Correlation between Oxidative Stress, Nutrition, and Cancer Initiation

 ,
, 
 and
and 
Abstract

1. Introduction
2. Correlation between Nutrition and Oxidative Stress
2.1. Nutrition Induces Oxidative Stress during Early Human Development
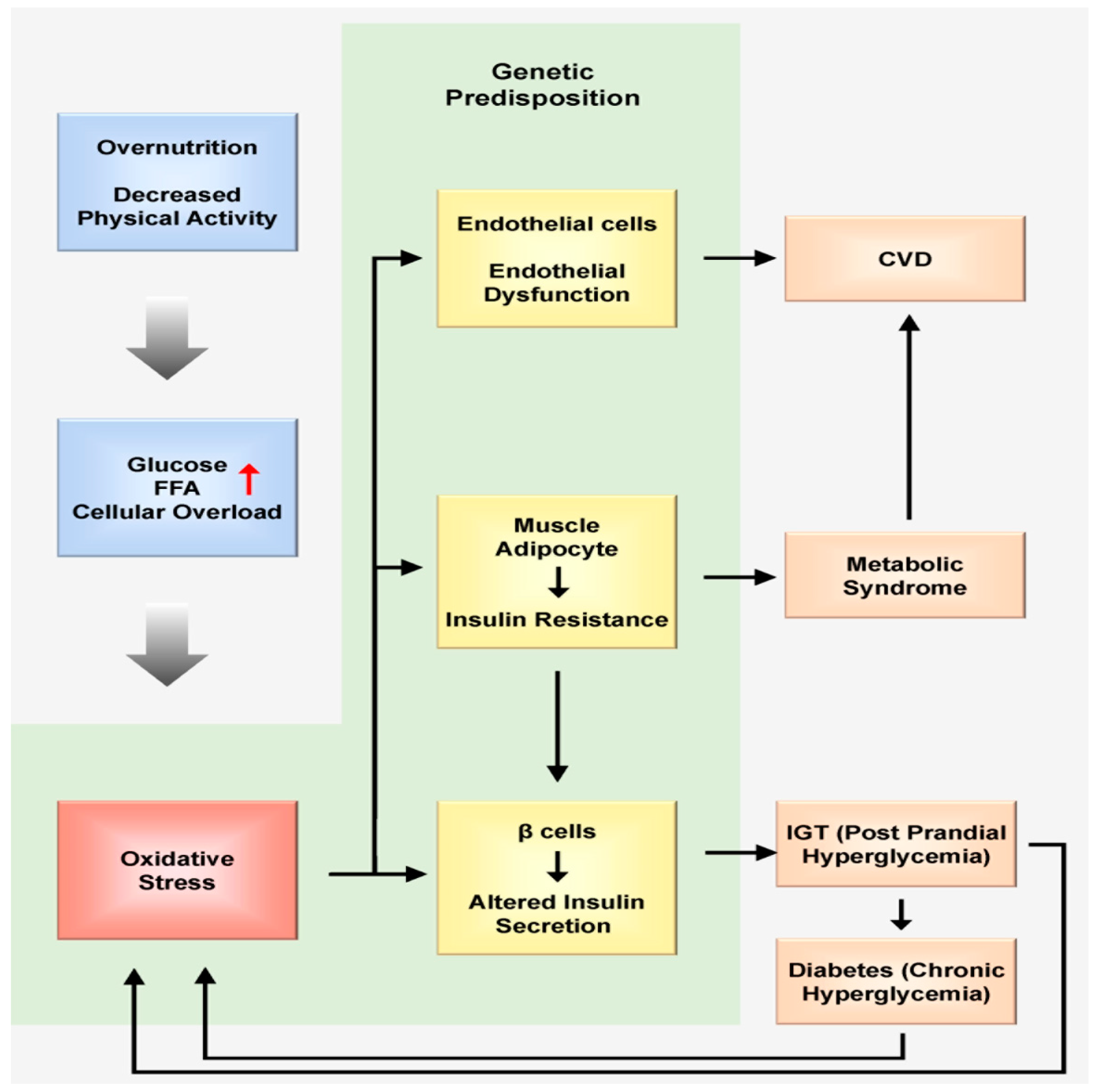
2.2. Nutrition Triggers Oxidative Stress at the Cellular Level
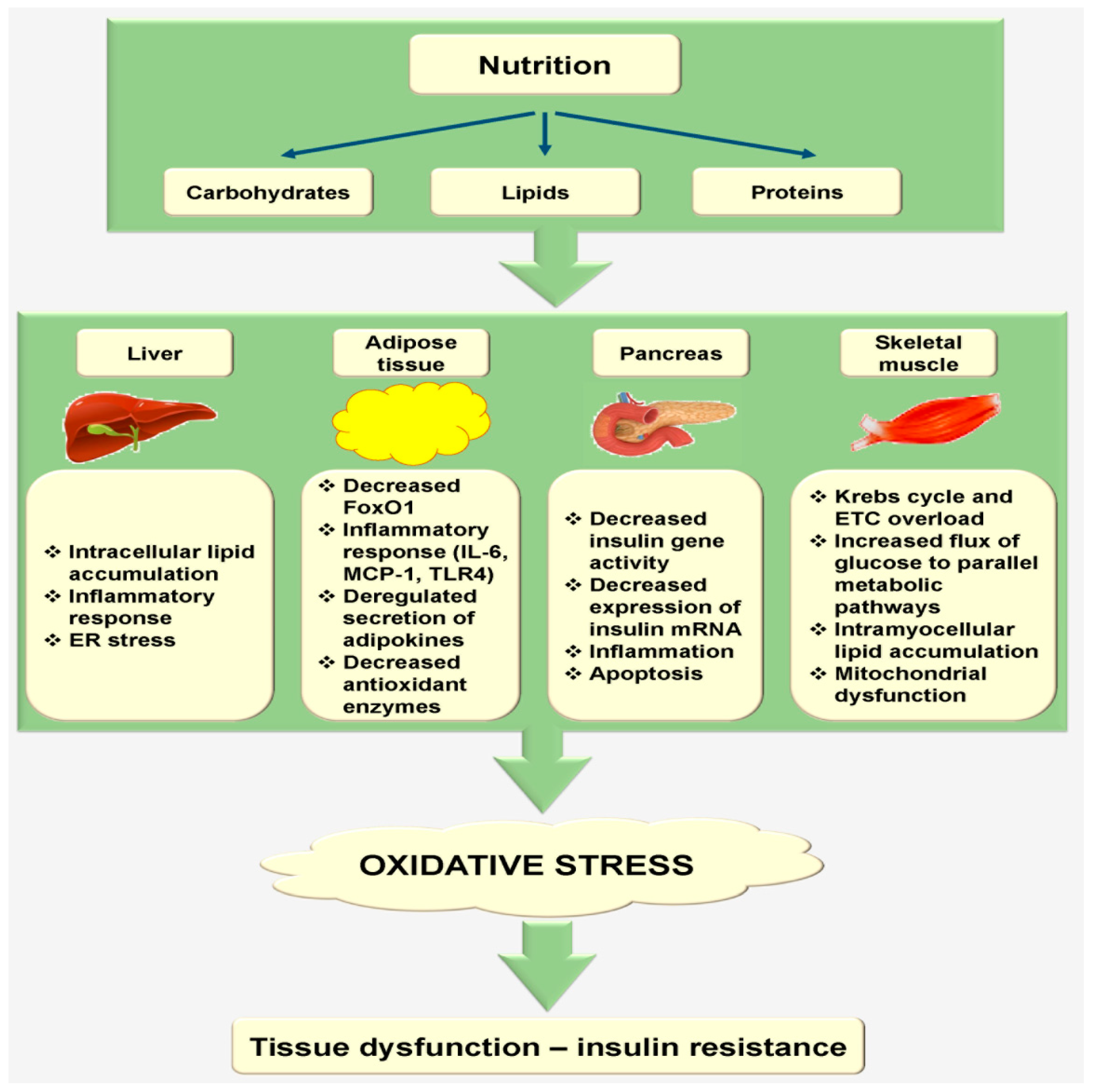
2.3. Nutrition Increases Oxidative Stress during Tissue Metabolism
2.3.1. Liver
2.3.2. Adipose Tissue
2.3.3. Pancreas
2.3.4. Skeletal Muscle
2.4. Nutrition Induces Oxidative Homeostasis
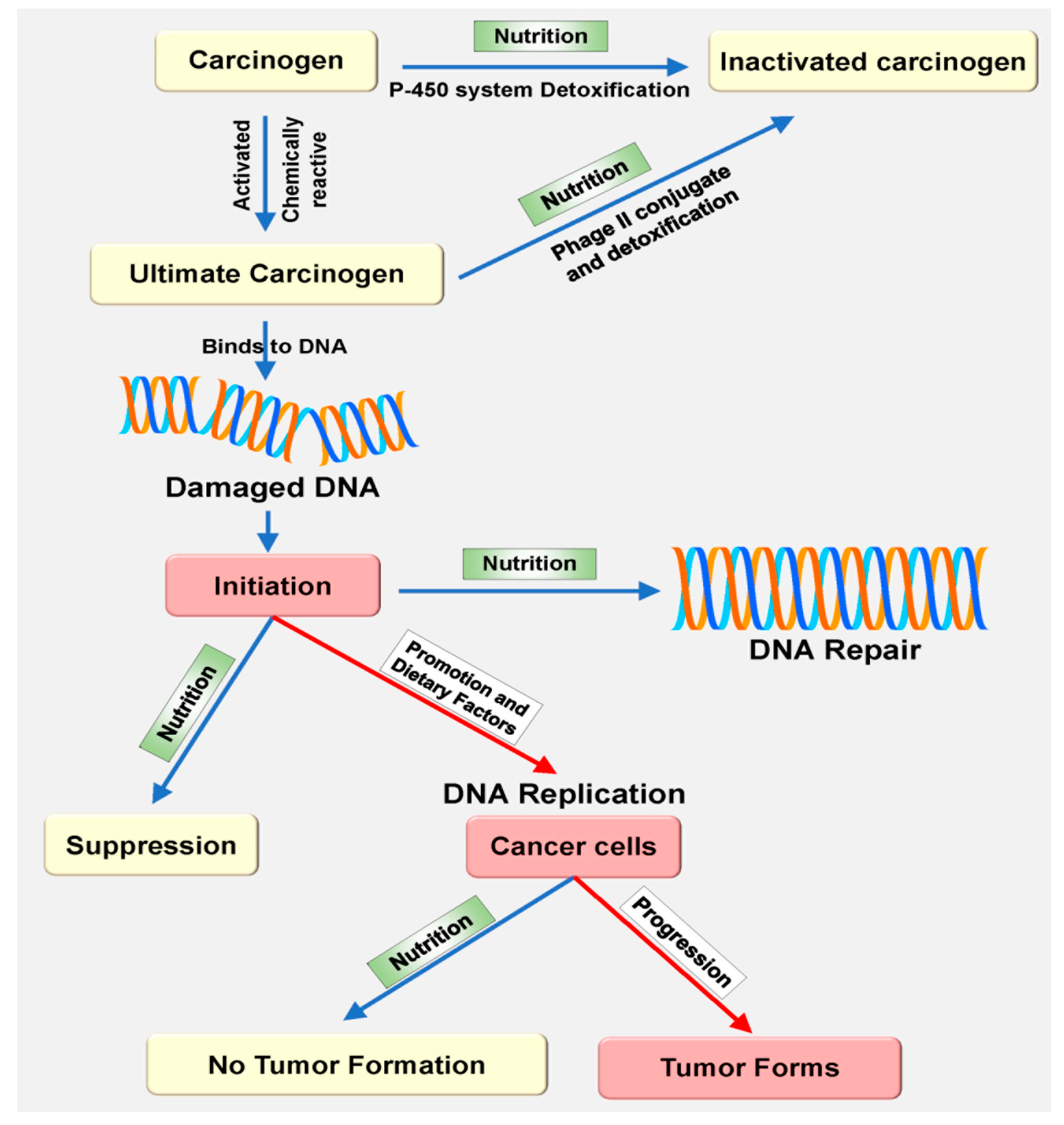
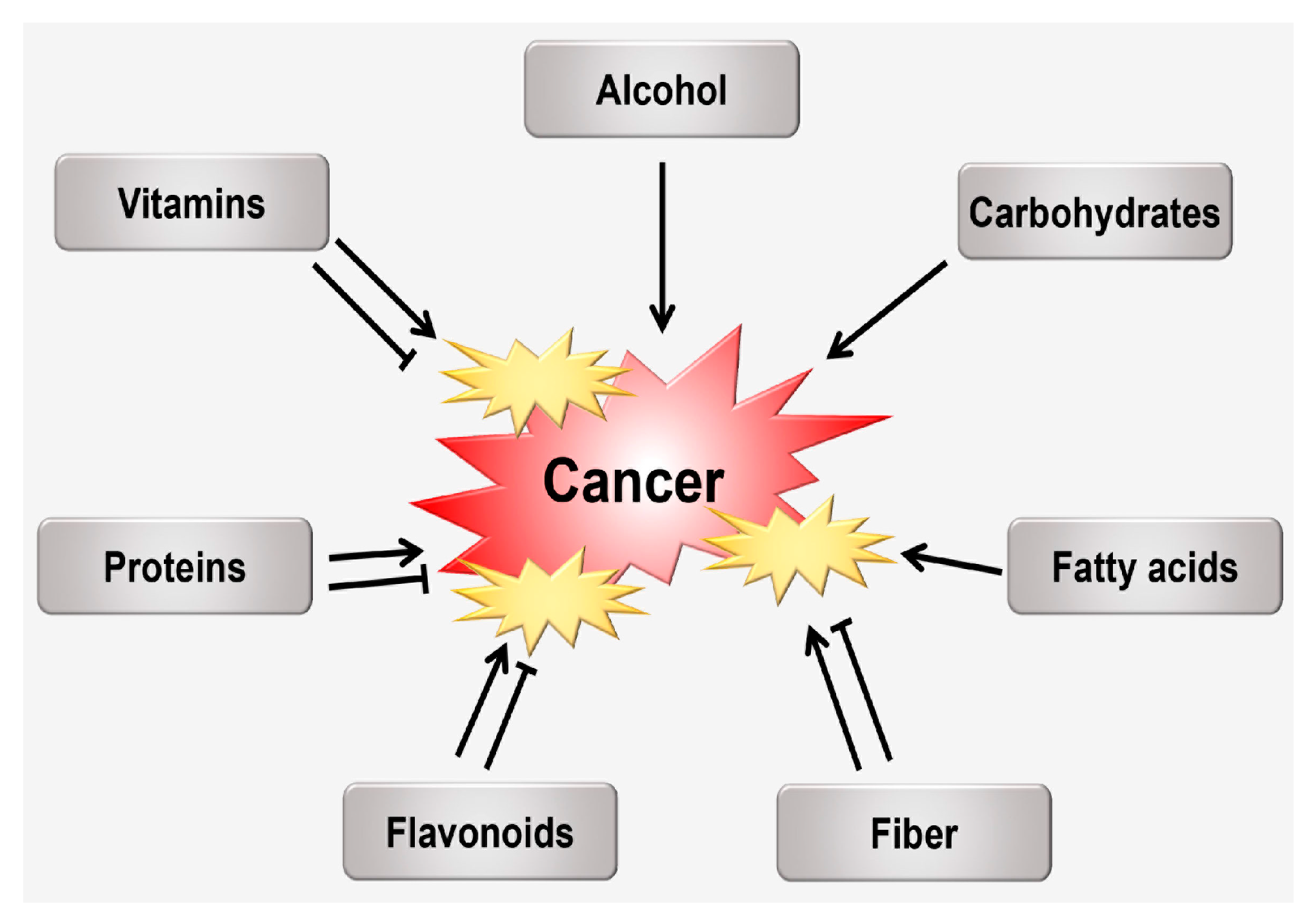
3. The Relationship between Nutrition and Oxidative Stress Following Carcinogenesis
3.1. Alcohol
3.2. Carbohydrates
3.3. Fatty acids (FAs)
3.4. Fiber
3.5. Flavonoids
3.6. Proteins
3.7. Vitamins
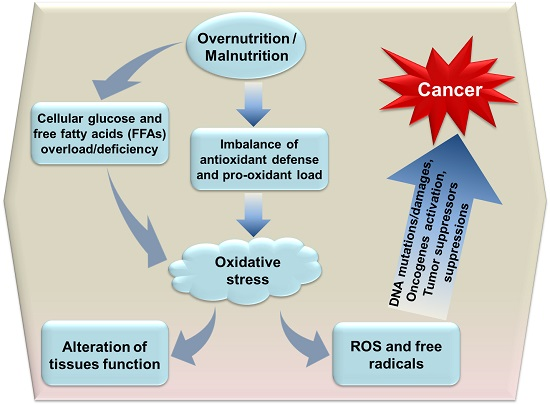
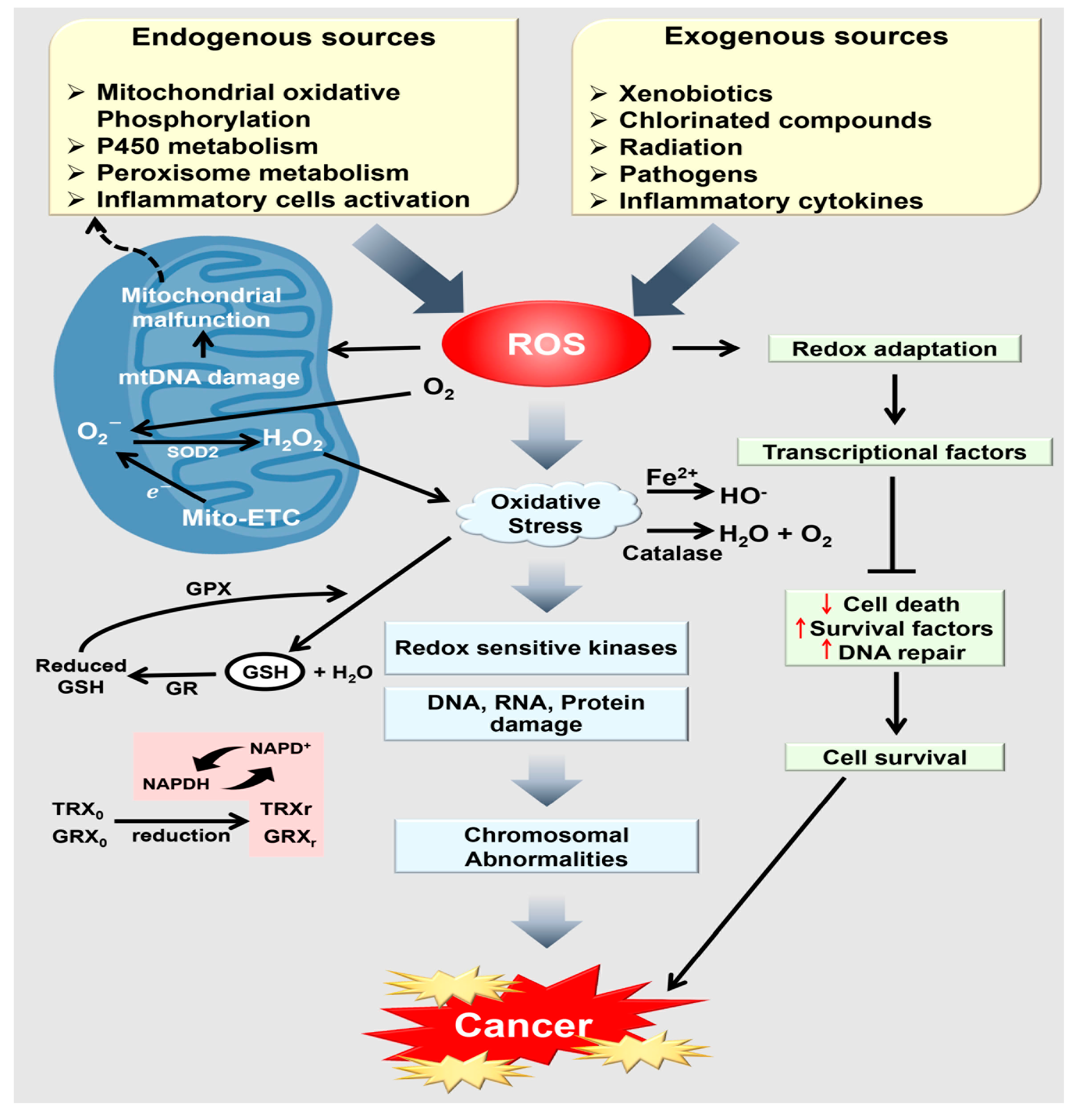
4. The Association between Oxidative Stress and Cancer Progression
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| 8-OHdG | 8-OH deoxyguanosine |
| AGE | advanced glycation end product |
| ATP | adenosine triphosphate |
| BMI | body mass index |
| COX-2 | cyclooxygenase 2 |
| CPT-1 | carnitine palmitoyltransferase-1 |
| CRP | C-reactive protein |
| CSC | cancer stem cells |
| CVD | cardiovascular disease |
| DAG | diacylglycerols |
| DHA | docosahexaenoic acid |
| EFA | Essential fatty acids |
| EPA | eicosapentaenoic acid |
| EPIC | European Prospective Investigation into Cancer and Nutrition |
| ER: | endoplasmic reticulum |
| ERK | extracellular signal-regulated kinase |
| ETC: | electron transport chain |
| FFA | free fatty acids |
| FoxO1 | Forkhead box protein O1 |
| GI | glycemic indexes |
| GLUT4 | glucose transporter type 4 |
| GPX | glutathione peroxidase |
| GR | glutathione reductase |
| GRXo | glutaredoxin (oxidized) |
| GRXr | glutaredoxin (reduced) |
| GSHr | glutathione (reduced) |
| HDL | high-density lipoproteins |
| IARC | International Agency for Research on Cancer |
| IGT | impaired glucose tolerance |
| IKKα | IκB kinase α |
| IKKβ | IκB kinase β |
| IL- 6 | Interleukin 6 |
| IκBα | inhibitor κBα |
| JNK | c-Jun N-terminal kinase |
| MAPK | mitogen-activated protein kinase |
| MCP-1 | Monocyte chemoattractant protein-1 |
| MDA | malondialdehyde |
| Mito-ETC | mitochondrial electron transport chain |
| MNC | mononuclear cells |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| NF-κB | nuclear factor κB |
| NO | nitric oxide |
| NOX4 | NADPH oxidase 4 |
| NRF2 | nuclear factor erythroid 2-related factor 2 |
| PAI-1 | plasminogen activator inhibitor-1 |
| PCOS | polycystic ovarian syndrome |
| PKC | protein kinase C |
| PMNL | polymorphonuclear leukocytes |
| PTEN | phosphatase and tensin homolog |
| PTP | protein tyrosine phosphatase |
| ROS | reactive oxygen species |
| SOD | superoxide dismutase |
| TAG | triacylglycerol |
| TLR4: | Toll-like receptor 4 |
| TRXo | thioredoxin (oxidized) |
| TRXr | thioredoxin (reduced) |
| VLDL | very low-density lipoproteins |
| WCRF | World Cancer Research Fund |
| WHI | Women’s Health Initiative |
| WHO | World Health Organization |
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Mosby, T.T.; Cosgrove, M.; Sarkardei, S.; Platt, K.L.; Kaina, B. Nutrition in Adult and Childhood Cancer: Role of Carcinogens and Anti-carcinogens. Anticancer Res. 2012, 32, 4171–4192. [Google Scholar] [PubMed]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global Cancer Statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Troselj, K.G.; Gueraud, F.; Glavan, T.M.; Pierre, F.; Zarkovic, N. A Review on Food-Associated Carcinogenesis. In Food Toxicology; CRC Press: Boca Raton, FL, USA, 2016; pp. 35–56. [Google Scholar]
- Diamanti-Kandarakis, E.; Papalou, O.; Kandaraki, E.A.; Kassi, G. MECHANISMS IN ENDOCRINOLOGY Nutrition as a mediator of oxidative stress in metabolic and reproductive disorders in women. Eur. J. Endocrinol. 2017, 176, R79–R99. [Google Scholar] [CrossRef] [PubMed]
- Commoner, B.; Townsend, J.; Pake, G.E. Free radicals in biological materials. Nature 1954, 174, 689–691. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Reactive oxygen species in living systems: Source, biochemistry, and role in human disease. Am. J. Med. 1991, 91, S14–S22. [Google Scholar] [CrossRef]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Oxidative stress, nutrition and health. Experimental strategies for optimization of nutritional antioxidant intake in humans. Free Radic. Res. 1996, 25, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Stahl, W.; Sevanian, A. Nutritional, dietary and postprandial oxidative stress. J. Nutr. 2005, 135, 969–972. [Google Scholar] [PubMed]
- Forcados, G.E.; Chinyere, C.N.; Shu, M.L. Acalypha wilkesiana: Therapeutic and toxic potential. J. Med. Surg. Pathol. 2016, 1, 122. [Google Scholar]
- Alpay, M.; Backman, L.R.F.; Cheng, X.D.; Dukel, M.; Kim, W.J.; Ai, L.B.; Brown, K.D. Oxidative stress shapes breast cancer phenotype through chronic activation of ATM-dependent signaling. Breast Cancer Res. Treat. 2015, 151, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Pisoschi, A.M.; Pop, A. The role of antioxidants in the chemistry of oxidative stress: A review. Eur. J. Med. Chem. 2015, 97, 55–74. [Google Scholar] [CrossRef] [PubMed]
- Dalle-Donne, I.; Rossi, R.; Colombo, R.; Giustarini, D.; Milzani, A. Biomarkers of oxidative damage in human disease. Clin. Chem. 2006, 52, 601–623. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.; Aponte-Mellado, A.; Premkumar, B.J.; Shaman, A.; Gupta, S. The effects of oxidative stress on female reproduction: A review. Reprod. Biol. Endocrinol. 2012, 10, 49. [Google Scholar] [CrossRef] [PubMed]
- Tangvarasittichai, S. Oxidative stress, insulin resistance, dyslipidemia and type 2 diabetes mellitus. World J. Diabetes 2015, 6, 456–480. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.D.; Cai, Q.; Shu, X.O.; Nechuta, S.J. The role of biomarkers of oxidative stress in breast cancer risk and prognosis: A systematic review of the epidemiologic literature. J. Women’s Health 2017, 26, 467–482. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Li, L.; Gao, G.; Wei, G.; Zheng, Y.; Wang, C.; Gao, N.; Zhao, Y.; Deng, J.; Chen, H. Elevation of GPRC5A expression in colorectal cancer promotes tumor progression through VNN-1 induced oxidative stress. Int. J. Cancer 2017, 140, 2734–2747. [Google Scholar] [CrossRef] [PubMed]
- Saijo, H.; Hirohashi, Y.; Torigoe, T.; Horibe, R.; Takaya, A.; Murai, A.; Kubo, T.; Kajiwara, T.; Tanaka, T.; Shionoya, Y.; et al. Plasticity of lung cancer stem-like cells is regulated by the transcription factor HOXA5 that is induced by oxidative stress. Oncotarget 2016, 7, 50043–50056. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.P.; Li, Z.N.; Ye, Y.S.; Xie, L.J.; Li, W. Oxidative stress and liver cancer: Etiology and therapeutic targets. Oxidative Med. Cell. Longev. 2016, 2016, 7891574. [Google Scholar] [CrossRef] [PubMed]
- Oh, B.; Figtree, G.; Costa, D.; Eade, T.; Hruby, G.; Lim, S.; Elfiky, A.; Martine, N.; Rosenthal, D.; Clarke, S.; et al. Oxidative stress in prostate cancer patients: A systematic review of case control studies. Prostate Int. 2016, 4, 71–87. [Google Scholar] [CrossRef] [PubMed]
- Saed, G.M.; Diamond, M.P.; Fletcher, N.M. Updates of the role of oxidative stress in the pathogenesis of ovarian cancer. Gynecol. Oncol. 2017, 145, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Jaroonwitchawan, T.; Chaicharoenaudomrung, N.; Natnkaew, J.; Noisa, P. Curcumin attenuates paraquat-induced cell death in human neuroblastoma cells through modulating oxidative stress and autophagy. Neurosci. Lett. 2017, 636, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Forcados, G.E.; James, D.B.; Sallau, A.B.; Muhammad, A.; Mabeta, P. Oxidative stress and carcinogenesis: Potential of phytochemicals in breast cancer therapy. Nutr. Cancer 2017, 69, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Matsui, A.; Ikeda, T.; Enomoto, K.; Hosoda, K.; Nakashima, H.; Omae, K.; Watanabe, M.; Hibi, T.; Kitajima, M. Increased formation of oxidative DNA damage, 8-hydroxy-2′-deoxyguanosine, in human breast cancer tissue and its relationship to GSTP1 and COMT genotypes. Cancer Lett. 2000, 151, 87–95. [Google Scholar] [CrossRef]
- Sova, H.; Jukkola-Vuorinen, A.; Puistola, U.; Kauppila, S.; Karihtala, P. 8-Hydroxydeoxyguanosine: A new potential independent prognostic factor in breast cancer. Br. J. Cancer 2010, 102, 1018–1023. [Google Scholar] [CrossRef] [PubMed]
- Aldini, G.; Dalle-Donne, I.; Facino, R.M.; Milzani, A.; Carini, M. Intervention strategies to inhibit protein carbonylation by lipoxidation-derived reactive carbonyls. Med. Res. Rev. 2007, 27, 817–868. [Google Scholar] [CrossRef] [PubMed]
- Skurk, T.; Alberti-Huber, C.; Herder, C.; Hauner, H. Relationship between adipocyte size and adipokine expression and secretion. J. Clin. Endocrinol. Metab. 2007, 92, 1023–1033. [Google Scholar] [CrossRef] [PubMed]
