
Friction-Induced Mitochondrial Dysregulation Contributes to Joint Deterioration in Prg4 Knockout Mice


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
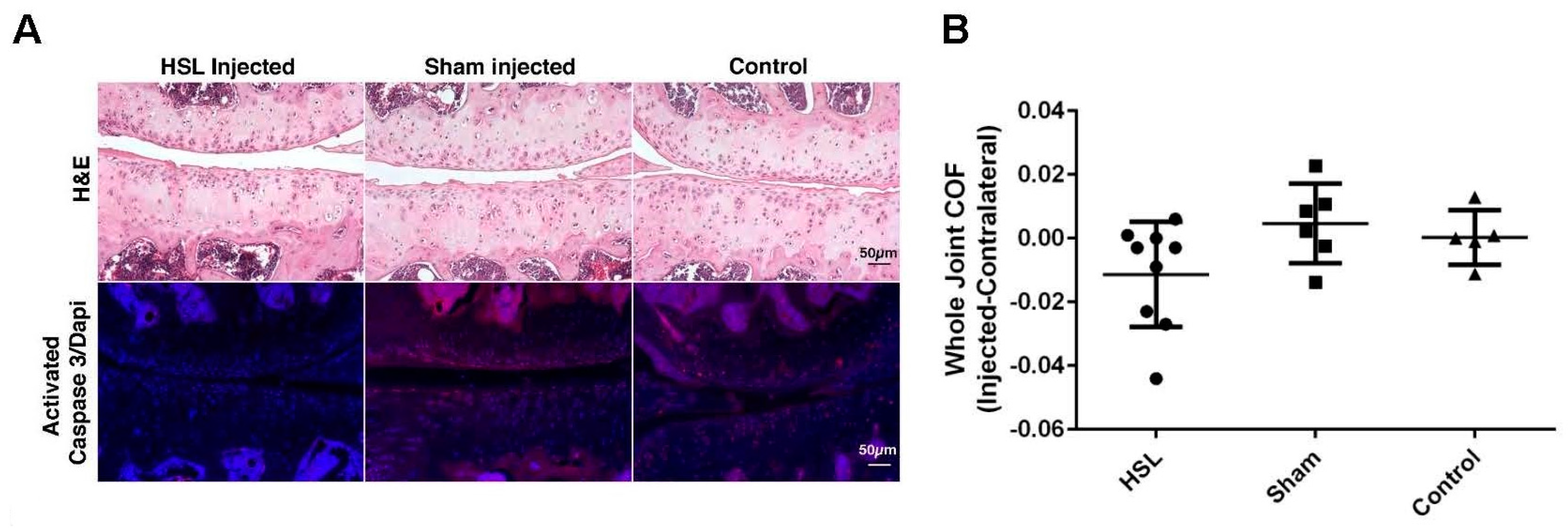
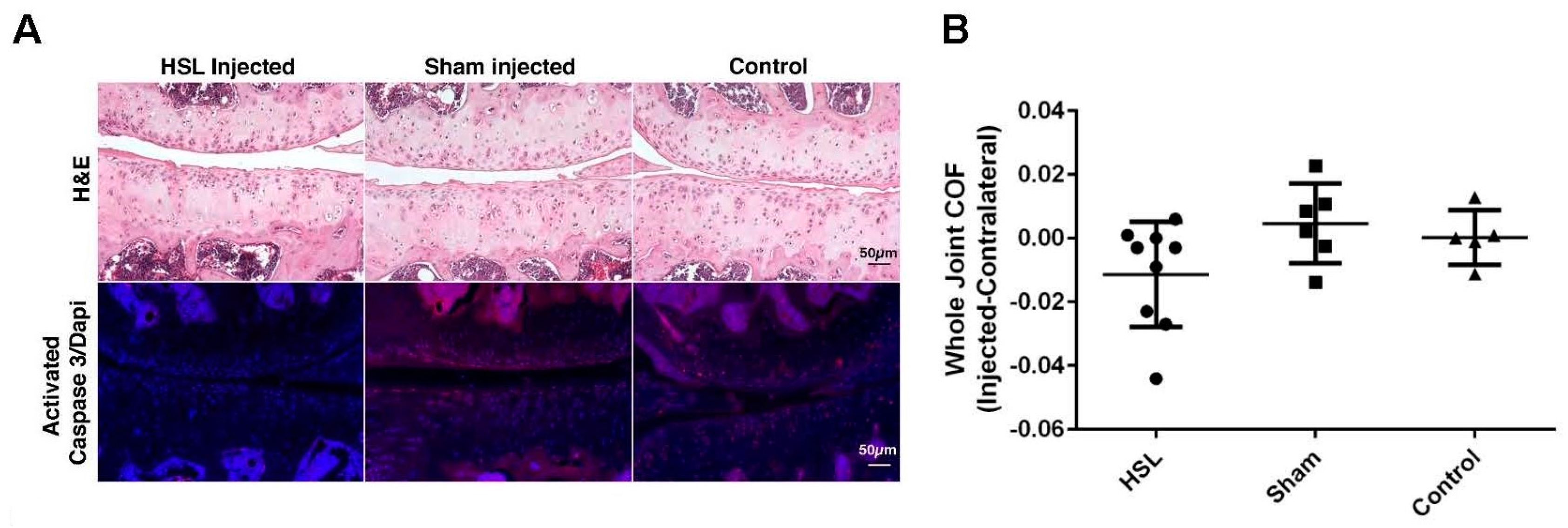
2.1. Whole Joint COF of Prg4−/− Mice Following Intra-Articular PRG4 Compared to Sham Injected and Control Mice
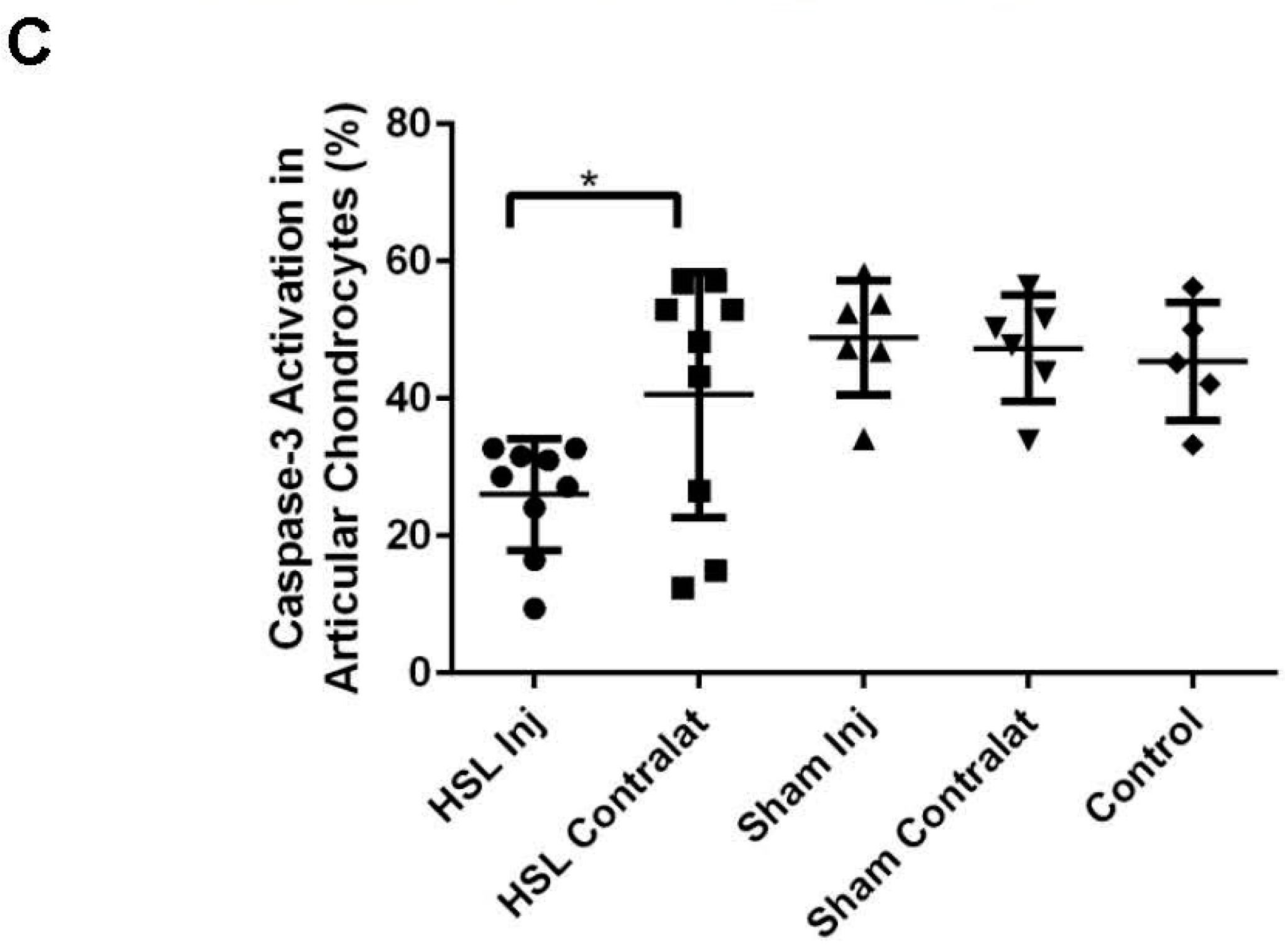
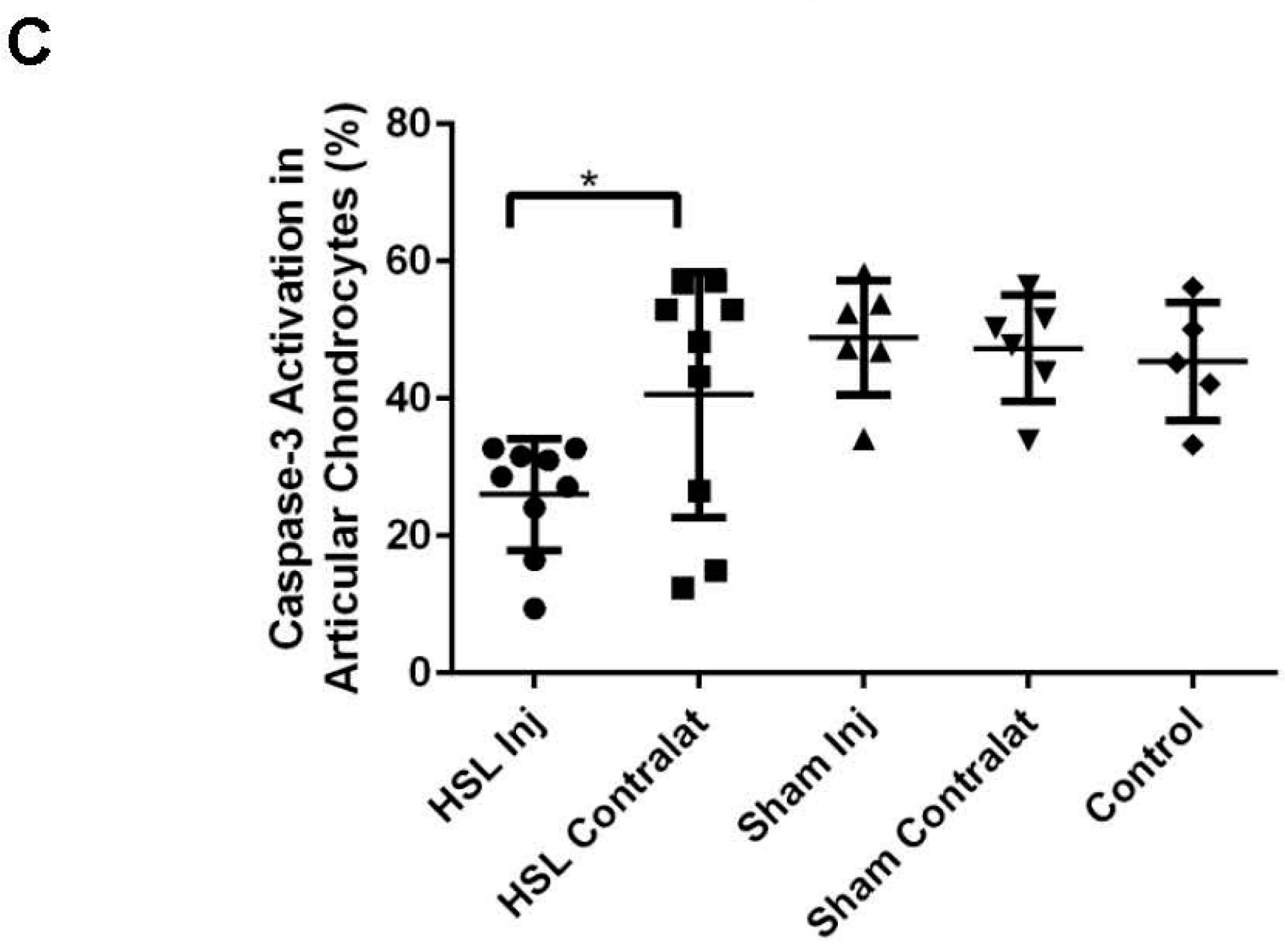
2.2. Caspase-3 Activation of Prg4−/− Mice Following Intra-Articular PRG4
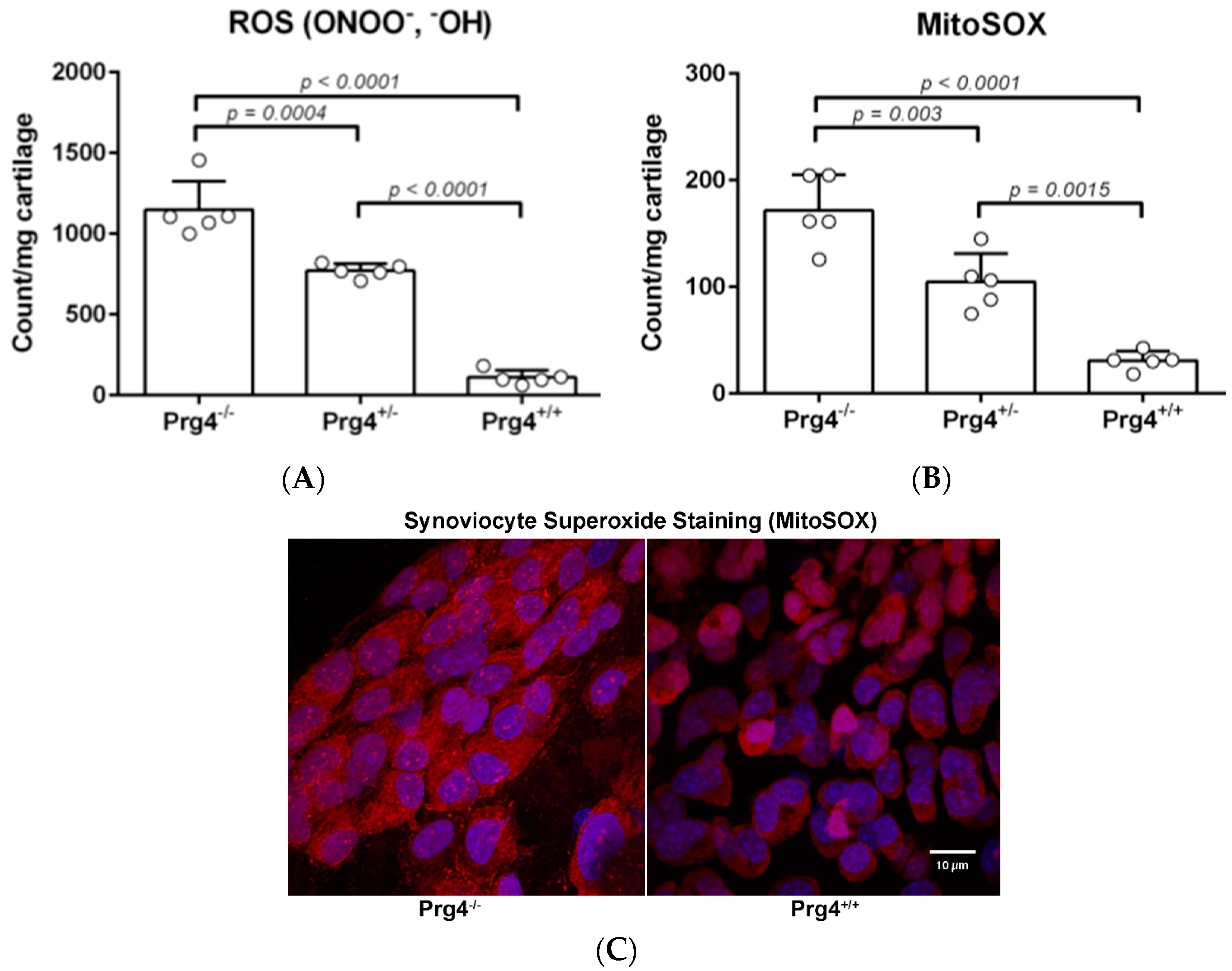
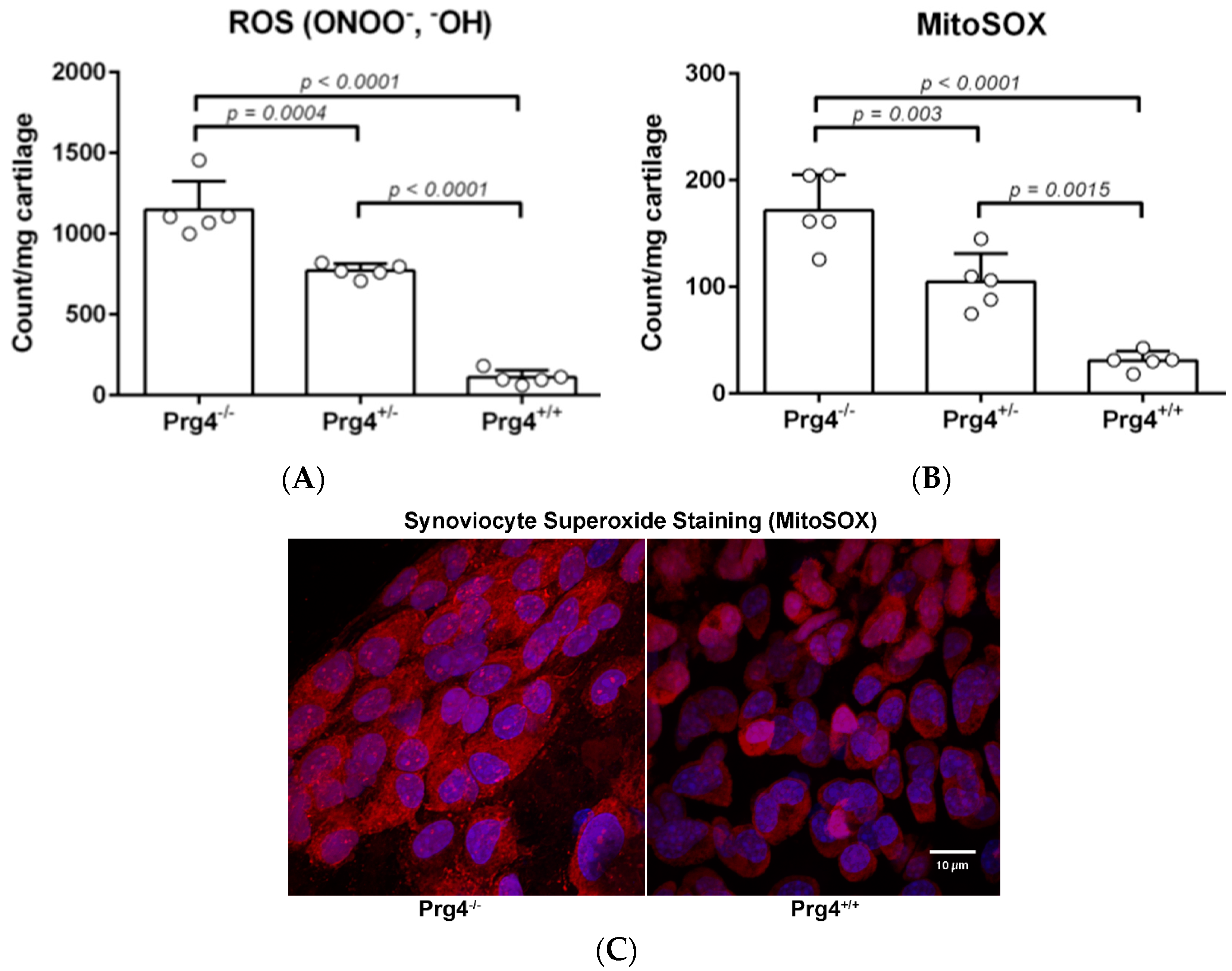
2.3. Peroxynitrite (PN) Assay in Prg4 Mutant Mice Femoral Head Cartilage
2.4. Superoxide (MitoSOX) Assay in Prg4 Mutant Mice Synoviocytes
2.5. Caspase-8 Activity in Chondrocytes and Synoviocytes from Prg4 Mutant Mice
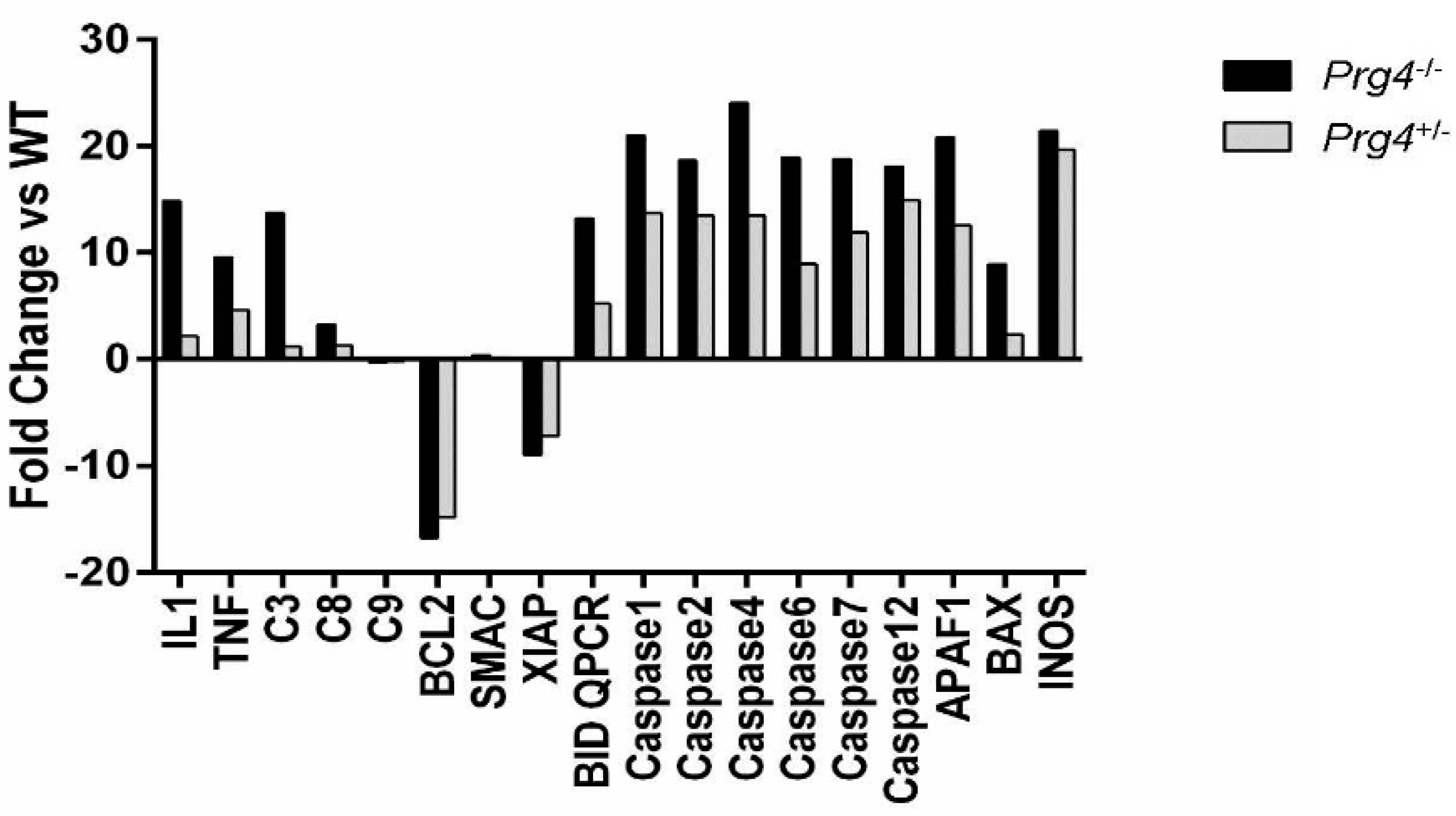
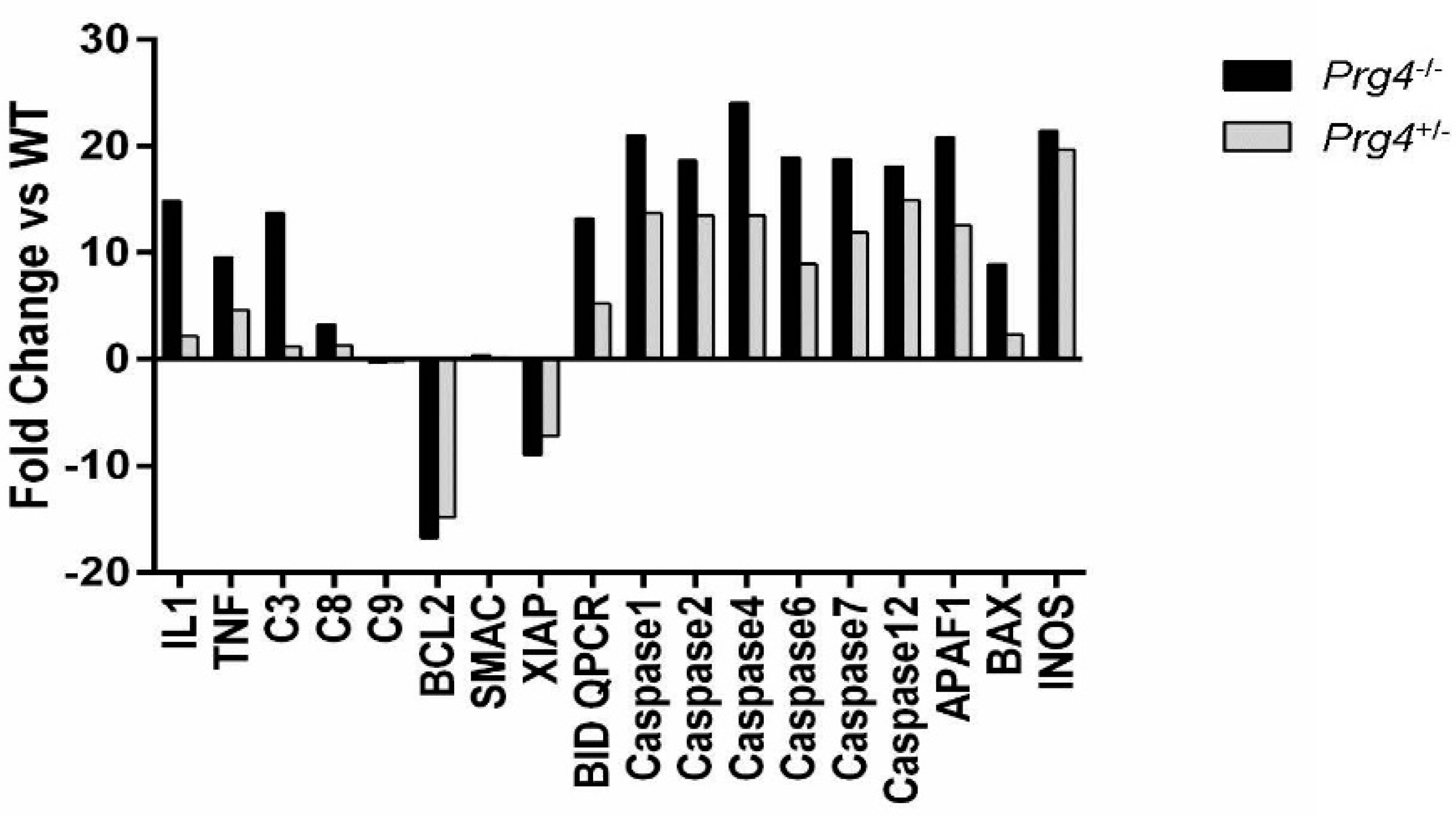
2.6. Real Time PCR of mRNA Recovered from Cartilage of Prg4 Mutant Mouse Joints
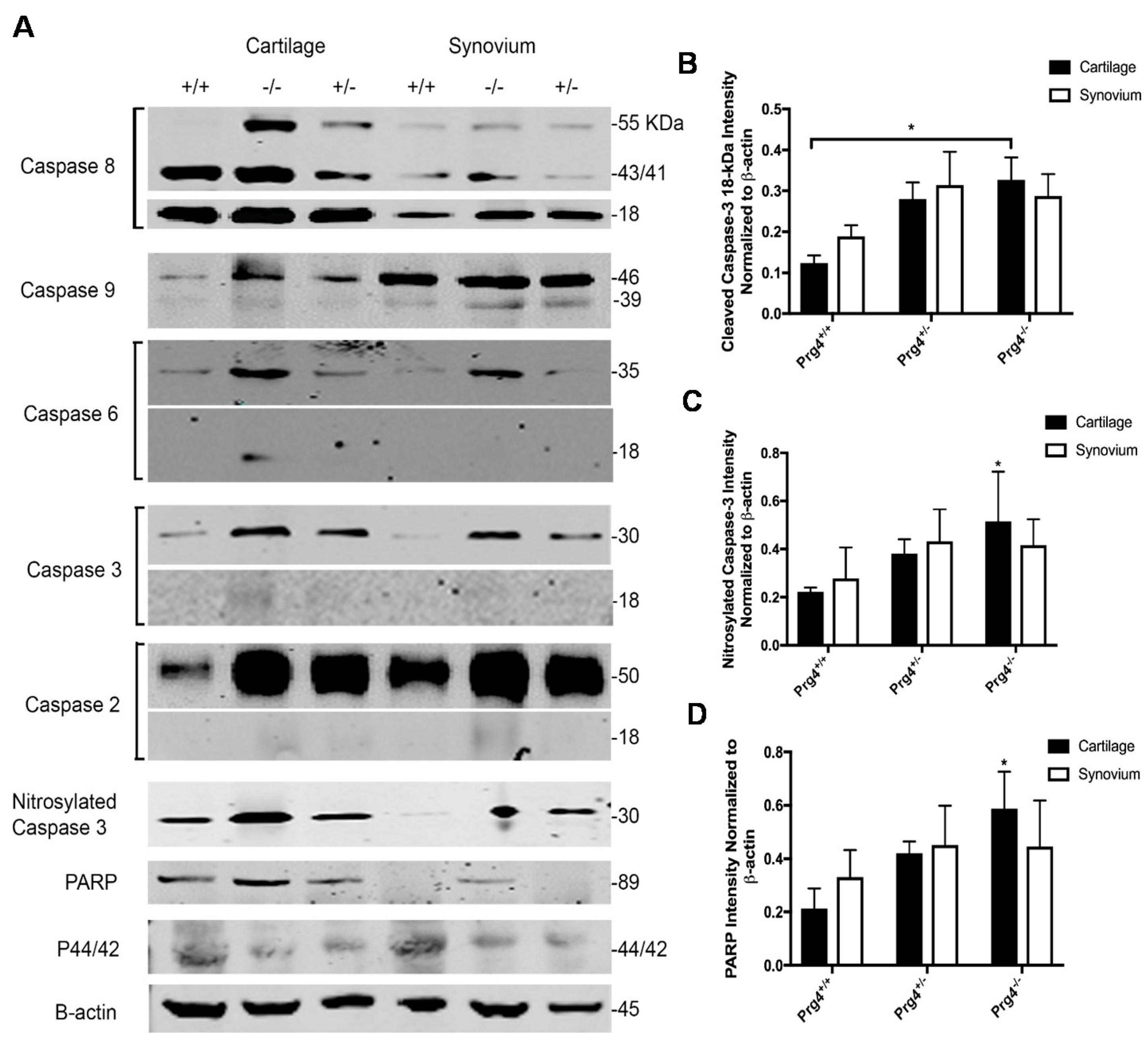
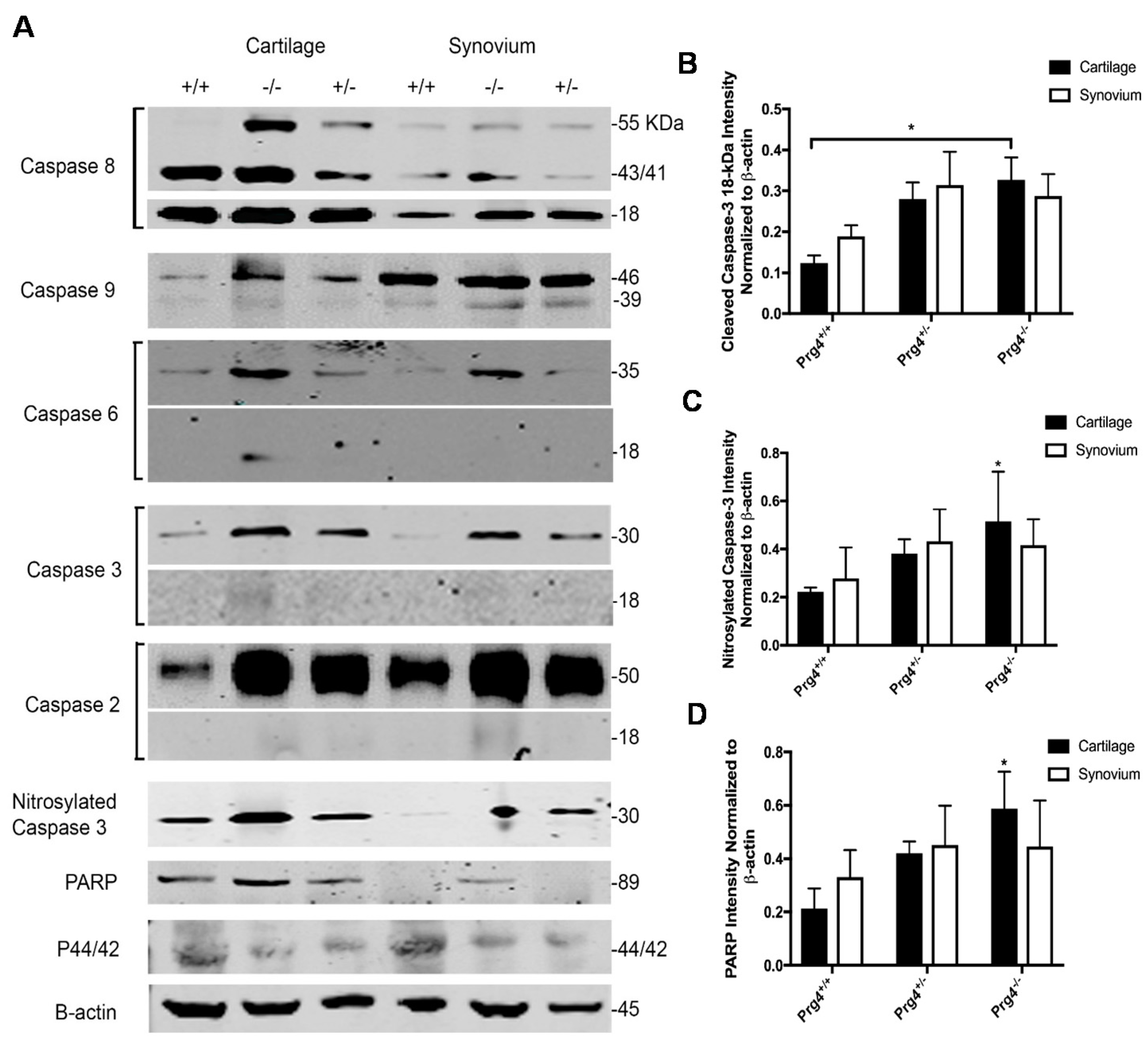
2.7. Western Blots and Densitometry
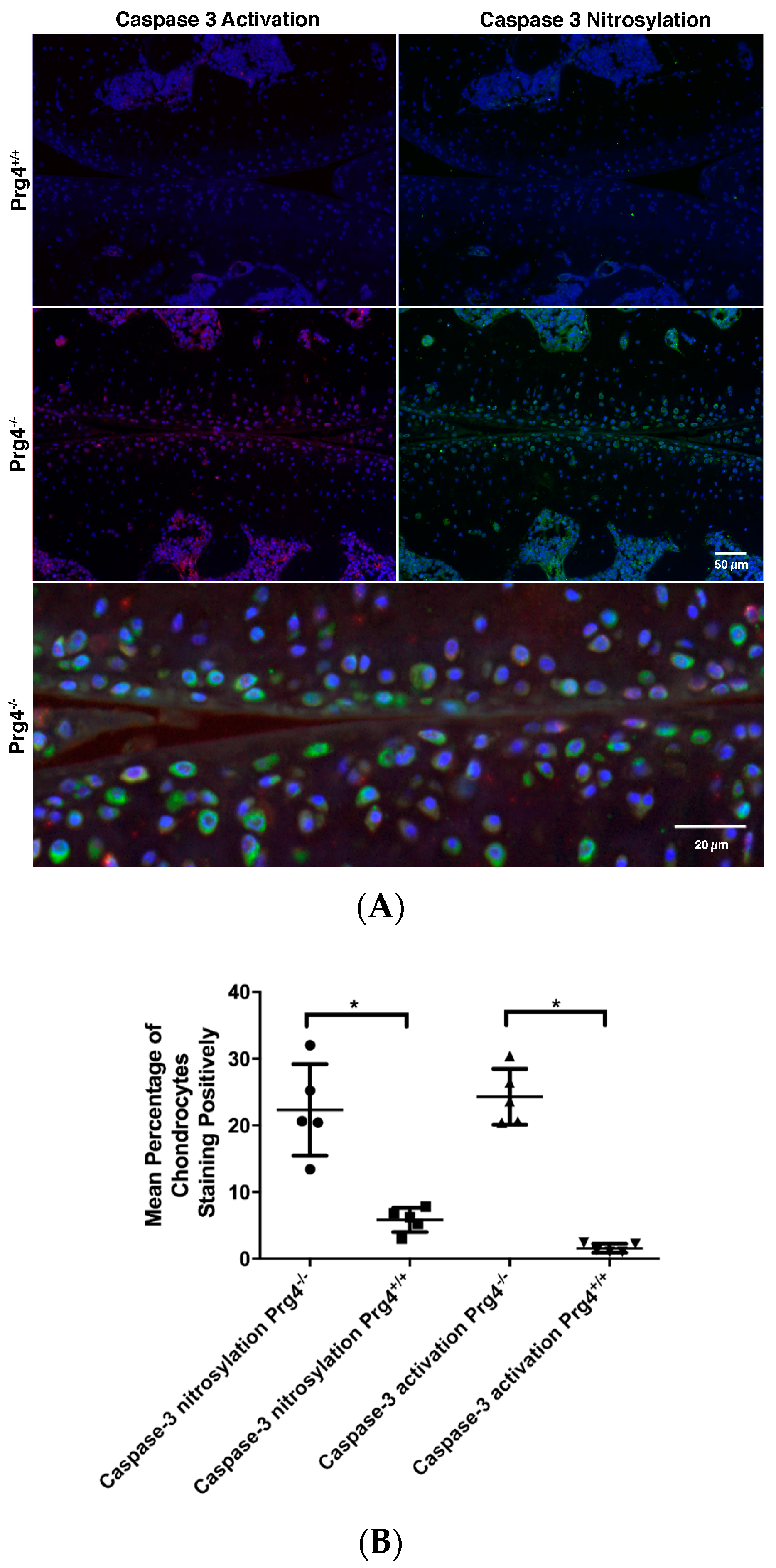
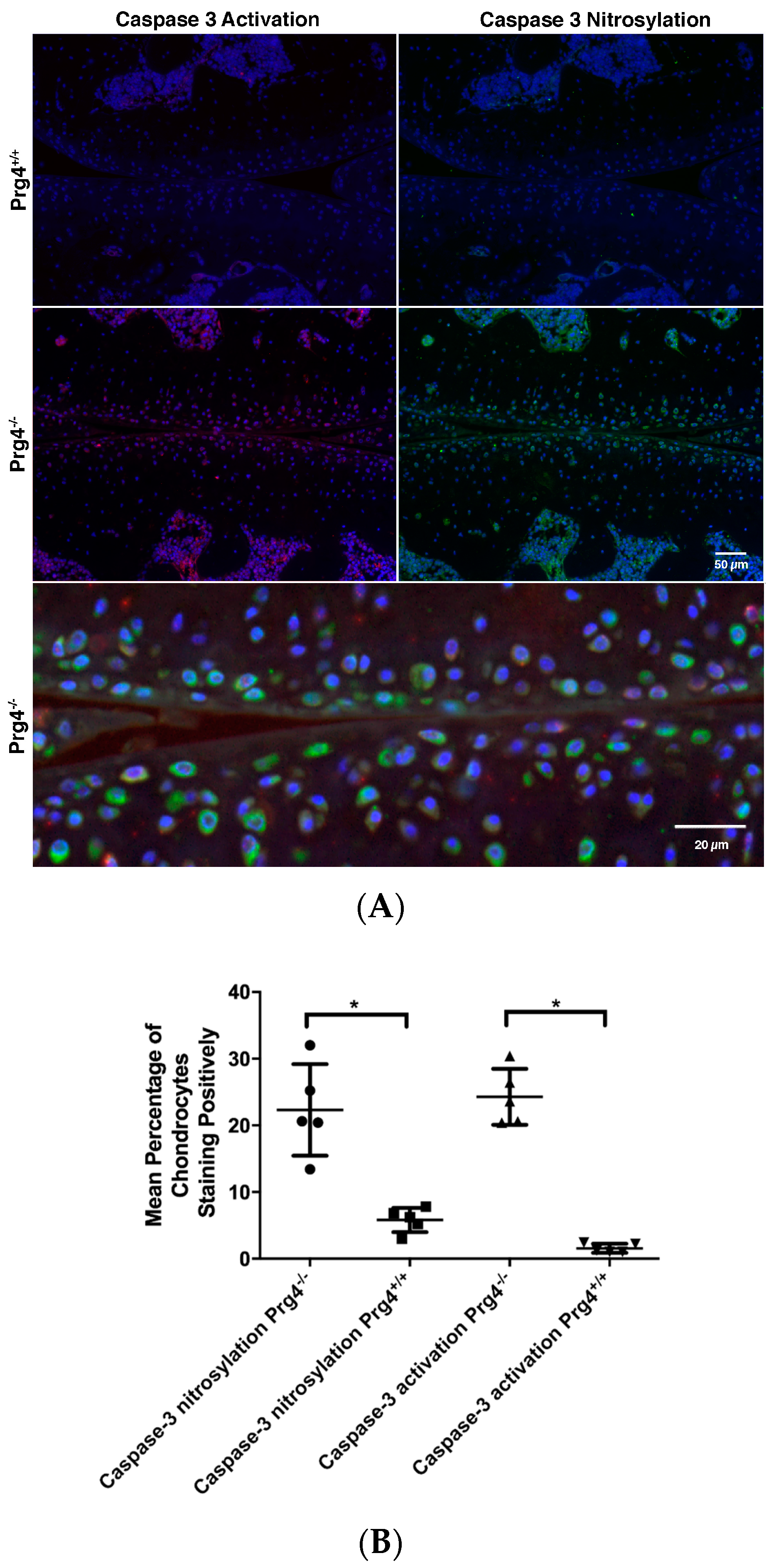
2.8. Immunohistochemistry of Nitrosyl-Cys163 Caspase-3 Neoepitope in Cartilage from Prg4−/− Mouse Joints
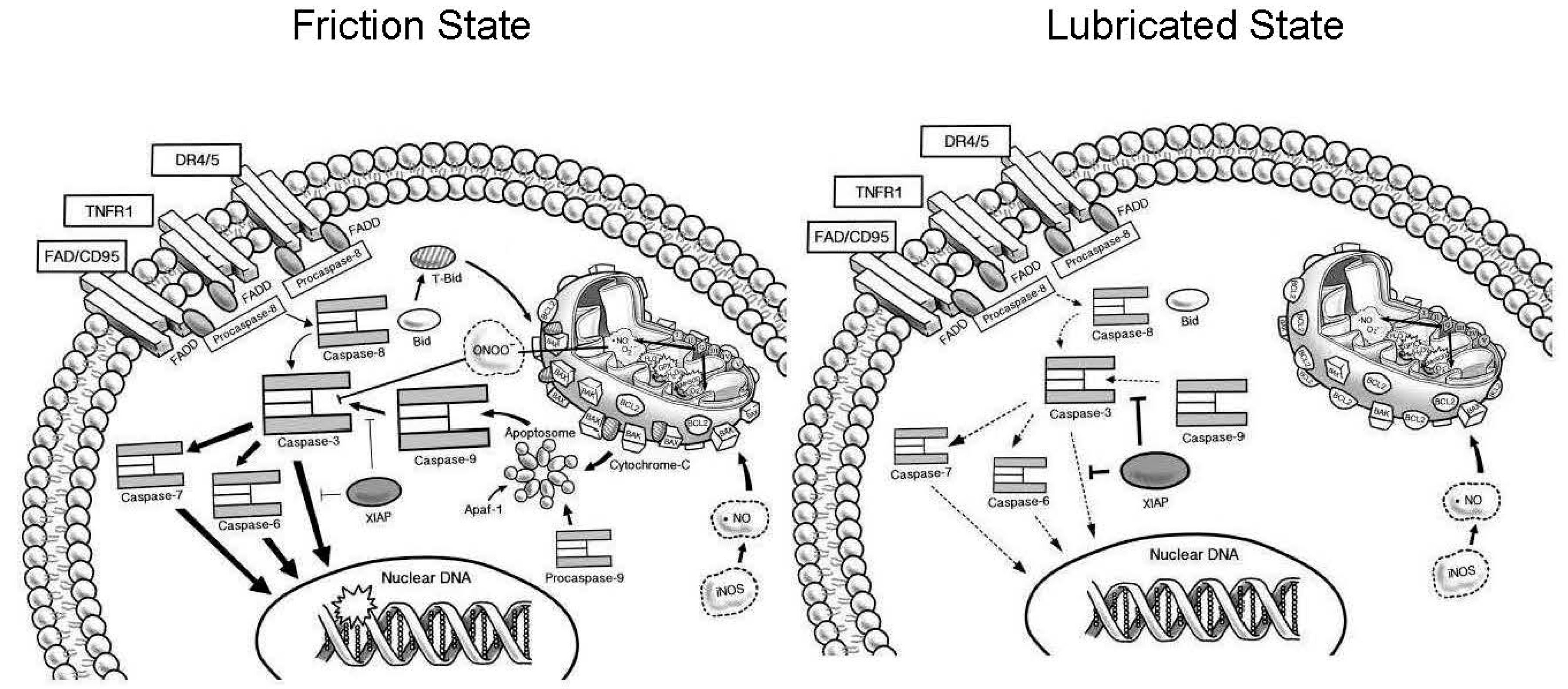
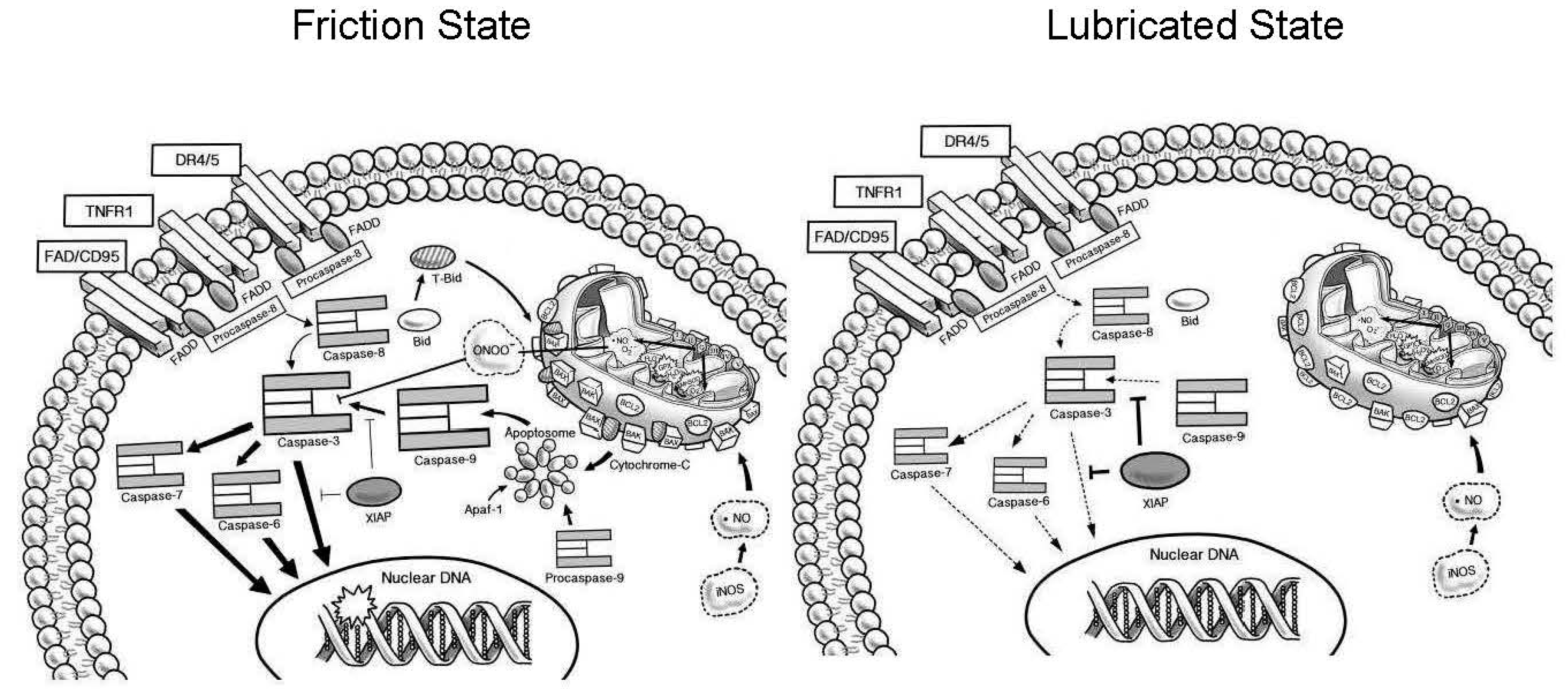
3. Discussion
4. Materials and Methods
4.1. Prg4 Mutant Mice
4.2. Tribosupplementation In Vivo of Prg4−/− Mouse Knees
4.3. Whole Joint Coefficient of Friction (COF) Measurement
4.4. Tissue Harvest for Cells, Protein and mRNA
4.5. Quantitative Real-Time Polymerase Chain Reaction
4.6. Measurement of ROS and Peroxynitrite
4.7. Measurement of MitoSOX
4.8. Imaging of Synoviocytes Stained with MitoSOX
4.9. Caspase-8 Activity Quantification
4.10. Tissue Processing and Hematoxylin and Eosin Staining
4.11. Immunodetection of Caspase Proteases and Signal Transduction Pathways
4.12. Blot Imaging and Densitometric Analysis
4.13. Immunohistochemistry for Activated Caspase-3 and Nitrosylated Cys163 Caspase-3
