Mutations of Pre-mRNA Splicing Regulatory Elements: Are Predictions Moving Forward to Clinical Diagnostics?


Abstract
1. Introduction
2. Predictions on SREs
3. Efficiency of SRE Predictions
4. Exons Susceptibility to the Splicing Defects Due to SREs Changes
5. Future Approaches at Evaluating the Effects of Splicing Disruption
6. Clinical Significance of Splicing Aberrations
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CERES | Composite exonic regulatory element of splicing |
| circRNA | Circular RNA |
| CLIP | Cross-linking immunoprecipitation |
| ESE | Exonic splicing enhancer |
| ESS | Exonic splicing silencer |
| HSF | Human Splicing Finder |
| ISE | Intronic splicing enhancer |
| ISS | Intronic splicing silencer |
| PTC | Premature termination codon |
| RESCUE | Relative Enhancer and Silencer Classification by Unanimous Enrichment |
| SELEX | Functional systematic evolution of ligands by exponential enrichment |
| SNP | Single nucleotide polymorphism |
| SPANR | Splicing-based Analysis of Variants |
| SRE | Splicing regulatory element |
References
- Sun, H.; Chasin, L.A. Multiple splicing defects in an intronic false exon. Mol. Cell. Biol. 2000, 20, 6414–6425. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.-D.; Ares, M. Context-dependent control of alternative splicing by RNA-binding proteins. Nat. Rev. Genet. 2014, 15, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Rio, D.C. Mechanisms and Regulation of Alternative Pre-mRNA Splicing. Annu. Rev. Biochem. 2015, 84, 291–323. [Google Scholar] [CrossRef] [PubMed]
- Baralle, D.; Buratti, E. RNA splicing in human disease and in the clinic. Clin. Sci. 2017, 131, 355–368. [Google Scholar] [CrossRef] [PubMed]
- Goren, A.; Ram, O.; Amit, M.; Keren, H.; Lev-Maor, G.; Vig, I.; Pupko, T.; Ast, G. Comparative analysis identifies exonic splicing regulatory sequences--The complex definition of enhancers and silencers. Mol. Cell 2006, 22, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Pagani, F.; Stuani, C.; Tzetis, M.; Kanavakis, E.; Efthymiadou, A.; Doudounakis, S.; Casals, T.; Baralle, F.E. New type of disease causing mutations: The example of the composite exonic regulatory elements of splicing in CFTR exon 12. Hum. Mol. Genet. 2003, 12, 1111–1120. [Google Scholar] [CrossRef] [PubMed]
- Greene, J.; Baird, A.-M.; Brady, L.; Lim, M.; Gray, S.G.; McDermott, R.; Finn, S.P. Circular RNAs: Biogenesis, Function and Role in Human Diseases. Front. Mol. Biosci. 2017, 4. [Google Scholar] [CrossRef] [PubMed]
- Ashwal-Fluss, R.; Meyer, M.; Pamudurti, N.R.; Ivanov, A.; Bartok, O.; Hanan, M.; Evantal, N.; Memczak, S.; Rajewsky, N.; Kadener, S. circRNA Biogenesis Competes with Pre-mRNA Splicing. Mol. Cell 2014, 56, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Kramer, M.C.; Liang, D.; Tatomer, D.C.; Gold, B.; March, Z.M.; Cherry, S.; Wilusz, J.E. Combinatorial control of Drosophila circular RNA expression by intronic repeats, hnRNPs, and SR proteins. Genes Dev. 2015, 29, 2168–2182. [Google Scholar] [CrossRef] [PubMed]