- Montezano, A.C.; Dulak-Lis, M.; Tsiropoulou, S.; Harvey, A.; Briones, A.M.; Touyz, R.M. Oxidative stress and human hypertension: Vascular mechanisms, biomarkers, and novel therapies. Can. J. Cardiol. 2015, 31, 631–641. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.T.; Tian, X.Y.; Huang, Y. Endothelial dysfunction in diabetes and hypertension: Cross talk in RAS, BMP4, and ROS-dependent COX-2-derived prostanoids. J. Cardiovasc. Pharmacol. 2013, 61, 204–214. [Google Scholar] [CrossRef] [PubMed]
- WHO. Diet, nutrition and the prevention of chronic diseases. In Proceedings of the WHO/FAO Expert Consultation on Diet, Nutrition and the Prevention of Chronic Diseases, Geneva, Switzerland, 28 January–1 February 2002. [Google Scholar]
- Hernanz, R.; Briones, A.M.; Salaices, M.; Alonso, M.J. New roles for old pathways? A circuitous relationship between reactive oxygen species and cyclo-oxygenase in hypertension. Clin. Sci. 2014, 126, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Ward, N.C.; Hodgson, J.M.; Puddey, I.B.; Mori, T.A.; Beilin, L.J.; Croft, K.D. Oxidative stress in human hypertension: Association with antihypertensive treatment, gender, nutrition, and lifestyle. Free Radic. Biol. Med. 2004, 36, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Houstis, N.; Rosen, E.D.; Lander, E.S. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature 2006, 440, 944–948. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-S. Impact of maternal diet on the epigenome during in utero life and the developmental programming of diseases in childhood and adulthood. Nutrients 2015, 7, 9492–9507. [Google Scholar] [CrossRef] [PubMed]
- Barker, D.J.; Osmond, C.; Winter, P.; Margetts, B.; Simmonds, S.J. Weight in infancy and death from ischaemic heart disease. Lancet 1989, 334, 577–580. [Google Scholar] [CrossRef]
- Barker, D.J. Maternal nutrition, fetal nutrition, and disease in later life. Nutr. J. 1997, 13, 807–813. [Google Scholar] [CrossRef]
- Armitage, J.A.; Taylor, P.D.; Poston, L. Experimental models of developmental programming: Consequences of exposure to an energy rich diet during development. J. Physiol. 2005, 565, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Gamborg, M.; Byberg, L.; Rasmussen, F.; Andersen, P.K.; Baker, J.L.; Bengtsson, C.; Canoy, D.; Drøyvold, W.; Eriksson, J.G.; Forsén, T. Birth weight and systolic blood pressure in adolescence and adulthood: Meta-regression analysis of sex-and age-specific results from 20 Nordic studies. Am. J. Epidemiol. 2007, 166, 634–645. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.P.; Al-Hasan, Y. Impact of oxidative stress in fetal programming. J. Pregnancy 2012, 2012, 582748. [Google Scholar] [CrossRef] [PubMed]
- Aiken, C.E.; Tarry-Adkins, J.L.; Penfold, N.C.; Dearden, L.; Ozanne, S.E. Decreased ovarian reserve, dysregulation of mitochondrial biogenesis, and increased lipid peroxidation in female mouse offspring exposed to an obesogenic maternal diet. FASEB J. 2016, 30, 1548–1556. [Google Scholar] [CrossRef] [PubMed]
- Saad, M.I.; Abdelkhalek, T.M.; Haiba, M.M.; Saleh, M.M.; Hanafi, M.Y.; Tawfik, S.H.; Kamel, M.A. Maternal obesity and malnourishment exacerbate perinatal oxidative stress resulting in diabetogenic programming in F1 offspring. J. Endocrinol. Investig. 2016, 39, 643–655. [Google Scholar] [CrossRef] [PubMed]
- Fetoui, H.; Garoui, M.; Zeghal, N. Protein restriction in pregnant- and lactating rats-induced oxidative stress and hypohomocysteinaemia in their offspring. J. Anim. Physiol. Anim. Nutr. 2009, 93, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.; Simmons, R.A. Maternal Antioxidant Supplementation Prevents Adiposity in the Offspring of Western Diet-Fed Rats. Diabetes 2010, 59, 3058–3065. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, P.; Hamouda, W.; Garg, R.; Aljada, A.; Ghanim, H.; Dandona, P. Glucose challenge stimulates reactive oxygen species (ROS) generation by leucocytes. J. Clin. Endocrinol. Metab. 2000, 85, 2970–2973. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, P.; Ghanim, H.; Hamouda, W.; Aljada, A.; Garg, R.; Dandona, P. Both lipid and protein intakes stimulate increased generation of reactive oxygen species by polymorphonuclear leukocytes and mononuclear cells. Am. J. Clin. Nutr. 2002, 75, 767–772. [Google Scholar] [PubMed]
- Aljada, A.; Mohanty, P.; Ghanim, H.; Abdo, T.; Tripathy, D.; Chaudhuri, A.; Dandona, P. Increase in intranuclear nuclear factor kappaB and decrease in inhibitor kappaB in mononuclear cells after a mixed meal: Evidence for a proinflammatory effect. Am. J. Clin. Nutr. 2004, 79, 682–690. [Google Scholar] [PubMed]
- Ceriello, A.; Motz, E. Is oxidative stress the pathogenic mechanism underlying insulin resistance, diabetes, and cardiovascular disease? The common soil hypothesis revisited. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 816–823. [Google Scholar] [CrossRef] [PubMed]
- Wallace, J.P.; Johnson, B.; Padilla, J.; Mather, K. Postprandial lipaemia, oxidative stress and endothelial function: A review. Int. J. Clin. Pract. 2010, 64, 389–403. [Google Scholar] [CrossRef] [PubMed]
- Dandona, P.; Mohanty, P.; Ghanim, H.; Aljada, A.; Browne, R.; Hamouda, W.; Prabhala, A.; Afzal, A.; Garg, R. The suppressive effect of dietary restriction and weight loss in the obese on the generation of reactive oxygen species by leukocytes, lipid peroxidation, and protein carbonylation. J. Clin. Endocrinol. Metab. 2001, 86, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Dandona, P.; Mohanty, P.; Hamouda, W.; Ghanim, H.; Aljada, A.; Garg, R.; Kumar, V. Inhibitory effect of a two day fast on reactive oxygen species (ROS) generation by leucocytes and plasma ortho-tyrosine and meta-tyrosine concentrations. J. Clin. Endocrinol. Metab. 2001, 86, 2899–2902. [Google Scholar] [CrossRef] [PubMed]
- Rizza, W.; Veronese, N.; Fontana, L. What are the roles of calorie restriction and diet quality in promoting healthy longevity? Ageing Res. Rev. 2014, 13, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Bloomer, R.J.; Fisher-Wellman, K.H. Systemic oxidative stress is increased to a greater degree in young, obese women following consumption of a high fat meal. Oxidative Med. Cell. Longev. 2009, 2, 19–25. [Google Scholar] [CrossRef]
- Goto, C.; Nishioka, K.; Umemura, T.; Jitsuiki, D.; Sakagutchi, A.; Kawamura, M.; Chayama, K.; Yoshizumi, M.; Higashi, Y. Acute moderate-intensity exercise induces vasodilation through oxide bioavailiability an increase in nitric in humans. Am. J. Hypertens. 2007, 20, 825–830. [Google Scholar] [CrossRef] [PubMed]
- Elosua, R.; Molina, L.; Fito, M.; Arquer, A.; Sanchez-Queseda, J.L.; Covas, M.I.; Ordonez-Llanos, J.; Marrugat, J. Response of oxidative stress biomarkers to a 16-week aerobic physical activity program, and to acute physical activity, in healthy young men and women. Atherosclerosis 2003, 167, 327–334. [Google Scholar] [CrossRef]
- Bloomer, R.J.; Ferebee, D.E.; Fisher-Wellman, K.H.; Quindry, J.C.; Schilling, B.K. Postprandial oxidative stress: Influence of sex and exercise training status. Med. Sci. Sports Exerc. 2009, 41, 2111–2119. [Google Scholar] [CrossRef] [PubMed]
- Droge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [PubMed]
- Uribarri, J.; Stirban, A.; Sander, D.; Cai, W.; Negrean, M.; Buenting, C.E.; Koschinsky, T.; Vlassara, H. Single oral challenge by advanced glycation end products acutely impairs endothelial function in diabetic and nondiabetic subjects. Diabetes Care 2007, 30, 2579–2582. [Google Scholar] [CrossRef] [PubMed]
- Diamanti-Kandarakis, E.; Katsikis, I.; Piperi, C.; Alexandraki, K.; Panidis, D. Effect of long-term orlistat treatment on serum levels of advanced glycation end-products in women with polycystic ovary syndrome. Clin. Endocrinol. 2007, 66, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Muoio, D.M.; Newgard, C.B. Mechanisms of disease: Molecular and metabolic mechanisms of insulin resistance and β-cell failure in type 2 diabetes. Nat. Rev. Mol. Cell Biol. 2008, 9, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Liu, Z.X.; Qu, X.; Elder, B.D.; Bilz, S.; Befroy, D.; Romanelli, A.J.; Shulman, G.I. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J. Biol. Chem. 2004, 279, 32345–32353. [Google Scholar] [CrossRef] [PubMed]
- Soardo, G.; Donnini, D.; Domenis, L.; Catena, C.; De Silvestri, D.; Cappello, D.; Dibenedetto, A.; Carnelutti, A.; Bonasia, V.; Pagano, C.; et al. Oxidative stress is activated by free fatty acids in cultured human hepatocytes. Metab. Syndr. Relat. Disord. 2011, 9, 397–401. [Google Scholar] [CrossRef] [PubMed]
- Lara-Castro, C.; Garvey, W.T. Intracellular lipid accumulation in liver and muscle and the insulin resistance syndrome. Endocrinol. Metab. Clin. N. Am. 2008, 37, 841–856. [Google Scholar] [CrossRef] [PubMed]
- Le Lay, S.; Simard, G.; Martinez, M.C.; Andriantsitohaina, R. Oxidative stress and metabolic pathologies: From an adipocentric point of view. Oxidative Med. Cell. Longev. 2014, 2014, 908539. [Google Scholar] [CrossRef] [PubMed]
