Hypoxia Inducible Factor-2 Alpha and Prolinhydroxylase 2 Polymorphisms in Patients with Acute Respiratory Distress Syndrome (ARDS)

Abstract
:
1. Introduction
2. Results
2.1. Hypoxia Inducible Factor-2α (C/G SNP [ch2: 46441523(hg18)]) Polymorphism
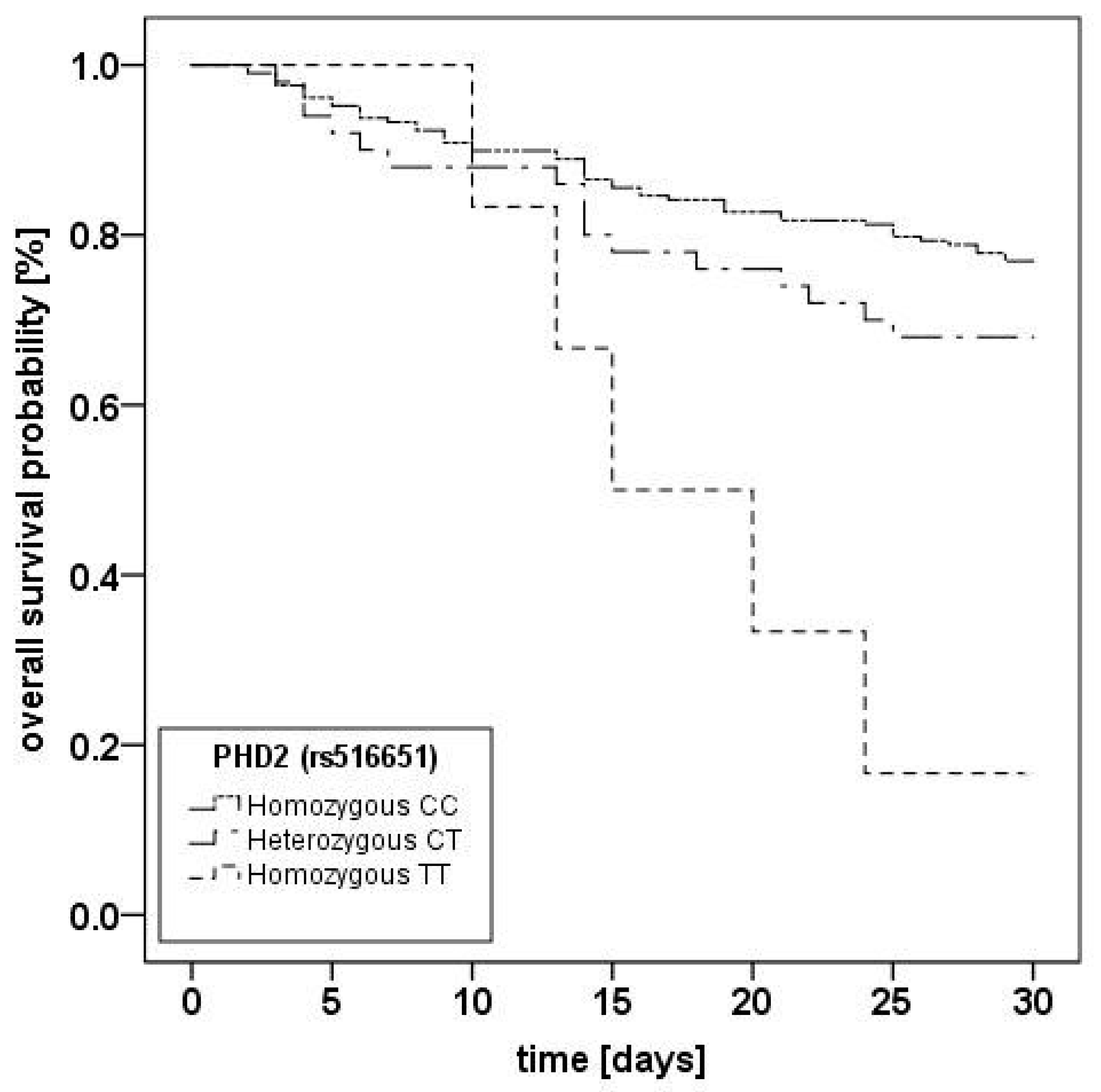
2.2. Prolylhydroxlase 2 (C/T; rs516651) Polymorphism
2.3. Prolylhydroxylase 2 (T/C; rs480902) Polymorphism
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Samplings
4.3. Genotyping
4.4. Statistical Analyses
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fujishima, S. Pathophysiology and biomarkers of acute respiratory distress syndrome. J. Intensive Care 2014, 2, 32. [Google Scholar] [CrossRef] [PubMed]
- Baron, R.M.; Levy, B.D. Recent advances in understanding and treating ARDS. F1000Research 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Ware, L.B.; Zhao, Z.; Koyama, T.; May, A.K.; Matthay, M.A.; Lurmann, F.W.; Balmes, J.R.; Calfee, C.S. Long-Term Ozone Exposure Increases the Risk of Developing the Acute Respiratory Distress Syndrome. Am. J. Respir. Crit. Care Med. 2016, 193, 1143–1150. [Google Scholar] [CrossRef] [PubMed]
- Ranieri, V.M.; Rubenfeld, G.D.; Thompson, B.T.; Ferguson, N.D.; Caldwell, E.; Fan, E.; Camporota, L.; Slutsky, A.S. Acute respiratory distress syndrome: The Berlin Definition. JAMA 2012, 307, 2526–2533. [Google Scholar] [PubMed]
- Sommer, N.; Dietrich, A.; Schermuly, R.T.; Ghofrani, H.A.; Gudermann, T.; Schulz, R.; Seeger, W.; Grimminger, F.; Weissmann, N. Regulation of hypoxic pulmonary vasoconstriction: Basic mechanisms. Eur. Respir. J. 2008, 32, 1639–1651. [Google Scholar] [CrossRef] [PubMed]
- Frede, S.; Stockmann, C.; Winning, S.; Freitag, P.; Fandrey, J. Hypoxia-inducible factor (HIF) 1 α accumulation and HIF target gene expression are impaired after induction of endotoxin tolerance. J. Immun. 2009, 182, 6470–6476. [Google Scholar] [CrossRef] [PubMed]
- Eltzschig, H.K.; Carmeliet, P. Hypoxia and inflammation. N. Engl. J. Med. 2011, 364, 656–665. [Google Scholar] [PubMed]
- Fandrey, J. Erythropoiesis--once more HIF! Blood 2008, 112, 931–932. [Google Scholar] [CrossRef] [PubMed]
- Frede, S.; Berchner-Pfannschmidt, U.; Fandrey, J. Regulation of hypoxia-inducible factors during inflammation. Methods Enzymol. 2007, 435, 405–419. [Google Scholar] [PubMed]