- Conn, S.J.; Pillman, K.A.; Toubia, J.; Conn, V.M.; Salmanidis, M.; Phillips, C.A.; Roslan, S.; Schreiber, A.W.; Gregory, P.A.; Goodall, G.J. The RNA Binding Protein Quaking Regulates Formation of circRNAs. Cell 2015, 160, 1125–1134. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Reckman, Y.J.; Aufiero, S.; van den Hoogenhof, M.M.; van der Made, I.; Beqqali, A.; Koolbergen, D.R.; Rasmussen, T.B.; Van Der Velden, J.; Creemers, E.E.; et al. RBM20 Regulates Circular RNA Production From the Titin Gene. Circ. Res. 2016, 119, 996–1003. [Google Scholar] [CrossRef] [PubMed]
- Errichelli, L.; Dini Modigliani, S.; Laneve, P.; Colantoni, A.; Legnini, I.; Capauto, D.; Rosa, A.; De Santis, R.; Scarfò, R.; Peruzzi, G.; et al. FUS affects circular RNA expression in murine embryonic stem cell-derived motor neurons. Nat. Commun. 2017, 8, 14741. [Google Scholar] [CrossRef] [PubMed]
- Di Giacomo, D.; Gaildrat, P.; Abuli, A.; Abdat, J.; Frébourg, T.; Tosi, M.; Martins, A. Functional Analysis of a Large set of BRCA2 exon 7 Variants Highlights the Predictive Value of Hexamer Scores in Detecting Alterations of Exonic Splicing Regulatory Elements: HUMAN MUTATION. Hum. Mutat. 2013, 34, 1547–1557. [Google Scholar] [CrossRef] [PubMed]
- Julien, P.; Miñana, B.; Baeza-Centurion, P.; Valcárcel, J.; Lehner, B. The complete local genotype–phenotype landscape for the alternative splicing of a human exon. Nat. Commun. 2016, 7, 11558. [Google Scholar] [CrossRef] [PubMed]
- Soukarieh, O.; Gaildrat, P.; Hamieh, M.; Drouet, A.; Baert-Desurmont, S.; Frébourg, T.; Tosi, M.; Martins, A. Exonic Splicing Mutations Are More Prevalent than Currently Estimated and Can Be Predicted by Using In Silico Tools. PLoS Genet. 2016, 12, e1005756. [Google Scholar] [CrossRef] [PubMed]
- Houdayer, C.; Dehainault, C.; Mattler, C.; Michaux, D.; Caux-Moncoutier, V.; Pagès-Berhouet, S.; d’Enghien, C.D.; Laugé, A.; Castera, L.; Gauthier-Villars, M.; et al. Evaluation of in silico splice tools for decision-making in molecular diagnosis. Hum. Mutat. 2008, 29, 975–982. [Google Scholar] [CrossRef] [PubMed]
- Houdayer, C.; Caux-Moncoutier, V.; Krieger, S.; Barrois, M.; Bonnet, F.; Bourdon, V.; Bronner, M.; Buisson, M.; Coulet, F.; Gaildrat, P.; et al. Guidelines for splicing analysis in molecular diagnosis derived from a set of 327 combined in silico/in vitro studies on BRCA1 and BRCA2 variants. Hum. Mutat. 2012, 33, 1228–1238. [Google Scholar] [CrossRef] [PubMed]
- Kergourlay, V.; Raї, G.; Blandin, G.; Salgado, D.; Béroud, C.; Lévy, N.; Krahn, M.; Bartoli, M. Identification of Splicing Defects Caused by Mutations in the Dysferlin Gene. Hum. Mutat. 2014, 35, 1532–1541. [Google Scholar] [CrossRef] [PubMed]
- Grodecká, L.; Hujová, P.; Kramárek, M.; Kršjaková, T.; Kováčová, T.; Vondrášková, K.; Ravčuková, B.; Hrnčířová, K.; Souček, P.; Freiberger, T. Systematic analysis of splicing defects in selected primary immunodeficiencies-related genes. Clin. Immunol. 2017, 180, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Baralle, D.; Lucassen, A.; Buratti, E. Missed threads. The impact of pre-mRNA splicing defects on clinical practice. EMBO Rep. 2009, 10, 810–816. [Google Scholar] [CrossRef] [PubMed]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Buratti, E.; Baralle, M.; Baralle, F.E. From single splicing events to thousands: The ambiguous step forward in splicing research. Brief Funct. Genom. 2013, 12, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.X.; Zhang, M.; Krainer, A.R. Identification of functional exonic splicing enhancer motifs recognized by individual SR proteins. Genes Dev. 1998, 12, 1998–2012. [Google Scholar] [CrossRef] [PubMed]
