Diabetes and Wound Angiogenesis

Abstract
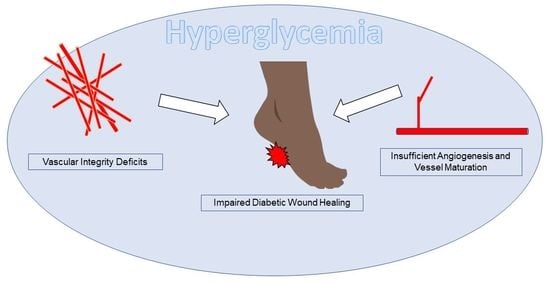
:
1. Introduction
2. The Anatomy and Maintenance of Blood Vessels
3. Vasculogenesis and Angiogenesis
4. Angiogenesis in Wound Healing
5. Diabetes: An Altered Angiogenic State
6. Diabetes and Wound Angiogenesis
7. Current Therapies to Ameliorate Diabetic Wound Healing
8. Limitations of Current Knowledge and Future Directions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| α-SMA | α-smooth muscle actin |
| Ang | angiopoietin |
| CDC | Centers for Disease Control and Prevention |
| db/db | genetically diabetic mouse model |
| DIO | diet-induced obesity |
| DN | diabetic nephropathy |
| DFU | diabetic foot ulcer |
| DM2 | diabetes mellitus type II |
| EC | endothelial cell |
| ECM | extracellular matrix |
| EGF | epidermal growth factor |
| EPC | endothelial progenitor cell |
| FGF | fibroblast growth factor |
| HBOT | hyperbaric oxygen therapy |
| HFD | high fat diet |
| HIF | hypoxia inducible factor |
| MAPK | mitogen-activated protein kinase |
| NG2 | neural/glial antigen-2 |
| PDGF | platelet derived growth factor |
| PDGFR | platelet derived growth factor receptor |
| PEDF | pigment epithelium derived factor |
| RGS5 | regulator of G-protein signaling 5 |
| SERPIN | serine protease inhibitor |
| Shh | sonic hedgehog |
| SPRY2 | sprouty homolog 2 |
| STZ | streptozotocin |
| Tie2 | angiopoietin-2 receptor |
| TGFβ | transforming growth factor β |
| TNF-α | tumor necrosis factor-α |
| VEGF | vascular endothelial growth factor |
References
- Centers for Disease Control and Prevention. National Diabetes Statistical Report: Estimates of Diabetes and Its Burden in the United States, 2014; US Department of Health and Human Services: Atlanta, GA, USA, 2014.
- Nathan, D.M. Long-Term Complications of Diabetes Mellitus. N. Engl. J. Med. 1993, 328, 1676–1685. [Google Scholar] [CrossRef] [PubMed]
- Alavi, A.; Sibbald, R.G.; Mayer, D.; Goodman, L.; Botros, M.; Armstrong, D.G.; Woo, K.; Boeni, T.; Ayello, E.A.; Kirsner, R.S. Diabetic foot ulcers: Part I. pathophysiology and prevention. J. Am. Acad. Dermatol. 2014, 70, 1.e1–1.e18. [Google Scholar] [CrossRef] [PubMed]
- Jeffcoate, W.J.; Harding, K.G. Diabetic foot ulcers. Lancet 2003, 361, 1545–1551. [Google Scholar] [CrossRef]
- Martins-Mendes, D.; Monteiro-Soares, M.; Boyko, E.J.; Ribeiro, M.; Barata, P.; Lima, J.; Soares, R. The independent contribution of diabetic foot ulcer on lower extremity amputation and mortality risk. J. Diabetes Complicat. 2013, 28, 632–638. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.C.; Driver, V.R.; Wrobel, J.S.; Armstrong, D.G. Foot ulcers in the diabetic patient, prevention and treatment. Vasc. Health Risk Manag. 2007, 3, 65–76. [Google Scholar] [PubMed]
- Noor, S.; Zubair, M.; Ahmad, J. Diabetic foot ulcer—A review on pathophysiology, classification and microbial etiology. Diabetes Metab. Syndr. Clin. Res. Rev. 2015, 9, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Prompers, L.; Schaper, N.; Apelqvist, J.; Edmonds, M.; Jude, E.; Mauricio, D.; Uccioli, L.; Urbancic, V.; Bakker, K.; Holstein, P.; et al. Prediction of outcome in individuals with diabetic foot ulcers: Focus on the differences between individuals with and without peripheral arterial disease. The EURODIALE Study. Diabetologia 2008, 51, 747–755. [Google Scholar] [CrossRef] [PubMed]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- Pittman, R.N. Regulation of Tissue Oxygenation. Morgan Claypool Life Sci. 2011, 3, 100. [Google Scholar] [CrossRef]
