerability. Toxicol. Rep. 2015, 2, 450–460. [Google Scholar] [CrossRef]
- Umeda, K.; Kotake, Y.; Miyara, M.; Ishida, K.; Sanoh, S.; Ohta, S. Methoxychlor and fenvalerate induce neuronal death by reducing GluR2 expression. J. Toxicol. Sci. 2016, 41, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Ishida, K.; Tsuyama, Y.; Sanoh, S.; Ohta, S.; Kotake, Y. Perfluorooctane sulfonate induces neuronal vulnerability by decreasing GluR2 expression. Arch. Toxicol. 2017, 91, 885–895. [Google Scholar] [CrossRef] [PubMed]
- Myers, S.J.; Peters, J.; Huang, Y.; Comer, M.B.; Barthel, F.; Dingledine, R. Transcriptional regulation of the GluR2 gene: Neural-specific expression, multiple promoters, and regulatory elements. J. Neurosci. 1998, 18, 6723–6739. [Google Scholar] [PubMed]
- Virbasius, J.V.; Scarpulla, R.C. Activation of the human mitochondrial transcription factor a gene by nuclear respiratory factors: A potential regulatory link between nuclear and mitochondrial gene expression in organelle biogenesis. Proc. Natl. Acad. Sci. USA 1994, 91, 1309–1313. [Google Scholar] [CrossRef] [PubMed]
- Scarpulla, R.C. Nuclear control of respiratory gene expression in mammalian cells. J. Cell. Biochem. 2006, 97, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Dhar, S.S.; Ongwijitwat, S.; Wong-Riley, M.T. Nuclear respiratory factor 1 regulates all ten nuclear-encoded subunits of cytochrome c oxidase in neurons. J. Biol. Chem. 2008, 283, 3120–3129. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef]
- Vercauteren, K.; Pasko, R.A.; Gleyzer, N.; Marino, V.M.; Scarpulla, R.C. PGC-1-related coactivator: Immediate early expression and characterization of a CREB/NRF-1 binding domain associated with cytochrome c promoter occupancy and respiratory growth. Mol. Cell. Biol. 2006, 26, 7409–7419. [Google Scholar] [CrossRef] [PubMed]
- Kjøller, C.; Diemer, N.H. GluR2 protein synthesis and metabolism in rat hippocampus following transient ischemia and ischemic tolerance induction. Neurochem. Int. 2000, 37, 7–15. [Google Scholar] [CrossRef]
- Gugneja, S.; Scarpulla, R.C. Serine phosphorylation within a concise amino-terminal domain in nuclear respiratory factor 1 enhances DNA binding. J. Biol. Chem. 1997, 272, 18732–18739. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.J.; Scarpulla, R.C. Interaction of nuclear factors with multiple sites in the somatic cytochrome c promoter. Characterization of upstream NRF-1, ATF, and intron Sp1 recognition sequences. J. Biol. Chem. 1989, 264, 14361–14368. [Google Scholar] [PubMed]
- Dhar, S.S.; Liang, H.L.; Wong-Riley, M.T. Nuclear respiratory factor 1 co-regulates AMPA glutamate receptor subunit 2 and cytochrome c oxidase: Tight coupling of glutamatergic transmission and energy metabolism in neurons. J. Neurochem. 2009, 108, 1595–1606. [Google Scholar] [CrossRef] [PubMed]
- Suske, G. The Sp-family of transcription factors. Gene 1999, 238, 291–300. [Google Scholar] [CrossRef]
- Borges, K.; Dingledine, R. Functional organization of the GluR1 glutamate receptor promoter. J. Biol. Chem. 2001, 276, 25929–25938. [Google Scholar] [CrossRef] [PubMed]
- Borges, K.; Myers, S.J.; Zhang, S.; Dingledine, R. Activity of the rat GluR4 promoter in transfected cortical neurons and glia. J. Neurochem. 2003, 86, 1162–1173. [Google Scholar] [CrossRef] [PubMed]
- Schoenherr, C.J.; Anderson, D.J. The neuron-restrictive silencer factor (NRSF): A coordinate repressor of multiple neuron-specific genes. Science 1995, 267, 1360–1363. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Grooms, S.Y.; Bennett, M.V.; Zukin, R.S. The AMPAR subunit GluR2: Still front and center-stage. Brain Res. 2000, 886, 190–207. [Google Scholar] [CrossRef]