- Cartegni, L. ESEfinder: A web resource to identify exonic splicing enhancers. Nucleic Acids Res. 2003, 31, 3568–3571. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Romfo, C.M.; Nilsen, T.W.; Green, M.R. Functional recognition of the 3′ splice site AG by the splicing factor U2AF35. Nature 1999, 402, 832–835. [Google Scholar] [CrossRef] [PubMed]
- Desmet, F.-O.; Hamroun, D.; Lalande, M.; Collod-Beroud, G.; Claustres, M.; Beroud, C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009, 37, e67. [Google Scholar] [CrossRef] [PubMed]
- Lastella, P.; Surdo, N.C.; Resta, N.; Guanti, G.; Stella, A. In silico and in vivo splicing analysis of MLH1 and MSH2 missense mutations shows exon- and tissue-specific effects. BMC Genom. 2006, 7, 243. [Google Scholar] [CrossRef] [PubMed]
- ElSharawy, A.; Hundrieser, B.; Brosch, M.; Wittig, M.; Huse, K.; Platzer, M.; Becker, A.; Simon, M.; Rosenstiel, P.; Schreiber, S.; et al. Systematic evaluation of the effect of common SNPs on pre-mRNA splicing. Hum. Mutat. 2009, 30, 625–632. [Google Scholar] [CrossRef] [PubMed]
- kConFab Investigators; Whiley, P.J.; Pettigrew, C.A.; Brewster, B.L.; Walker, L.C.; Spurdle, A.B.; Brown, M.A. Effect of BRCA2 sequence variants predicted to disrupt exonic splice enhancers on BRCA2transcripts. BMC Med. Genet. 2010, 11, 80. [Google Scholar] [CrossRef] [PubMed]
- Woolfe, A.; Mullikin, J.C.; Elnitski, L. Genomic features defining exonic variants that modulate splicing. Genome Biol. 2010, 11, R20. [Google Scholar] [CrossRef] [PubMed]
- Gaildrat, P.; Krieger, S.; Di Giacomo, D.; Abdat, J.; Révillion, F.; Caputo, S.; Vaur, D.; Jamard, E.; Bohers, E.; Ledemeney, D.; et al. Multiple sequence variants of BRCA2 exon 7 alter splicing regulation. J. Med. Genet. 2012, 49, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Ke, S.; Shang, S.; Kalachikov, S.M.; Morozova, I.; Yu, L.; Russo, J.J.; Ju, J.; Chasin, L.A. Quantitative evaluation of all hexamers as exonic splicing elements. Genome Res. 2011, 21, 1360–1374. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Rolish, M.E.; Yeo, G.; Tung, V.; Mawson, M.; Burge, C.B. Systematic identification and analysis of exonic splicing silencers. Cell 2004, 119, 831–845. [Google Scholar] [CrossRef] [PubMed]
- Erkelenz, S.; Theiss, S.; Otte, M.; Widera, M.; Peter, J.O.; Schaal, H. Genomic HEXploring allows landscaping of novel potential splicing regulatory elements. Nucleic Acids Res. 2014, 42, 10681–10697. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.H.-F. Computational definition of sequence motifs governing constitutive exon splicing. Genes Dev. 2004, 18, 1241–1250. [Google Scholar] [CrossRef] [PubMed]
