Chrysin Attenuates VCAM-1 Expression and Monocyte Adhesion in Lipopolysaccharide-Stimulated Brain Endothelial Cells by Preventing NF-κB Signaling


Abstract
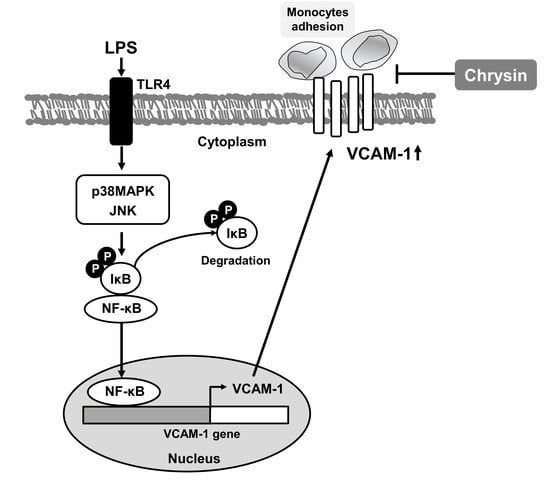
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
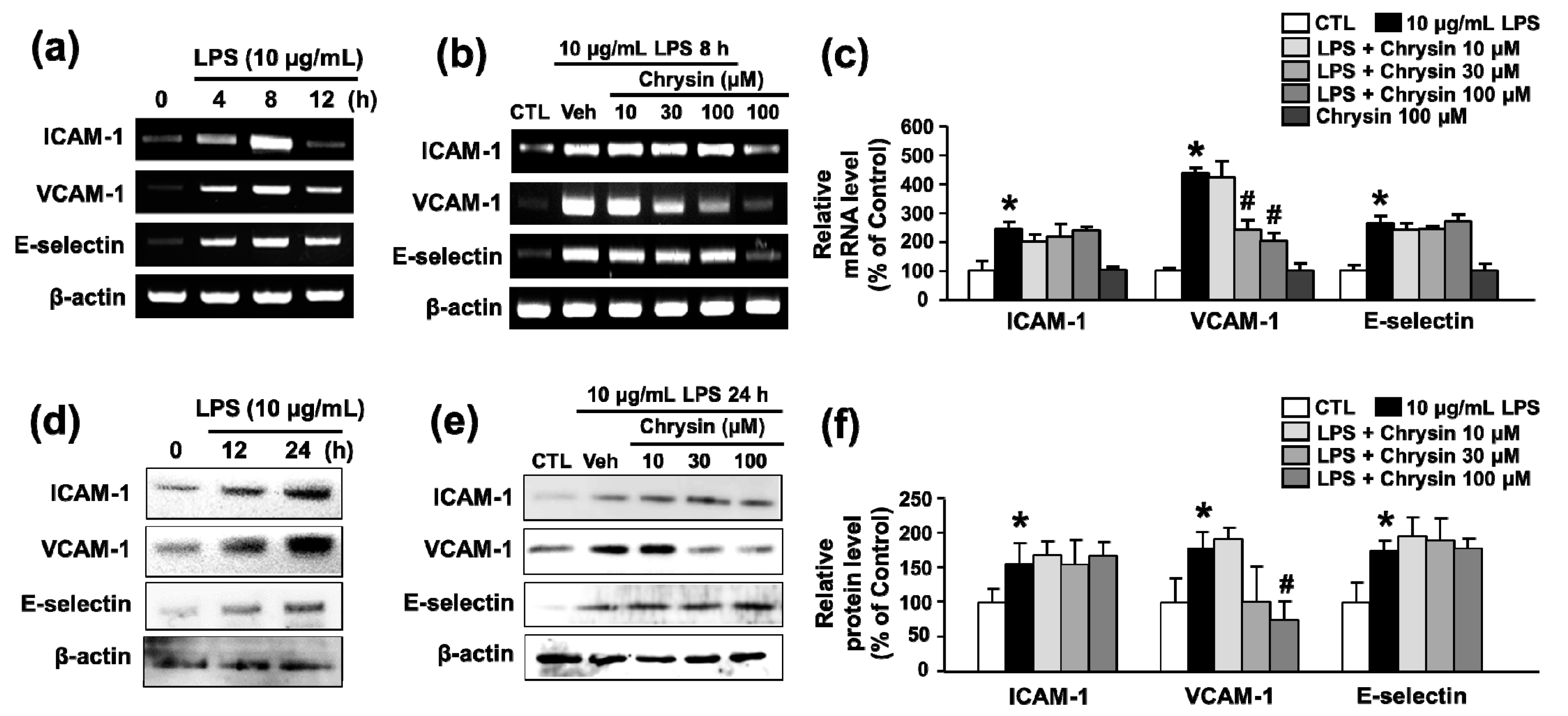
2.1. Chrysin Inhibits LPS-Induced VCAM-1 mRNA Expression in bEnd.3 Cells
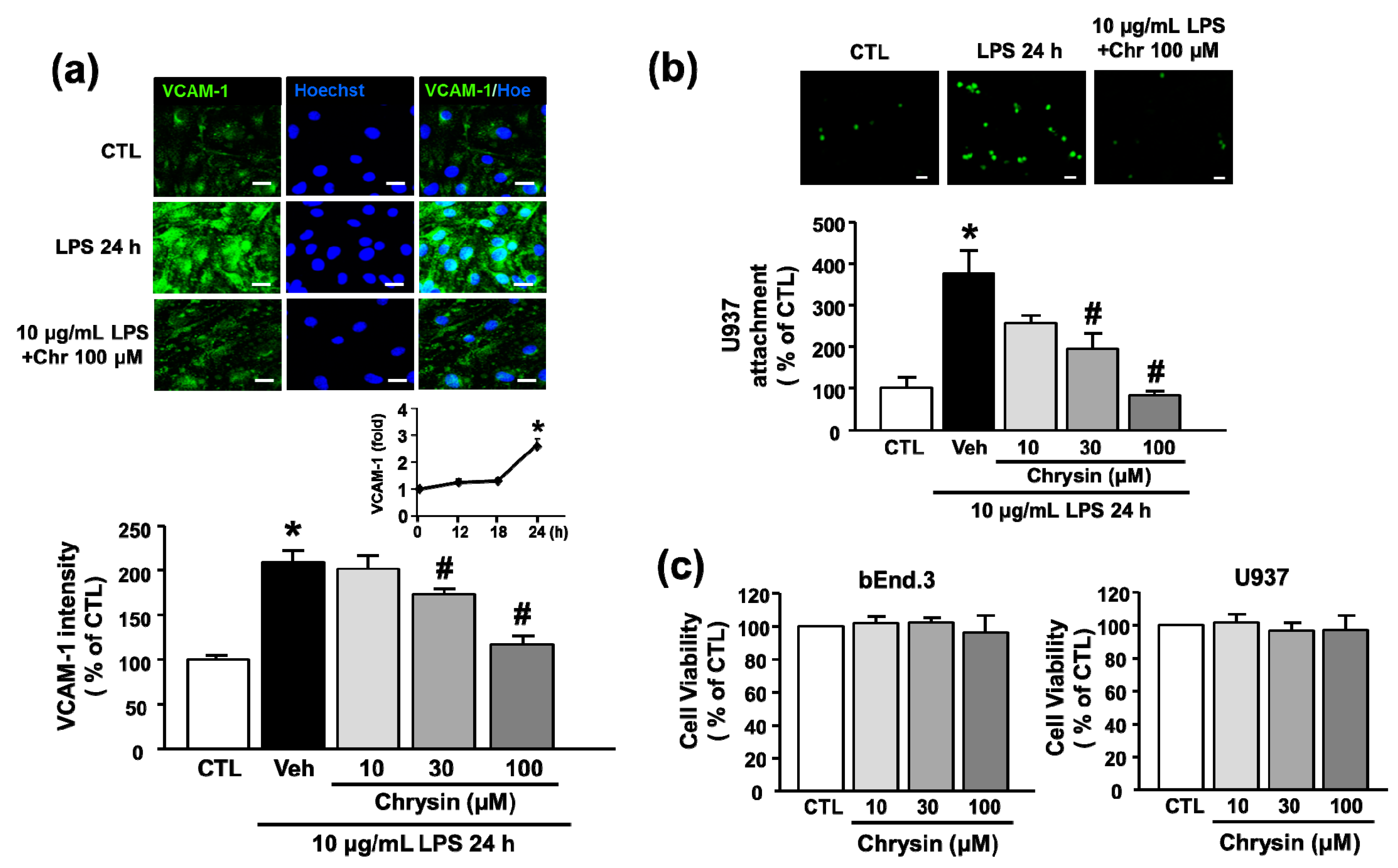
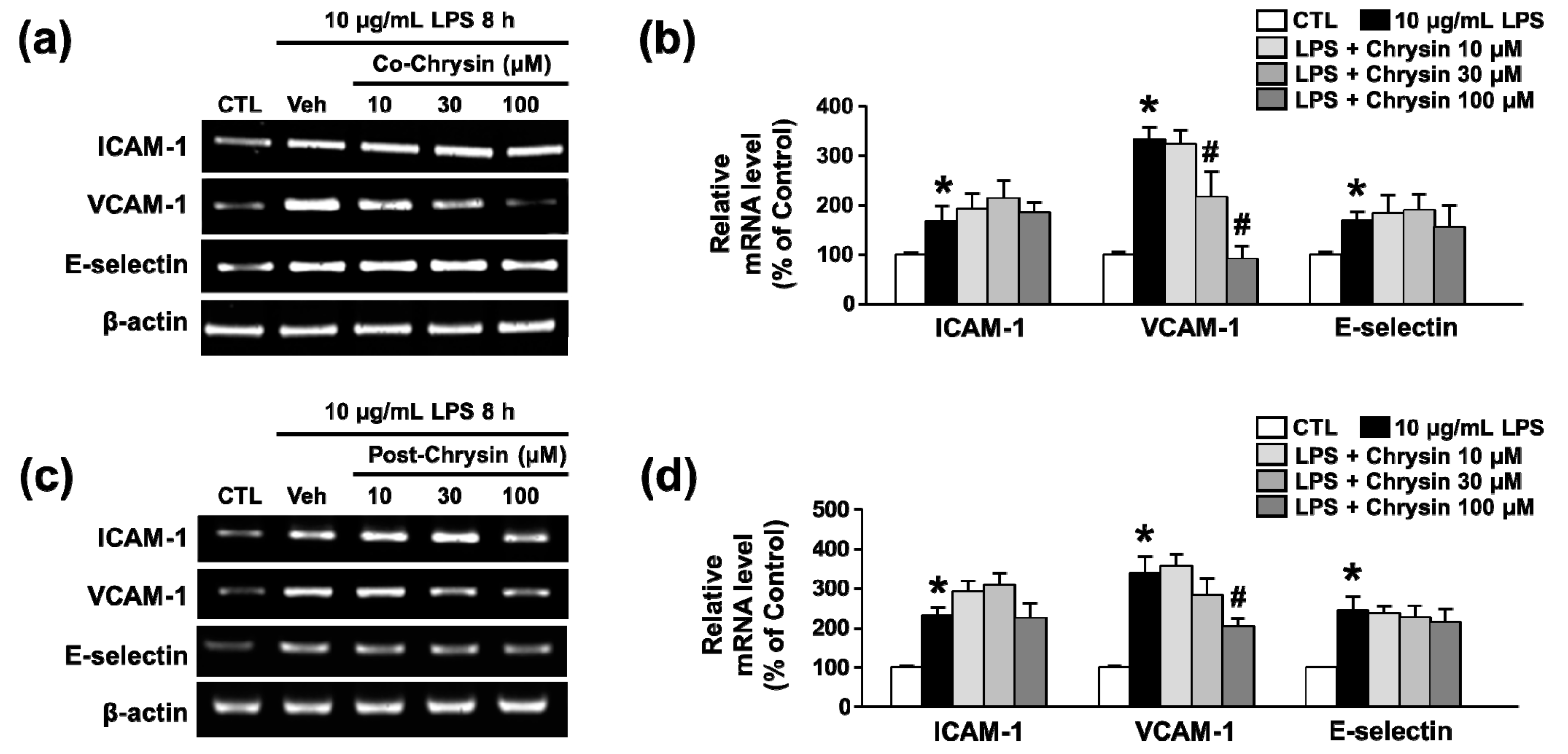
2.2. Chrysin Inhibits LPS-Induced VCAM-1 Protein Expression and Monocyte Attachment in bEnd.3 Cells
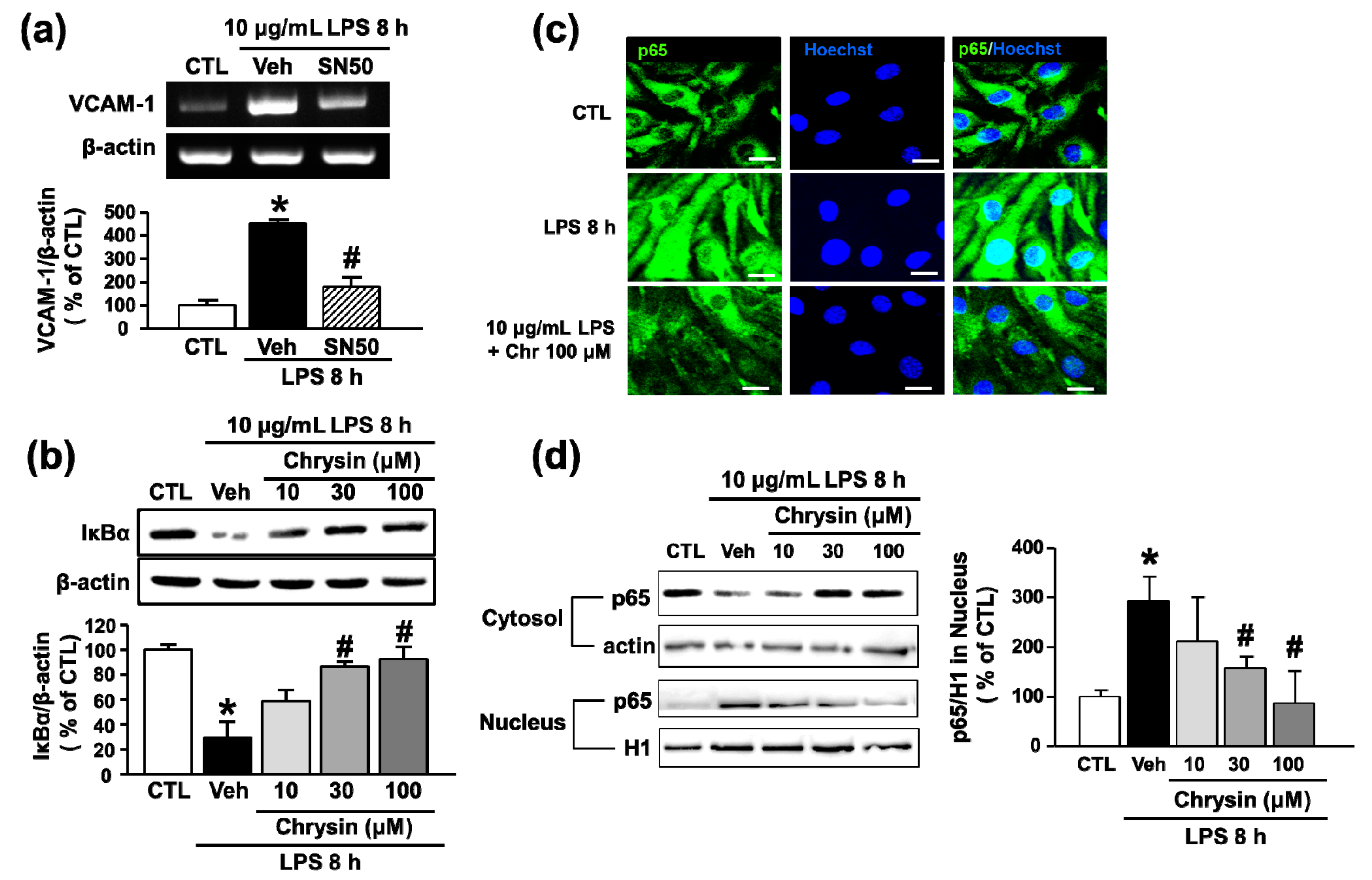
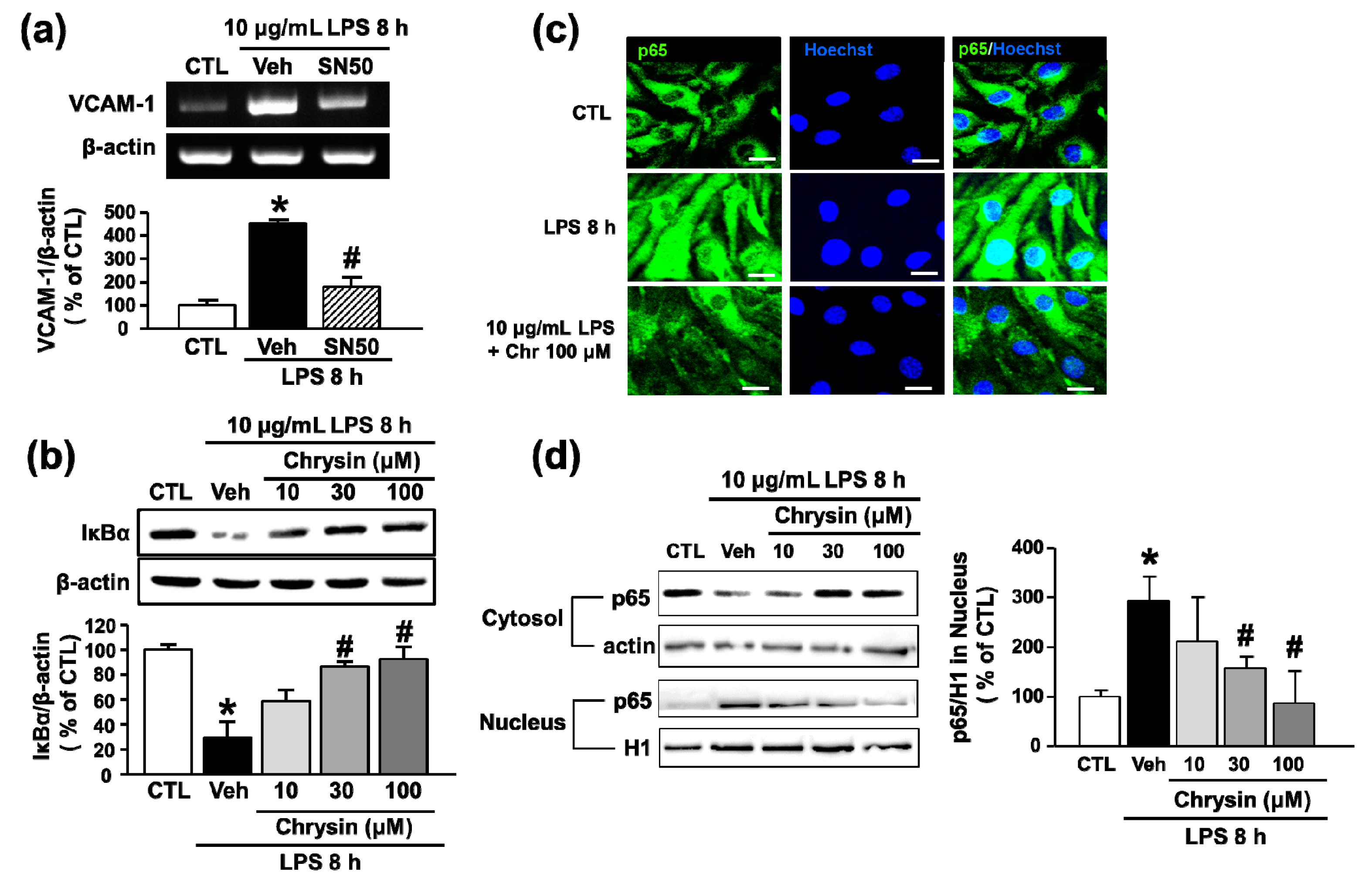
2.3. Chrysin Inhibits LPS-Induced VCAM-1 Expression by Blocking NF-κB Translocation in bEnd.3 Cells
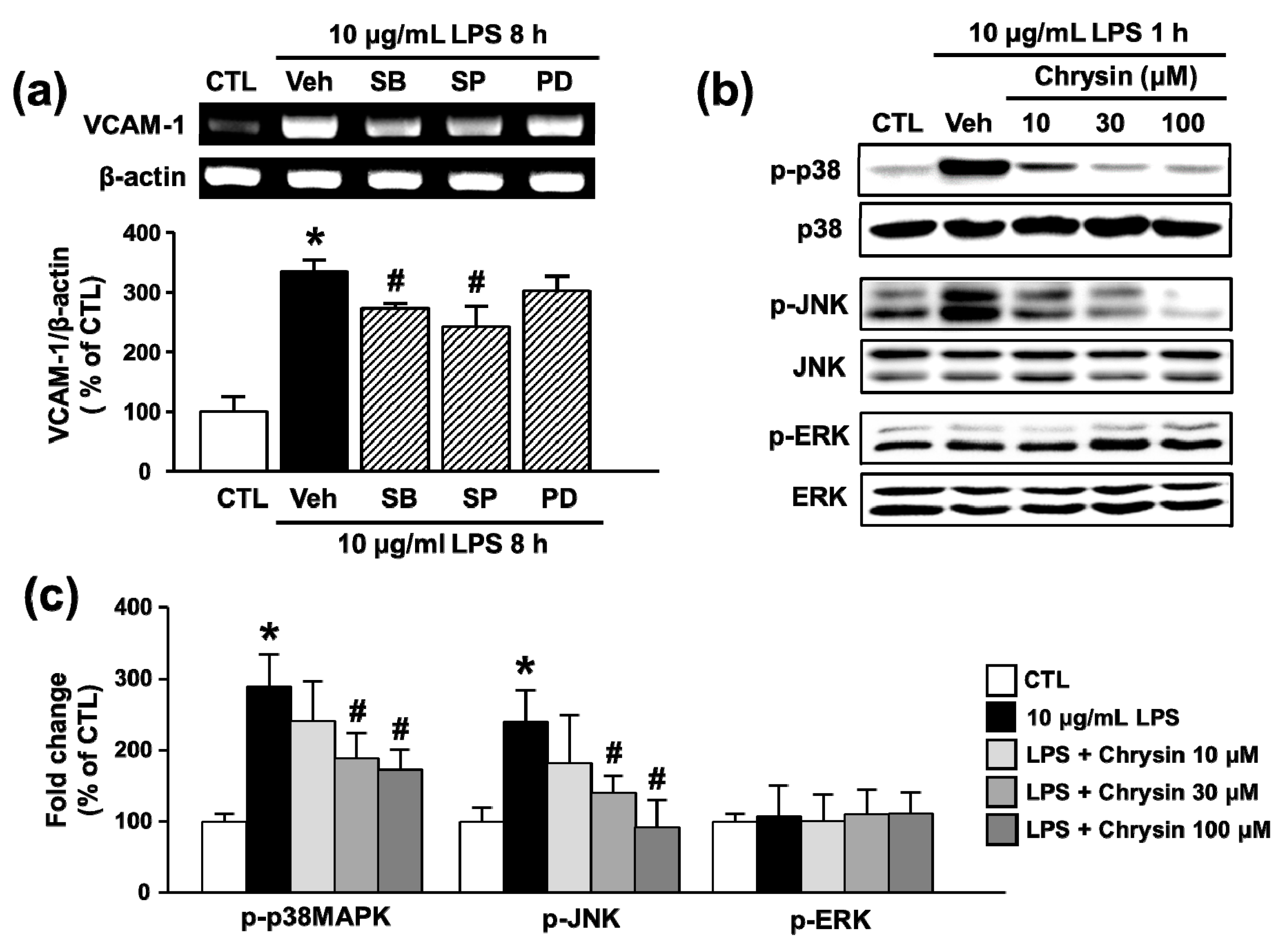
2.4. Chrysin Attenuates LPS-Induced VCAM-1 Expression by Inhibiting p38MAPK and JNK Phosphorylation in bEnd.3 Cells
2.5. Chrysin Attenuates LPS-Induced VCAM-1 Expression by Inhibiting p38MAPK and JNK Phosphorylation in bEnd.3 Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Reverse-Transcription Polymerase Chain Reaction (RT-PCR)
4.3. Preparation of Cytosolic or Nuclear Extracts
4.4. Western Blotting
4.5. Immunocytochemistry
4.6. Adhesion Assay
4.7. 3-[4,5-Dimethyl-Triazolyl-2]2,5-Diphenyl Tetrazolium Bromide (MTT) Assay
4.8. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| VCAM-1 | Vascular Cell Adhesion Molecule-1 |
| LPS | Lipopolysaccharide |
| NF-κB | Nuclear Factor-κB |
| MAPK | Mitogen-Activated Protein Kinase |
| CMECs | Cerebral Microvascular Endothelial Cells |
| BBB | Blood-Brain Barrier |
| MS | Multiple Sclerosis |
References
- Gendelman, H.E. Neural immunity: Friend or foe? J. Neurovirol. 2002, 8, 474–479. [Google Scholar] [CrossRef] [PubMed]