- Kusminski, C.M.; Scherer, P.E. Mitochondrial dysfunction in white adipose tissue. Trends Endocrinol. Metab. 2012, 23, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Mahadev, K.; Motoshima, H.; Wu, X.D.; Ruddy, J.M.; Arnold, R.S.; Cheng, G.J.; Lambeth, J.D.; Goldstein, B.J. The NAD(P)H oxidase homolog Nox4 modulates insulin-stimulated generation of H2O2 and plays an integral role in insulin signal transduction. Mol. Cell. Biol. 2004, 24, 1844–1854. [Google Scholar] [CrossRef] [PubMed]
- Han, C.Y.; Umemoto, T.; Omer, M.; Den Hartigh, L.J.; Chiba, T.; Leboeuf, R.; Buller, C.L.; Sweet, I.R.; Pennathur, S.; Abel, E.D.; et al. NADPH Oxidase-derived reactive oxygen species increases expression of monocyte chemotactic factor genes in cultured adipocytes. J. Biol. Chem. 2012, 287, 10379–10393. [Google Scholar] [CrossRef] [PubMed]
- Herieka, M.; Erridge, C. High-fat meal induced postprandial inflammation. Mol. Nutr. Food Res. 2014, 58, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Magne, J.; Mariotti, F.; Fischer, R.; Mathe, V.; Tome, D.; Huneau, J.F. Early postprandial low-grade inflammation after high-fat meal in healthy rats: Possible involvement of visceral adipose tissue. J. Nutr. Biochem. 2010, 21, 550–555. [Google Scholar] [CrossRef] [PubMed]
- Travers, R.L.; Motta, A.C.; Betts, J.A.; Thompson, D. Adipose tissue metabolic and inflammatory responses to a mixed meal in lean, overweight and obese men. Eur. J. Nutr. 2017, 56, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Teno, C.; Perez-Martinez, P.; Delgado-Lista, J.; Yubero-Serrano, E.M.; Garcia-Rios, A.; Marin, C.; Gomez, P.; Jimenez-Gomez, Y.; Camargo, A.; Rodriguez-Cantalejo, F.; et al. Dietary fat modifies the postprandial inflammatory state in subjects with metabolic syndrome: The LIPGENE study. Mol. Nutr. Food Res. 2012, 56, 854–865. [Google Scholar] [CrossRef] [PubMed]
- Meneses, M.E.; Camargo, A.; Perez-Martinez, P.; Delgado-Lista, J.; Cruz-Teno, C.; Jimenez-Gomez, Y.; Paniagua, J.A.; Gutierrez-Mariscal, F.M.; Tinahones, F.J.; Vidal-Puig, A.; et al. Postprandial inflammatory response in adipose tissue of patients with metabolic syndrome after the intake of different dietary models. Mol. Nutr. Food Res. 2011, 55, 1759–1770. [Google Scholar] [CrossRef] [PubMed]
- Donath, M.Y. Targeting inflammation in the treatment of type 2 diabetes: Time to start. Nat. Rev. Drug Discov. 2014, 13, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Investig. 2004, 114, 1752–1761. [Google Scholar] [CrossRef] [PubMed]
- Murdolo, G.; Piroddi, M.; Luchetti, F.; Tortoioli, C.; Canonico, B.; Zerbinati, C.; Galli, F.; Iuliano, L. Oxidative stress and lipid peroxidation by-products at the crossroad between adipose organ dysregulation and obesity-linked insulin resistance. Biochimie 2013, 95, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Maechler, P.; Jornot, L.; Wollheim, C.B. Hydrogen peroxide alters mitochondrial activation and insulin secretion in pancreatic β cells. J. Biol. Chem. 1999, 274, 27905–27913. [Google Scholar] [CrossRef] [PubMed]
- Tiedge, M.; Lortz, S.; Drinkgern, J.; Lenzen, S. Relation between antioxidant enzyme gene expression and antioxidative defense status of insulin-producing cells. Diabetes 1997, 46, 1733–1742. [Google Scholar] [CrossRef] [PubMed]
- Jacqueminet, S.; Briaud, I.; Rouault, C.; Reach, G.; Poitout, V. Inhibition of insulin gene expression by long-term exposure of pancreatic β cells to palmitate is dependent on the presence of a stimulatory glucose concentration. Metab. Clin. Exp. 2000, 49, 532–536. [Google Scholar] [CrossRef]
- Sakai, K.; Matsumoto, K.; Nishikawa, T.; Suefuji, M.; Nakamaru, K.; Hirashima, Y.; Kawashima, J.; Shirotani, T.; Ichinose, K.; Brownlee, M.; et al. Mitochondrial reactive oxygen species reduce insulin secretion by pancreatic β-cells. Biochem. Biophys. Res. Commun. 2003, 300, 216–222. [Google Scholar] [CrossRef]
- Nowotny, K.; Jung, T.; Hohn, A.; Weber, D.; Grune, T. Advanced glycation end products and oxidative stress in type 2 diabetes mellitus. Biomolecules 2015, 5, 194–222. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A.; Gunnarsson, R.; Bjorkman, O.; Olsson, M.; Wahren, J. Effects of insulin on peripheral and splanchnic glucose metabolism in noninsulin-dependent (type II) diabetes mellitus. J. Clin. Investig. 1985, 76, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Dimitriadis, G.; Mitrou, P.; Lambadiari, V.; Maratou, E.; Raptis, S.A. Insulin effects in muscle and adipose tissue. Diabetes Res. Clin. Pract. 2011, 93, S52–S59. [Google Scholar] [CrossRef]
- Galgani, J.E.; Moro, C.; Ravussin, E. Metabolic flexibility and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E1009–E1017. [Google Scholar] [CrossRef] [PubMed]
- Corpeleijn, E.; Saris, W.H.M.; Blaak, E.E. Metabolic flexibility in the development of insulin resistance and type 2 diabetes: Effects of lifestyle. Obes. Rev. 2009, 10, 178–193. [Google Scholar] [CrossRef] [PubMed]
- Pagel-Langenickel, I.; Bao, J.J.; Pang, L.Y.; Sack, M.N. The role of mitochondria in the pathophysiology of skeletal muscle insulin resistance. Endocr. Rev. 2010, 31, 25–51. [Google Scholar] [CrossRef] [PubMed]
- Heilbronn, L.K.; de Jonge, L.; Frisard, M.I.; DeLany, J.P.; Larson-Meyer, D.E.; Rood, J.; Nguyen, T.; Martin, C.K.; Volaufova, J.; Most, M.M.; et al. Effect of 6-month calorie restriction on biomarkers of longevity, metabolic adaptation, and oxidative stress in overweight individuals: A randomized controlled trial. J. Am. Med. Assoc. 2006, 295, 1539–1548. [Google Scholar] [CrossRef] [PubMed]
- Ceriello, A.; Esposito, K.; La Sala, L.; Pujadas, G.; De Nigris, V.; Testa, R.; Bucciarelli, L.; Rondinelli, M.; Genovese, S. The protective effect of the Mediterranean diet on endothelial resistance to GLP-1 in type 2 diabetes: A preliminary report. Cardiovasc. Diabetol. 2014, 13, 140. [Google Scholar] [CrossRef] [PubMed]
- Vineis, P.; Wild, C.P. Global cancer patterns: Causes and prevention. Lancet 2014, 383, 549–557. [Google Scholar] [CrossRef]
- World Cancer Research Fund/American Institute for Cancer Research. Food, Nutrition, Physical Activity, and the Prevention of Cancer: A Global Perspective; American Institute for Cancer Research: Washington, DC, USA, 2007. [Google Scholar]
- World Cancer Research Fund/American Institute for Cancer Research. Breast Cancer 2010 Report: Food, Nutrition, Physical Activity, and the Prevention of Breast Cancer; American Institute for Cancer Research: Washington, DC, USA, 2010. [Google Scholar]
- World Cancer Research Fund/American Institute for Cancer Research. Colorectal Cancer 2011 Report: Food, Nutrition, Physical Activity, and the Prevention of Colorectal Cancer; American Institute for Cancer Research: Washington, DC, USA, 2011. [Google Scholar]
- World Cancer Research Fund/American Institute for Cancer Research. Pancreatic Cancer 2012 Report: Food, Nutrition, Physical Activity, and the Prevention of Pancreatic Cancer; American Institute for Cancer Research: Washington, DC, USA, 2012. [Google Scholar]
- World Cancer Research Fund/American Institute for Cancer Research. Endometrial Cancer 2013 Report: Food, Nutrition, Physical Activity, and the Prevention of Endometrial Cancer; American Institute for Cancer Research: Washington, DC, USA, 2013. [Google Scholar]
- World Cancer Research Fund/American Institute for Cancer Research. Ovarian Cancer 2014 Report: Food, Nutrition, Physical Activity, and the Prevention of Ovarian Cancer; American Institute for Cancer Research: Washington, DC, USA, 2014. [Google Scholar]
- Ames, B.N.; Wakimoto, P. Are vitamin and mineral deficiencies a major cancer risk? Nat. Rev. Cancer 2002, 2, 694–704. [Google Scholar] [CrossRef] [PubMed]
- Brash, D.E.; Havre, P. New careers for antioxidants. Proc. Natl. Acad. Sci. USA 2002, 99, 13969–13971. [Google Scholar] [CrossRef] [PubMed]
- Mathers, J.; Coxhead, J.; Tyson, J. Nutrition and DNA repair-potential molecular mechanisms of action. Curr. Cancer Drug Targets 2007, 7, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Shimada, T.; Fujii-Kuriyama, Y. Metabolic activation of polycyclic aromatic hydrocarbons to carcinogens by cytochromes P450 1A1 and 1B1. Cancer Sci. 2004, 95, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Shimada, T.; Inoue, K.; Suzuki, Y.; Kawai, T.; Azuma, E.; Nakajima, T.; Shindo, M.; Kurose, K.; Sugie, A.; Yamagishi, Y.; et al. Arylhydrocarbon receptor-dependent induction of liver and lung cytochromes P450 1A1, 1A2, and 1B1 by polycyclic aromatic hydrocarbons and polychlorinated biphenyls in genetically engineered C57BL/6J mice. Carcinogenesis 2002, 23, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Puga, A.; Ma, C.; Marlowe, J.L. The aryl hydrocarbon receptor cross-talks with multiple signal transduction pathways. Biochem. Pharmacol. 2009, 77, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Androutsopoulos, V.P.; Tsatsakis, A.M.; Spandidos, D.A. Cytochrome P450 CYP1A1: Wider roles in cancer progression and prevention. BMC Cancer 2009, 9, 187. [Google Scholar] [CrossRef] [PubMed]