4.14. Quantification of Active Caspase-3 and Nitrosylated Cys163 Caspase-3
4.15. Statistical Analyses
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CACP | Camptodactyl-arthropathy-coxa vara-pericarditis |
| ROS | Reactive oxygen species |
| COF | Coefficient of friction |
| OA | Osteoarthritis |
| IA | Intra-articular |
| HSL | Human synoviocyte lubricin (PRG4) |
| PN | Peroxynitrite |
| O−2 | Superoxide |
| NO | Nitric oxide |
| mRNA | Messenger RNA |
| ONOO− | Peroxynitrite |
| −OH | Hydroxyl radical |
| DAPI | 4,6-Diamidino-2-phenylindole, dihydrochloride |
| EDTA | Ethylenediaminetetraacetic acid |
| EGTA | Ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid |
| PMSF | Phenylmethylsulfonyl fluoride |
| H&E | Hematoxylin and eosin |
| Prg4 | Murine lubricin gene |
| PRG4 | Purified lubricin protein (Proteoglycan 4) |
References
- Hill, A.; Waller, K.A.; Cui, Y.; Allen, J.M.; Smits, P.; Zhang, L.X.; Ayturk, U.M.; Hann, S.; Lessard, S.G.; Zurakowski, D.; et al. Lubricin restoration in a mouse model of congenital deficiency. Arthritis Rheumatol. 2015, 67, 3070–3081. [Google Scholar] [CrossRef] [PubMed]
- Marcelino, J.; Carpten, J.D.; Suwairi, W.M.; Gutierrez, O.M.; Schwartz, S.; Robbins, C.; Sood, R.; Makalowska, I.; Baxevanis, A.; Johnstone, B.; et al. CACP, encoding a secreted proteoglycan, is mutated in camptodactyly-arthropathy-coxa vara-pericarditis syndrome. Nat. Genet. 1999, 23, 319–322. [Google Scholar] [PubMed]
- Bahabri, S.A.; Suwairi, W.M.; Laxer, R.M.; Polinkovsky, A.; Dalaan, A.A.; Warman, M.L. The camptodactyly-arthropathy-coxa vara-pericarditis syndrome: Clinical features and genetic mapping to human chromosome 1. Arthritis Rheumatol. 1998, 41, 730–735. [Google Scholar] [CrossRef]
- Rhee, D.K.; Marcelino, J.; Baker, M.; Gong, Y.; Smits, P.; Lefebvre, V.; Jay, G.D.; Stewart, M.; Wang, H.; Warman, M.L.; et al. The secreted glycoprotein lubricin protects cartilage surfaces and inhibits synovial cell overgrowth. J. Clin. Investig. 2005, 115, 622–631. [Google Scholar] [CrossRef] [PubMed]