- Galley, H.F.; Webster, N.R. Physiology of the endothelium. Br. J. Anaesth. 2004, 93, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Aird, W.C. Endothelium in health and disease. Pharmacol. Rep. 2008, 60, 139–143. [Google Scholar] [PubMed]
- Caporali, A.; Martello, A.; Miscianinov, V.; Maselli, D.; Vono, R.; Spinetti, G. Contribution of pericyte paracrine regulation of the endothelium to angiogenesis. Pharmacol. Ther. 2017, 171, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Shepro, D.; Morel, N.M. Pericyte physiology. FASEB J. 1993, 7, 1031–1038. [Google Scholar] [PubMed]
- Armulik, A.; Genové, G.; Betsholtz, C. Pericytes: Developmental, Physiological, and Pathological Perspectives, Problems, and Promises. Dev. Cell 2011, 21, 193–215. [Google Scholar] [CrossRef] [PubMed]
- Armulik, A.; Abramsson, A.; Betsholtz, C. Endothelial/pericyte interactions. Circ. Res. 2005, 97, 512–523. [Google Scholar] [CrossRef] [PubMed]
- Geevarghese, A.; Herman, I.M. Pericyte-endothelial crosstalk: Implications and opportunities for advanced cellular therapies. Transl. Res. 2014, 163, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Dulmovits, B.M.; Herman, I.M. Microvascular remodeling and wound healing: A role for pericytes. Int. J. Biochem. Cell Biol. 2012, 44, 1800–1812. [Google Scholar] [CrossRef] [PubMed]
- Mills, S.J.; Cowin, A.J.; Kaur, P. Pericytes, mesenchymal stem cells and the wound healing process. Cells 2013, 2, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, C.G.M.; Nieuweboer, F.E.; Pei, J.Y.; Xu, Y.J.; Burgisser, P.; Van Mulligen, E.; El Azzouzi, H.; Duncker, D.J.; Verhaar, M.C.; Cheng, C. The complex mural cell: Pericyte function in health and disease. Int. J. Cardiol. 2015, 190, 75–89. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, V.S.; Shiwen, X.; Bostrom, M.; Leoni, P.; Muddle, J.; Ivarsson, M.; Gerdin, B.; Denton, C.P.; Bou-Gharios, G.; Black, C.M.; et al. Platelet-derived growth factor-β receptor activation is essential for fibroblast and pericyte recruitment during cutaneous wound healing. Am. J. Pathol. 2006, 169, 2254–2265. [Google Scholar] [CrossRef] [PubMed]
- Hellström, M.; Gerhardt, H.; Kalén, M.; Li, X.; Eriksson, U.; Wolburg, H.; Betsholtz, C. Lack of pericytes leads to endothelial hyperplasia and abnormal vascular morphogenesis. J. Cell Biol. 2001, 152, 543–553. [Google Scholar] [CrossRef]
- Abramsson, A.; Berlin, Ö.; Papayan, H.; Paulin, D.; Shani, M.; Betsholtz, C. Analysis of mural cell recruitment to tumor vessels. Circulation 2002, 105, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.N.; Tozawa, Y.; Deutsch, U.; Wolburg Buchholz, K.; Fujiwara, Y.; Gendron Maguire, M.; Gridley, T.; Wolburg, H.; Risau, W.; Qin, Y. Distinct roles of the receptor tyrosine kinases Tie-1 and Tie-2 in blood vessel formation. Nature 1995, 376, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Sundberg, C.; Kowanetz, M.; Brown, L.F.; Detmar, M.; Dvorak, H.F. Stable expression of angiopoietin-1 and other markers by cultured pericytes: Phenotypic similarities to a subpopulation of cells in maturing vessels during later stages of angiogenesis in vivo. Lab. Investig. 2002, 82, 387–401. [Google Scholar] [CrossRef] [PubMed]
- Kolte, D.; McClung, J.A.; Aronow, W.S. Vasculogenesis and Angiogenesis. In Translational Research in Coronary Artery Disease: Pathophysiology to Treatment; Aronow, W.S., McClung, J.A., Eds.; Academic Press: Cambridge, MA, USA, 2015; pp. 49–65. [Google Scholar]