- Percy, M.J.; Zhao, Q.; Flores, A.; Harrison, C.; Lappin, T.R.J.; Maxwell, P.H.; McMullin, M.F.; Lee, F.S. A family with erythrocytosis establishes a role for prolyl hydroxylase domain protein 2 in oxygen homeostasis. Proc. Natl. Acad. Sci. USA 2006, 103, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Schafer, S.T.; Frede, S.; Winning, S.; Bick, A.; Roshangar, P.; Fandrey, J.; Peters, J.; Adamzik, M. Hypoxia-inducible factor and target gene expression are decreased in patients with sepsis: Prospective observational clinical and cellular studies. Anesthesiology 2013, 118, 1426–1436. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Sun, M.G.; Matro, J.; Huynh, T.T.; Rahimpour, S.; Prchal, J.T.; Lechan, R.; Lonser, R.; Pacak, K.; Zhuang, Z. Novel HIF2A mutations disrupt oxygen sensing, leading to polycythemia, paragangliomas, and somatostatinomas. Blood 2013, 121, 2563–2566. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Z.; Yang, C.; Lorenzo, F.; Merino, M.; Fojo, T.; Kebebew, E.; Popovic, V.; Stratakis, C.A.; Prchal, J.T.; Pacak, K. Somatic HIF2A gain-of-function mutations in paraganglioma with polycythemia. N. Engl. J. Med. 2012, 367, 922–930. [Google Scholar] [CrossRef] [PubMed]
- Labrousse-Arias, D.; Castillo-Gonzalez, R.; Rogers, N.M.; Torres-Capelli, M.; Barreira, B.; Aragones, J.; Cogolludo, A.; Isenberg, J.S.; Calzada, M.J. HIF-2α -mediated induction of pulmonary thrombospondin-1 contributes to hypoxia-driven vascular remodelling and vasoconstriction. Cardiovasc. Res. 2016, 109, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Buroker, N.E.; Ning, X.-H.; Zhou, Z.-N.; Li, K.; Cen, W.-J.; Wu, X.-F.; Zhu, W.-Z.; Scott, C.R.; Chen, S.-H. EPAS1 and EGLN1 associations with high altitude sickness in Han and Tibetan Chinese at the Qinghai-Tibetan Plateau. Blood Cells Mol. Dis. 2012, 49, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Gale, D.P.; Harten, S.K.; Reid, C.D.L.; Tuddenham, E.G.D.; Maxwell, P.H. Autosomal dominant erythrocytosis and pulmonary arterial hypertension associated with an activating HIF2 α mutation. Blood 2008, 112, 919–921. [Google Scholar] [CrossRef] [PubMed]
