Capsaicin Induces Autophagy and Apoptosis in Human Nasopharyngeal Carcinoma Cells by Downregulating the PI3K/AKT/mTOR Pathway

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
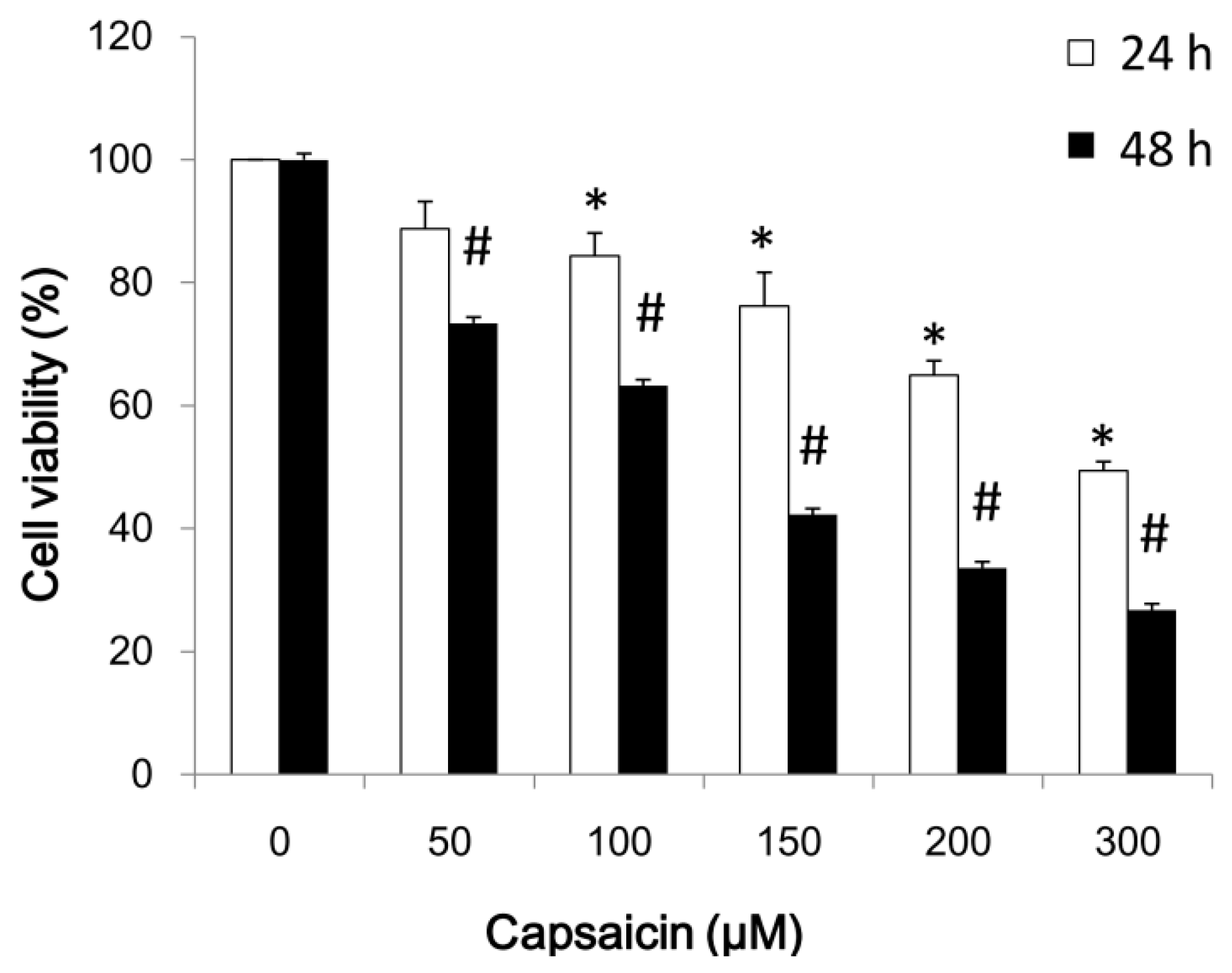
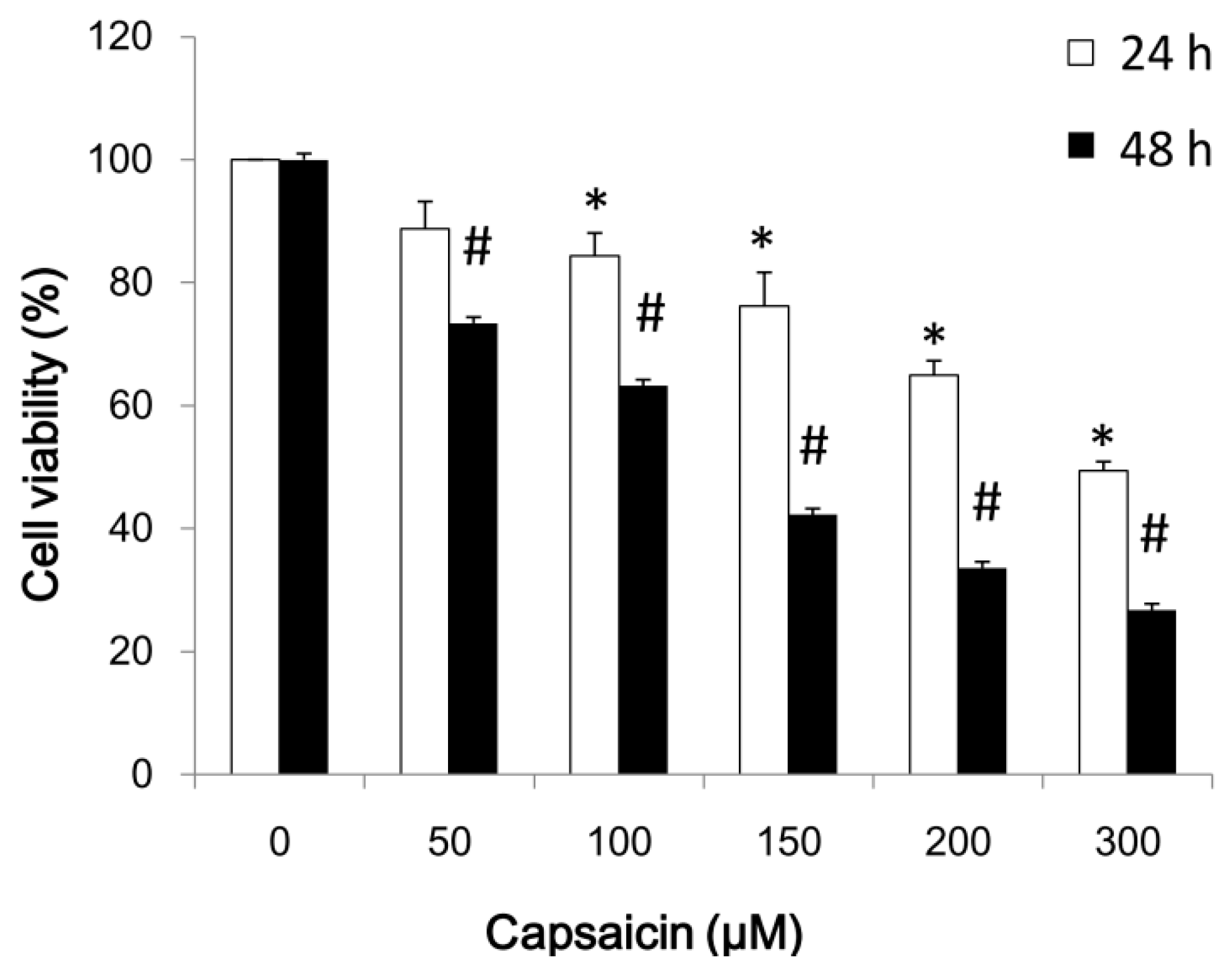
2.1. Capsaicin Inhibits NPC-TW01 Cell Proliferation
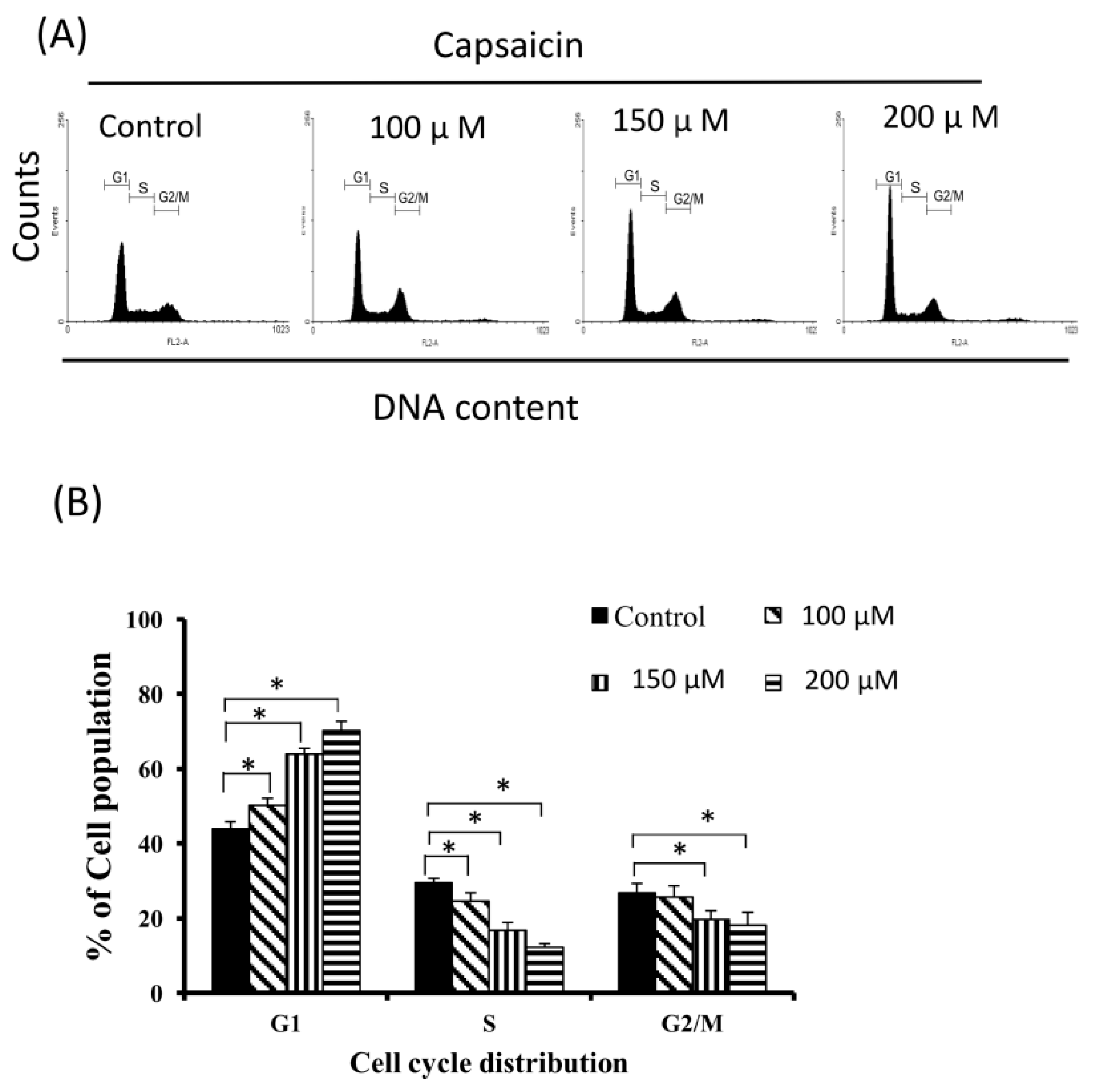
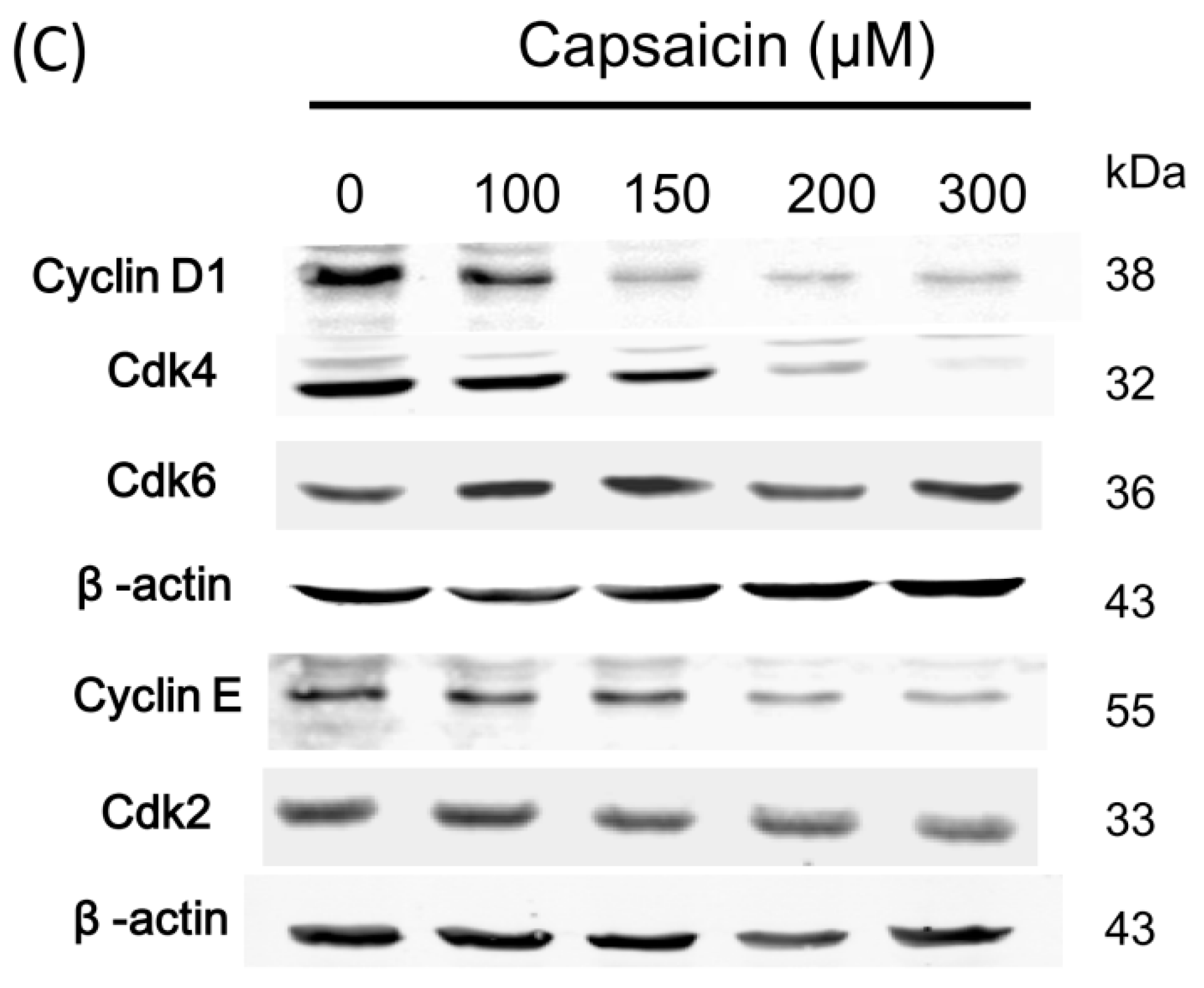
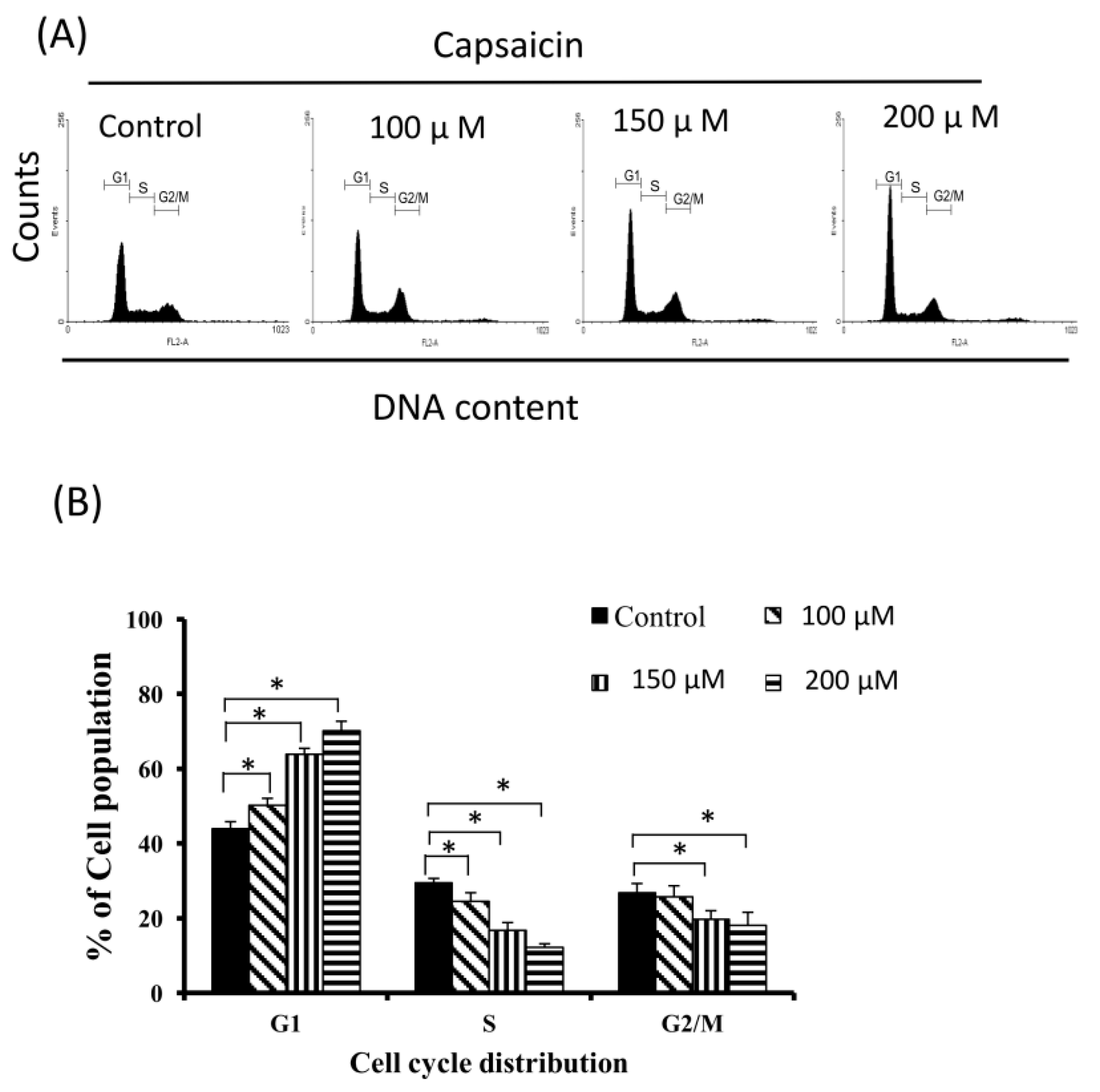
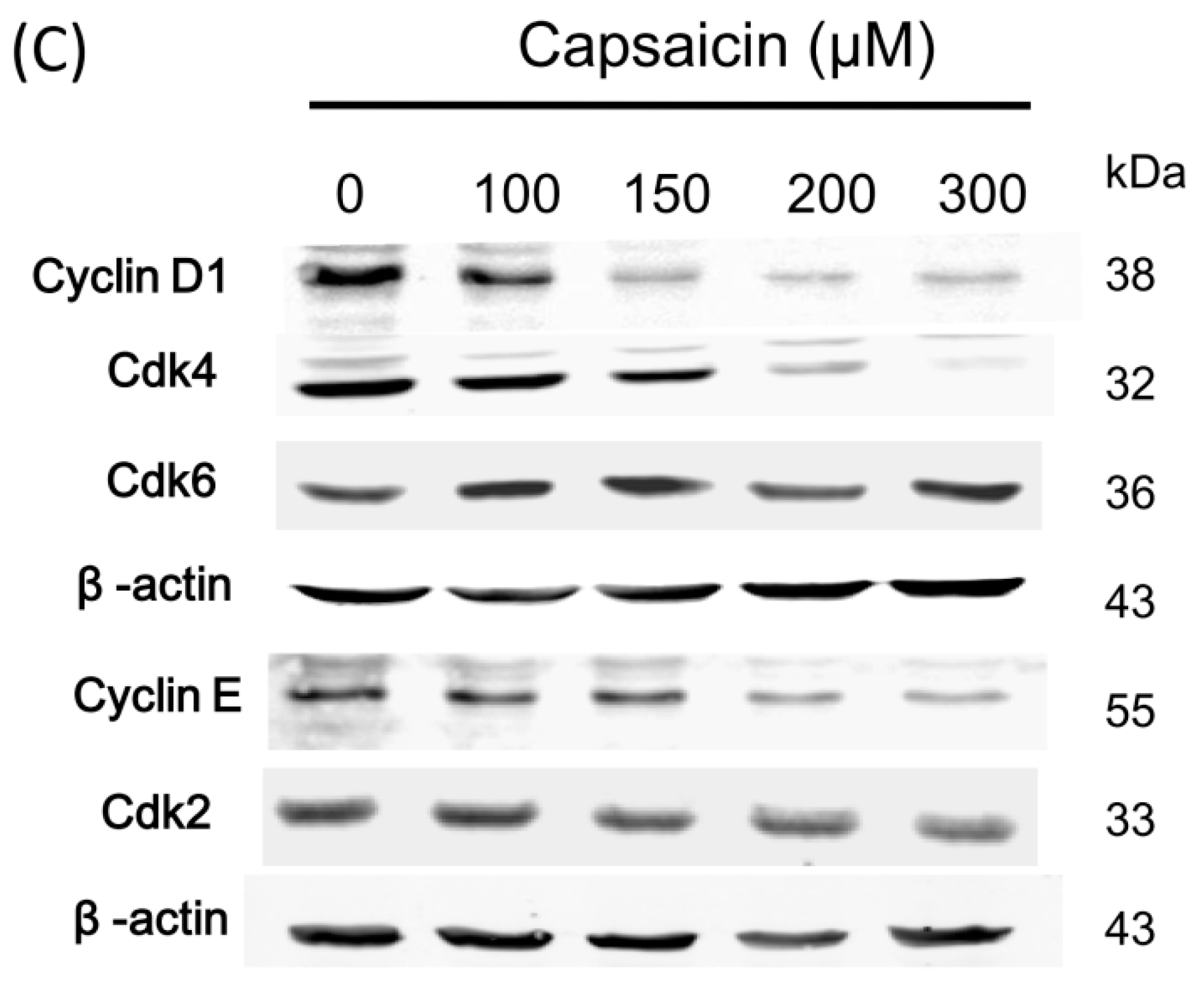
2.2. Capsaicin-Induced Cell Cycle Arrest in G1 Phase in NPC-TW01 Cells
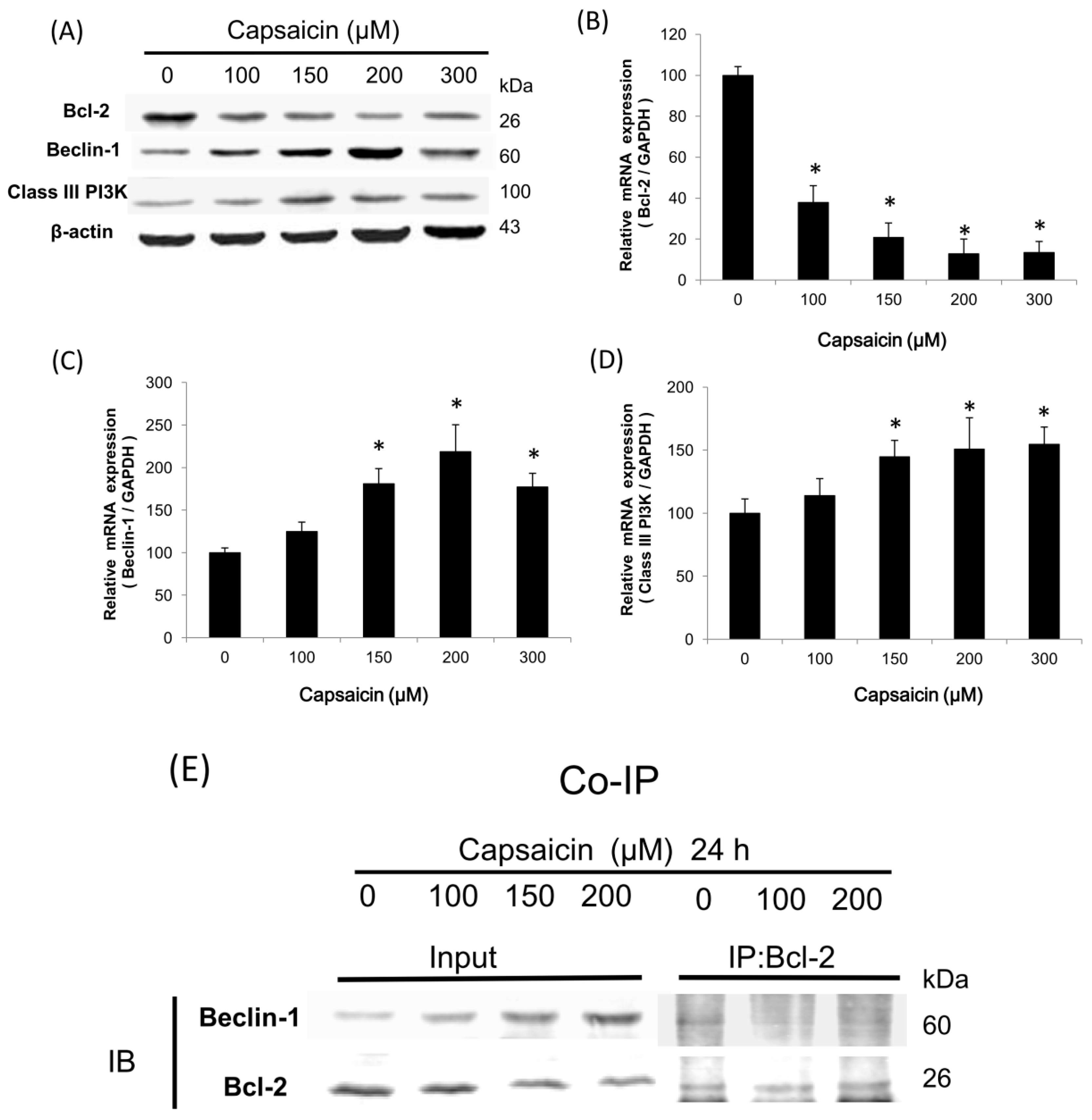
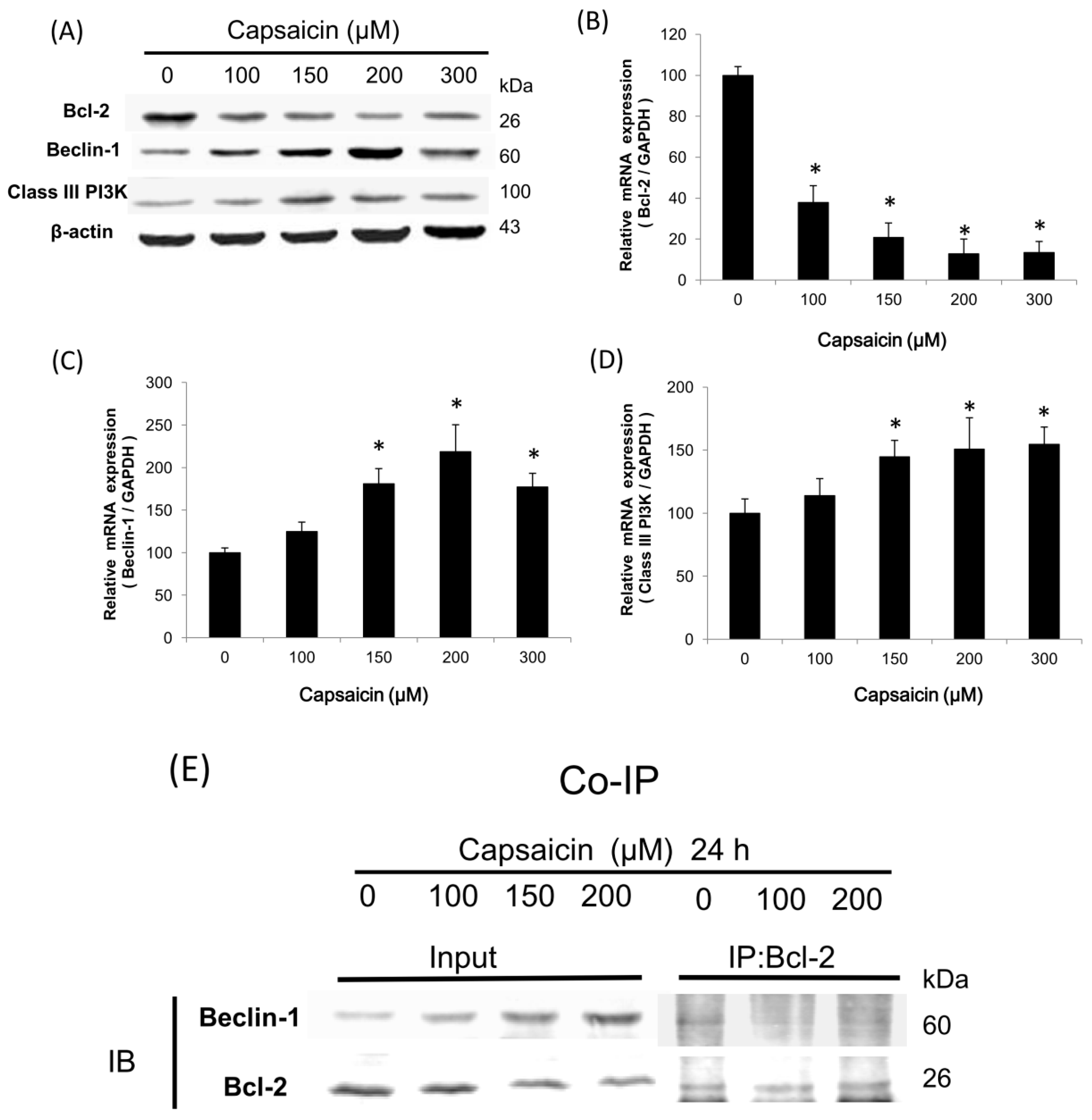
2.3. Capsaicin Induces the Class III PI3K/Beclin-1/Bcl-2 Signaling Pathway to Contribute to Autophagy Activation in NPC-TW01 Cells
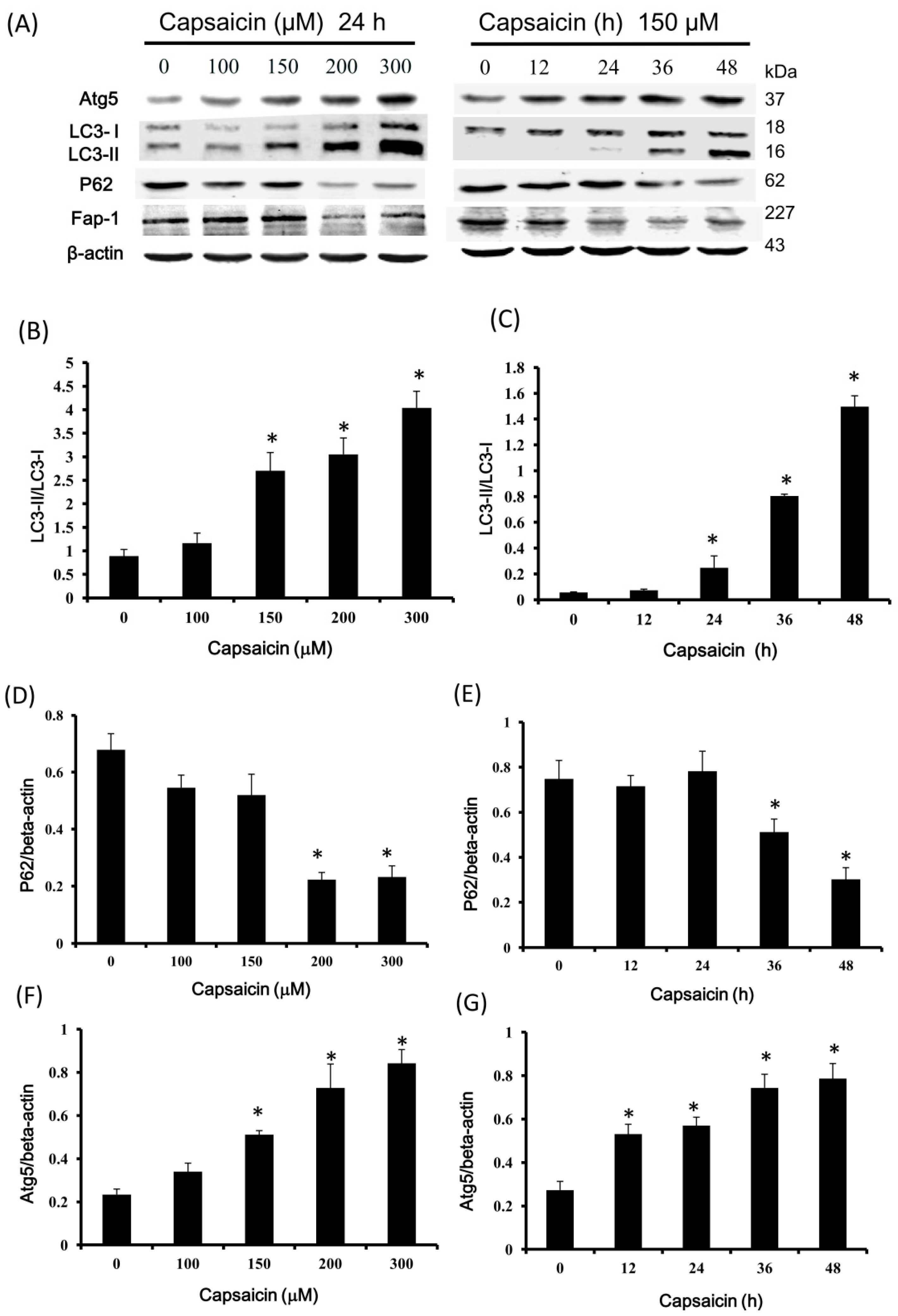
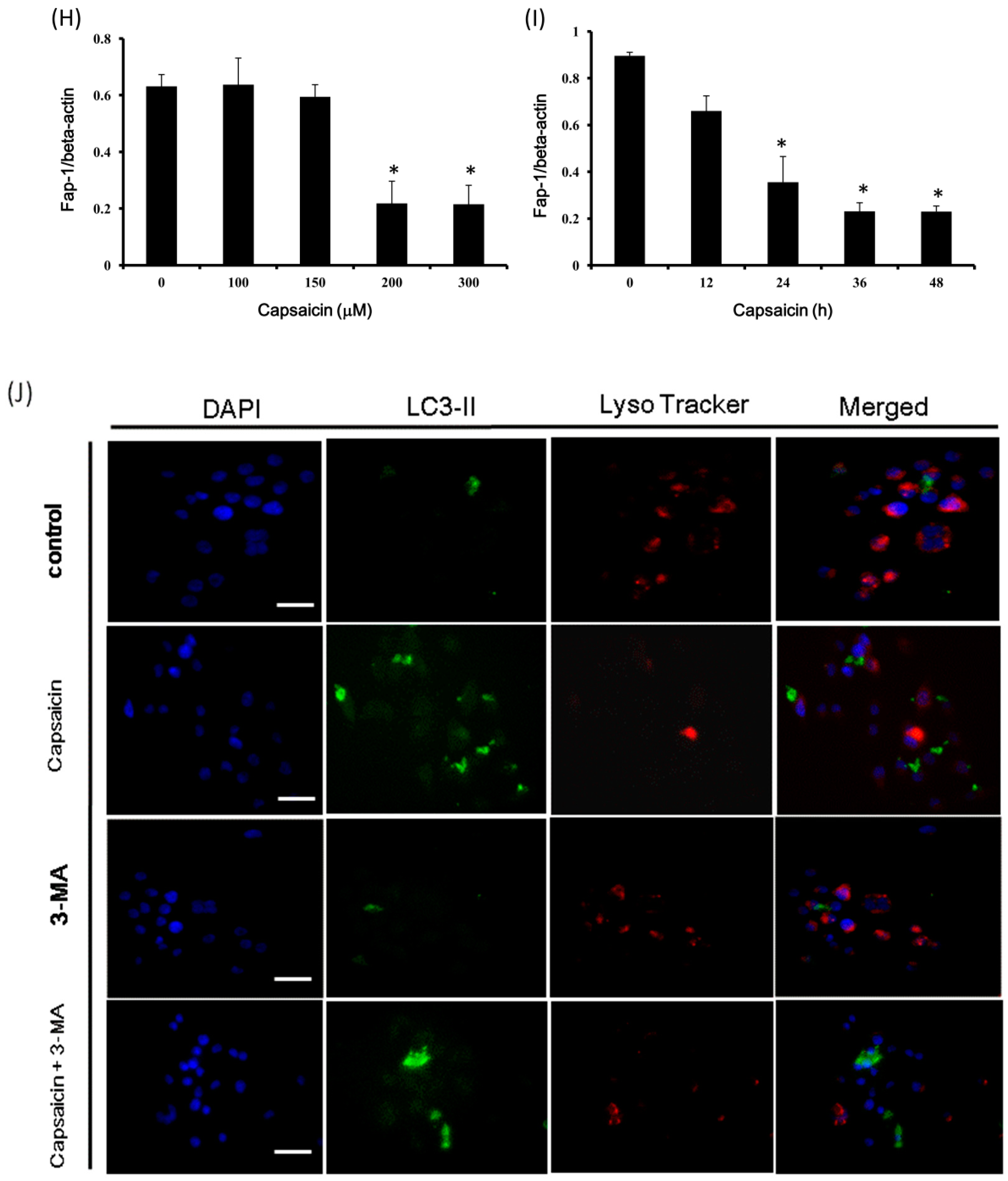
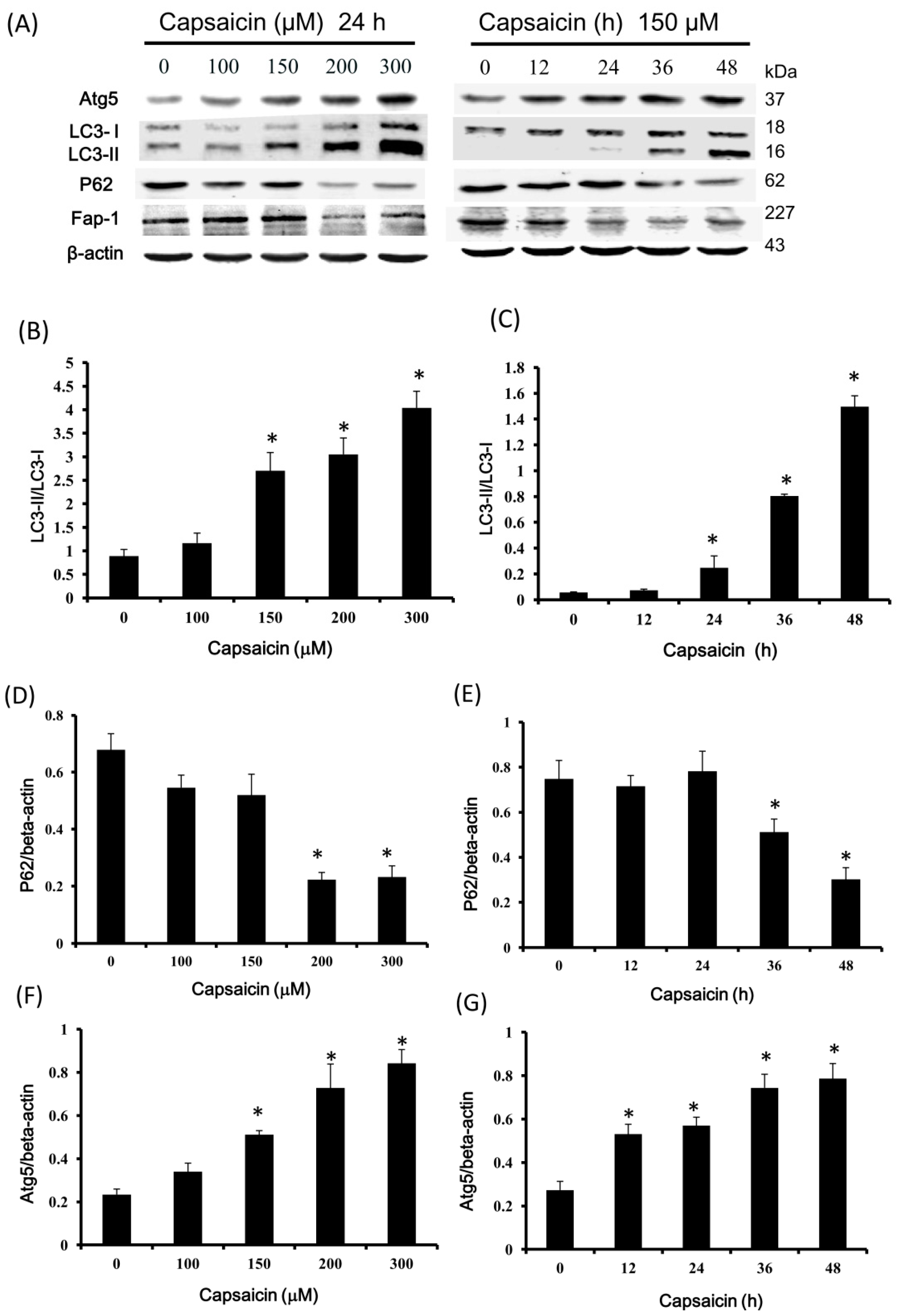
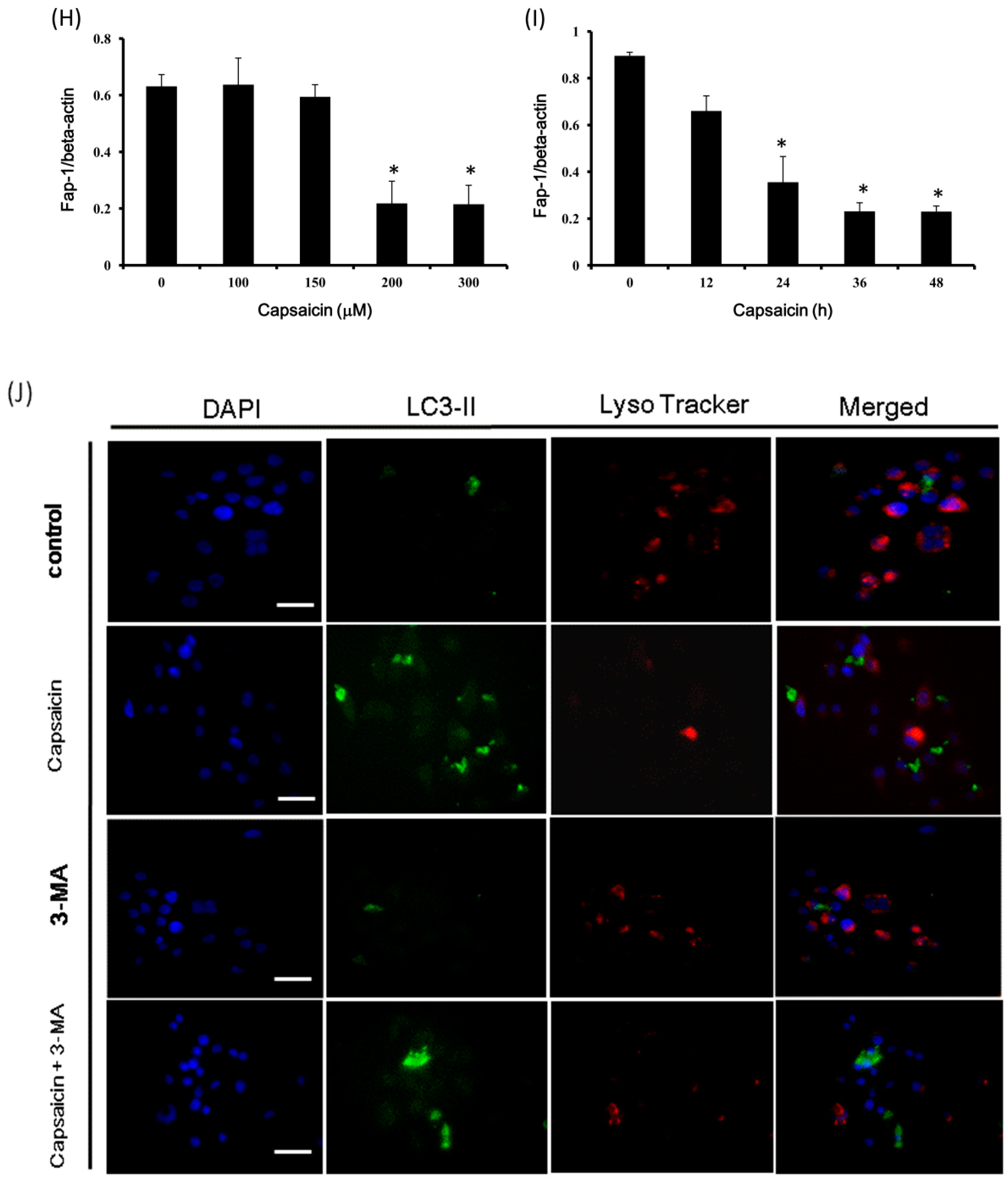
2.4. Capsaicin Upregulates LC3-II and Atg5 Expression and Downregulates p62 and Fap-1 Expression in NPC-TW01 Cells
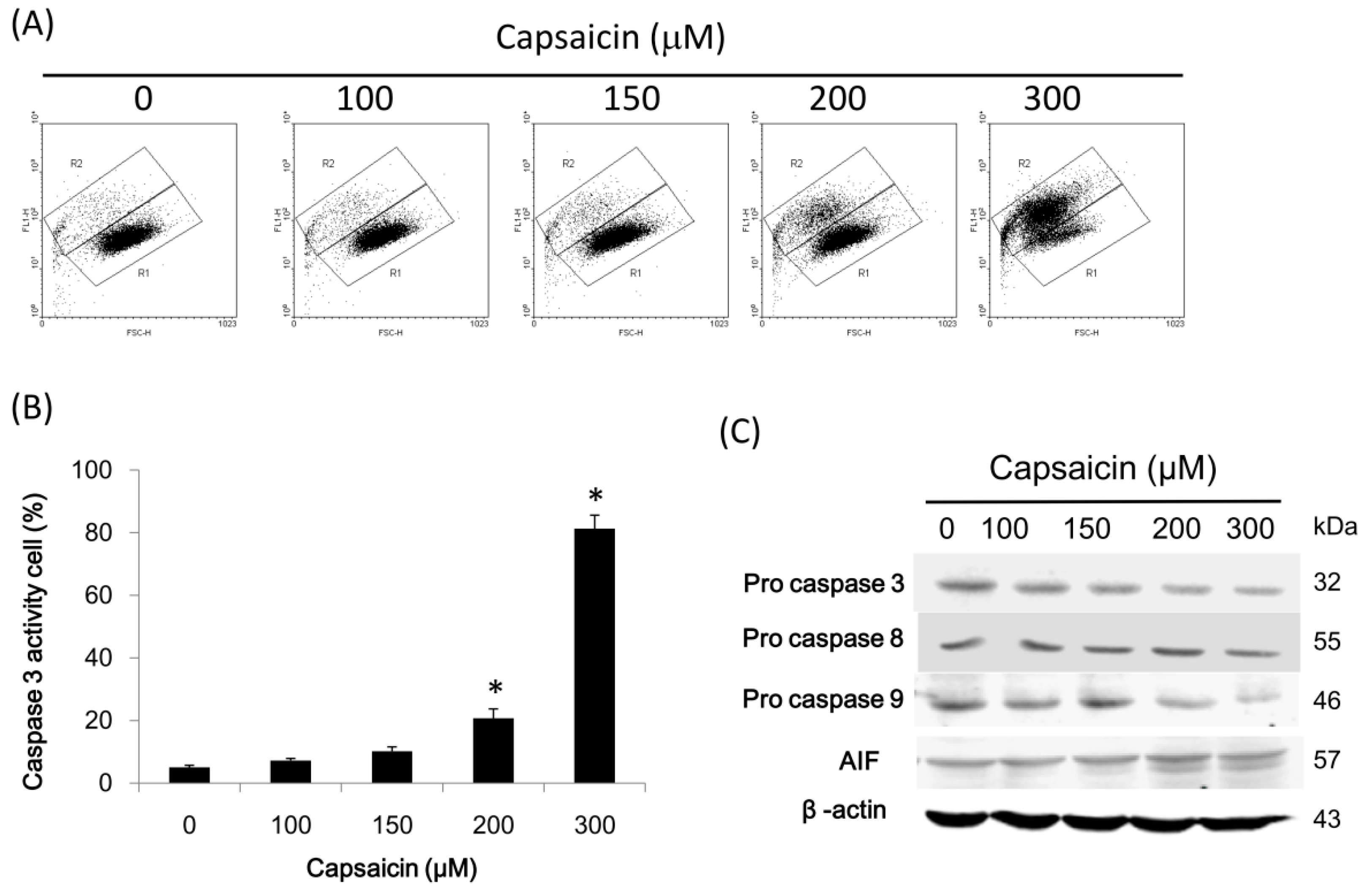
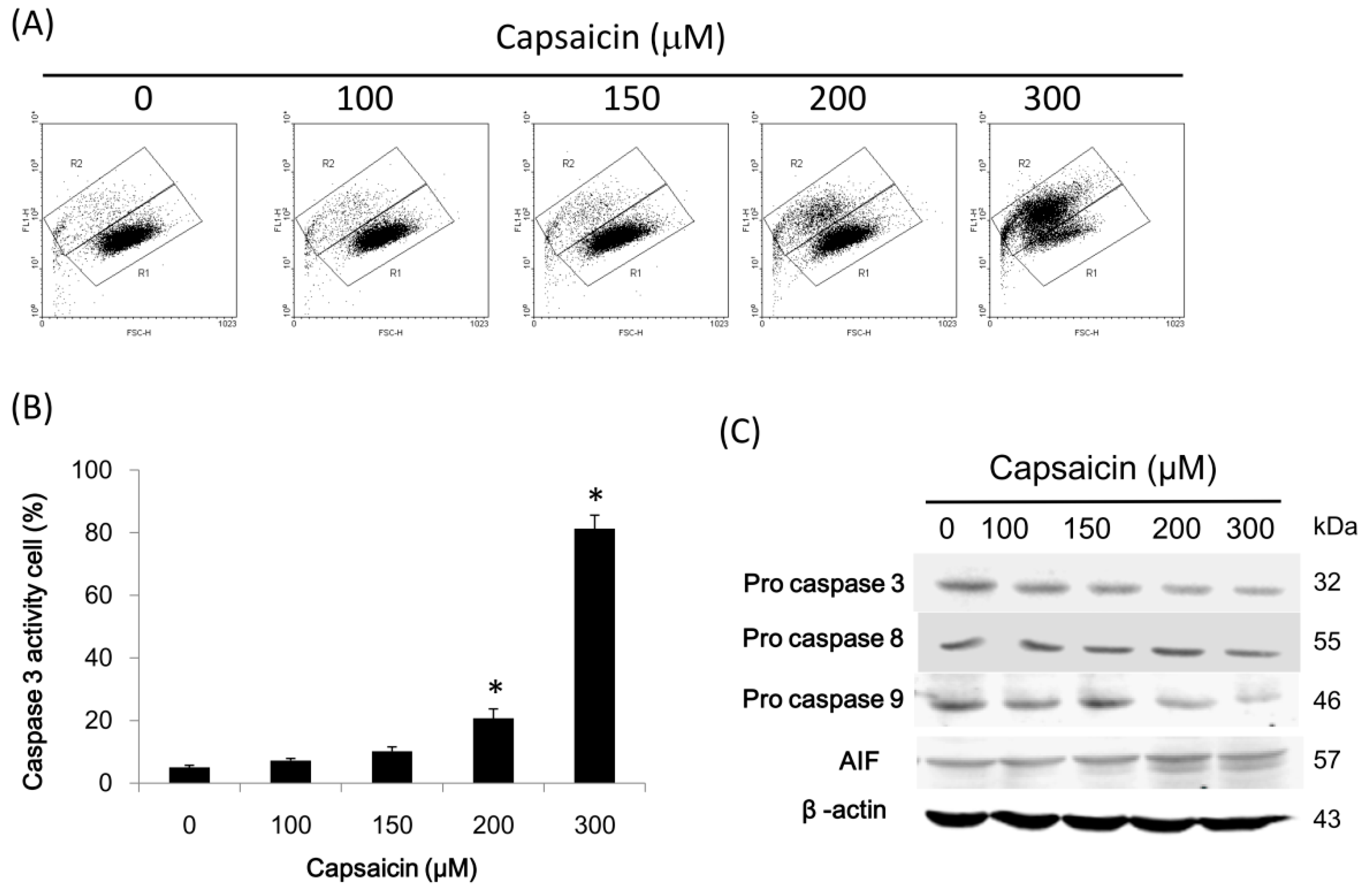
2.5. Capsaicin Induces Caspase-3 Activity in NPC-TW01 Cells
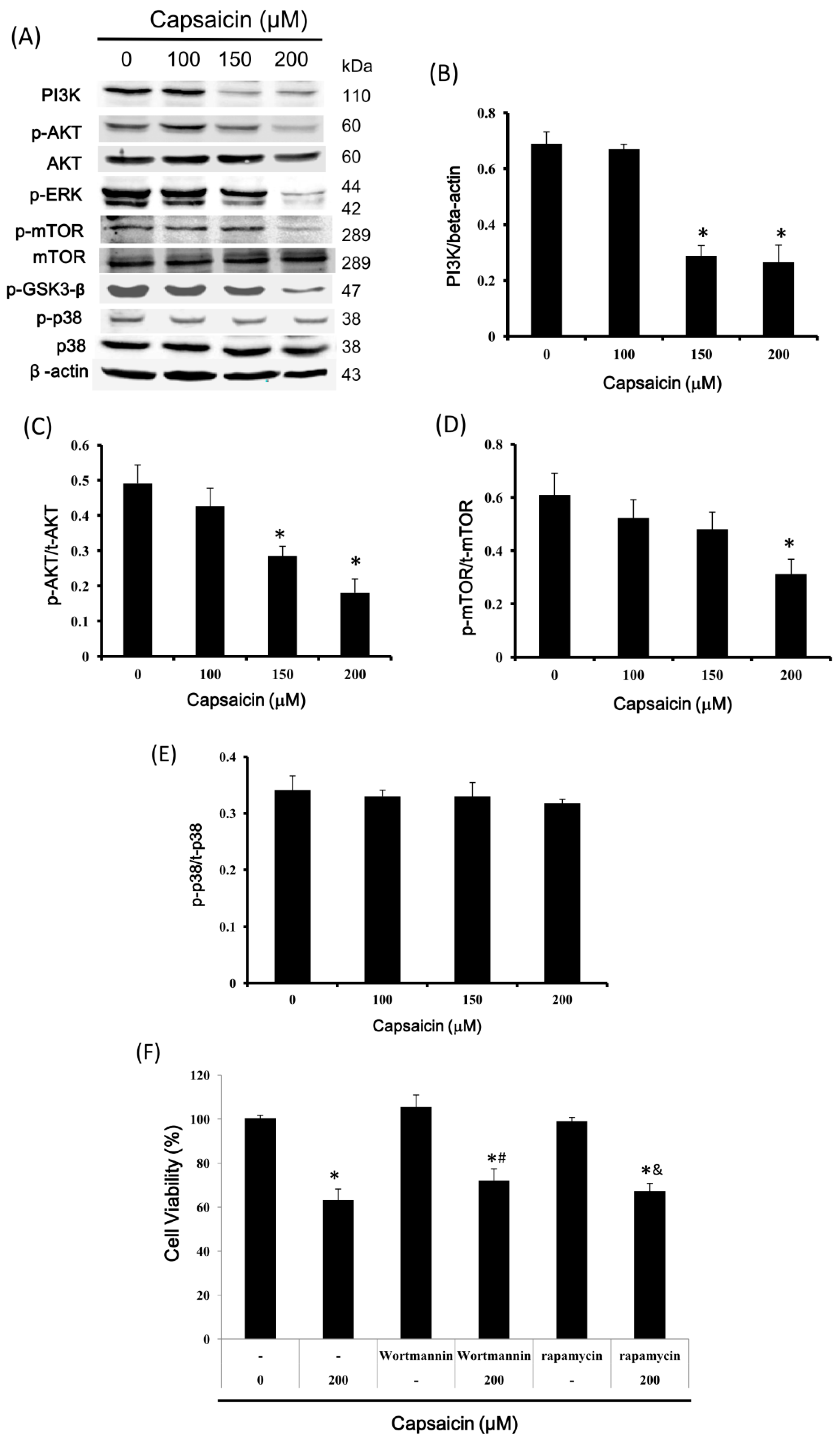
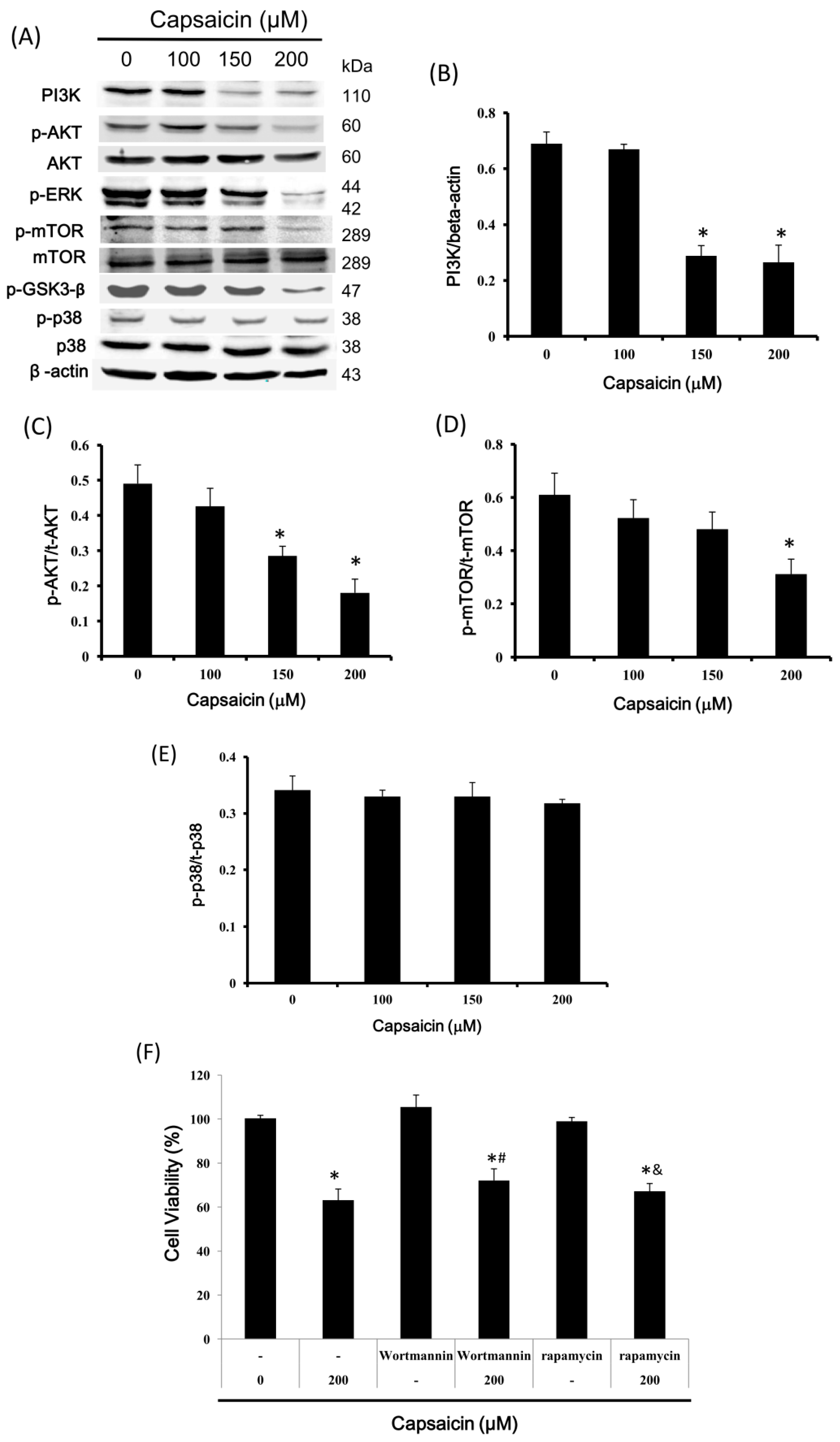
2.6. Capsaicin Inhibits PI3K Expression and the Phosphorylation of Downstream Effectors of the PI3K/Akt/mTOR Pathway in NPC-TW01 Cells
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cells
4.3. Cell Proliferation Assays
4.4. Cell Cycle Distribution Analysis
4.5. Quantitative Real-Time Reverse Transcriptase PCR
4.6. Western Blotting
4.7. Co-Immunoprecipitation
4.8. Immunofluorescence Staining
4.9. Senescence-Associated β-Galactosidase Staining
4.10. Caspase-3 Activity Assay
4.11. Statistical Analysis
5. Conclusions
Acknowledgments
Authors Contributions
Conflicts of Interest
Abbreviations
| NPC | Nasopharyngeal carcinoma |