- Jay, G.D.; Torres, J.R.; Rhee, D.K.; Helminen, H.J.; Hytinnen, M.M.; Cha, C.J.; Elsaid, K.; Kim, K.S.; Cui, Y.; Warman, M.L. Association between friction and wear in diarthrodial joints lacking lubricin. Arthritis Rheumatol. 2007, 56, 3662–3669. [Google Scholar] [CrossRef] [PubMed]
- Jay, G.D.; Elsaid, K.A.; Kelly, K.A.; Anderson, S.C.; Zhang, L.; Teeple, E.; Waller, K.; Fleming, B.C. Prevention of cartilage degeneration and gait asymmetry by lubricin tribosupplementation in the rat following anterior cruciate ligament transection. Arthritis Rheumatol. 2012, 64, 1162–1171. [Google Scholar] [CrossRef] [PubMed]
- Waller, K.A.; Chin, K.E.; Jay, G.D.; Zhang, L.X.; Teeple, E.; McAllister, S.; Badger, G.J.; Schmidt, T.A.; Fleming, B.C. Intra-articular recombinant human proteoglycan 4 mitigates cartilage damage after destabilization of the medial meniscus in the Yucatan Minipig. Am. J. Sports Med. 2017, 45, 1512–1521. [Google Scholar] [CrossRef] [PubMed]
- Karamchedu, N.P.; Tofte, J.N.; Waller, K.A.; Zhang, L.X.; Patel, T.K.; Jay, G.D. Superficial zone cellularity is deficient in mice lacking lubricin: A stereoscopic analysis. Arthritis Res. Ther. 2015, 18, 64. [Google Scholar] [CrossRef] [PubMed]
- Elsaid, K.A.; Machan, J.T.; Waller, K.; Fleming, B.C.; Jay, G.D. The impact of anterior cruciate ligament injury on lubricin metabolism and the effect of inhibiting tumor necrosis factor α on chondroprotection in an animal model. Arthritis Rheumatol. 2009, 60, 2997–3006. [Google Scholar] [CrossRef] [PubMed]
- Jay, G.D.; Fleming, B.C.; Watkins, B.A.; McHugh, K.A.; Anderson, S.C.; Zhang, L.X.; Teeple, E.; Waller, K.A.; Elsaid, K.A. Prevention of cartilage degeneration and restoration of chondroprotection by lubricin tribosupplementation in the rat following anterior cruciate ligament transection. Arthritis Rheumatol. 2010, 62, 2382–2391. [Google Scholar] [CrossRef] [PubMed]