- Baan, R.; Straif, K.; Grosse, Y.; Secretan, B.; El Ghissassi, F.; Bouvard, V.; Altieri, A.; Cogliano, V.; WHO International Agency for Research on Cancer Monograph Working Group. Carcinogenicity of alcoholic beverages. Lancet Oncol. 2007, 8, 292–293. [Google Scholar] [CrossRef]
- Savolainen, V.; Liesto, K.; Männikkö, A.; Penttilä, A.; Karhunen, P. Alcohol consumption and alcoholic liver disease: Evidence of a threshold level of effects of ethanol. Alcohol. Clin. Exp. Res. 1993, 17, 1112–1117. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Cederbaum, A.I. Oxidative stress and alcoholic liver disease. Semin. Liver Dis. 2009, 29, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Sabitha, K.; Venugopal, B.; Rafi, M.; Ramana, K. Role of antioxidant enzymes in glucose and lipid metabolism in association with obesityand type 2 diabetes. Am. J. Med. Sci. Med. 2014, 2, 21–24. [Google Scholar] [CrossRef]
- Fang, J.-L.; Vaca, C.E. Detection of DNA adducts of acetaldehyde in peripheral white blood cells of alcohol abusers. Carcinogenesis 1997, 18, 627–632. [Google Scholar] [CrossRef] [PubMed]
- Praud, D.; Rota, M.; Rehm, J.; Shield, K.; Zatoński, W.; Hashibe, M.; La Vecchia, C.; Boffetta, P. Cancer incidence and mortality attributable to alcohol consumption. Int. J. Cancer 2016, 138, 1380–1387. [Google Scholar] [CrossRef] [PubMed]
- Gavin, D.P.; Kusumo, H.; Zhang, H.; Guidotti, A.; Pandey, S.C. Role of Growth arrest and DNA damage-inducible, β in alcohol-drinking behaviors. Alcohol. Clin. Exp. Res. 2016, 40, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Chuang, S.C.; La Vecchia, C.; Boffetta, P. Liver cancer: Descriptive epidemiology and risk factors other than HBV and HCV infection. Cancer Lett. 2009, 286, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Druesne-Pecollo, N.; Tehard, B.; Mallet, Y.; Gerber, M.; Norat, T.; Hercberg, S.; Latino-Martel, P. Alcohol and genetic polymorphisms: Effect on risk of alcohol-related cancer. Lancet Oncol. 2009, 10, 173–180. [Google Scholar] [CrossRef]
- Deshpande, N.; Kandi, S.; Muddeshwar, M.; Ramana, K.V. Effect of alcohol consumption and oxidative stress and its role in dna damage. Am. J. Biomed. Res. 2014, 2, 7–10. [Google Scholar] [CrossRef]
- Wang, Y.; Millonig, G.; Nair, J.; Patsenker, E.; Stickel, F.; Mueller, S.; Bartsch, H.; Seitz, H.K. Ethanol-induced cytochrome P4502E1 causes carcinogenic etheno-DNA lesions in alcoholic liver disease. Hepatology 2009, 50, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Brooks, P.J.; Zakhari, S. Moderate alcohol consumption and breast cancer in women: From epidemiology to mechanisms and interventions. Alcohol. Clin. Exp. Res. 2013, 37, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, R.; Orsini, N.; Mignone, L.; Saji, S.; Wolk, A. Alcohol intake and risk of breast cancer defined by estrogen and progesterone receptor status—A meta-analysis of epidemiological studies. Int. J. Cancer 2008, 122, 1832–1841. [Google Scholar] [CrossRef] [PubMed]
- Zuccolo, L.; Lewis, S.J.; Donovan, J.L.; Hamdy, F.C.; Neal, D.E.; Smith, G.D. Alcohol consumption and PSA-detected prostate cancer risk—A case-control nested in the ProtecT study. Int. J. Cancer 2013, 132, 2176–2185. [Google Scholar] [CrossRef] [PubMed]
- Sawada, N.; Inoue, M.; Iwasaki, M.; Sasazuki, S.; Yamaji, T.; Shimazu, T.; Tsugane, S. Alcohol and smoking and subsequent risk of prostate cancer in Japanese men: The Japan Public Health Center-based prospective study. Int. J. Cancer 2014, 134, 971–978. [Google Scholar] [CrossRef] [PubMed]
- Watters, J.L.; Park, Y.; Hollenbeck, A.; Schatzkin, A.; Albanes, D. Alcoholic beverages and prostate cancer in a prospective US cohort study. Am. J. Epidemiol. 2010, 172, 773–780. [Google Scholar] [CrossRef] [PubMed]
- Chao, C.; Haque, R.; Van Den Eeden, S.K.; Caan, B.J.; Poon, K.Y.; Quinn, V.P. Red wine consumption and risk of prostate cancer: The California men’s health study. Int. J. Cancer 2010, 126, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Breslow, R.A.; Chen, C.M.; Graubard, B.I.; Mukamal, K.J. Prospective study of alcohol consumption quantity and frequency and cancer-specific mortality in the US population. Am. J. Epidemiol. 2011, 174, 1044–1053. [Google Scholar] [CrossRef] [PubMed]
- Middleton Fillmore, K.; Chikritzhs, T.; Stockwell, T.; Bostrom, A.; Pascal, R. Alcohol use and prostate cancer: A meta-analysis. Mol. Nutr. Food Res. 2009, 53, 240–255. [Google Scholar] [CrossRef] [PubMed]
- Rota, M.; Scotti, L.; Turati, F.; Tramacere, I.; Islami, F.; Bellocco, R.; Negri, E.; Corrao, G.; Boffetta, P.; La Vecchia, C. Alcohol consumption and prostate cancer risk: A meta-analysis of the dose–risk relation. Eur. J. Cancer Prev. 2012, 21, 350–359. [Google Scholar] [CrossRef] [PubMed]
- Shahedi, K.; Pandol, S.J.; Hu, R. Oxidative stress and alcoholic pancreatitis. J. Gastroenterol. Hepatol. Res. 2013, 2, 335–342. [Google Scholar]
- Kandi, S.; Deshpande, N.; Pinnelli, V.B.K.; Devaki, R.; Rao, P.; Ramana, K. Alcoholism and its role in the development of oxidative stress and DNA damage: An Insight. Am. J. Med. Sci. Med. 2014, 2, 64–66. [Google Scholar] [CrossRef]
- Cunningham, C.C.; Bailey, S.M. Ethanol consumption and liver mitochondria function. Neurosignals 2001, 10, 271–282. [Google Scholar] [CrossRef]
- Arranz, S.; Chiva-Blanch, G.; Valderas-Martínez, P.; Medina-Remón, A.; Lamuela-Raventós, R.M.; Estruch, R. Wine, beer, alcohol and polyphenols on cardiovascular disease and cancer. Nutrients 2012, 4, 759–781. [Google Scholar] [CrossRef] [PubMed]
- Varela-Rey, M.; Woodhoo, A.; Martinez-Chantar, M.-L.; Mato, J.M.; Lu, S.C. Alcohol, DNA methylation, and cancer. Alcohol Res. 2013, 35, 25. [Google Scholar] [PubMed]
- Wei-Chuan, T.; Yi-Heng, L.; Chih-Chan, L.; Ting-Hsing, C.; Jyh-Hong, C. Effects of oxidative stress on endothelial function after a high-fat meal. Clin. Sci. 2004, 106, 315–319. [Google Scholar]
- Gregersen, S.; Samocha-Bonet, D.; Heilbronn, L.; Campbell, L. Inflammatory and oxidative stress responses to high-carbohydrate and high-fat meals in healthy humans. J. Nutr. Metab. 2012, 2012, 238056. [Google Scholar] [CrossRef] [PubMed]
- Michels, K.B.; Mohllajee, A.P.; Roset-Bahmanyar, E.; Beehler, G.P.; Moysich, K.B. Diet and breast cancer: A review of the prospective observational studies. Cancer 2007, 109, 2712–2749. [Google Scholar] [CrossRef] [PubMed]
- Shikany, J.M.; Redden, D.T.; Neuhouser, M.L.; Chlebowski, R.T.; Rohan, T.E.; Simon, M.S.; Liu, S.; Lane, D.S.; Tinker, L. Dietary glycemic load, glycemic index, and carbohydrate and risk of breast cancer in the Women’s Health Initiative. Nutr. Cancer 2011, 63, 899–907. [Google Scholar] [CrossRef] [PubMed]
- Amador-Licona, N.; Díaz-Murillo, T.A.; Gabriel-Ortiz, G.; Pacheco-Moises, F.P.; Pereyra-Nobara, T.A.; Guízar-Mendoza, J.M.; Barbosa-Sabanero, G.; Orozco-Aviña, G.; Moreno-Martínez, S.C.; Luna-Montalbán, R. Omega 3 fatty acids supplementation and oxidative stress in HIV-seropositive patients. A clinical trial. PLoS ONE 2016, 11, e0151637. [Google Scholar] [CrossRef] [PubMed]
- Assies, J.; Mocking, R.J.; Lok, A.; Ruhé, H.G.; Pouwer, F.; Schene, A.H. Effects of oxidative stress on fatty acid-and one-carbon-metabolism in psychiatric and cardiovascular disease comorbidity. Acta Psychiatr. Scand. 2014, 130, 163–180. [Google Scholar] [CrossRef] [PubMed]
- Bieniek, J.; Childress, C.; Swatski, M.D.; Yang, W. COX-2 inhibitors arrest prostate cancer cell cycle progression by down/regulation of kinetochore/ centromere proteins. Prostate 2014, 74, 999–1011. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Hu, S.; Cui, Y.; Sun, M.-K.; Xie, F.; Zhang, Q.; Jin, J. Saturated fatty acids up-regulate COX-2 expression in prostate epithelial cells via toll-like receptor 4/NF-κB signaling. Inflammation 2014, 37, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.N.; Suburu, J.; Chen, H.Q.; Chen, Y.Q. Mechanisms of Omega-3 polyunsaturated fatty acids in prostate cancer prevention. Biomed. Res. Int. 2013, 2013, 824563. [Google Scholar] [CrossRef] [PubMed]
- Larsson, S.C.; Kumlin, M.; Ingelman-Sundberg, M.; Wolk, A. Dietary long-chain n-3 fatty acids for the prevention of cancer: A review of potential mechanisms. Am. J. Clin. Nutr. 2004, 79, 935–945. [Google Scholar] [PubMed]
- Sánchez, D.; Quiñones, M.; Moulay, L.; Muguerza, B.; Miguel, M.; Aleixandre, A. Soluble fiber-enriched diets improve inflammation and oxidative stress biomarkers in Zucker fatty rats. Pharmacol. Res. 2011, 64, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Nance, S.A.; A’ja, V.D.; Gwathmey, T.M.; Hairston, K.G. Soluble dietary fiber in obesity-associated inflammation and oxidative stress in African American women. FASEB J. 2017, 31, 434.2. [Google Scholar]
- Belobrajdic, D.P.; Lam, Y.Y.; Mano, M.; Wittert, G.A.; Bird, A.R. Cereal based diets modulate some markers of oxidative stress and inflammation in lean and obese Zucker rats. Nutr. Metab. 2011, 8, 27. [Google Scholar] [CrossRef] [PubMed]