- Tan, Q.; Kerestes, H.; Percy, M.J.; Pietrofesa, R.; Chen, L.; Khurana, T.S.; Christofidou-Solomidou, M.; Lappin, T.R.J.; Lee, F.S. Erythrocytosis and pulmonary hypertension in a mouse model of human HIF2A gain of function mutation. J. Biol. Chem. 2013, 288, 17134–17144. [Google Scholar] [CrossRef] [PubMed]
- Neff, M.J. The epidemiology and definition of the acute respiratory distress syndrome. Respir. Care Clin. N. Am. 2003, 9, 273–282. [Google Scholar] [CrossRef]
- Webb, J.D.; Coleman, M.L.; Pugh, C.W. Hypoxia, hypoxia-inducible factors (HIF), HIF hydroxylases and oxygen sensing. Cell. Mol. Life Sci. 2009, 66, 3539–3554. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Aguila, H.L.; Parikh, N.S.; Li, X.; Lamothe, K.; Duan, L.-J.; Takeda, H.; Lee, F.S.; Fong, G.-H. Regulation of adult erythropoiesis by prolyl hydroxylase domain proteins. Blood 2008, 111, 3229–3235. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Cowan, A.; Fong, G.-H. Essential role for prolyl hydroxylase domain protein 2 in oxygen homeostasis of the adult vascular system. Circulation 2007, 116, 774–781. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, M.; Lamb, J.; Ebert, B.L.; Lynch, M.; Neil, C.; Schmidt, E.; Golub, T.R.; Iliopoulos, O. The connectivity map links iron regulatory protein-1-mediated inhibition of hypoxia-inducible factor-2a translation to the anti-inflammatory 15-deoxy- β 12,14-prostaglandin J2. Cancer Res. 2010, 70, 3071–3079. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, S.K.; Shah, Y.M. Role of Intestinal HIF-2alpha in Health and Disease. Annu. Rev. Physiol. 2016, 78, 301–325. [Google Scholar] [CrossRef] [PubMed]
- Swenson, E.R.; Bartsch, P. High-altitude pulmonary edema. Compr. Physiol. 2012, 2, 2753–2773. [Google Scholar] [PubMed]
- Wu, A.L.; Xiong, Y.S.; Li, Z.Q.; Liu, Y.G.; Quan, Q.; Wu, L.J. Correlation between single nucleotide polymorphisms in hypoxia-related genes and susceptibility to acute high-altitude pulmonary edema. Gene Mol. Res. 2015, 14, 11562–11572. [Google Scholar] [CrossRef] [PubMed]
- Yi, X.; Liang, Y.; Huerta-Sanchez, E.; Jin, X.; Cuo, Z.X.P.; Pool, J.E.; Xu, X.; Jiang, H.; Vinckenbosch, N.; Korneliussen, T.S.; et al. Sequencing of 50 human exomes reveals adaptation to high altitude. Science 2010, 329, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Le Gall, J.R.; Lemeshow, S.; Saulnier, F. A new Simplified Acute Physiology Score (SAPS II) based on a European/North American multicenter study. JAMA 1993, 270, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Aegerter, P.; Boumendil, A.; Retbi, A.; Minvielle, E.; Dervaux, B.; Guidet, B. SAPS II revisited. Intensive Care Med. 2005, 31, 416–423. [Google Scholar] [CrossRef] [PubMed]