- Geudens, I.; Gerhardt, H. Coordinating cell behaviour during blood vessel formation. Development 2011, 138, 4569–4583. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, L.P.; Grazul-Bilska, A.T.; Redmer, D.A. Angiogenesis in the female reproductive organs: Pathological implications. Int. J. Exp. Pathol. 2002, 83, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Fukumura, D.A.I.; Jain, R.K. Imaging angiogenesis and the microenvironment. APMIS 2008, 116, 695–715. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat. Med. 1995, 1, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases. Nature 2000, 407, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P. Angiogenesis in life, disease and medicine. Nature 2005, 438, 932–936. [Google Scholar] [CrossRef] [PubMed]
- Papetti, M.; Herman, I.M. Mechanisms of normal and tumor-derived angiogenesis. AJP Cell Physiol. 2002, 282, C947–C970. [Google Scholar] [CrossRef] [PubMed]
- Kerbel, R.S. Tumor angiogenesis. N. Engl. J. Med. 2008, 358, 2039–2049. [Google Scholar] [CrossRef] [PubMed]
- Nissen, N.N.; Polverini, P.J.; Koch, A.E.; Volin, M.V.; Gamelli, R.L.; DiPietro, L.A. Vascular endothelial growth factor mediates angiogenic activity during the proliferative phase of wound healing. Am. J. Pathol. 1998, 152, 1445–1452. [Google Scholar] [PubMed]
- Fowler, M.J. Microvascular and macrovascular complications of diabetes. Clin. Diabetes 2011, 29, 116–122. [Google Scholar] [CrossRef]
- Guo, S.; DiPietro, L.A. Factors affecting wound healing. J. Dent. Res. 2010, 89, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Demidova-Rice, T.N.; Durham, J.T.; Herman, I.M. Wound healing angiogenesis: Innovations and challenges in acute and chronic wound healing. Adv. Wound Care 2012, 1, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Tonnesen, M.G.; Feng, X.; Clark, R.A.F. Angiogenesis in wound healing. J. Investig. Dermatol. Symp. Proc. 2000, 5, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Tandara, A.A.; Mustoe, T.A. Oxygen in wound healing—More than a nutrient. World J. Surg. 2004, 28, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Turabelidze, A.; Dipietro, L.A. Inflammation and Wound Healing. In Oral Wound Healing: Cell Biology and Clinical Management; Larjava, H., Ed.; John Wiley and Sons: Hoboken, NJ, USA, 2013; pp. 39–56. [Google Scholar]
- Koh, T.J.; DiPietro, L.A. Inflammation and wound healing: The role of the macrophage. Expert Rev. Mol. Med. 2011, 13, e23. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Marti, G.P.; Wei, X.; Zhang, X.; Zhang, H.; Liu, Y.V.; Semenza, G.L.; Harmon, J.W. Age-dependent Impairment of HIF-1α̣ expression in diabetic mice: Correction with electroporation-facilitated gene therapy increases wound healing, angiogenesis, and circulating angiogenic cells. J. Cell Physiol. 2008, 217, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Eming, S.A.; Brachvogel, B.; Odorisio, T.; Koch, M. Regulation of angiogenesis: Wound healing as a model. Prog. Histochem. Cytochem. 2007, 42, 115–170. [Google Scholar] [CrossRef] [PubMed]
- Broughton, G.; Janis, J.E.; Attinger, C.E. The basic science of wound healing. Plast. Reconstr. Surg. 2006, 117, 12S–34S. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.E.; Wilgus, T.A. Vascular endothelial growth factor and angiogenesis in the regulation of cutaneous wound repair. Adv. Wound Care 2014, 3, 647–661. [Google Scholar] [CrossRef] [PubMed]