- Fairbrother, W.G.; Yeo, G.W.; Yeh, R.; Goldstein, P.; Mawson, M.; Sharp, P.A.; Burge, C.B. RESCUE-ESE identifies candidate exonic splicing enhancers in vertebrate exons. Nucleic Acids Res. 2004, 32, W187–W190. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.Y.; Alipanahi, B.; Lee, L.J.; Bretschneider, H.; Merico, D.; Yuen, R.K.C.; Hua, Y.; Gueroussov, S.; Najafabadi, H.S.; Hughes, T.R.; et al. The human splicing code reveals new insights into the genetic determinants of disease. Science 2015, 347, 1254806. [Google Scholar] [CrossRef] [PubMed]
- Piva, F.; Giulietti, M.; Burini, A.B.; Principato, G. SpliceAid 2: A database of human splicing factors expression data and RNA target motifs. Hum. Mutat. 2012, 33, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Fairbrother, W.G. Predictive Identification of Exonic Splicing Enhancers in Human Genes. Science 2002, 297, 1007–1013. [Google Scholar] [CrossRef] [PubMed]
- Castello, A.; Fischer, B.; Frese, C.; Horos, R.; Alleaume, A.-M.; Foehr, S.; Curk, T.; Krijgsveld, J.; Hentze, M. Comprehensive Identification of RNA-Binding Domains in Human Cells. Mol. Cell 2016, 63, 696–710. [Google Scholar] [CrossRef] [PubMed]
- Raponi, M.; Kralovicova, J.; Copson, E.; Divina, P.; Eccles, D.; Johnson, P.; Baralle, D.; Vorechovsky, I. Prediction of single-nucleotide substitutions that result in exon skipping: Identification of a splicing silencer in BRCA1 exon 6. Hum. Mutat. 2011, 32, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S.; Hall, E.; Ast, G. SROOGLE: Webserver for integrative, user-friendly visualization of splicing signals. Nucleic Acids Res. 2009, 37, W189–W192. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Li, W.-H.; Krainer, A.R.; Zhang, M.Q. RNA landscape of evolution for optimal exon and intron discrimination. Proc. Natl. Acad. Sci. USA 2008, 105, 5797–5802. [Google Scholar] [CrossRef] [PubMed]
- Stadler, M.B.; Shomron, N.; Yeo, G.W.; Schneider, A.; Xiao, X.; Burge, C.B. Inference of Splicing Regulatory Activities by Sequence Neighborhood Analysis. PLoS Genet. 2006, 2, e191. [Google Scholar] [CrossRef] [PubMed]
- Aissat, A.; de Becdelièvre, A.; Golmard, L.; Vasseur, C.; Costa, C.; Chaoui, A.; Martin, N.; Costes, B.; Goossens, M.; Girodon, E.; et al. Combined Computational-Experimental Analyses of CFTR Exon Strength Uncover Predictability of Exon-Skipping Level. Hum. Mutat. 2013, 34, 873–881. [Google Scholar] [CrossRef] [PubMed]
- Voelker, R.B.; Berglund, J.A. A comprehensive computational characterization of conserved mammalian intronic sequences reveals conserved motifs associated with constitutive and alternative splicing. Genome Res. 2007, 17, 1023–1033. [Google Scholar] [CrossRef] [PubMed]
- Yeo, G.W.; Van Nostrand, E.L.; Nostrand, E.L.V.; Liang, T.Y. Discovery and analysis of evolutionarily conserved intronic splicing regulatory elements. PLoS Genet. 2007, 3, e85. [Google Scholar]
- Van der Klift, H.M.; Jansen, A.M.L.; van der Steenstraten, N.; Bik, E.C.; Tops, C.M.J.; Devilee, P.; Wijnen, J.T. Splicing analysis for exonic and intronic mismatch repair gene variants associated with Lynch syndrome confirms high concordance between minigene assays and patient RNA analyses. Mol. Genet. Genom. Med. 2015, 3, 327–345. [Google Scholar] [CrossRef] [PubMed]