- Qian, L.; Flood, P.M.; Hong, J.S. Neuroinflammation is a key player in Parkinson's disease and a prime target for therapy. J. Neural Transm. 2010, 117, 971–979. [Google Scholar] [CrossRef] [PubMed]
- Semmler, A.; Hermann, S.; Mormann, F.; Weberpals, M.; Paxian, S.A.; Okulla, T.; Schafers, M.; Kummer, M.P.; Klockgether, T.; Heneka, M.T. Sepsis causes neuroinflammation and concomitant decrease of cerebral metabolism. J. Neuroinflamm. 2008, 5, 38. [Google Scholar] [CrossRef] [PubMed]
- Coles, A.J.; Compston, D.A.; Selmaj, K.W.; Lake, S.L.; Moran, S.; Margolin, D.H.; Norris, K.; Tandon, P.K. Alemtuzumab vs. interferon β-1a in early multiple sclerosis. N. Engl. J. Med. 2008, 359, 1786–1801. [Google Scholar] [PubMed]
- Yednock, T.A.; Cannon, C.; Fritz, L.C.; Sanchez-Madrid, F.; Steinman, L.; Karin, N. Prevention of experimental autoimmune encephalomyelitis by antibodies against α4β1 integrin. Nature 1992, 356, 63–66. [Google Scholar] [CrossRef] [PubMed]
- Serizawa, F.; Patterson, E.; Potter, R.F.; Fraser, D.D.; Cepinskas, G. Pre-treatment of human cerebrovascular endothelial cells with CO-releasing molecule-3 interferes with JNK/AP-1 signaling and suppresses LPS-induced pro-adhesive phenotype. Microcirculation 2015, 22, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Ritter, L.S.; Orozco, J.A.; Coull, B.M.; McDonagh, P.F.; Rosenblum, W.I. Leukocyte accumulation and hemodynamic changes in the cerebral microcirculation during early reperfusion after stroke. Stroke 2000, 31, 1153–1161. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.D.; Zhang, H.L.; Qin, Z.H. Inflammatory mechanism in ischemic neuronal injury. Neurosci. Bull. 2006, 22, 171–182. [Google Scholar] [PubMed]