- Szpaderska, A.M.; Zuckerman, J.D.; DiPietro, L.A. Differential injury responses in oral mucosal and cutaneous wounds. J. Dent. Res. 2003, 82, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Greenhalgh, D.G. The role of apoptosis in wound healing. Int. J. Biochem. Cell Biol. 1998, 30, 1019–1030. [Google Scholar] [CrossRef]
- Dimmeler, S.; Zeiher, A.M. Endothelial cell apoptosis in angiogenesis and vessel regression. Circ. Res. 2000, 87, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.R.; Silva, E.A.; Yuen, W.W.; Mooney, D.J. Spatio-temporal VEGF and PDGF delivery patterns blood vessel formation and maturation. Pharm. Res. 2007, 24, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Hirschi, K.K.; D’Amore, P.A. Pericytes in the microvasculature. Cardiovasc. Res. 1996, 32, 687–698. [Google Scholar] [CrossRef]
- Allt, G.; Lawrenson, J.G. Pericytes: Cell biology and pathology. Cells Tissues Organs 2001, 169, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Lindblom, P.; Gerhardt, H.; Liebner, S.; Abramsson, A.; Enge, M.; Hellström, M.; Bäckström, G.; Fredriksson, S.; Landegren, U.; Nyström, H.C.; et al. Endothelial PDGF-B retention is required for proper investment of pericytes in the microvessel wall. Genes Dev. 2003, 17, 1835–1840. [Google Scholar] [CrossRef] [PubMed]
- Pries, A.R.; Secomb, T.W. Making Microvascular Networks Work: Angiogenesis, Remodeling, and Pruning. Physiology 2014, 29, 446–455. [Google Scholar] [CrossRef] [PubMed]
- Wietecha, M.S.; Cerny, W.L.; Dipietro, L.A. Mechanisms of vessel regression: Toward an understanding of the resolution of angiogenesis. Curr. Top. Microbiol. Immunol. 2013, 367, 3–32. [Google Scholar] [PubMed]
- Filleur, S.; Nelius, T.; De Riese, W.; Kennedy, R.C. Characterization of PEDF: A multi-functional serpin family protein. J. Cell. Biochem. 2009, 106, 769–775. [Google Scholar] [CrossRef] [PubMed]
- Broadhead, M.L.; Becerra, S.P.; Choong, P.F.M.; Dass, C.R. The applied biochemistry of PEDF and implications for tissue homeostasis. Growth Factors 2010, 28, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, C.; He, T.; Mao, J.; Li, C.; Lyu, J.; Meng, Q.H. Metformin inhibits prostate cancer cell proliferation, migration, and tumor growth through upregulation of PEDF expression. Cancer Biol. Ther. 2016, 17, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Becerra, S.P.; Notario, V. The effects of PEDF on cancer biology: Mechanisms of action and therapeutic potential. Nat. Rev. Cancer 2013, 13, 258–271. [Google Scholar] [CrossRef] [PubMed]
- Wietecha, M.S.; Krol, M.J.; DiPietro, L.A. PEDF functions as an endogenous anti-angiogenic during dermal wound repair. Wound Repair Regen. 2012, 20, A45. [Google Scholar]
- Wietecha, M.S.; Król, M.J.; Michalczyk, E.R.; Chen, L.; Gettins, P.G.; DiPietro, L.A. Pigment epithelium-derived factor as a multifunctional regulator of wound healing. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H812–H826. [Google Scholar] [CrossRef] [PubMed]
- Wietecha, M.S.; Chen, L.; Ranzer, M.J.; Anderson, K.; Ying, C.; Patel, T.B.; DiPietro, L.A. Sprouty2 downregulates angiogenesis during mouse skin wound healing. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H459–H467. [Google Scholar] [CrossRef] [PubMed]
- Fagiani, E.; Christofori, G. Angiopoietins in angiogenesis. Cancer Lett. 2013, 328, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Staton, C.A.; Valluru, M.; Hoh, L.; Reed, M.W.R.; Brown, N.J. Angiopoietin-1, angiopoietin-2 and Tie-2 receptor expression in human dermal wound repair and scarring. Br. J. Dermatol. 2010, 163, 920–927. [Google Scholar] [CrossRef] [PubMed]