- Královicová, J.; Vorechovsky, I. Global control of aberrant splice-site activation by auxiliary splicing sequences: Evidence for a gradient in exon and intron definition. Nucleic Acids Res. 2007, 35, 6399–6413. [Google Scholar] [CrossRef] [PubMed]
- Flanigan, K.M.; Dunn, D.M.; von Niederhausern, A.; Soltanzadeh, P.; Howard, M.T.; Sampson, J.B.; Swoboda, K.J.; Bromberg, M.B.; Mendell, J.R.; Taylor, L.E.; et al. Nonsense mutation-associated Becker muscular dystrophy: Interplay between exon definition and splicing regulatory elements within the DMD gene. Hum. Mutat. 2011, 32, 299–308. [Google Scholar] [CrossRef] [PubMed]
- De Conti, L.; Baralle, M.; Buratti, E. Exon and intron definition in pre-mRNA splicing. Wiley Interdiscip. Rev. RNA 2013, 4, 49–60. [Google Scholar] [PubMed]
- Gutierrez-Arcelus, M.; Ongen, H.; Lappalainen, T.; Montgomery, S.B.; Buil, A.; Yurovsky, A.; Bryois, J.; Padioleau, I.; Romano, L.; Planchon, A.; et al. Tissue-Specific Effects of Genetic and Epigenetic Variation on Gene Regulation and Splicing. PLoS Genet. 2015, 11, e1004958. [Google Scholar]
- Baralle, M.; Skoko, N.; Knezevich, A.; De Conti, L.; Motti, D.; Bhuvanagiri, M.; Baralle, D.; Buratti, E.; Baralle, F.E. NF1 mRNA biogenesis: Effect of the genomic milieu in splicing regulation of the NF1 exon 37 region. FEBS Lett. 2006, 580, 4449–4456. [Google Scholar] [CrossRef] [PubMed]
- Mine, M.; Brivet, M.; Touati, G.; Grabowski, P.; Abitbol, M.; Marsac, C. Splicing Error in E1α Pyruvate Dehydrogenase mRNA Caused by Novel Intronic Mutation Responsible for Lactic Acidosis and Mental Retardation. J. Biol. Chem. 2003, 278, 11768–11772. [Google Scholar] [PubMed]
- Moseley, C.T.; Mullis, P.E.; Prince, M.A.; Phillips, J.A. An Exon Splice Enhancer Mutation Causes Autosomal Dominant GH Deficiency. J. Clin. Endocrinol. Metab. 2002, 87, 847–852. [Google Scholar] [CrossRef] [PubMed]
- Dhir, A.; Buratti, E. Alternative splicing: Role of pseudoexons in human disease and potential therapeutic strategies: Pseudoexons in human disease. FEBS J. 2010, 277, 841–855. [Google Scholar] [CrossRef] [PubMed]
- Divina, P.; Kvitkovicova, A.; Buratti, E.; Vorechovsky, I. Ab initio prediction of mutation-induced cryptic splice-site activation and exon skipping. Eur. J. Hum. Genet. 2009, 17, 759–765. [Google Scholar] [PubMed]
- Sharma, N.; Sosnay, P.R.; Ramalho, A.S.; Douville, C.; Franca, A.; Gottschalk, L.B.; Park, J.; Lee, M.; Vecchio-Pagan, B.; Raraigh, K.S.; et al. Experimental Assessment of Splicing Variants Using Expression Minigenes and Comparison with In Silico Predictions. Hum. Mutat. 2014, 35, 1249–1259. [Google Scholar] [PubMed]
- Walker, L.C.; Whiley, P.J.; Houdayer, C.; Hansen, T.V.O.; Vega, A.; Santamarina, M.; Blanco, A.; Fachal, L.; Southey, M.C.; Lafferty, A.; et al. Evaluation of a 5-Tier Scheme Proposed for Classification of Sequence Variants Using Bioinformatic and Splicing Assay Data: Inter-Reviewer Variability and Promotion of Minimum Reporting Guidelines. Hum. Mutat. 2013, 34, 1424–1431. [Google Scholar] [PubMed]