| STATs | Signal transducer and activator of transcription protein family |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase |
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| DMSO | Dimethyl sulfoxide |
| DMEM | Dulbecco’s Modified Eagle’s Medium |
| FBS | Fetal bovine serum |
| PBS | Phosphate-buffered saline |
| PVDF | Polyvinylidene fluoride |
| 3-MA | 3-methyladenine |
| 3-methyladenine | Propidium iodide |
| CDK | Cyclin-dependent kinase |
| Atg5 | Autophagy protein 5 |
| AIF | Apoptosis inducing factor |
| qRT-PCR | Quantitative real-time reverse transcription polymerase chain reaction |
| Co-IP | Co-immunoprecipitation |
| DAPI | 4′,6-diamidino-2-phenylindole |
| DMF | N,N-dimethylformamide |
References
- Xu, T.; Tang, J.; Gu, M.; Liu, L.; Wei, W.; Yang, H. Recurrent nasopharyngeal carcinoma: A clinical dilemma and challenge. Curr. Oncol. 2013, 20, e406–e419. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.W.; Lin, J.C.; Ng, W.T. Current management of nasopharyngeal cancer. Semin. Radiat. Oncol. 2012, 22, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Loyo, M.; Brait, M.; Kim, M.S.; Ostrow, K.L.; Jie, C.C.; Chuang, A.Y.; Califano, J.A.; Liegeois, N.J.; Begum, S.; Westra, W.H.; et al. A survey of methylated candidate tumor suppressor genes in nasopharyngeal carcinoma. Int. J. Cancer 2011, 128, 1393–1403. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.M.; Tsai, W.L.; Go, S.F.; Ho, M.W.; Wu, J.M.; Wang, C.J.; Su, C.Y.; Chen, W.C.; Huang, E.Y. Implications of quantitative tumor and nodal regression rates for nasopharyngeal carcinomas after 45 Gy of radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 2001, 50, 961–969. [Google Scholar] [CrossRef]