- Maisonpierre, P.C. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science 1997, 277, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Brudno, Y.; Ennett-Shepard, A.B.; Chen, R.R.; Aizenberg, M.; Mooney, D.J. Enhancing microvascular formation and vessel maturation through temporal control over multiple pro-angiogenic and pro-maturation factors. Biomaterials 2013, 34, 9201–9209. [Google Scholar] [CrossRef] [PubMed]
- Hickey, M.M.; Simon, M.C. Regulation of Angiogenesis by hypoxia and hypoxia-inducible factors. Curr. Top. Dev. Biol. 2006, 76, 217–257. [Google Scholar] [PubMed]
- Jain, R.K. Molecular regulation of vessel maturation. Nat. Med. 2003, 9, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Gurtner, G.C.; Werner, S.; Barrandon, Y.; Longaker, M.T. Wound repair and regeneration. Nature 2008, 453, 314–321. [Google Scholar] [CrossRef] [PubMed]
- DiPietro, L.A. Angiogenesis and wound repair: When enough is enough. J. Leukoc. Biol. 2016, 100, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Altabas, V. Diabetes, endothelial dysfunction, and vascular repair: What should a diabetologist keep his eye on? Int. J. Endocrinol. 2015, 2015, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Piconi, L.; Quagliaro, L.; Assaloni, R.; Da Ros, R.; Maier, A.; Zuodar, G.; Ceriello, A. Constant and intermittent high glucose enhances endothelial cell apoptosis through mitochondrial superoxide overproduction. Diabetes Metab. Res. Rev. 2006, 22, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.Q.; Liu, X.F.; Chin, L.K.; Liu, A.Q.; Luo, K.Q. Study of endothelial cell apoptosis using fluorescence resonance energy transfer (FRET) biosensor cell line with hemodynamic microfluidic chip system. Lab Chip 2013, 13, 2693–2700. [Google Scholar] [CrossRef] [PubMed]
- McClung, J.A.; Naseer, N.; Saleem, M.; Rossi, G.P.; Weiss, M.B.; Abraham, N.G.; Kappas, A. Circulating endothelial cells are elevated in patients with type 2 diabetes mellitus independently of HbA1c. Diabetologia 2005, 48, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Kota, S.; Meher, L.; Jammula, S.; Kota, S.; Krishna, S.V.S.; Modi, K. Aberrant angiogenesis: The gateway to diabetic complications. Indian J. Endocrinol. Metab. 2012, 16, 918. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, T.; Yamagishi, S.I.; Inagaki, Y.; Amano, S.; Takeuchi, M.; Kikuchi, S.; Ohno, S.; Yoshimura, A. Incadronate disodium inhibits advanced glycation end products-induced angiogenesis in vitro. Biochem. Biophys. Res. Commun. 2002, 297, 419–424. [Google Scholar] [CrossRef]
- McGinn, S.; Saad, S.; Poronnik, P.; Pollock, C.A. High glucose-mediated effects on endothelial cell proliferation occur via p38 MAP kinase. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E708–E717. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.E.; Vranes, D.; Youssef, S.; Stacker, S.A.; Cox, A.J.; Rizkalla, B.; Casley, D.J.; Bach, L.A.; Kelly, D.J.; Gilbert, R.E. Increased renal expression of vascular endothelial growth factor (VEGF) and its receptor VEGFR-2 in experimental diabetes. Diabetes 1999, 48, 2229–2239. [Google Scholar] [CrossRef] [PubMed]
- Simons, M. Angiogenesis, arteriogenesis, and diabetes: Paradigm reassessed? J. Am. Coll. Cardiol. 2005, 46, 835–837. [Google Scholar] [CrossRef] [PubMed]
- Dinh, T.; Veves, A. Microcirculation of the Diabetic Foot. Curr. Pharm. Des. 2005, 11, 2301–2309. [Google Scholar] [CrossRef] [PubMed]