- Thompson, B.A.; Spurdle, A.B.; Plazzer, J.-P.; Greenblatt, M.S.; Akagi, K.; Al-Mulla, F.; Bapat, B.; Bernstein, I.; Capellá, G.; den Dunnen, J.T.; et al. Application of a 5-tiered scheme for standardized classification of 2,360 unique mismatch repair gene variants in the InSiGHT locus-specific database. Nat. Genet. 2014, 46, 107–115. [Google Scholar] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Spurdle, A.B.; Couch, F.J.; Hogervorst, F.B.L.; Radice, P.; Sinilnikova, O.M. Prediction and assessment of splicing alterations: Implications for clinical testing. Hum. Mutat. 2008, 29, 1304–1313. [Google Scholar] [CrossRef] [PubMed]
- Fraile-Bethencourt, E.; Díez-Gómez, B.; Velásquez-Zapata, V.; Acedo, A.; Sanz, D.J.; Velasco, E.A. Functional classification of DNA variants by hybrid minigenes: Identification of 30 spliceogenic variants of BRCA2 exons 17 and 18. PLoS Genet. 2017, 13, e1006691. [Google Scholar] [CrossRef] [PubMed]
- Petkovic, V.; Godi, M.; Lochmatter, D.; Eblé, A.; Flück, C.E.; Robinson, I.C.; Mullis, P.E. Growth Hormone (GH)-Releasing Hormone Increases the Expression of the Dominant-Negative GH Isoform in Cases of Isolated GH Deficiency due to GH Splice-Site Mutations. Endocrinology 2010, 151, 2650–2658. [Google Scholar] [CrossRef] [PubMed]
- Suñé-Pou, M.; Prieto-Sánchez, S.; Boyero-Corral, S.; Moreno-Castro, C.; El Yousfi, Y.; Suñé-Negre, J.; Hernández-Munain, C.; Suñé, C. Targeting Splicing in the Treatment of Human Disease. Genes 2017, 8, 87. [Google Scholar] [CrossRef] [PubMed]
- Sardone, V.; Zhou, H.; Muntoni, F.; Ferlini, A.; Falzarano, M. Antisense Oligonucleotide-Based Therapy for Neuromuscular Disease. Molecules 2017, 22, 563. [Google Scholar] [CrossRef] [PubMed]
- Tian, N.; Li, J.; Shi, J.; Sui, G. From General Aberrant Alternative Splicing in Cancers and Its Therapeutic Application to the Discovery of an Oncogenic DMTF1 Isoform. Int. J. Mol. Sci. 2017, 18, 191. [Google Scholar] [CrossRef] [PubMed]
- Havens, M.A.; Hastings, M.L. Splice-switching antisense oligonucleotides as therapeutic drugs. Nucleic Acids Res. 2016, 44, 6549–6563. [Google Scholar] [CrossRef] [PubMed]


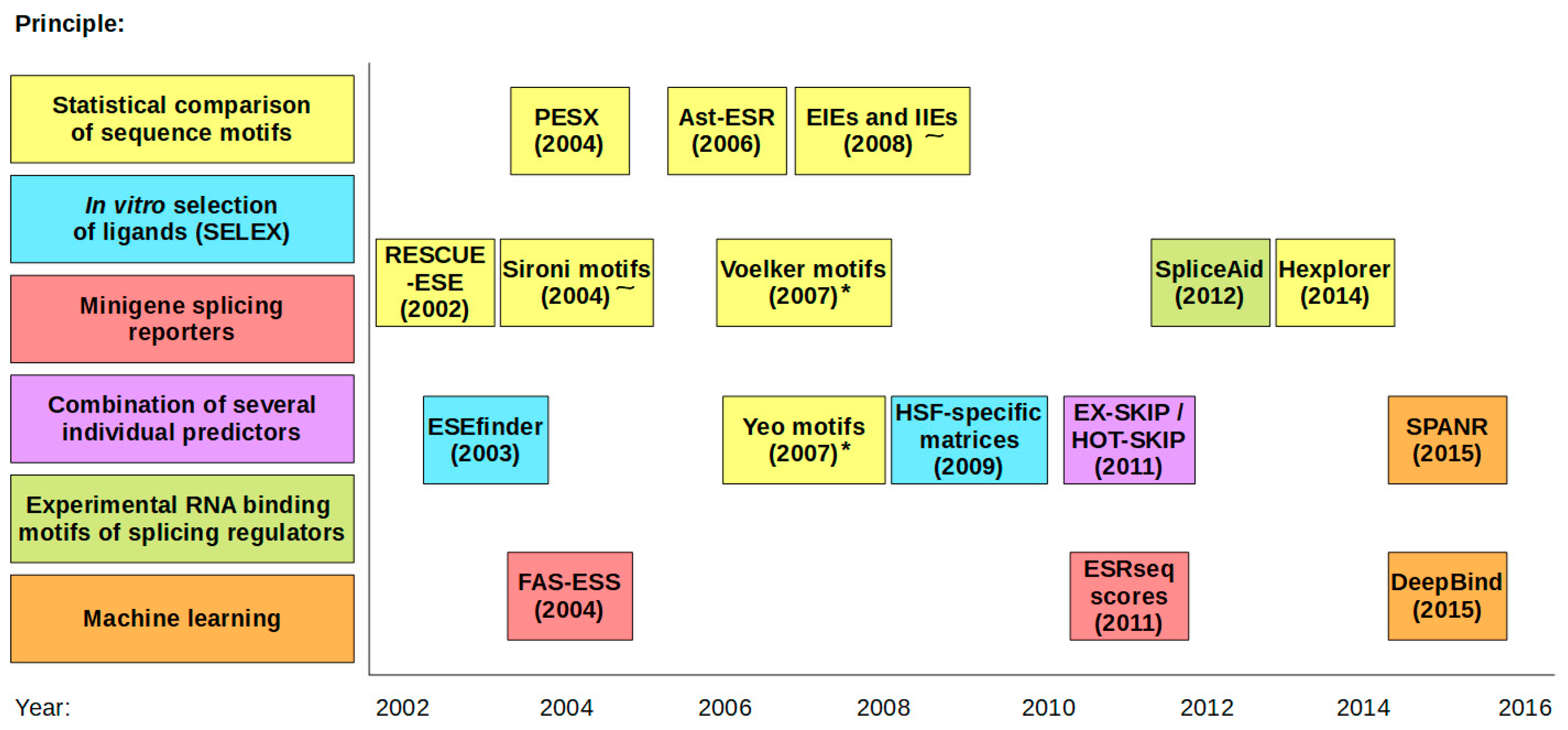{kind=link}
| Prediction Tool | Principle | Website | Reference | Evaluation |
|---|---|---|---|---|
| ESE-finder | SELEX (in vitro selection of ligands) | http://krainer01.cshl.edu/cgi-bin/tools/ESE3/esefinder.cgi?process=home | [24] | [15,19,27,28,29,30,31] |
| ESRseq | testing of all possible k-mers for positive and negative splicing influences, based on QUEPASA method | [32] | [13,15,19] | |
| FAS-ESS | analysis of random sequence silencing properties in the minigene settings | http://genes.mit.edu/fas-ess/ | [33] | [30] |
| Hexplorer | statistical comparison of hexamer sequence motifs | http://nar.oxfordjournals.org/content/early/2014/08/21/nar.gku736/suppl/DC1 | [34] | [15,19] |
| PESX | statistical comparison of octamer sequence motifs | http://cubio.biology.columbia.edu/pesx/pesx/ | [35] | [19,27] |
| RESCUE-ESE | statistical comparison of hexamer sequence motifs | http://genes.mit.edu/burgelab/rescue-ese/ | [36] | [19,27,30,31] |
| SPANR | splicing code, machine learning | http://tools.genes.toronto.edu/ | [37] | [15] |