- Hwang, C.F.; Chien, C.Y.; Huang, S.C.; Yin, Y.F.; Huang, C.C.; Fang, F.M.; Tsai, H.T.; Su, L.J.; Chen, C.H. Fibulin-3 is associated with tumour progression and a poor prognosis in nasopharyngeal carcinomas and inhibits cell migration and invasion via suppressed AKT activity. J. Pathol. 2010, 222, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Zhang, C.Z.; Ng, T.B.; Wong, J.H.; Pan, W.L.; Ye, X.J.; Chan, Y.S.; Fong, W.P. Momordica charantia lectin, a type ii ribosome inactivating protein, exhibits antitumor activity toward human nasopharyngeal carcinoma cells in vitro and in vivo. Cancer Prev. Res. 2012, 5, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Chen, Q.Y.; Liu, H.; Tang, L.Q.; Mai, H.Q. Emerging treatment options for nasopharyngeal carcinoma. Drug Des. Devel. Ther. 2013, 7, 37–52. [Google Scholar] [PubMed]
- Fattori, V.; Hohmann, M.S.; Rossaneis, A.C.; Pinho-Ribeiro, F.A.; Verri, W.A. Capsaicin: Current Understanding of Its Mechanisms and Therapy of Pain and Other Pre-Clinical and Clinical Uses. Molecules 2016, 21, 844. [Google Scholar] [CrossRef] [PubMed]
- Mankowski, C.; Patel, S.; Trueman, D.; Bentley, A.; Poole, C. Cost-Effectiveness of Capsaicin 8% Patch Compared with Pregabalin for the Treatment of Patients with Peripheral Neuropathic Pain in Scotland. PLoS ONE 2016, 11, e0150973. [Google Scholar] [CrossRef] [PubMed]
- Haanpaa, M.; Cruccu, G.; Nurmikko, T.J.; McBride, W.T.; Docu Axelarad, A.; Bosilkov, A.; Chambers, C.; Ernault, E.; Abdulahad, A.K. Capsaicin 8% patch versus oral pregabalin in patients with peripheral neuropathic pain. Eur. J. Pain 2016, 20, 316–328. [Google Scholar] [CrossRef] [PubMed]
- Ludy, M.J.; Moore, G.E.; Mattes, R.D. The effects of capsaicin and capsiate on energy balance: Critical review and meta-analyses of studies in humans. Chem. Senses 2012, 37, 103–121. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, J.A.; Davis, B.P.; Picard, J.K.; Cooper, J.P.; Zheng, S.; Levin, L.S. A randomized, double-blind, parallel trial comparing capsaicin nasal spray with placebo in subjects with a significant component of nonallergic rhinitis. Ann. Allergy Asthma Immunol. 