- Teeple, E.; Elsaid, K.A.; Jay, G.D.; Zhang, L.; Badger, G.J.; Akelman, M.; Bliss, T.F.; Fleming, B.C. Effects of supplemental intra-articular lubricin and hyaluronic acid on the progression of posttraumatic arthritis in the anterior cruciate ligament-deficient rat knee. Am. J. Sports Med. 2011, 39, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Flannery, C.R.; Zollner, R.; Corcoran, C.; Jones, A.R.; Root, A.; Rivera-Bermudez, M.A.; Blanchet, T.; Gleghorn, J.P.; Bonassar, L.J.; Bendele, A.M.; et al. Prevention of cartilage degeneration in a rat model of osteoarthritis by intraarticular treatment with recombinant lubricin. Arthritis Rheumatol. 2009, 60, 840–847. [Google Scholar] [CrossRef] [PubMed]
- Hughes, L.C.; Archer, C.W.; Ap Gwynn, I. The ultrastructure of mouse articular cartilage: Collagen orientation and implications for tissue functionality. A polarised light and scanning electron microscope study and review. Eur. Cell Mater. 2005, 9, 68–84. [Google Scholar] [CrossRef] [PubMed]
- Waller, K.A.; Zhang, L.X.; Elsaid, K.A.; Fleming, B.C.; Warman, M.L.; Jay, G.D. Role of lubricin and boundary lubrication in the prevention of chondrocyte apoptosis. Proc. Natl. Acad. Sci. USA 2013, 110, 5852–5857. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.C.; Burris, D.L. An analytical model to predict interstitial lubrication of cartilage in migrating contact areas. J. Biomech. 2014, 47, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Caligaris, M.; Ateshian, G.A. Effects of sustained interstitial fluid pressurization under migrating contact area, and boundary lubrication by synovial fluid, on cartilage friction. Osteoarthr. Cartil. 2008, 16, 1220–1227. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, J.B.; Jin, M.; Chai, D.H.; Siparsky, P.; Fanning, P.; Grodzinsky, A.J. Shear- and compression-induced chondrocyte transcription requires MAPK activation in cartilage explants. J. Biol. Chem. 2008, 283, 6735–6743. [Google Scholar] [CrossRef] [PubMed]