- Dhar, S.S.; Wong-Riley, M.T. Coupling of energy metabolism and synaptic transmission at the transcriptional level: Role of nuclear respiratory factor 1 in regulating both cytochrome c oxidase and NMDA glutamate receptor subunit genes. J. Neurosci. 2009, 29, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Stridh, H.; Fava, E.; Single, B.; Nicotera, P.; Orrenius, S.; Leist, M. Tributyltin-induced apoptosis requires glycolytic adenosine trisphosphate production. Chem. Res. Toxicol. 1999, 12, 874–882. [Google Scholar] [CrossRef] [PubMed]
- Von Ballmoos, C.; Brunner, J.; Dimroth, P. The ion channel of F-ATP synthase is the target of toxic organotin compounds. Proc. Natl. Acad. Sci. USA 2004, 101, 11239–11244. [Google Scholar] [CrossRef] [PubMed]
- Izumi, H.; Ohta, R.; Nagatani, G.; Ise, T.; Nakayama, Y.; Nomoto, M.; Kohno, K. p300/CBP-associated factor (P/CAF) interacts with nuclear respiratory factor-1 to regulate the UDP-N-acetyl-α-d-galactosamine: Polypeptide N-acetylgalactosaminyltransferase-3 gene. Biochem. J. 2003, 373, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Li, Z.; Lu, Y.; Du, R.; Katiyar, S.; Yang, J.; Fu, M.; Leader, J.E.; Quong, A.; Novikoff, P.M.; et al. Cyclin D1 repression of nuclear respiratory factor 1 integrates nuclear DNA synthesis and mitochondrial function. Proc. Natl. Acad. Sci. USA 2006, 103, 11567–11572. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.T.; Chen, H.I.; Chiou, R.J.; Chen, C.Y.; Huang, A.M. A novel function of transcription factor α-Pal/NRF-1: Increasing neurite outgrowth. Biochem. Biophys. Res. Commun. 2005, 334, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Huo, L.; Scarpulla, R.C. Mitochondrial DNA instability and peri-implantation lethality associated with targeted disruption of nuclear respiratory factor 1 in mice. Mol. Cell. Biol. 2001, 21, 644–654. [Google Scholar] [CrossRef] [PubMed]
- Larsson, N.G.; Wang, J.; Wilhelmsson, H.; Oldfors, A.; Rustin, P.; Lewandoski, M.; Barsh, G.S.; Clayton, D.A. Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat. Genet. 1998, 18, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Ishida, K.; Kotake, Y.; Sanoh, S.; Ohta, S. Lead-induced ERK activation is mediated by GluR2 non-containing AMPA receptor in cortical neurons. Biol. Pharm. Bull. 2017, 40, 303–309. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ishida, K.; Aoki, K.; Takishita, T.; Miyara, M.; Sakamoto, S.; Sanoh, S.; Kimura, T.; Kanda, Y.; Ohta, S.; Kotake, Y. Low-Concentration Tributyltin Decreases GluR2 Expression via Nuclear Respiratory Factor-1 Inhibition. Int. J. Mol. Sci. 2017, 18, 1754. https://doi.org/10.3390/ijms18081754
Ishida K, Aoki K, Takishita T, Miyara M, Sakamoto S, Sanoh S, Kimura T, Kanda Y, Ohta S, Kotake Y. Low-Concentration Tributyltin Decreases GluR2 Expression via Nuclear Respiratory Factor-1 Inhibition. International Journal of Molecular Sciences. 2017; 18(8):1754. https://doi.org/10.3390/ijms18081754
Chicago/Turabian StyleIshida, Keishi, Kaori Aoki, Tomoko Takishita, Masatsugu Miyara, Shuichiro Sakamoto, Seigo Sanoh, Tomoki Kimura, Yasunari Kanda, Shigeru Ohta, and Yaichiro Kotake. 2017. "Low-Concentration Tributyltin Decreases GluR2 Expression via Nuclear Respiratory Factor-1 Inhibition" International Journal of Molecular Sciences 18, no. 8: 1754. https://doi.org/10.3390/ijms18081754
APA StyleIshida, K., Aoki, K., Takishita, T., Miyara, M., Sakamoto, S., Sanoh, S., Kimura, T., Kanda, Y., Ohta, S., & Kotake, Y. (2017). Low-Concentration Tributyltin Decreases GluR2 Expression via Nuclear Respiratory Factor-1 Inhibition. International Journal of Molecular Sciences, 18(8), 1754. https://doi.org/10.3390/ijms18081754