- Mirza, R.; Koh, T.J. Dysregulation of monocyte/macrophage phenotype in wounds of diabetic mice. Cytokine 2011, 56, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Michaels, J.; Churgin, S.S.; Blechman, K.M.; Greives, M.R.; Aarabi, S.; Galiano, R.D.; Gurtner, G.C. db/db mice exhibit severe wound-healing impairments compared with other murine diabetic strains in a silicone-splinted excisional wound model. Wound Repair Regen. 2007, 15, 665–670. [Google Scholar] [CrossRef] [PubMed]
- Khanna, S.; Biswas, S.; Shang, Y.; Collard, E.; Azad, A.; Kauh, C.; Bhasker, V.; Gordillo, G.M.; Sen, C.K.; Roy, S. Macrophage dysfunction impairs resolution of inflammation in the wounds of diabetic mice. PLoS ONE 2010, 5, e9539. [Google Scholar] [CrossRef] [PubMed]
- Seitz, O.; Schürmann, C.; Hermes, N.; Müller, E.; Pfeilschifter, J.; Frank, S.; Goren, I. Wound healing in mice with high-fat diet- or ob gene-induced diabetes-obesity syndromes: A comparative study. Exp. Diabetes Res. 2010, 2010. [Google Scholar] [CrossRef] [PubMed]
- Galiano, R.D.; Tepper, O.M.; Pelo, C.R.; Bhatt, K.A.; Callaghan, M.; Bastidas, N.; Bunting, S.; Steinmetz, H.G.; Gurtner, G.C. Topical vascular endothelial growth factor accelerates diabetic wound healing through increased angiogenesis and by mobilizing and recruiting bone marrow-derived cells. Am. J. Pathol. 2004, 164, 1935–1947. [Google Scholar] [CrossRef]
- Qi, W.; Yang, C.; Dai, Z.; Che, D.; Feng, J.; Mao, Y.; Cheng, R.; Wang, Z.; He, X.; Zhou, T.; et al. High levels of pigment epithelium-derived factor in diabetes impair wound healing through suppression of Wnt signaling. Diabetes 2015, 64, 1407–1419. [Google Scholar] [CrossRef] [PubMed]
- Isidori, A.M.; Venneri, M.A.; Fiore, D. Angiopoietin-1 and Angiopoietin-2 in metabolic disorders: Therapeutic strategies to restore the highs and lows of angiogenesis in diabetes. J. Endocrinol. Investig. 2016, 39, 1235–1246. [Google Scholar] [CrossRef] [PubMed]
- Kampfer, H.; Pfeilschifter, J.; Frank, S. Expressional Regulation of Angiopoietin-1 and -2 and the Tie-1 and -2 receptor tyrosine kinases during cutaneous wound healing: A comparative study of normal and impaired repair. Lab. Investig. 2001, 81, 361–373. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Yu, T.; Liu, Y.; Chen, X.; Zhang, X. Topical Application of insulin accelerates vessel maturation of wounds by regulating angiopoietin-1 in diabetic mice. Int. J. Low. Extrem. Wounds 2015, 14, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Balaji, S.; Han, N.; Moles, C.; Shaaban, A.F.; Bollyky, P.L.; Crombleholme, T.M.; Keswani, S.G. Angiopoietin-1 improves endothelial progenitor cell-dependent neovascularization in diabetic wounds. Surgery 2015, 158, 846–856. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zgheib, C.; Hu, J.; Wu, W.; Zhang, L.; Liechty, K.W. The role of microRNA-15b in the impaired angiogenesis in diabetic wounds. Wound Repair Regen. 2014, 22, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Icli, B.; Nabzdyk, C.S.; Lujan-Hernandez, J.; Cahill, M.; Auster, M.E.; Wara, A.K.M.; Sun, X.; Ozdemir, D.; Giatsidis, G.; Orgill, D.P.; et al. Regulation of impaired angiogenesis in diabetic dermal wound healing by microRNA-26a. J. Mol. Cell. Cardiol. 2016, 91, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.C.; Roy, S.; Khanna, S.; Sen, C.K. Downregulation of endothelial MicroRNA-200b supports cutaneous wound angiogenesis by desilencing GATA binding protein 2 and vascular endothelial growth factor receptor 2. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1372–1382. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.M.; Tao, J.; Chen, D.D.; Cai, J.J.; Irani, K.; Wang, Q.; Yuan, H.; Chen, A.F. MicroRNA miR-27b rescues bone marrow-derived angiogenic cell function and accelerates wound healing in type 2 diabetes mellitus. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Hellberg, C.; Östman, A.; Heldin, C.-H. PDGF and Vessel Maturation. Recent Results Cancer Res. 2010, 180, 103–114. [Google Scholar] [PubMed]