- Brouillette, M.J.; Ramakrishnan, P.S.; Wagner, V.M.; Sauter, E.E.; Journot, B.J.; McKinley, T.O.; Martin, J.A. Strain-dependent oxidant release in articular cartilage originates from mitochondria. Biomech. Model. Mechanobiol. 2014, 13, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Blanco, F.J.; Rego, I.; Ruiz-Romero, C. The role of mitochondria in osteoarthritis. Nat. Rev. Rheumatol. 2011, 7, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Wong, B.L.; Bae, W.C.; Chun, J.; Gratz, K.R.; Lotz, M.; Sah, R.L. Biomechanics of cartilage articulation: Effects of lubrication and degeneration on shear deformation. Arthritis Rheumatol. 2008, 58, 2065–2074. [Google Scholar] [CrossRef] [PubMed]
- Al-Sharif, A.; Jamal, M.; Zhang, L.X.; Larson, K.; Schmidt, T.A.; Jay, G.D.; Elsaid, K.A. Lubricin/Proteoglycan 4 Binding to CD44 Receptor: A mechanism of the suppression of proinflammatory cytokine-induced synoviocyte proliferation by Lubricin. Arthritis Rheumatol. 2015, 67, 1503–1513. [Google Scholar] [CrossRef] [PubMed]
- Nugent, G.E.; Schmidt, T.A.; Schumacher, B.L.; Voegtline, M.S.; Bae, W.C.; Jadin, K.D.; Sah, R.L. Static and dynamic compression regulate cartilage metabolism of PRoteoGlycan 4 (PRG4). Biorheology 2006, 43, 191–200. [Google Scholar] [PubMed]
- Grad, S.; Lee, C.R.; Wimmer, M.A.; Alini, M. Chondrocyte gene expression under applied surface motion. Biorheology 2006, 43, 259–269. [Google Scholar] [PubMed]
- Larson, K.M.; Zhang, L.; Elsaid, K.A.; Schmidt, T.A.; Fleming, B.C.; Badger, G.J.; Jay, G.D. Reduction of friction by recombinant human proteoglycan 4 in IL-1α stimulated bovine cartilage explants. J. Orthop. Res. 2017, 35, 580–589. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, H.; Kozhemyakina, E.; Hung, H.H.; Grodzinsky, A.J.; Lassar, A.B. Mechanical motion promotes expression of Prg4 in articular cartilage via multiple CREB-dependent, fluid flow shear stress-induced signaling pathways. Genes Dev. 2014, 28, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Carter, D.R.; Wong, M. Modelling cartilage mechanobiology. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2003, 358, 1461–1471. [Google Scholar] [CrossRef] [PubMed]