2011, 107, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Ciabatti, P.G.; D’Ascanio, L. Intranasal Capsicum spray in idiopathic rhinitis: A randomized prospective application regimen trial. Acta. Otolaryngol. 2009, 129, 367–371. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Laviada, I.; Rodríguez-Henche, N. The potential antitumor effects of capsaicin. Prog. Drug Res. 2014, 68, 181–208. [Google Scholar] [PubMed]
- Chou, C.C.; Wu, Y.C.; Wang, Y.F.; Chou, M.J.; Kuo, S.J.; Chen, D.R. Capsaicin-induced apoptosis in human breast cancer MCF-7 cells through caspase independent pathway. Oncol. Rep. 2009, 21, 665–671. [Google Scholar] [PubMed]
- Thoennissen, N.H.; O’Kelly, J.; Lu, D.; Iwanski, G.B.; La, D.T.; Abbassi, S.; Leiter, A.; Karlan, B.; Mehta, R.; Koeffler, H.P. Capsaicin causes cell-cycle arrest and apoptosis in ER-positive and -negative breast cancer cells by modulating the EGFR/HER-2 pathway. Oncogene 2010, 29, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.M.; Pyo, J.O.; Kim, G.Y.; Yu, R.; Han, I.S.; Ju, S.A.; Kim, W.H.; Kim, B.S. Capsaicin induces apoptosis by generating reactive oxygen species and disrupting mitochondrial transmembrane potential in human colon cancer cell lines. Cell Mol. Biol. Lett. 2009, 14, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.M.; Hwang, J.T.; Kwak, D.W.; Lee, Y.K.; Park, O.J. Involvement of AMPK signaling cascade in capsaicin-induced apoptosis of HT-29 colon cancer cells. Ann. N. Y. Acad. Sci. 2007, 1095, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.; Lin, J.P.; Yang, J.S.; Chou, S.T.; Chen, S.C.; Lin, Y.T.; Lin, H.L.; Chung, J.G. Capsaicin induced cell cycle arrest and apoptosis in human esophagus epidermoid carcinoma CE 81T/VGH cells through the elevation of intracellular reactive oxygen species and Ca2+ productions and caspase-3 activation. Mutat. Res. 2006, 601, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.P.; Chen, J.C.; Wu, C.C.; Chen, C.T.; Tang, N.Y.; Ho, Y.T.; Lo, C.; Lin, J.P.; Chung, J.G.; Lin, J.G. Capsaicin-induced apoptosis in human hepatoma HepG2 cells. Anticancer Res. 2009, 29, 165–174. [Google Scholar] [PubMed]