- Beer, H.D.; Longaker, M.T.; Werner, S. Reduced expression of PDGF and PDGF receptors during impaired wound healing. J.Investig. Dermatol. 1997, 109, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.L.; Breeden, M.P.; Greenhalgh, D.G. PDGF and TGF-α act synergistically to improve wound healing in the genetically diabetic mouse. J. Surg. Res. 1994, 56, 562–570. [Google Scholar] [CrossRef] [PubMed]
- Drela, E.; Stankowska, K.; Kulwas, A.; Rość, D. Endothelial progenitor cells in diabetic foot syndrome. Adv. Clin. Exp. Med. 2012, 21, 249–254. [Google Scholar] [PubMed]
- Kolluru, G.K.; Bir, S.C.; Kevil, C.G. Endothelial dysfunction and diabetes: Effects on angiogenesis, vascular remodeling, and wound healing. Int. J. Vasc. Med. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Tamarat, R.; Silvestre, J.-S.; Le Ricousse-Roussanne, S.; Barateau, V.; Lecomte-Raclet, L.; Clergue, M.; Duriez, M.; Tobelem, G.; Lévy, B.I. Impairment in ischemia-induced neovascularization in diabetes: Bone marrow mononuclear cell dysfunction and therapeutic potential of placenta growth factor treatment. Am. J. Pathol. 2004, 164, 457–466. [Google Scholar] [CrossRef]
- Fiorina, P.; Pietramaggiori, G.; Scherer, S.S.; Jurewicz, M.; Mathews, J.C.; Vergani, A.; Thomas, G.; Orsenigo, E.; Staudacher, C.; La Rosa, S.; et al. The mobilization and effect of endogenous bone marrow progenitor cells in diabetic wound healing. Cell Transplant. 2010, 19, 1369–1381. [Google Scholar] [CrossRef] [PubMed]
- Asai, J.; Takenaka, H.; Kusano, K.F.; Ii, M.; Luedemann, C.; Curry, C.; Eaton, E.; Iwakura, A.; Tsutsumi, Y.; Hamada, H.; et al. Topical sonic hedgehog gene therapy accelerates wound healing in diabetes by enhancing endothelial progenitor cell-mediated microvascular remodeling. Circulation 2006, 113, 2413–2424. [Google Scholar] [CrossRef] [PubMed]
- Sangiorgi, S.; Manelli, A.; Reguzzoni, M.; Ronga, M.; Protasoni, M.; Dell’Orbo, C. The cutaneous microvascular architecture of human diabetic toe studied by corrosion casting and scanning electron microscopy analysis. Anat. Rec. 2010, 293, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Urao, N.; Okonkwo, U.A.; Fang, M.M.; Zhuang, Z.W.; Koh, T.J.; DiPietro, L.A. MicroCT angiography detects vascular formation and regression in skin wound healing. Microvasc. Res. 2016, 106, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Okonkwo, O.U.; Chen, L.; Modilevsky, B.; Zhao, Y.; DiPietro, L.A. Vascular Maturity and Integrity in Diabetic skin wounds. In Wound Repair and Regeneration; Davidson, J.M., Ed.; Wound Healing Society: San Diego, CA, USA, 2017; pp. w7–w8. [Google Scholar]
- Tiaka, E.K.; Papanas, N.; Manolakis, A.C.; Maltezos, E. The role of hyperbaric oxygen in the treatment of diabetic foot ulcers. Angiology 2011, 63, 302–314. [Google Scholar] [CrossRef] [PubMed]