- Hocker, A.; Rabeling, M.; Bick, A.; Cox, L.; Kreuzer, M.; Engler, A.; Walstein, K.; Bachmann, H.S.; Jockel, K.-H.; Eisele, L.; et al. Hypoxia inducible factor-1 alpha and prolinhydroxlase 2 polymorphisms in patients with severe sepsis: A prospective observational trial. BMC Anesthesiol. 2016, 16, 61. [Google Scholar] [CrossRef] [PubMed]
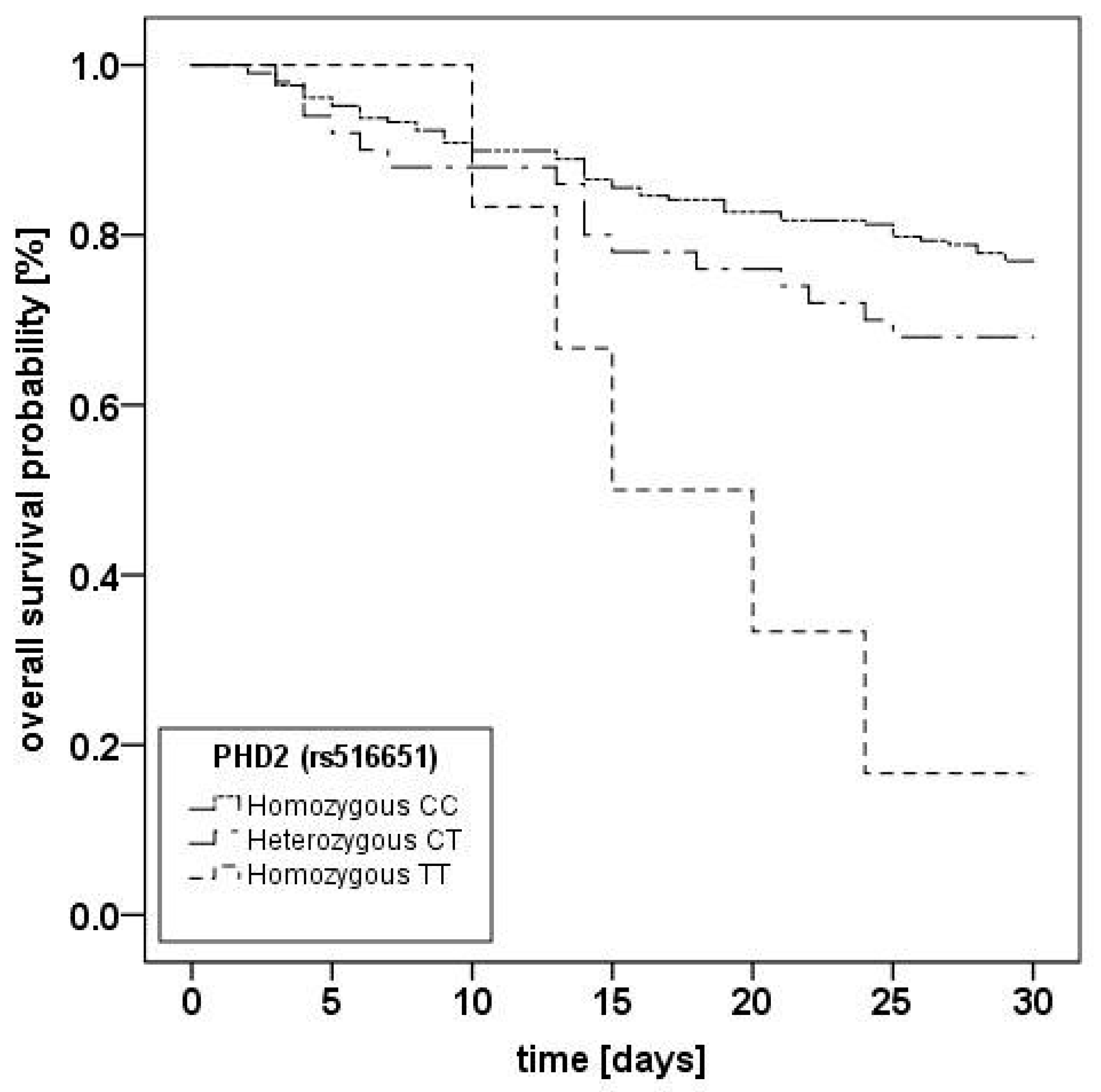

{kind=link}
{kind=link}
| Genotype (n) | rs516651 | rs516651 | rs516651 | p Value | rs480902 | rs480902 | rs480902 | p Value | HIF-2α | HIF-2α | p Value |
|---|---|---|---|---|---|---|---|---|---|---|---|
| CC (n = 208) | CT (n = 50) | TT (n = 6) | n = 264 | CC (n = 142) | CT (n = 119) | TT (n = 10) | n = 271 | CC (n = 264) | CG (n = 1) | n = 265 | |
| Gender (women/men; n; %) | 89/119 (42.8/57.2) | 18/32 (36/64) | 0/6 (0/100) | 0.084 ** | 54/88 (38/62) | 48/71 (40/60) | 8/2 (80/20) | 0.033 ** | 107 /157 (59.5/40.5) | 1/0 (100/0) | 0.410 |
| Age (years; median; Q1; Q3) # | 44 (33–55) | 45 (35–57) | 65.5 (56–71) | 0.011 * | 45 (34–57) | 43 (32–54) | 43 (34–50) | 0.290 * | 44 (33–56) | 23 | 0.178 |
| Height (cm; median; Q1; Q3) # | 175 (165–180) | 178 (168–185) | 178 (171–185) | 0.160 * | 175 (167–180) | 175 (165–183) | 170 (163–175) | 0.190 * | 175 (165–180) | 192 | 0.098 |
| Body weight (kg; median; Q1; Q3) # | 85 (70–90) | 80 (70–94) | 80 (70–90) | 0.644 * | 85 (70–90) | 85 (70–93) | 78 (65–80) | 0.322 * | 85 (70–90) | 86 | 0.782 |
| Body mass index (kg·m−2; median; Q1; Q3) # | 26.7 (23.4–30.5) | 26.3 (23.9–29.4) | 24.2 (23.5–26.3) | 0.389 * | 26.1 (23.4–29.4) | 27.3 (23.9–31.2) | 26.4 (21.9–28.5) | 0.436 * | 26.3 (23.9–30.1) | 23.3 | 0.322 |
| Mean arterial blood pressure (mmHg; median; Q1; Q3) # | 80 (72–87) | 83 (75–88) | 86 (68–98) | 0.472 * | 80 (72–87) | 81 (73–87) | 95 (87–121) | 0.046 * | 110 (90–125) | - | - |
| Mean systolic arterial blood pressure (mmHg; median; Q1; Q3) # | 120 (110–130) | 120 (115–130) | 136 (128–150) | 0.126 * | 120 (110–129) | 120 (110–130) | 121 (115–145) | 0.330 * | 80 (72–87) | - | - |