- Vanwanseele, B.; Eckstein, F.; Knecht, H.; Stussi, E.; Spaepen, A. Knee cartilage of spinal cord-injured patients displays progressive thinning in the absence of normal joint loading and movement. Arthritis Rheumatol. 2002, 46, 2073–2078. [Google Scholar] [CrossRef] [PubMed]
- D’Lima, D.D.; Hashimoto, S.; Chen, P.C.; Colwell, C.W., Jr.; Lotz, M.K. Human chondrocyte apoptosis in response to mechanical injury. Osteoarthr. Cartil. 2001, 9, 712–719. [Google Scholar] [CrossRef] [PubMed]
- D’Lima, D.D.; Hashimoto, S.; Chen, P.C.; Lotz, M.K.; Colwell, C.W., Jr. Cartilage injury induces chondrocyte apoptosis. J. Bone Jt. Surg. Am. 2001, 83, 19–21. [Google Scholar] [CrossRef] [PubMed]
- Elsaid, K.A.; Zhang, L.; Waller, K.; Tofte, J.; Teeple, E.; Fleming, B.C.; Jay, G.D. The impact of forced joint exercise on lubricin biosynthesis from articular cartilage following ACL transection and intra-articular lubricin’s effect in exercised joints following ACL transection. Osteoarthr. Cartil. 2012, 20, 940–948. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.S.; Kim, H.A. Chondrocyte apoptosis in the pathogenesis of osteoarthritis. Int. J. Mol. Sci. 2015, 16, 26035–26054. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Johnson, T.S.; Thomas, G.L.; Watson, P.F.; Wagner, B.; Furness, P.N.; El Nahas, A.M. A shift in the Bax/Bcl-2 balance may activate caspase-3 and modulate apoptosis in experimental glomerulonephritis. Kidney Int. 2002, 62, 1301–1313. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S. Shifting the balance of mitochondrial apoptosis: Therapeutic perspectives. Front. Oncol. 2012, 2, 121. [Google Scholar] [CrossRef] [PubMed]
- Mebratu, Y.; Tesfaigzi, Y. How ERK1/2 activation controls cell proliferation and cell death: Is subcellular localization the answer? Cell Cycle 2009, 8, 1168–1175. [Google Scholar] [CrossRef] [PubMed]
- Stanton, L.A.; Underhill, T.M.; Beier, F. MAP kinases in chondrocyte differentiation. Dev. Biol. 2003, 263, 165–175. [Google Scholar] [CrossRef]