- Diniz, Y.S.; Cicogna, A.C.; Padovani, C.R.; Silva, M.D.; Faine, L.A.; Galhardi, C.M.; Rodrigues, H.G.; Novelli, E.L. Dietary restriction and fibre supplementation: Oxidative stress and metabolic shifting for cardiac health. Can. J. Physiol. Pharmacol. 2003, 81, 1042–1048. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.Y.; He, K.; Wang, P.Y.; Qin, L.Q. Dietary fiber intake and risk of breast cancer: A meta-analysis of prospective cohort studies. Am. J. Clin. Nutr. 2011, 94, 900–905. [Google Scholar] [CrossRef] [PubMed]
- Aune, D.; Chan, D.S.M.; Greenwood, D.C.; Vieira, A.R.; Rosenblatt, D.A.N.; Vieira, R.; Norat, T. Dietary fiber and breast cancer risk: A systematic review and meta-analysis of prospective studies. Ann. Oncol. 2012, 23, 1394–1402. [Google Scholar] [CrossRef] [PubMed]
- Romaneiro, S.; Parekh, N. Dietary fiber intake and colorectal cancer risk: Weighing the evidence from epidemiologic studies. Top. Clin. Nutr. 2012, 27, 41–47. [Google Scholar] [CrossRef]
- Lottenberg, A.M.P.; Fan, P.L.T.; Buonacorso, V. Effects of dietary fiber intake on inflammation in chronic diseases. Einstein 2010, 8, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Mukai, R.; Terao, J. Role of dietary flavonoids in oxidative stress and prevention of muscle atrophy. J. Phys. Fit. Sport Med. 2013, 2, 385–392. [Google Scholar] [CrossRef]
- Costa Marques, T.H.; Santos De Melo, C.H.; De Carvalho, F.; Rusbene, B.; Costa, L.M.; De Souza, A.A.; David, J.M.; De Lima David, J.P.; De Freitas, R.M. Phytochemical profile and qualification of biological activity of an isolated fraction of Bellis perennis. Biol. Res. 2013, 46, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Yokomizo, A.; Moriwaki, M. Effects of uptake of flavonoids on oxidative stress induced by hydrogen peroxide in human intestinal Caco-2 cells. Biosci. Biotechnol. Biochem. 2006, 70, 1317–1324. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.Y.; Cui, J.; Zhang, Y.; Wang, Z.L.; Chong, T.; Wang, Z.M. Isoflavones and prostate cancer: A review of some critical issues. Chin. Med. J. 2016, 129, 341–347. [Google Scholar] [PubMed]
- Tse, G.; Eslick, G.D. Soy and isoflavone consumption and risk of gastrointestinal cancer: A systematic review and meta-analysis. Eur. J. Nutr. 2016, 55, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Nagata, C. Factors to consider in the association between soy isoflavone intake and breast cancer risk. J. Epidemiol. 2010, 20, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Stubert, J.; Gerber, B. Isoflavones—Mechanism of action and impact on breast cancer risk. Breast Care 2009, 4, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Nakata, T.; Kuzumaki, T. Effect of flavonoids on cell cycle progression in prostate cancer cells. Cancer Lett. 2002, 176, 17–23. [Google Scholar] [CrossRef]
- Ramos, S. Effects of dietary flavonoids on apoptotic pathways related to cancer chemoprevention. J. Nutr. Biochem. 2007, 18, 427–442. [Google Scholar] [CrossRef] [PubMed]
- Petzke, K.J.; Elsner, A.; Proll, J.; Thielecke, F.; Metges, C.C. Long-term high protein intake does not increase oxidative stress in rats. J. Nutr. 2000, 130, 2889–2896. [Google Scholar] [PubMed]
- Ezraty, B.; Gennaris, A.; Barras, F.; Collet, J.-F. Oxidative stress, protein damage and repair in bacteria. Nat. Rev. Microbiol. 2017, 15, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Levine, M.E.; Suarez, J.A.; Brandhorst, S.; Balasubramanian, P.; Cheng, C.-W.; Madia, F.; Fontana, L.; Mirisola, M.G.; Guevara-Aguirre, J.; Wan, J. Low protein intake is associated with a major reduction in IGF-1, cancer, and overall mortality in the 65 and younger but not older population. Cell Metab. 2014, 19, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.D.; Weed, D.L.; Miller, P.E.; Mohamed, M.A. Red meat and colorectal cancer: A quantitative update on the state of the epidemiologic science. J. Am. Coll. Nutr. 2015, 34, 521–543. [Google Scholar] [CrossRef] [PubMed]
- Olson, J.A. Carotenoids and Vitamin A: An Overview. In Lipid-Soluble Antioxidants: Biochemistry and Clinical Applications; Ong, A.S.H., Packer, L., Eds.; Birkhäuser Basel: Basel, Switzerland, 1992; pp. 178–192. [Google Scholar]
- Meydani, M. Protective role of dietary vitamin E on oxidative stress in aging. Age 1992, 15, 89–93. [Google Scholar] [CrossRef]
- Huang, X.Y.; Gao, Y.S.; Zhi, X.S.; Ta, N.; Jiang, H.; Zheng, J.M. Association between vitamin A, retinol and carotenoid intake and pancreatic cancer risk: Evidence from epidemiologic studies. Sci. Rep. 2016, 6, 38936. [Google Scholar] [CrossRef] [PubMed]
- Gong, Z.; Holly, E.A.; Bracci, P.M. Intake of folate, vitamins B6, B12 and methionine and risk of pancreatic cancer in a large population-based case–control study. Cancer Causes Control 2009, 20, 1317–1325. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Kou, J.T.; Han, D.D.; Li, P.; Zhang, D.; Wu, Q.; He, Q. Association between vitamin C intake and the risk of pancreatic cancer: A meta-analysis of observational studies. Sci. Rep. 2015, 5, 13973. [Google Scholar] [CrossRef] [PubMed]
- Cadeau, C.; Fournier, A.; Mesrine, S.; Clavel-Chapelon, F.; Fagherazzi, G.; Boutron-Ruault, M.C. Postmenopausal breast cancer risk and interactions between body mass index, menopausal hormone therapy use, and vitamin D supplementation: Evidence from the E3N cohort. Int. J. Cancer 2016, 139, 2193–2200. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.J.; Liu, X.D.; Lu, Q.; Tang, T.Q.; Yang, Z.Y. Vitamin E intake and pancreatic cancer risk: A meta-analysis of observational studies. Med. Sci. Monit. 2015, 21, 1249–1255. [Google Scholar] [PubMed]
- Garland, C.F.; Garland, F.C.; Gorham, E.D.; Lipkin, M.; Newmark, H.; Mohr, S.B.; Holick, M.F. The role of vitamin D in cancer prevention. Am. J. Public Health 2006, 96, 252–261. [Google Scholar] [CrossRef] [PubMed]
- Brand-Miller, J.C. Postprandial glycemia, glycemic index, and the prevention of type 2 diabetes. Am. J. Clin. Nutr. 2004, 80, 243–244. [Google Scholar] [PubMed]
- Turati, F.; Galeone, C.; Gandini, S.; Augustin, L.S.; Jenkins, D.J.; Pelucchi, C.; La Vecchia, C. High glycemic index and glycemic load are associated with moderately increased cancer risk. Mol. Nutr. Food Res. 2015, 59, 1384–1394. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Giovannucci, E.; Lee, J.E. Glycaemic index and glycaemic load in relation to risk of diabetes-related cancers: A meta-analysis. Br. J. Nutr. 2012, 108, 1934–1947. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Wu, Y.; Xu, J.; Ding, K.; Shan, X.; Xia, D. Association between dietary carbohydrate intake, glycemic index and glycemic load, and risk of gastric cancer. Eur. J. Nutr. 2017, 56, 1169–1177. [Google Scholar] [CrossRef] [PubMed]
- Melkonian, S.C.; Daniel, C.R.; Ye, Y.; Pierzynski, J.A.; Roth, J.A.; Wu, X. Glycemic index, glycemic load, and lung cancer risk in non-hispanic whites. Cancer Epidemiol. Biomark. Prev. 2016, 25, 532–539. [Google Scholar] [CrossRef] [PubMed]
- Gordin, D.; Groop, P.-H. Aspects of Hyperglycemia Contribution to arterial stiffness and cardiovascular complications in patients with type 1 diabetes. J. Diabetes Sci. Technol. 2016, 10, 1059–1064. [Google Scholar] [CrossRef] [PubMed]
- Ceriello, A. The post-prandial state and cardiovascular disease: Relevance to diabetes mellitus. Diabetes Metab. Res. Rev. 2000, 16, 125–132. [Google Scholar] [CrossRef]
- Vanessa Fiorentino, T.; Prioletta, A.; Zuo, P.; Folli, F. Hyperglycemia-induced oxidative stress and its role in diabetes mellitus related cardiovascular diseases. Curr. Pharm. Des. 2013, 19, 5695–5703. [Google Scholar] [CrossRef]
- Tang, W.H.; Martin, K.A.; Hwa, J. Aldose reductase, oxidative stress, and diabetic mellitus. Front. Pharmacol. 2012, 3, 87. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Block, G.; Norkus, E.P.; Morrow, J.D.; Dietrich, M.; Hudes, M. Relations of glycemic index and glycemic load with plasma oxidative stress markers. Am. J. Clin. Nutr. 2006, 84, 70–76. [Google Scholar] [PubMed]
- Bajaj, S.; Khan, A. Antioxidants and diabetes. Indian J. Endocrinol. Metab. 2012, 16, S267–S271. [Google Scholar] [PubMed]
- Choi, S.-W.; Benzie, I.F.; Ma, S.-W.; Strain, J.; Hannigan, B.M. Acute hyperglycemia and oxidative stress: Direct cause and effect? Free Radic. Biol. Med. 2008, 44, 1217–1231. [Google Scholar] [CrossRef] [PubMed]
- De Kreutzenberg, S.V.; Fadini, G.P.; Boscari, F.; Rossi, E.; Guerra, S.; Sparacino, G.; Cobelli, C.; Ceolotto, G.; Bottero, M.; Avogaro, A. Impaired hemodynamic response to meal intake in insulin-resistant subjects: An impedance cardiography approach. Am. J. Clin. Nutr. 2011, 93, 926–933. [Google Scholar] [CrossRef] [PubMed]
- Romieu, I.; Ferrari, P.; Rinaldi, S.; Slimani, N.; Jenab, M.; Olsen, A.; Tjonneland, A.; Overvad, K.; Boutron-Ruault, M.-C.; Lajous, M. Dietary glycemic index and glycemic load and breast cancer risk in the European Prospective Investigation into Cancer and Nutrition (EPIC). Am. J. Clin. Nutr. 2012, 96, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Wirfalt, E.; McTaggart, A.; Pala, V.; Gullberg, B.; Frasca, G.; Panico, S.; Bueno-de-Mesquita, H.B.; Peeters, P.H.; Engeset, D.; Skeie, G.; et al. Food sources of carbohydrates in a European cohort of adults. Public Health Nutr. 2002, 5, 1197–1215. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, C.E.; Keast, D.R.; Fulgoni, V.L.; Nicklas, T.A. Food sources of energy and nutrients among adults in the US: NHANES 2003–2006. Nutrients 2012, 4, 2097–2120. [Google Scholar] [CrossRef] [PubMed]