- Kaya, A.; Aydin, F.; Altay, T.; Karapinar, L.; Ozturk, H.; Karakuzu, C. Can major amputation rates be decreased in diabetic foot ulcers with hyperbaric oxygen therapy? Int. Orthop. 2009, 33, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Stoekenbroek, R.M.; Santema, T.B.; Legemate, D.A.; Ubbink, D.T.; Van Den Brink, A.; Koelemay, M.J.W. Hyperbaric oxygen for the treatment of diabetic foot ulcers: A systematic review. Eur. J. Vasc. Endovasc. Surg. 2014, 47, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.J.; An, S.; Choi, S.; Nam, K.; Jung, H.S.; Yoon, C.S.; Ko, J.H.; Jun, H.J.; Kim, T.K.; Jung, S.J.; et al. Effective healing of diabetic skin wounds by using nonviral gene therapy based on minicircle vascular endothelial growth factor DNA and a cationic dendrimer. J. Gene Med. 2012, 14, 272–278. [Google Scholar] [CrossRef] [PubMed]
- Bao, P.; Kodra, A.; Tomic-canic, M.; Golinko, M.S.; Ehrlich, H.P.; Brem, H. The role of vascular endothelial groth factor in wound healing. J. Surg. Res. 2010, 153, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Barrientos, S.; Brem, H.; Stojadinovic, O.; Tomic-Canic, M. Clinical application of growth factors and cytokines in wound healing. Wound Repair Regen. 2014, 22, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Steed, D.L. Clinical evaluation of recombinant human platelet-derived growth factor for the treatment of lower extremity diabetic ulcers. J. Vasc. Surg. 1995, 21, 71–81. [Google Scholar] [CrossRef]
- Smiell, J.M.; Wieman, T.J.; Steed, D.L.; Perry, B.H.; Sampson, A.R.; Schwab, B.H. Efficacy and safety of becaplermin (recombinant human platelet-derived growth factor-BB)in patients with nonhealing, lower extremity diabetic ulcers: A combined analysis of four randomized studies. Wound Repair Regen. 1999, 7, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Embil, J.M.; Papp, K.; Sibbald, G.; Tousignant, J.; Smiell, J.M.; Wong, B.; Lau, C.Y. Recombinant human platelet-derived growth factor-BB (becaplermin) for healing chronic lower extremity diabetic ulcers: An open-label clinical evaluation of efficacy. Wound Repair Regen. 2000, 8, 162–168. [Google Scholar] [CrossRef] [PubMed]
- De Pascale, M.R.; Sommese, L.; Casamassimi, A.; Napoli, C. Platelet derivatives in regenerative medicine: An update. Transfus. Med. Rev. 2017, 29, 52–61. [Google Scholar] [CrossRef] [PubMed]
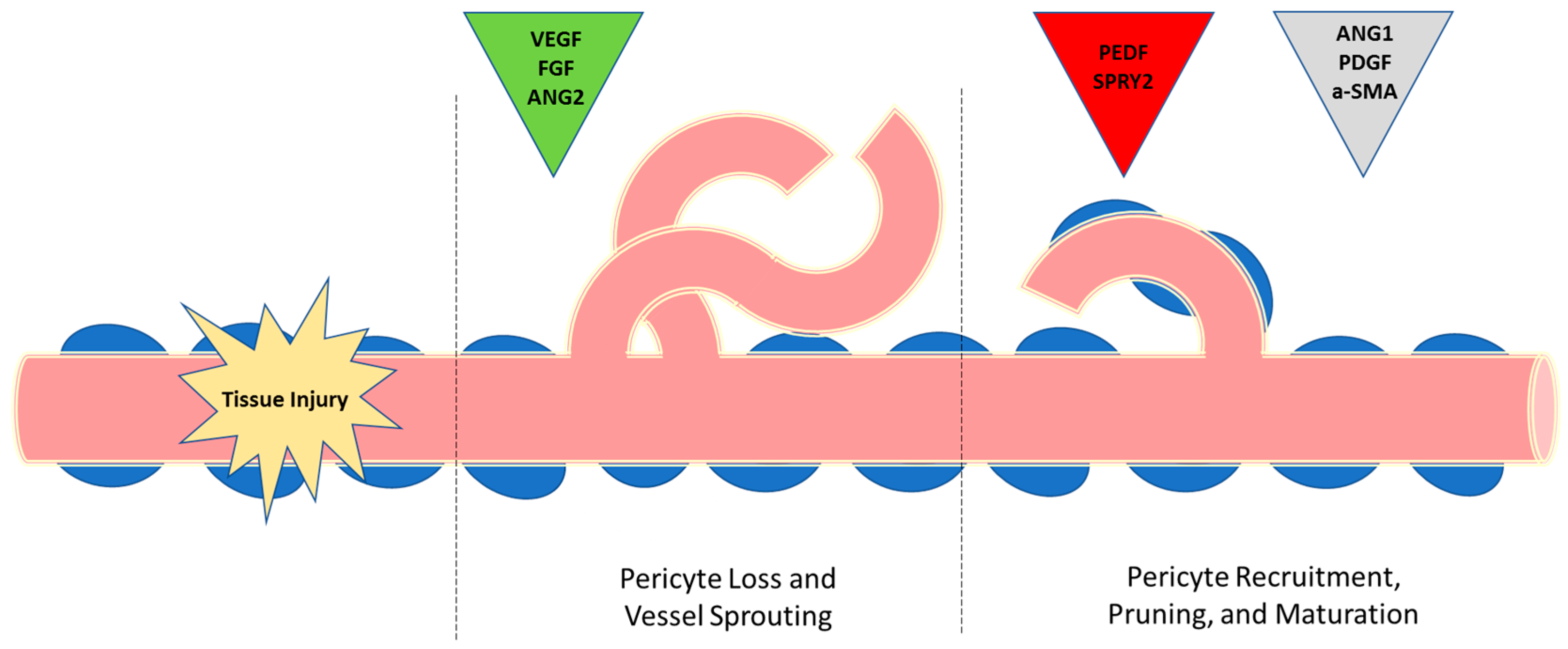

{kind=link}
{kind=link}
| Event | Diabetes-Associated Changes | References |
|---|---|---|
| Normal Quiescent Capillary Bed | Microangiopathies, loss of pericytes | [80] |
| Proangiogenic Stimulus in Wounds | Decreased response to hypoxia, decreased production of pro-angiogenic factors, impaired receptor function, miRNA misregulation, macrophage dysfunction | [43,81,83,85,90,92] |
| Angiogenic Response in Wounds | Blunted, miRNA misregulation, decreased in endothelial progenitor cells | [91,93,94,101,102] |
| Capillary Pruning and Maturation during Wound Resolution | Not yet well studied, but altered production of anti-angiogenic factors reported | [17,86,89] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Okonkwo, U.A.; DiPietro, L.A. Diabetes and Wound Angiogenesis. Int. J. Mol. Sci. 2017, 18, 1419. https://doi.org/10.3390/ijms18071419
Okonkwo UA, DiPietro LA. Diabetes and Wound Angiogenesis. International Journal of Molecular Sciences. 2017; 18(7):1419. https://doi.org/10.3390/ijms18071419
Chicago/Turabian StyleOkonkwo, Uzoagu A., and Luisa A. DiPietro. 2017. "Diabetes and Wound Angiogenesis" International Journal of Molecular Sciences 18, no. 7: 1419. https://doi.org/10.3390/ijms18071419
APA StyleOkonkwo, U. A., & DiPietro, L. A. (2017). Diabetes and Wound Angiogenesis. International Journal of Molecular Sciences, 18(7), 1419. https://doi.org/10.3390/ijms18071419