- Sinha, K.; Das, J.; Pal, P.B.; Sil, P.C. Oxidative stress: The mitochondria-dependent and mitochondria-independent pathways of apoptosis. Arch. Toxicol. 2013, 87, 1157–1180. [Google Scholar] [CrossRef] [PubMed]
- Hardin, J.A.; Cobelli, N.; Santambrogio, L. Consequences of metabolic and oxidative modifications of cartilage tissue. Nat. Rev. Rheumatol. 2015, 11, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Buckwalter, J.A.; Anderson, D.D.; Brown, T.D.; Tochigi, Y.; Martin, J.A. The roles of mechanical stresses in the pathogenesis of osteoarthritis: Implications for treatment of joint injuries. Cartilage 2013, 4, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Yudoh, K.; van Nguyen, T.; Nakamura, H.; Hongo-Masuko, K.; Kato, T.; Nishioka, K. Potential involvement of oxidative stress in cartilage senescence and development of osteoarthritis: Oxidative stress induces chondrocyte telomere instability and downregulation of chondrocyte function. Arthritis Res. Ther. 2005, 7, R380–R391. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Pinto, J.T.; Deng, H.; Richie, J.P., Jr. Inhibition of caspase-3 activity and activation by protein glutathionylation. Biochem. Pharmacol. 2008, 75, 2234–2244. [Google Scholar] [CrossRef] [PubMed]
- Mabley, J.G.; Liaudet, L.; Pacher, P.; Southan, G.J.; Groves, J.T.; Salzman, A.L.; Szabo, C. Part II: Beneficial effects of the peroxynitrite decomposition catalyst FP15 in murine models of arthritis and colitis. Mol. Med. 2002, 8, 581–590. [Google Scholar] [PubMed]
- Ndengele, M.M.; Cuzzocrea, S.; Esposito, E.; Mazzon, E.; Di Paola, R.; Matuschak, G.M.; Salvemini, D. Cyclooxygenases 1 and 2 contribute to peroxynitrite-mediated inflammatory pain hypersensitivity. FASEB J. 2008, 22, 3154–3164. [Google Scholar] [CrossRef] [PubMed]
- Drewniak, E.I.; Jay, G.D.; Fleming, B.C.; Zhang, L.; Warman, M.L.; Crisco, J.J. Cyclic loading increases friction and changes cartilage surface integrity in lubricin-mutant mouse knees. Arthritis Rheumatol. 