| Heart rate (min−1; median. Q1; Q3) # | 107 (90–123) | 113 (99–130) | 106 (77–148) | 0.648 * | 110 (95–120) | 110 (90–132) | 100 (82–114) | 0.569 * | 120 (110–130) | 110 | 0.334 |
| Mean pulmonary arterial pressure (mmHg; median; Q1; Q3) # | 35.5 (31–40) | 33 (28–39) | 38.5 (34–42) | 0.261 * | 35 (30–41) | 35 (29–40) | 38 (35–45) | 0.416 * | 35.5 (30–41) | 29 | 0.321 |
| Lower airway pressure (median; Q1; Q3) # | 18 (15–20) | 18 (14–20) | 16 (10–18.5) | 0.468 * | 18 (15–20) | 18 (15–20) | 15 (10–20) | 0.359 * | 18 (15–20) | 22 | 0.126 |
| Horovitz ratio (median; Q1; Q3) # | 108 (73–195) | 95 (72–172) | 146 (98–193) | 0.614 * | 110 (77–201) | 106 (68–178) | 96 (77–139) | 0.517 * | 107 (73–187) | 115 | 0.642 |
| Creatinin serum concentration (mg·dL−1; median; Q1; Q3) # | 1.42 (1.00–2.43) | 1.27 (0.96–1.89) | 1.34 (1.16–1.48) | 0.413 * | 1.48 (1.01–2.17) | 1.33 (1.00–2.75) | 1.00 (0.82–1.76) | 0.274 * | 1.4 (1–2.4) | 1.11 | 0.225 |
| Dialysis (yes/no; %) | 115/76 (60.2/39.8) | 21/24 (53.3/46.7) | 5/1 (83.3/16.7) | 0.114 ** | 81/46 (63.8/36.2) | 61/51 (54.5/45.5) | 5/5 (50/50) | 0.288 ** | 145/98 (59.7/40.3) | 0/1 (0/100) | 0.759 |
| Infectious variables | |||||||||||
| White blood cell count (109·L−1; median; Q1; Q3) # | 14.2 (8.9–21.9) | 13.4 (9.4–22.2) | 9.8 (8.8–15.6) | 0.517 * | 14.5 (9.2–22.9) | 13.4 (8.8–21.1) | 12.8 (8.6–22.3) | 0.785 * | 13.8 (8.8–21.8) | 16.4 | 0.357 |
| Procalcitonin serum concentration (µg·L−1; median; Q1; Q3) # | 4.48 (0.69–28.70) | 2.56 (0.85–11.9) | 0.71 (0.29–30.13) | 0.801 * | 2.21 (0.60–16.67) | 5.38 (0.93–37.38) | 5.34 (1.01–68.55) | 0.139 * | 3.78 (0.69–26.4) | - | - |
| C-reactive protein concentration (g·L−1; median; Q1; Q3) # | 19.9 (13.3–27.5) | 17.4 (7.1–28.3) | 18.4 (12.8–37.3) | 0.435 * | 18.4 (11.50–24.8) | 20.7 (12.8–32.7) | 19.0 (8.3–24.1) | 0.286 * | 19.6 (11.8–27.8) | 28.2 | 0.967 |
| Disease severity | |||||||||||
| SAPS II ## (mean ± SD) + | 42.5 (30–58) | 39.5 (30–60) | 41.5 (32–71) | 0.836 * | 43 (31–61) | 42 (30–60) | 27 (19–42) | 0.099 * | 43 (31–60) | 42 | 0.482 |
| SOFA ++ (mean ± SD) + | 14 (11–20) | 13 (10–16) | 13.5 (10–20) | 0.368 * | 15 (12–22) | 13 (10–18) | 14 (11–22) | 0.095 * | 14 (11–20) | 12 | 0.433 |