- Chen, X.; Tan, M.; Xie, Z.; Feng, B.; Zhao, Z.; Yang, K.; Hu, C.; Liao, N.; Wang, T.; Chen, D.; et al. Inhibiting ROS-STAT3-dependent autophagy enhanced capsaicin-induced apoptosis in human hepatocellular carcinoma cells. Free Radic. Res. 2016, 50, 744–755. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, A.M.; Malagarie-Cazenave, S.; Olea, N.; Vara, D.; Chiloeches, A.; Díaz-Laviada, I. Apoptosis induced by capsaicin in prostate PC-3 cells involves ceramide accumulation, neutral sphingomyelinase, and JNK activation. Apoptosis 2007, 12, 2013–2024. [Google Scholar] [CrossRef] [PubMed]
- Le, T.D.; Jin, D.; Rho, S.R.; Kim, M.S.; Yu, R.; Yoo, H. Capsaicin-induced apoptosis of FaDu human pharyngeal squamous carcinoma cells. Yonsei. Med. J. 2012, 53, 834–841. [Google Scholar] [CrossRef] [PubMed]
- Ip, S.W.; Lan, S.H.; Lu, H.F.; Huang, A.C.; Yang, J.S.; Lin, J.P.; Huang, H.Y.; Lien, J.C.; Ho, C.C.; Chiu, C.F.; et al. Capsaicin mediates apoptosis in human nasopharyngeal carcinoma NPC-TW039 cells through mitochondrial depolarization and endoplasmic reticulum stress. Hum. Exp. Toxicol. 2012, 31, 539–559. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg-Lerner, A.; Kimchi, A. The paradox of autophagy and its implication in cancer etiology and therapy. Apoptosis 2009, 14, 376–391. [Google Scholar] [CrossRef] [PubMed]
- Zois, C.E.; Koukourakis, M.I. Radiation-induced autophagy in normal and cancer cells: Towards novel cytoprotection and radiosensitization policies? Autophagy 2009, 5, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Cheong, H.; Klionsky, D.J. Biochemical methods to monitor autophagy-related processes in yeast. Methods Enzymol. 2008, 451, 1–26. [Google Scholar] [PubMed]