| SpliceAid2 | database of in vitro proved splicing factors binding sites | www.introni.it/spliceaid.html | [38] |
| Prediction Tool | Included Tools | Website | Reference | Evaluation |
|---|---|---|---|---|
| EX-SKIP | PESE and PESS [35], FAS-ESS [33], RESCUE-ESE [36], EIEs and IIEs [43], NI-ESE and NI-ESS [44] | http://ex-skip.img.cas.cz/ | [41] | [13,15,19,31,45] |
| HOT-SKIP | PESE and PESS, FAS-ESS, RESCUE-ESE, EIEs and IIEs, NI-ESE and NI-ESS | http://hot-skip.img.cas.cz/ | [41] | |
| Human Splicing Finder (HSF) | ESE-finder [24], RESCUE-ESE, PESE and PESS, EIEs and IIEs, FAS-ESS and ESS decamers [33], Exonic splicing regulatory sequences [5], HSF- specific matrices for Tra2-β, 9G8 and hnRNP A1 [26] | http://www.umd.be/HSF3/index.html | [26] | [15] |
| Sroogle | ESE-finder, RESCUE-ESE, FAS-ESS, PESE and PESS, other SRE predictions according to Voelker [46], Yeo [47], Goren [5] | http://sroogle.tau.ac.il/ | [42] |
| Gene | cDNA Variant | Exon | Effect on Exon Skipping | ΔESRseq (−0.5) | Hexplorer: ΔHZEI (−0.5) | EX-SKIP: ESS/ESE mut/wt (1) |
|---|---|---|---|---|---|---|
| BRCA1 | c.5123C > A | 18 | increased | −2.574 | -10.85 | 1.05 |
| c.5434C > G | 23 | increased | 0.558 | −1.28 | 1.09 | |
| c.5453A > G | 23 | increased | −2.176 | −15.02 | 1.43 | |
| c.5096G > A | 18 | none | −1.731 | −0.22 | 0.93 | |
| c.5116G > A | 18 | none | 1.582 | 2.67 | 0.86 | |
| c.5411T > A | 23 | none | 0.66 | 4.33 | 0.83 | |
| BRCA2 | c.231T > G | 3 | increased | 1.65 | 4.76 | 0.97 |
| c.439C > T | 5 | increased | −2.69 | −13.06 | 1.77 | |
| c.7992T > A | 18 | increased | −1.11 | 0.00 | 1.00 | |
| c.8257_8259delCTT | 18 | increased | −0.51 | 0.52 | 0.98 | |
| c.9234C > T | 24 | increased | −1.24 | −12.07 | 1.12 | |
| c.223 > C | 3 | none | 0.37 | −11.19 | 0.97 | |
| c.433_435delGTT | 5 | none | −0.23 | 6.46 | 0.51 | |
| c.7994A > G | 18 | none | −1.21 | 0.24 | 1.02 | |
| c.8182G > A | 18 | none | −1.65 | 0.00 | 0.98 | |
| c.9216G > 1 | 24 | none | −2.88 | −10.94 | 1.04 | |
| NF1 | c.557A > T | 5 | increased | −2.43 | −12.21 | 1.25 |
| c.528T > A | 5 | none | 1.17 | 0.70 | 0.90 | |
| DMD | c.5287C > T | 37 | increased | −0.70 | −16.84 | 1.16 |
| c.5308A > T | 37 | none | −0.05 | −2.85 | 1.05 | |
| True calls | 70.0% | 70.0% | 70.0% | |||
| Sensitivity | 80.0% | 70.0% | 70.0% | |||
| Specificity | 60.0% | 70.0% | 70.0% |
| Class | Observation | Reference | |
|---|---|---|---|
| 5: pathogenic | • | assay on mRNA from patients tissue samples | [59] 1 [60] |
| AND | no wt transcript detected from variant allele | ||
| AND | aberrant transcripts introduce PTC or deletion disrupting functional domain | ||
| OR deletion disrupting protein conformation | only in [60] | ||
| OR | damaging effect on the gene or gene product (extent not specified) | [61] | |
| AND | other lines of evidence supporting variant pathogenicity 2 (stronger than for class 4) | ||
| • | lab assays based on mRNA (e.g., minigenes) | [60,61] | |
| AND | variant-specific abrogated function (extent not specified) | ||
| AND | additional frequency/co-segregation/clinical data, additional molecular/mechanistic evidences from other sources, supporting variant pathogenicity (stronger than for class 4) | ||