- Varela-López, A.; Quiles, J.L.; Cordero, M.; Giampieri, F.; Bullón, P. Oxidative stress and dietary fat type in relation to periodontal disease. Antioxidants 2015, 4, 322–344. [Google Scholar] [CrossRef] [PubMed]
- Guéraud, F.; Taché, S.; Steghens, J.-P.; Milkovic, L.; Borovic-Sunjic, S.; Zarkovic, N.; Gaultier, E.; Naud, N.; Héliès-Toussaint, C.; Pierre, F. Dietary polyunsaturated fatty acids and heme iron induce oxidative stress biomarkers and a cancer promoting environment in the colon of rats. Free Radic. Biol. Med. 2015, 83, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, R.B.; Hernandez, P.S. Diet and cancer: Risk factors and epidemiological evidence. Maturitas 2014, 77, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Dinwiddie, M.T.; Terry, P.D.; Whelan, J.; Patzer, R.E. Omega-3 fatty acid consumption and prostate cancer: A review of exposure measures and results of epidemiological studies. J. Am. Coll. Nutr. 2016, 35, 452–468. [Google Scholar] [CrossRef] [PubMed]
- Mocellin, M.C.; Camargo, C.Q.; Nunes, E.A.; Fiates, G.M.R.; Trindade, E.B.S.M. A systematic review and meta-analysis of the n-3 polyunsaturated fatty acids effects on inflammatory markers in colorectal cancer. Clin. Nutr. 2016, 35, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Bassett, J.K.; Hodge, A.M.; English, D.R.; MacInnis, R.J.; Giles, G.G. Plasma phospholipids fatty acids, dietary fatty acids, and breast cancer risk. Cancer Causes Control 2016, 27, 759–773. [Google Scholar] [CrossRef] [PubMed]
- MacLean, C.H.; Newberry, S.J.; Mojica, W.A.; Khanna, P.; Issa, A.M.; Suttorp, M.J.; Lim, Y.W.; Traina, S.B.; Hilton, L.; Garland, R.; et al. Effects of omega-3 fatty acids on cancer risk—A systematic review. JAMA J. Am. Med. Assoc. 2006, 295, 403–415. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.M.; Gann, P.H.; Giovannucci, E.L. Role of diet in prostate cancer development and progression. J. Clin. Oncol. 2005, 23, 8152–8160. [Google Scholar] [CrossRef] [PubMed]
- Eser, P.O.; Vanden Heuvel, J.P.; Araujo, J.; Thompson, J.T. Marine-and plant-derived Omega-3 fatty acids differentially regulate prostate cancer cell proliferation. Mol. Clin. Oncol. 2013, 1, 444–452. [Google Scholar] [PubMed]
- McCarty, M.F.; Lavie, C.J.; O’Keefe, J.H. Omega-3 and prostate cancer: Examining the pertinent evidence. Mayo Clin. Proc. 2014, 89, 444. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Zhang, H.; Liu, J.; Lau, C.W.; Liu, P.; Chen, Z.Y.; Lee, H.K.; Tipoe, G.L.; Ho, H.M.; Yao, X. Cyclooxygenase-2-dependent oxidative stress mediates palmitate-induced impairment of endothelium-dependent relaxations in mouse arteries. Biochem. Pharmacol. 2014, 91, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Hori, S.; Butler, E.; McLoughlin, J. Prostate cancer and diet: Food for thought? BJU Int. 2011, 107, 1348–1359. [Google Scholar] [CrossRef] [PubMed]
- Shapira, N. Nutritional approach to sun protection: A suggested complement to external strategies. Nutr. Rev. 2010, 68, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Barone, M.; Lofano, K.; De Tullio, N.; Licinio, R.; Albano, F.; Di Leo, A. Dietary, endocrine, and metabolic factors in the development of colorectal cancer. J. Gastrointest. Cancer 2012, 43, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Chua, M.E.; Sio, M.C.; Sorongon, M.C.; Dy, J.S. Relationship of dietary intake of omega-3 and omega-6 Fatty acids with risk of prostate cancer development: A meta-analysis of prospective studies and review of literature. Prostate Cancer 2012, 2012, 826254. [Google Scholar] [CrossRef] [PubMed]
- Szymanski, K.M.; Wheeler, D.C.; Mucci, L.A. Fish consumption and prostate cancer risk: A review and meta-analysis. Am. J. Clin. Nutr. 2010, 92, 1223–1233. [Google Scholar] [CrossRef] [PubMed]
- Aune, D.; Chan, D.S.; Lau, R.; Vieira, R.; Greenwood, D.C.; Kampman, E.; Norat, T. Dietary fibre, whole grains, and risk of colorectal cancer: Systematic review and dose-response meta-analysis of prospective studies. BMJ 2011, 343, d6617. [Google Scholar] [CrossRef] [PubMed]
- Mileo, A.M.; Miccadei, S. Polyphenols as modulator of oxidative stress in cancer disease: New therapeutic strategies. Oxidative Med. Cell. Longev. 2016, 2016, 6475624. [Google Scholar] [CrossRef] [PubMed]
- Luo, K.-W.; Ko, C.-H.; Yue, G.G.-L.; Lee, J.K.-M.; Li, K.-K.; Lee, M.; Li, G.; Fung, K.-P.; Leung, P.-C.; Bik-San Lau, C. Green tea (Camellia sinensis) extract inhibits both the metastasis and osteolytic components of mammary cancer 4T1 lesions in mice. J. Nutr. Biochem. 2014, 25, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Norat, T.; Aune, D.; Chan, D.; Romaguera, D. Fruits and vegetables: Updating the epidemiologic evidence for the WCRF/AICR lifestyle recommendations for cancer prevention. In Advances in Nutrition and Cancer; Springer: Berlin, Germany, 2014; pp. 35–50. [Google Scholar]
- Willcox, J.K.; Ash, S.L.; Catignani, G.L. Antioxidants and prevention of chronic disease. Crit. Rev. Food Sci. Nutr. 2004, 44, 275–295. [Google Scholar] [CrossRef] [PubMed]
- Hussain, T.; Tan, B.; Yin, Y.; Blachier, F.; Tossou, M.C.B.; Rahu, N. Oxidative stress and inflammation: What polyphenols can do for us? Oxidative Med. Cell. Longev. 2016, 2016, 7432797. [Google Scholar] [CrossRef] [PubMed]
- De Souza, P.L.; Russell, P.J.; Kearsley, J.H.; Howes, L.G. Clinical pharmacology of isoflavones and its relevance for potential prevention of prostate cancer. Nutr. Rev. 2010, 68, 542–555. [Google Scholar] [CrossRef] [PubMed]
- Messina, M.J. Emerging evidence on the role of soy in reducing prostate cancer risk. Nutr. Rev. 2003, 61, 117–131. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, F.H.; Li, Y.W. Soy isoflavones and cancer prevention. Cancer Investig. 2003, 21, 744–757. [Google Scholar] [CrossRef]
- Yan, L.; Spitznagel, E.L. Soy consumption and prostate cancer risk in men: A revisit of a meta-analysis. Am. J. Clin. Nutr. 2009, 89, 1155–1163. [Google Scholar] [CrossRef] [PubMed]
- Saxena, A.; Dhillon, V.S.; Shahid, M.; Khalil, H.S.; Rani, M.; Das, T.P.; Hedau, S.; Hussain, A.; Naqvi, R.A.; Deo, S.V.S.; et al. GSTP1 methylation and polymorphism increase the risk of breast cancer and the effects of diet and lifestyle in breast cancer patients. Exp. Ther. Med. 2012, 4, 1097–1103. [Google Scholar] [PubMed]
- Frassetto, L.A.; Todd, K.M.; Morris, R.C., Jr.; Sebastian, A. Estimation of net endogenous noncarbonic acid production in humans from diet potassium and protein contents. Am. J. Clin. Nutr. 1998, 68, 576–583. [Google Scholar] [PubMed]
- Welbourne, T.C. Acid-base balance and plasma glutamine concentration in man. Eur. J. Appl. Physiol. Occup. Physiol. 1980, 45, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Gu, C.; Shi, Y.; Le, G. Effect of dietary protein level and origin on the redox status in the digestive tract of mice. Int. J. Mol. Sci. 2008, 9, 464–475. [Google Scholar] [CrossRef] [PubMed]
- Montonen, J.; Boeing, H.; Fritsche, A.; Schleicher, E.; Joost, H.-G.; Schulze, M.B.; Steffen, A.; Pischon, T. Consumption of red meat and whole-grain bread in relation to biomarkers of obesity, inflammation, glucose metabolism and oxidative stress. Eur. J. Nutr. 2013, 52, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Wu, G. Amino acids: Metabolism, functions, and nutrition. Amino Acids 2009, 37, 1–17. [Google Scholar] [CrossRef] [PubMed]
- De Carvalho, A.M.; de Oliveira, A.A.F.; de Melo Loureiro, A.P.; Gattás, G.J.F.; Fisberg, R.M.; Marchioni, D.M. Arginine intake is associated with oxidative stress in a general population. Nutrition 2017, 33, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, L.R. Meat and cancer. Meat Sci. 2010, 84, 308–313. [Google Scholar] [CrossRef] [PubMed]
- Dodson, S.; Baracos, V.E.; Jatoi, A.; Evans, W.J.; Cella, D.; Dalton, J.T.; Steiner, M.S. Muscle wasting in cancer cachexia: Clinical implications, diagnosis, and emerging treatment strategies. Annu. Rev. Med. 2011, 62, 265–279. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, A.M.; Song, M.Y.; Zhang, X.H.; Pan, A.; Wang, M.L.; Fuchs, C.S.; Le, N.; Chan, A.T.; Willett, W.C.; Ogino, S.; et al. Processed and unprocessed red meat and risk of colorectal cancer: Analysis by tumor location and modification by time. PLoS ONE 2015, 10, e0135959. [Google Scholar] [CrossRef] [PubMed]
- Anand, S.S.; Hawkes, C.; de Souza, R.J.; Mente, A.; Dehghan, M.; Nugent, R.; Zulyniak, M.A.; Weis, T.; Bernstein, A.M.; Krauss, R.M.; et al. Food consumption and its impact on cardiovascular disease: Importance of solutions focused on the globalized food system: A report from the workshop convened by the world heart federation. J. Am. Coll. Cardiol. 2015, 66, 1590–1614. [Google Scholar] [CrossRef] [PubMed]
- Ollberding, N.J.; Wilkens, L.R.; Henderson, B.E.; Kolonel, L.N.; Le Marchand, L. Meat consumption, heterocyclic amines and colorectal cancer risk: The Multiethnic Cohort Study. Int. J. Cancer 2012, 131, E1125–E1133. [Google Scholar] [CrossRef] [PubMed]