- Tang, C.; Xue, H.L.; Bai, C.L.; Fu, R. Regulation of adhesion molecules expression in TNF-α-stimulated brain microvascular endothelial cells by tanshinone IIA: Involvement of NF-κB and ROS generation. Phytother. Res. 2011, 25, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, J.B. The adhesion molecule ICAM-1 and its regulation in relation with the blood-brain barrier. J. Neuroimmunol. 2002, 128, 58–68. [Google Scholar] [CrossRef]
- Cardenas, M.; Marder, M.; Blank, V.C.; Roguin, L.P. Antitumor activity of some natural flavonoids and synthetic derivatives on various human and murine cancer cell lines. Bioorg. Med. Chem. 2006, 14, 2966–2971. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Chow, M.P.; Huang, W.C.; Lin, Y.C.; Chang, Y.J. Flavonoids inhibit tumor necrosis factor-α-induced up-regulation of intercellular adhesion molecule-1 (ICAM-1) in respiratory epithelial cells through activator protein-1 and nuclear factor-κB: Structure-activity relationships. Mol. Pharmacol. 2004, 66, 683–693. [Google Scholar] [PubMed]
- Middleton, E.; Kandaswami, C.; Theoharides, T.C. The effects of plant flavonoids on mammalian cells: Implications for inflammation, heart disease, and cancer. Pharmacol. Rev. 2000, 52, 673–751. [Google Scholar] [PubMed]
- Mani, R.; Natesan, V.; Arumugam, R. Neuroprotective effect of chrysin on hyperammonemia mediated neuroinflammatory responses and altered expression of astrocytic protein in the hippocampus. Biomed. Pharmacother. 2017, 88, 762–769. [Google Scholar] [CrossRef] [PubMed]