| 4: probably pathogenic | • | assay on mRNA from patients tissue samples | [61] |
| AND | damaging effect on the gene or gene product (extent not specified) | ||
| AND | other lines of evidence supporting variant pathogenicity (milder than for class 5) | ||
| • | lab assays based on mRNA (e.g., minigenes) | [60,61] | |
| AND | variant-specific abrogated function (extent not specified) | ||
| AND | additional frequency/co-segregation/clinical data, additional molecular/mechanistic evidences from other sources, supporting variant pathogenicity (milder than for class 5) | ||
| • | minigene assays | [48] | |
| AND | complete aberrant and frameshifting effect/ deletion of a functional domain effect | ||
| 3: uncertain pathogenicity | all variants that do not fall into other classes | [59] | |
| e.g., | aberrant transcripts produce deletion not disrupting known functional domains | ||
| e.g., | change in the level of alternative transcripts, at least some of which do not introduce PTC or protein-disrupting deletion | ||
| e.g., | leaky aberrant splicing | ||
| e.g., | contradictory benign and pathogenic criteria | [61] | |
| 2: likely not pathogenic | • | assay on mRNA from patients tissue samples | [59,60,61] |
| AND | no associated mRNA aberration detected | ||
| AND | analysis including NMD inhibition | only [60] | |
| AND | other lines of evidence disproving variant pathogenicity | only [61] | |
| • | lab assays based on mRNA (e.g., minigenes) | [60,61] | |
| AND | variant-specific proficient function | ||
| AND | additional frequency/co-segregation/clinical data, additional molecular/mechanistic evidences from other sources, disproving variant pathogenicity (milder than for class 1) | ||
| 1: not pathogenic | • | lab assays based on mRNA (e.g., minigenes) | [60,61] |
| AND | variant-specific proficient function | ||
| AND | additional frequency/co-segregation/clinical data, additional molecular/mechanistic evidences from other sources, disproving variant pathogenicity (stronger than for class 2) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grodecká, L.; Buratti, E.; Freiberger, T. Mutations of Pre-mRNA Splicing Regulatory Elements: Are Predictions Moving Forward to Clinical Diagnostics? Int. J. Mol. Sci. 2017, 18, 1668. https://doi.org/10.3390/ijms18081668
Grodecká L, Buratti E, Freiberger T. Mutations of Pre-mRNA Splicing Regulatory Elements: Are Predictions Moving Forward to Clinical Diagnostics? International Journal of Molecular Sciences. 2017; 18(8):1668. https://doi.org/10.3390/ijms18081668
Chicago/Turabian StyleGrodecká, Lucie, Emanuele Buratti, and Tomáš Freiberger. 2017. "Mutations of Pre-mRNA Splicing Regulatory Elements: Are Predictions Moving Forward to Clinical Diagnostics?" International Journal of Molecular Sciences 18, no. 8: 1668. https://doi.org/10.3390/ijms18081668
APA StyleGrodecká, L., Buratti, E., & Freiberger, T. (2017). Mutations of Pre-mRNA Splicing Regulatory Elements: Are Predictions Moving Forward to Clinical Diagnostics? International Journal of Molecular Sciences, 18(8), 1668. https://doi.org/10.3390/ijms18081668