- Beresford, S.A.A.; Johnson, K.C.; Ritenbaugh, C.; Lasser, N.L.; Snetselaar, L.G.; Black, H.R.; Anderson, G.L.; Assaf, A.R.; Bassford, T.; Bowen, D.; et al. Low-fat dietary pattern and risk of colorectal cancer—The Women’s Health Initiative randomized controlled dietary modification trial. J. Am. Med. Assoc. 2006, 295, 643–654. [Google Scholar] [CrossRef] [PubMed]
- Schatzkin, A.; Lanza, E.; Corle, D.; Lance, P.; Iber, F.; Caan, B.; Shike, M.; Weissfeld, J.; Burt, R.; Cooper, M.R.; et al. Lack of effect of a low-fat, high-fiber diet on the recurrence of colorectal adenomas. N. Engl. J. Med. 2000, 342, 1149–1155. [Google Scholar] [CrossRef] [PubMed]
- Lanza, E.; Schatzkin, A.; Daston, C.; Corle, D.; Freedman, L.; Ballard-Barbash, R.; Caan, B.; Lance, P.; Marshall, J.; Iber, F.; et al. Implementation of a 4-y, high-fiber, high-fruit-and-vegetable, low-fat dietary intervention: Results of dietary changes in the Polyp Prevention Trial. Am. J. Clin. Nutr. 2001, 74, 387–401. [Google Scholar] [PubMed]
- Trapp, D.; Knez, W.; Sinclair, W. Could a vegetarian diet reduce exercise-induced oxidative stress? A review of the literature. J. Sports Sci. 2010, 28, 1261–1268. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Espey, M.G.; Sun, A.Y.; Pooput, C.; Kirk, K.L.; Krishna, M.C.; Khosh, D.S.; Drisko, J.; Levine, M. Pharmacologic doses of ascorbate act as a prooxidant and decrease growth of aggressive tumor xenografts in mice. Proc. Natl. Acad. Sci. USA 2008, 105, 11105–11109. [Google Scholar] [CrossRef] [PubMed]
- Uetaki, M.; Tabata, S.; Nakasuka, F.; Soga, T.; Tomita, M. Metabolomic alterations in human cancer cells by vitamin C-induced oxidative stress. Sci. Rep. 2015, 5, 13896. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Espey, M.G.; Krishna, M.C.; Mitchell, J.B.; Corpe, C.P.; Buettner, G.R.; Shacter, E.; Levine, M. Pharmacologic ascorbic acid concentrations selectively kill cancer cells: Action as a pro-drug to deliver hydrogen peroxide to tissues. Proc. Natl. Acad. Sci. USA 2005, 102, 13604–13609. [Google Scholar] [CrossRef] [PubMed]
- Du, J.A.; Martin, S.M.; Levine, M.; Wagner, B.A.; Buettner, G.R.; Wang, S.H.; Taghiyev, A.F.; Du, C.B.; Knudson, C.M.; Cullen, J.J. Mechanisms of ascorbate-induced cytotoxicity in pancreatic cancer. Clin. Cancer Res. 2010, 16, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Yu, J.; Chalmers, B.; Drisko, J.; Yang, J.; Li, B.Y.; Chen, Q. Pharmacological ascorbate induces cytotoxicity in prostate cancer cells through ATP depletion and induction of autophagy. Anti-Cancer Drug 2012, 23, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Sharhar, S.; Normah, H.; Fatimah, A.; Fadilah, R.N.; Rohi, G.A.; Amin, I.; Cham, B.G.; Rizal, R.M.; Fairulnizal, M.N. Antioxidant intake and status, and oxidative stress in relation to breast cancer risk: A case-control study. Asian Pac. J. Cancer Prev. 2008, 9, 343–349. [Google Scholar] [PubMed]
- Xu, X.; Chen, J. One-carbon metabolism and breast cancer: An epidemiological perspective. J. Genet. Genom. 2009, 36, 203–214. [Google Scholar] [CrossRef]
- Lajous, M.; Lazcano-Ponce, E.; Hernandez-Avila, M.; Willett, W.; Romieu, I. Folate, vitamin B-6, and vitamin B-12 intake and the risk of breast cancer among Mexican women. Cancer Epidemiol. Biomark. Prev. 2006, 15, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Bassett, J.K.; Severi, G.; Hodge, A.M.; Baglietto, L.; Hopper, J.L.; English, D.R.; Giles, G.G. Dietary intake of B vitamins and methionine and prostate cancer incidence and mortality. Cancer Causes Control 2012, 23, 855–863. [Google Scholar] [CrossRef] [PubMed]
- Glauert, H.P.; Calfee-Mason, K.; Stemm, D.N.; Tharappel, J.C.; Spear, B.T. Dietary antioxidants in the prevention of hepatocarcinogenesis: A review. Mol. Nutr. Food Res. 2010, 54, 875–896. [Google Scholar] [CrossRef] [PubMed]
- Trottier, G.; Bostrom, P.J.; Lawrentschuk, N.; Fleshner, N.E. Nutraceuticals and prostate cancer prevention: A current review. Nat. Rev. Urol. 2010, 7, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, G.V.; Weinstein, S.J.; Stolzenberg-Solomon, R.Z. Is dietary fat, vitamin D, or folate associated with pancreatic cancer? Mol. Carcinog. 2012, 51, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.; de Mejia, E.G. Dietary factors and pancreatic cancer: The role of food bioactive compounds. Mol. Nutr. Food Res. 2011, 55, 58–73. [Google Scholar] [CrossRef] [PubMed]
- Oberley, L.W. Free radicals and diabetes. Free Radic. Biol. Med. 1988, 5, 113–124. [Google Scholar] [CrossRef]
- Niedzielska, E.; Smaga, I.; Gawlik, M.; Moniczewski, A.; Stankowicz, P.; Pera, J.; Filip, M. Oxidative Stress in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 4094–4125. [Google Scholar] [CrossRef] [PubMed]
- Sairazi, N.S.M.; Sirajudeen, K.; Asari, M.A.; Mummedy, S.; Muzaimi, M.; Sulaiman, S.A. Effect of tualang honey against KA-induced oxidative stress and neurodegeneration in the cortex of rats. BMC Complement. Altern. Med. 2017, 17, 31. [Google Scholar]
- Lindqvist, D.; Dhabhar, F.S.; James, S.J.; Hough, C.M.; Jain, F.A.; Bersani, F.S.; Reus, V.I.; Verhoeven, J.E.; Epel, E.S.; Mahan, L.; et al. Oxidative stress, inflammation and treatment response in major depression. Psychoneuroendocrinology 2017, 76, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Kallaur, A.P.; Reiche, E.M.V.; Oliveira, S.R.; Simao, A.N.C.; Pereira, W.L.D.J.; Alfieri, D.F.; Flauzino, T.; Proenca, C.D.; Lozovoy, M.A.B.; Kaimen-Maciel, D.R.; et al. Genetic, immune-inflammatory, and oxidative stress biomarkers as predictors for disability and disease progression in multiple sclerosis. Mol. Neurobiol. 2017, 54, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Carrier, A. Metabolic syndrome and oxidative stress: A complex relationship. Antioxid. Redox Signal. 2017, 26, 429–431. [Google Scholar] [CrossRef] [PubMed]
- Ratneswaran, A.; Sun, M.M.G.; Dupuis, H.; Sawyez, C.; Borradaile, N.; Beier, F. Nuclear receptors regulate lipid metabolism and oxidative stress markers in chondrocytes. J. Mol. Med. 2017, 95, 431–444. [Google Scholar] [CrossRef] [PubMed]
- Doppler, H.; Storz, P. Mitochondrial and Oxidative Stress-Mediated Activation of Protein Kinase D1 and its importance in Pancreatic Cancer. Front. Oncol. 2017, 7, 41. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.; Zuehlsdorff, T.; Cole, D.; Di Antonio, M.; Bohndiek, S. An Activatable Contrast Agent for Photoacoustic Imaging to Probe Oxidative Stress in Cancer. Proc. Physiol. Soc. 2016, 36, C06. [Google Scholar]
- Piskounova, E.; Agathocleous, M.; Murphy, M.; Hu, Z.P.; DeBerardinis, R.; Morrison, S. Oxidative stress limits metastasis of human melanoma cells. Cancer Res. 2016, 76, 2806. [Google Scholar] [CrossRef]
- Toyokuni, S. Oxidative stress as an iceberg in carcinogenesis and cancer biology. Arch. Biochem. Biophys. 2016, 595, 46–49. [Google Scholar] [CrossRef] [PubMed]
- Prasad, S.; Gupta, S.C.; Pandey, M.K.; Tyagi, A.K.; Deb, L. Oxidative stress and cancer: Advances and challenges. Oxidative Med. Cell. Longev. 2016, 2016, 5010423. [Google Scholar] [CrossRef] [PubMed]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef] [PubMed]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Gomez, M.; Kwak, M.-K.; Dolan, P.M.; Itoh, K.; Yamamoto, M.; Talalay, P.; Kensler, T.W. Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in Nrf2 transcription factor-deficient mice. Proc. Natl. Acad. Sci. USA 2001, 98, 3410–3415. [Google Scholar] [CrossRef] [PubMed]
- Iida, K.; Itoh, K.; Kumagai, Y.; Oyasu, R.; Hattori, K.; Kawai, K.; Shimazui, T.; Akaza, H.; Yamamoto, M. Nrf2 is essential for the chemopreventive efficacy of oltipraz against urinary bladder carcinogenesis. Cancer Res. 2004, 64, 6424–6431. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; McMahon, M. The double-edged sword of Nrf2: Subversion of redox homeostasis during the evolution of cancer. Mol. Cell 2006, 21, 732–734. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Roberts, J.R.; Apopa, P.L.; Kan, Y.W.; Ma, Q. Accelerated ovarian failure induced by 4-vinyl cyclohexene diepoxide in Nrf2 null mice. Mol. Cell. Biol. 2006, 26, 940–954. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.J.; Yuan, X.L.; Pan, Z.; Shen, G.X.; Kim, J.H.; Yu, S.W.; Khor, T.O.; Li, W.G.; Ma, J.J.; Kong, A.N.T. Mechanism of action of isothiocyanates: The induction of ARE-regulated genes is associated with activation of ERK and JNK and the phosphorylation and nuclear translocation of Nrf2. Mol. Cancer Ther. 2006, 5, 1918–1926. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q. Role of Nrf2 in Oxidative Stress and Toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; McKercher, S.R.; Lipton, S.A. Nrf2/ARE-mediated antioxidant actions of pro-electrophilic drugs. Free Radic. Biol. Med. 2013, 65, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, P.; Anathy, V.; Burch, P.M.; Weirather, K.; Lambeth, J.D.; Heintz, N.H. Redox-dependent expression of cyclin D1 and cell proliferation by Nox1 in mouse lung epithelial cells. Antioxid. Redox Signal. 2006, 8, 1447–1459. [Google Scholar] [CrossRef] [PubMed]