- Lotito, S.B.; Frei, B. Dietary flavonoids attenuate tumor necrosis factor α-induced adhesion molecule expression in human aortic endothelial cells. Structure-function relationships and activity after first pass metabolism. J. Biol. Chem. 2006, 281, 37102–37110. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.S.; Yang, E.J.; Shin, D.H.; Son, K.H.; Park, H.Y.; Lee, J.S. Effect of arazyme on the lipopolysaccharide induced inflammatory response in human endothelial cells. Mol. Med. Rep. 2014, 10, 1025–1029. [Google Scholar] [CrossRef] [PubMed]
- Grenon, S.M.; Aguado-Zuniga, J.; Hatton, J.P.; Owens, C.D.; Conte, M.S.; Hughes-Fulford, M. Effects of fatty acids on endothelial cells: Inflammation and monocyte adhesion. J. Surg. Res. 2012, 177, e35–e43. [Google Scholar] [CrossRef] [PubMed]
- Calzado, M.A.; Bacher, S.; Schmitz, M.L. NF-κB inhibitors for the treatment of inflammatory diseases and cancer. Curr. Med. Chem. 2007, 14, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Jeong, S.K.; Kim, S.R.; Bae, S.K.; Kim, W.S.; Jin, S.D.; Koo, T.H.; Jang, H.O.; Yun, I.; Kim, K.W.; Bae, M.K. Resveratrol inhibits Porphyromonas gingivalis lipopolysaccharide-induced endothelial adhesion molecule expression by suppressing NF-κB activation. Arch. Pharm. Res. 2009, 32, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.W.; Kim, P.H.; Lee, W.H.; Hirani, A.A. Interleukin-4, Oxidative Stress, Vascular Inflammation and Atherosclerosis. Biomol. Ther. 2010, 18, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Muller, W.A. Getting leukocytes to the site of inflammation. Vet. Pathol. 2013, 50, 7–22. [Google Scholar] [CrossRef] [PubMed]
- Elices, M.J.; Osborn, L.; Takada, Y.; Crouse, C.; Luhowskyj, S.; Hemler, M.E.; Lobb, R.R. VCAM-1 on activated endothelium interacts with the leukocyte integrin VLA-4 at a site distinct from the VLA-4/fibronectin binding site. Cell 1990, 60, 577–584. [Google Scholar] [CrossRef]