- Hong, Z.F.; Zhao, W.X.; Yin, Z.Y.; Xie, C.R.; Xu, Y.P.; Chi, X.Q.; Zhang, S.; Wang, X.M. Capsaicin enhances the drug sensitivity of cholangiocarcinoma through the Inhibition of chemotherapeutic-induced autophagy. PLoS ONE 2015, 10, e0121538. [Google Scholar] [CrossRef] [PubMed]
- Amantini, C.; Morelli, M.B.; Nabissi, M.; Cardinali, C.; Santoni, M.; Gismondi, A.; Santoni, G. Capsaicin triggers autophagic cell survival which drives epithelial mesenchymal transition and chemoresistance in bladder cancer cells in an Hedgehog-dependent manner. Oncotarget 2016, 7, 50180–50194. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Torres, Á.; Bort, A.; Morell, C.; Rodríguez-Henche, N.; Díaz-Laviada, I. The pepper’s natural ingredient capsaicin induces autophagy blockage in prostate cancer cells. Oncotarget 2016, 7, 1569–1583. [Google Scholar] [PubMed]
- Lewinska, A.; Jarosz, P.; Czech, J.; Rzeszutek, I.; Bielak-Zmijewska, A.; Grabowska, W.; Wnuk, M. Capsaicin-induced genotoxic stress does not promote apoptosis in A549 human lung and DU145 prostate cancer cells. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2015, 779, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, Y.; Ye, Y.C.; Shi, Q.F.; Chai, K.; Tashiro, S.; Onodera, S.; Ikejima, T. Activation of ERK-p53 and ERK-mediated phosphorylation of Bcl-2 are involved in autophagic cell death induced by the c-Met inhibitor SU11274 in human lung cancer A549 cells. J. Pharmacol. Sci. 2012, 118, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; Le Toumelin, G.; Criollo, A.; Rain, J.C.; Gautier, F.; Juin, P.; Tasdemir, E.; Pierron, G.; Troulinaki, K.; Tavernarakis, N.; et al. Functional and physical interaction between Bcl-Xl and a BH3-like domain in Beclin-1. EMBO. J. 2007, 26, 2527–2539. [Google Scholar] [CrossRef] [PubMed]