- Martindale, J.L.; Holbrook, N.J. Cellular response to oxidative stress: Signaling for suicide and survival. J. Cell. Physiol. 2002, 192, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Lu, M.S.; Zhang, Q.W. Chloride intracellular channel 1 regulates migration and invasion in gastric cancer by triggering the ROS-mediated p38 MAPK signaling pathway. Mol. Med. Rep. 2015, 12, 8041–8047. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Zeng, Y.; Liu, T.; Zhang, C.; Yu, P.W.; Hao, Y.X.; Luo, H.X.; Liu, G. Chloride intracellular channel 1 regulates colon cancer cell migration and invasion through ROS/ERK pathway. World J. Gastroenterol. 2014, 20, 2071–2078. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Nikulenkov, F.; Zawacka-Pankau, J.; Li, H.; Gabdoulline, R.; Xu, J.; Eriksson, S.; Hedström, E.; Issaeva, N.; Kel, A. ROS-dependent activation of JNK converts p53 into an efficient inhibitor of oncogenes leading to robust apoptosis. Cell Death Differ. 2014, 21, 612–623. [Google Scholar] [CrossRef] [PubMed]
- Shimura, T.; Sasatani, M.; Kamiya, K.; Kawai, H.; Inaba, Y.; Kunugita, N. Mitochondrial reactive oxygen species perturb AKT/cyclin D1 cell cycle signaling via oxidative inactivation of PP2A in lowdose irradiated human fibroblasts. Oncotarget 2016, 7, 3559–3570. [Google Scholar] [PubMed]
- Leslie, N.R.; Bennett, D.; Lindsay, Y.E.; Stewart, H.; Gray, A.; Downes, C.P. Redox regulation of PI 3-kinase signalling via inactivation of PTEN. EMBO J. 2003, 22, 5501–5510. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Rovira, I.I.; Finkel, T. Oxidants painting the cysteine chapel: Redox regulation of PTPs. Dev. Cell 2002, 2, 251–252. [Google Scholar] [CrossRef]
- Sullivan, L.B.; Chandel, N.S. Mitochondrial reactive oxygen species and cancer. Cancer Metab. 2014, 2, 17. [Google Scholar] [CrossRef] [PubMed]
- Harris, I.S.; Blaser, H.; Moreno, J.; Treloar, A.E.; Gorrini, C.; Sasaki, M.; Mason, J.M.; Knobbe, C.B.; Rufini, A.; Halle, M.; et al. PTPN12 promotes resistance to oxidative stress and supports tumorigenesis by regulating FOXO signaling. Oncogene 2014, 33, 1047–1054. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Zhang, Y.; Zheng, J.; Pan, J. Reactive oxygen species in cancer stem cells. Antioxid. Redox Signal. 2012, 16, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Diehn, M.; Cho, R.W.; Lobo, N.A.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.; Lam, J.S.; Ailles, L.E.; Wong, M.; et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009, 458, 780–783. [Google Scholar] [CrossRef] [PubMed]
- Schafer, Z.T.; Grassian, A.R.; Song, L.; Jiang, Z.; Gerhart-Hines, Z.; Irie, H.Y.; Gao, S.; Puigserver, P.; Brugge, J.S. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 2009, 461, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Dayem, A.A.; Choi, H.Y.; Kim, J.H.; Cho, S.G. Role of oxidative stress in stem, cancer, and cancer stem cells. Cancers 2010, 2, 859–884. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Dietary Components | Role in Oxidative Stress | Role in Carcinogenesis |
|---|---|---|---|
| 1 | Alcohol | ▪ Promotes ROS production while lowering cellular antioxidant levels, thereby altering homeostasis between pro- and anti-oxidants leading to oxidative stress in multiple tissues [123]. ▪ Increases ROS production and oxidative stress, and results in the accumulation of acetaldehyde [124]. ▪ alters mitochondrial function resulting in cellular death [125]. | ▪ Prominent carcinogen linked with several cancers [95]. ▪ Higher risk for esophageal cancer [95]. ▪ Highly associated with risks for breast tumors [115]. ▪ Alcohol intake and the genes involved in alcohol metabolism and their interaction increase the risk of breast cancer in post-menopausal women [126]. ▪ Chronic alcohol abuse can cause folate deficiency, which is a well-documented risk factor for breast cancer [127]. |
| 2 | Carbohydrates | ▪ Lead to increased oxidative stress, which has been associated with increased risk for atherosclerosis and related disorders [128]. ▪ High-carbohydrate meal may evoke a greater postprandial oxidative stress response [129]. | ▪ Could affect breast cancer influencing plasma levels of glucose and insulin, and insulin resistance [130]. ▪ Consuming foods with high insulinogenic content may increase the risk of breast cancer [131]. |
| 3 | Fatty acids (FAs) | ▪ Omega-3 FAs reduce oxidative stress [132]. ▪ FAs shorten in chain length and decrease unsaturation and peroxidation, while the 1-carbon cycle shifts from the methylation to the transsulfuration pathway [133]. | ▪ Established mechanism is an association between inflammatory pathways and the function of omega-3 and omega-6 FAs on the action of cyclooxygenase-2 (COX-2) in prostate cancer [134,135,136]. ▪ n-3 FAs, especially the long-chain polyunsaturated FAs, eicosapentaenoic acid and docosahexaenoic acid, present in fatty fish and fish oils inhibit carcinogenesis [137]. |
| 4 | Fiber | ▪ Could protect from oxidative stress [138]. ▪ Reduced levels of oxidative stress [139]. ▪ Elicited modest improvements in indices of oxidative stress and inflammation [140]. ▪ Dietary fiber supplementation, rather than energy intake and dietary restriction, appears to be the main process regarding oxidative stress in the cardiac tissue [141]. | ▪ An 11% decrease in breast cancer risk in individuals consuming a fiber-rich diet versus that in individuals consuming the lowest amount of fiber [142]. ▪ With up to a 25% reduction in cancer risk when ingesting around 12.6–33.1 g/day of fiber, or 17% reduction for consuming fiber 3 times a day [143,144]. ▪ It reduces the risk of developing some types of cancer [145]. |
| 5 | Flavonoids | ▪ Prevent disuse muscle atrophy by attenuating oxidative stress derived from mitochondrial dysfunction [146]. ▪ Have potential antioxidant actions by reacting with and inactivating O2−, oxygen lipid peroxide radicals, and/or stabilizing free radicals involved in the oxidative process by hydrogenation or complexing with oxidant species [147]. ▪ Have both a cytoprotective effect owing to ROS scavenging and cytotoxic effect caused by H2O2 generation [148]. | ▪ Isoflavones are the most well-known compounds that possess well-characterized anti-estrogenic activity; functions in intracellular steroid metabolism; and anti-angiogenic, anti-proliferative, and pro-apoptotic activities in various tumor cells [149,150,151]. ▪ Isoflavones consumption of 20 mg/day can decrease breast cancer risk by 29% compared to that by consumption of 5 mg/day [152]. ▪ Flavonoids are potent regulators of cyclin B and p21 required for cell cycle progression, which may play some roles in the prevention of carcinogenesis [153]. ▪ Flavonoids have emerged as potential chemopreventive candidates for cancer treatment, especially, by their ability to induce apoptosis [154]. |
| 6 | Proteins | ▪ Long-term intake of high protein diets did not increase variables of oxidative stress [155]. ▪ Become activated by oxidation and help bacteria to respond to oxidative stress [156]. | ▪ Protein-rich food (especially animal protein) could be associated with a higher risk of cancer [157]. ▪ Colorectal cancer progression occurs upon satisfactory consumption of animal protein [158]. |
| 7 | Vitamins | ▪ Vitamin A is rapidly oxidized in the presence of oxygen, transient metals, and light [159]. ▪ Vitamin E plays an important protective antioxidant role in elderly, particularly in conditions where oxidative stress and free radicals are potentiated [160]. | ▪ Numerous vitamins, including vitamin A, B, C, D, and E, have been implicated in the risk of cancer occurrence [161,162,163,164,165]. ▪ Intake or synthesis of vitamin D is associated with reduced incidence and death rates of colon, breast, prostate, and ovarian cancers [166]. |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saha, S.K.; Lee, S.B.; Won, J.; Choi, H.Y.; Kim, K.; Yang, G.-M.; Dayem, A.A.; Cho, S.-g. Correlation between Oxidative Stress, Nutrition, and Cancer Initiation. Int. J. Mol. Sci. 2017, 18, 1544. https://doi.org/10.3390/ijms18071544
Saha SK, Lee SB, Won J, Choi HY, Kim K, Yang G-M, Dayem AA, Cho S-g. Correlation between Oxidative Stress, Nutrition, and Cancer Initiation. International Journal of Molecular Sciences. 2017; 18(7):1544. https://doi.org/10.3390/ijms18071544
Chicago/Turabian StyleSaha, Subbroto Kumar, Soo Bin Lee, Jihye Won, Hye Yeon Choi, Kyeongseok Kim, Gwang-Mo Yang, Ahmed Abdal Dayem, and Ssang-goo Cho. 2017. "Correlation between Oxidative Stress, Nutrition, and Cancer Initiation" International Journal of Molecular Sciences 18, no. 7: 1544. https://doi.org/10.3390/ijms18071544
APA StyleSaha, S. K., Lee, S. B., Won, J., Choi, H. Y., Kim, K., Yang, G.-M., Dayem, A. A., & Cho, S.-g. (2017). Correlation between Oxidative Stress, Nutrition, and Cancer Initiation. International Journal of Molecular Sciences, 18(7), 1544. https://doi.org/10.3390/ijms18071544