- Hakkert, B.C.; Kuijpers, T.W.; Leeuwenberg, J.F.; van Mourik, J.A.; Roos, D. Neutrophil and monocyte adherence to and migration across monolayers of cytokine-activated endothelial cells: The contribution of CD18, ELAM-1, and VLA-4. Blood 1991, 78, 2721–2726. [Google Scholar] [PubMed]
- Klemke, M.; Weschenfelder, T.; Konstandin, M.H.; Samstag, Y. High affinity interaction of integrin α4β1 (VLA-4) and vascular cell adhesion molecule 1 (VCAM-1) enhances migration of human melanoma cells across activated endothelial cell layers. J. Cell. Physiol. 2007, 212, 368–374. [Google Scholar] [CrossRef] [PubMed]
- Garmy-Susini, B.; Jin, H.; Zhu, Y.; Sung, R.J.; Hwang, R.; Varner, J. Integrin α4β1-VCAM-1-mediated adhesion between endothelial and mural cells is required for blood vessel maturation. J. Clin. Investig. 2005, 115, 1542–1551. [Google Scholar] [CrossRef] [PubMed]
- Steinman, L. Blocking adhesion molecules as therapy for multiple sclerosis: Natalizumab. Nat. Rev. Drug Discov. 2005, 4, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Constantinescu, C.S.; Farooqi, N.; O'Brien, K.; Gran, B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br. J. Pharmacol. 2011, 164, 1079–1106. [Google Scholar] [CrossRef] [PubMed]
- Brocke, S.; Piercy, C.; Steinman, L.; Weissman, I.L.; Veromaa, T. Antibodies to CD44 and integrin α4, but not L-selectin, prevent central nervous system inflammation and experimental encephalomyelitis by blocking secondary leukocyte recruitment. Proc. Natl. Acad. Sci. USA 1999, 96, 6896–6901. [Google Scholar] [CrossRef] [PubMed]