| Hospital stay (d; mean ± SD) + | 22 (14–38) | 22 (14–34) | 18 (13–24) | 0.742 * | 24 (13–39) | 21 (14–33) | 18 (14–26) | 0.596 * | 22 (14–37) | 14 | 0.548 |
| 30-day mortality (%) | 48 (23) | 16 (32) | 5 (83.3) | 0.002 ** | 39 (27.5) | 32 (26.9) | 0 (0) | 0.158 ** | 70 (26.5) | 0 (0) | - |
| Covariables | HR (95% CI) | p Value |
|---|---|---|
| rs516651, CT vs. CC | 1.71 (0.89–3.29) | 0.11 |
| rs516651, TT vs. CC | 3.34 (1.09–10.22) | 0.034 |
| Age, per year | 1.01 (0.99–1.036) | 0.40 |
| Gender, male vs. female | 0.92 (0.49–1.74) | 0.80 |
| Dialysis, yes vs. no | 0.94 (0.44–2.01) | 0.87 |
| SAPS II, per point | 1.03 (1.01–1.04) | 0.0032 |
| SOFA | 1.01 (0.98–1.04) | 0.43 |
| PCT | 1.002 (1.00–1.003) | 0.038 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dötsch, A.; Eisele, L.; Rabeling, M.; Rump, K.; Walstein, K.; Bick, A.; Cox, L.; Engler, A.; Bachmann, H.S.; Jöckel, K.-H.; et al. Hypoxia Inducible Factor-2 Alpha and Prolinhydroxylase 2 Polymorphisms in Patients with Acute Respiratory Distress Syndrome (ARDS). Int. J. Mol. Sci. 2017, 18, 1266. https://doi.org/10.3390/ijms18061266
Dötsch A, Eisele L, Rabeling M, Rump K, Walstein K, Bick A, Cox L, Engler A, Bachmann HS, Jöckel K-H, et al. Hypoxia Inducible Factor-2 Alpha and Prolinhydroxylase 2 Polymorphisms in Patients with Acute Respiratory Distress Syndrome (ARDS). International Journal of Molecular Sciences. 2017; 18(6):1266. https://doi.org/10.3390/ijms18061266
Chicago/Turabian StyleDötsch, Annika, Lewin Eisele, Miriam Rabeling, Katharina Rump, Kai Walstein, Alexandra Bick, Linda Cox, Andrea Engler, Hagen S. Bachmann, Karl-Heinz Jöckel, and et al. 2017. "Hypoxia Inducible Factor-2 Alpha and Prolinhydroxylase 2 Polymorphisms in Patients with Acute Respiratory Distress Syndrome (ARDS)" International Journal of Molecular Sciences 18, no. 6: 1266. https://doi.org/10.3390/ijms18061266
APA StyleDötsch, A., Eisele, L., Rabeling, M., Rump, K., Walstein, K., Bick, A., Cox, L., Engler, A., Bachmann, H. S., Jöckel, K.-H., Adamzik, M., Peters, J., & Schäfer, S. T. (2017). Hypoxia Inducible Factor-2 Alpha and Prolinhydroxylase 2 Polymorphisms in Patients with Acute Respiratory Distress Syndrome (ARDS). International Journal of Molecular Sciences, 18(6), 1266. https://doi.org/10.3390/ijms18061266