- Oberstein, A.; Jeffrey, P.D.; Shi, Y. Crystal structure of the Bcl-Xl-Beclin 1 peptide complex: Beclin 1 is a novel BH3-only protein. J. Biol. Chem. 2007, 282, 13123–13132. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Kanaseki, T.; Mizushima, N.; Mizuta, T.; Arakawa-Kobayashi, S.; Thompson, C.B.; Tsujimoto, Y. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat. Cell. Biol. 2004, 6, 1221–1228. [Google Scholar] [CrossRef] [PubMed]
- Molinari, M. Cell cycle checkpoints and their inactivation in human cancer. Cell Prolif. 2000, 33, 261–274. [Google Scholar] [CrossRef] [PubMed]
- Schafer, K.A. The cell cycle: A review. Vet. Pathol. 1998, 35, 461–478. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012, 8, 445–544. [Google Scholar] [CrossRef] [PubMed]
- Saiki, S. Caffeine induces apoptosis by enhancement of autophagy via PI3K/Akt/mTOR/p70S6K inhibition. Autophagy 2011, 7, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.; Ryan, K.M. Autophagy chews Fap to promote apoptosis. Nat. Cell Biol. 2014, 16, 23–25. [Google Scholar] [CrossRef] [PubMed]
- Gump, J.M.; Staskiewicz, L.; Morgan, M.J.; Bamberg, A.; Riches, D.W.; Thorburn, A. Autophagy variation within a cell population determines cell fate through selective degradation of Fap-1. Nat. Cell Biol. 2014, 16, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, K.M.; Anderson, N.G. The protein kinase B/Akt signalling pathway in human malignancy. Cell Signal. 2002, 14, 381–395. [Google Scholar] [CrossRef]
- Hay, N.; Sonenberg, N. Upstream and downstream of mTOR. Genes Dev. 2004, 18, 1926–1945. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.W.; Mutter, R.W.; Cao, C.; Albert, J.M.; Freeman, M.; Hallahan, D.E.; Lu, B. Autophagy for cancer therapy through inhibition of pro-apoptotic proteins and mammalian target of rapamycin signaling. J. Biol. Chem. 2006, 281, 36883–36890. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; McPhee, C.K.; Zheng, L.; Mardones, G.A.; Rong, Y.; Peng, J.; Mi, N.; Zhao, Y.; Liu, Z.; Wan, F.; Hailey, D.W.; Oorschot, V.; Klumperman, J.; Baehrecke, E.H.; Lenardo, M.J. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010, 465, 942–946. [Google Scholar] [CrossRef] [PubMed]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA. 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.H.; Kim, Y.S.; Lim, S.C.; Hou, Y.F.; Chang, I.Y.; You, H.J. Dihydrocapsaicin (DHC), a saturated structural analog of capsaicin, induces autophagy in human cancer cells in a catalase-regulated manner. Autophagy 2008, 4, 1009–1019. [Google Scholar] [CrossRef] [PubMed]
- Lewinska, A.; Chochrek, P.; Smolag, K.; Rawska, E.; Wnuk, M. Oxidant-based anticancer activity of a novel synthetic analogue of capsaicin, capsaicin epoxide. Redox Rep. 2015, 20, 116–125. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, Y.-T.; Wang, H.-C.; Hsu, Y.-C.; Cho, C.-L.; Yang, M.-Y.; Chien, C.-Y. Capsaicin Induces Autophagy and Apoptosis in Human Nasopharyngeal Carcinoma Cells by Downregulating the PI3K/AKT/mTOR Pathway. Int. J. Mol. Sci. 2017, 18, 1343. https://doi.org/10.3390/ijms18071343
Lin Y-T, Wang H-C, Hsu Y-C, Cho C-L, Yang M-Y, Chien C-Y. Capsaicin Induces Autophagy and Apoptosis in Human Nasopharyngeal Carcinoma Cells by Downregulating the PI3K/AKT/mTOR Pathway. International Journal of Molecular Sciences. 2017; 18(7):1343. https://doi.org/10.3390/ijms18071343
Chicago/Turabian StyleLin, Yu-Tsai, Hung-Chen Wang, Yi-Chiang Hsu, Chung-Lung Cho, Ming-Yu Yang, and Chih-Yen Chien. 2017. "Capsaicin Induces Autophagy and Apoptosis in Human Nasopharyngeal Carcinoma Cells by Downregulating the PI3K/AKT/mTOR Pathway" International Journal of Molecular Sciences 18, no. 7: 1343. https://doi.org/10.3390/ijms18071343
APA StyleLin, Y.-T., Wang, H.-C., Hsu, Y.-C., Cho, C.-L., Yang, M.-Y., & Chien, C.-Y. (2017). Capsaicin Induces Autophagy and Apoptosis in Human Nasopharyngeal Carcinoma Cells by Downregulating the PI3K/AKT/mTOR Pathway. International Journal of Molecular Sciences, 18(7), 1343. https://doi.org/10.3390/ijms18071343