- Zamvil, S.; Nelson, P.; Trotter, J.; Mitchell, D.; Knobler, R.; Fritz, R.; Steinman, L. T-cell clones specific for myelin basic protein induce chronic relapsing paralysis and demyelination. Nature 1985, 317, 355–358. [Google Scholar] [CrossRef] [PubMed]
- Norman, M.U.; James, W.G.; Hickey, M.J. Differential roles of ICAM-1 and VCAM-1 in leukocyte-endothelial cell interactions in skin and brain of MRL/faslpr mice. J. Leukoc. Biol. 2008, 84, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Eppihimer, M.J.; Russell, J.; Langley, R.; Vallien, G.; Anderson, D.C.; Granger, D.N. Differential expression of platelet-endothelial cell adhesion molecule-1 (PECAM-1) in murine tissues. Microcirculation 1998, 5, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.S.; Kim, C.H.; Ha, T.S.; Ahn, H.Y. Inhibition of STAT3 phosphorylation by sulforaphane reduces adhesion molecule expression in vascular endothelial cell. Can. J. Physiol. Pharmacol. 2015, 94, 1220–1226. [Google Scholar] [CrossRef] [PubMed]
- Rafiee, L.; Hajhashemi, V.; Javanmard, S.H. Maprotiline inhibits LPS-induced expression of adhesion molecules (ICAM-1 and VCAM-1) in human endothelial cells. Res. Pharm. Sci. 2016, 11, 138–144. [Google Scholar] [PubMed]
- Giuliani, C.; Napolitano, G.; Bucci, I.; Montani, V.; Monaco, F. Nf-κB transcription factor: Role in the pathogenesis of inflammatory, autoimmune, and neoplastic diseases and therapy implications. Clin. Ther. 2001, 152, 249–253. [Google Scholar]
- Jeong, E.M.; Moon, C.H.; Kim, C.S.; Lee, S.H.; Baik, E.J.; Moon, C.K.; Jung, Y.S. Cadmium stimulates the expression of ICAM-1 via NF-κB activation in cerebrovascular endothelial cells. Biochem. Biophys. Res. Commun. 2004, 320, 887–892. [Google Scholar] [CrossRef] [PubMed]
- Eder, J. Tumour necrosis factor α and interleukin 1 signalling: Do MAPKK kinases connect it all? Trends Pharmacol. Sci. 1997, 18, 319–322. [Google Scholar] [CrossRef]
- Robinson, M.J.; Cobb, M.H. Mitogen-activated protein kinase pathways. Curr. Opin. Cell Biol. 1997, 9, 180–186. [Google Scholar] [CrossRef]
- Yang, C.M.; Luo, S.F.; Wang, C.C.; Chiu, C.T.; Chien, C.S.; Lin, C.C.; Hsiao, L.D. Tumour necrosis factor-α- and interleukin-1β-stimulated cell proliferation through activation of mitogen-activated protein kinase in canine tracheal smooth muscle cells. Br. J. Pharmacol. 2000, 130, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Kempe, S.; Kestler, H.; Lasar, A.; Wirth, T. NF-κB controls the global pro-inflammatory response in endothelial cells: Evidence for the regulation of a pro-atherogenic program. Nucleic Acids Res. 2005, 33, 5308–5319. [Google Scholar] [CrossRef] [PubMed]
- Krappmann, D.; Wegener, E.; Sunami, Y.; Esen, M.; Thiel, A.; Mordmuller, B.; Scheidereit, C. The IκB kinase complex and NF-κB act as master regulators of lipopolysaccharide-induced gene expression and control subordinate activation of AP-1. Mol. Cell. Biol. 2004, 24, 6488–6500. [Google Scholar] [CrossRef] [PubMed]
- Caruso, M.; Belloni, L.; Sthandier, O.; Amati, P.; Garcia, M.I. A4β1 integrin acts as a cell receptor for murine polyomavirus at the postattachment level. J. Virol. 2003, 77, 3913–3921. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.Y.; Rieckmann, P.; Kang, Y.S. The Changes of P-glycoprotein Activity by Interferon-gamma and Tumor Necrosis Factor-α in Primary and Immortalized Human Brain Microvascular Endothelial Cells. Biomol. Ther. 2012, 20, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Yu, F.; Cui, X.; Duan, J.; Wu, Q.; Nagarkatti, P.; Fan, D. Sparstolonin B suppresses lipopolysaccharide-induced inflammation in human umbilical vein endothelial cells. Arch. Pharm. Res. 2013, 36, 890–896. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.S.; Jeong, E.M.; Park, E.K.; Kim, Y.M.; Sohn, S.; Lee, S.H.; Baik, E.J.; Moon, C.H. Cadmium induces apoptotic cell death through p38 MAPK in brain microvessel endothelial cells. Eur. J. Pharmacol. 2008, 578, 11–18. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, B.K.; Lee, W.J.; Jung, Y.-S. Chrysin Attenuates VCAM-1 Expression and Monocyte Adhesion in Lipopolysaccharide-Stimulated Brain Endothelial Cells by Preventing NF-κB Signaling. Int. J. Mol. Sci. 2017, 18, 1424. https://doi.org/10.3390/ijms18071424
Lee BK, Lee WJ, Jung Y-S. Chrysin Attenuates VCAM-1 Expression and Monocyte Adhesion in Lipopolysaccharide-Stimulated Brain Endothelial Cells by Preventing NF-κB Signaling. International Journal of Molecular Sciences. 2017; 18(7):1424. https://doi.org/10.3390/ijms18071424
Chicago/Turabian StyleLee, Bo Kyung, Won Jae Lee, and Yi-Sook Jung. 2017. "Chrysin Attenuates VCAM-1 Expression and Monocyte Adhesion in Lipopolysaccharide-Stimulated Brain Endothelial Cells by Preventing NF-κB Signaling" International Journal of Molecular Sciences 18, no. 7: 1424. https://doi.org/10.3390/ijms18071424
APA StyleLee, B. K., Lee, W. J., & Jung, Y.-S. (2017). Chrysin Attenuates VCAM-1 Expression and Monocyte Adhesion in Lipopolysaccharide-Stimulated Brain Endothelial Cells by Preventing NF-κB Signaling. International Journal of Molecular Sciences, 18(7), 1424. https://doi.org/10.3390/ijms18071424