2012, 64, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Q.; Porreca, F.; Cuzzocrea, S.; Galen, K.; Lightfoot, R.; Masini, E.; Muscoli, C.; Mollace, V.; Ndengele, M.; Ischiropoulos, H.; Salvemini, D. A newly identified role for superoxide in inflammatory pain. J. Pharmacol. Exp. Ther. 2004, 309, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Kidd, B.L.; McDougall, J.J.; Inglis, J.J. Inflammatory mediators and nociception in osteoarthritis. In Pain in Osteoarthritis; David, H.-G.S., Felson, T., Eds.; Wiley-Blackwell: Hoboken, NJ, USA, 2009; pp. 55–72. [Google Scholar]
- Shalini, S.; Kumar, S. Caspase-2 and the oxidative stress response. Mol. Cell. Oncol. 2015, 2, e1004956. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Mani, S.B.; He, Y.; Hall, A.M.; Xu, L.; Li, Y.; Zurakowski, D.; Jay, G.D.; Warman, M.L. Induced superficial chondrocyte death reduces catabolic cartilage damage in murine posttraumatic osteoarthritis. J. Clin. Investig. 2016, 126, 2893–2902. [Google Scholar] [CrossRef] [PubMed]
- Elsaid, K.A.; Fleming, B.C.; Oksendahl, H.L.; Machan, J.T.; Fadale, P.D.; Hulstyn, M.J.; Shalvoy, R.M.; Jay, G.D. Decreased lubricin concentrations and markers of joint inflammation in synovial fluids from patients with anterior cruciate ligament injury. Arthritis Rheumatol. 2008, 58, 1707–1715. [Google Scholar] [CrossRef] [PubMed]
- Drewniak, E.I.; Jay, G.D.; Fleming, B.C.; Crisco, J.J. Comparison of two methods for calculating the frictional properties of articular cartilage using a simple pendulum and intact mouse knee joints. J. Biomech. 2009, 42, 1996–1999. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, R.; Kopacz, M.; Ateshian, G.A. Experimental verification of the role of interstitial fluid pressurization in cartilage lubrication. J. Orthop. Res. 2004, 22, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.J.; Lu, S.H.; Xu, B.; Yang, R.C.; Ren, Q.; Liu, B.; Li, B.; Lu, M.; Yan, F.Y.; Han, Z.B.; et al. Hemangiopoietin, a novel human growth factor for the primitive cells of both hematopoietic and endothelial cell lineages. Blood 2004, 103, 4449–4456. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.C.; Berkelman, T.; Yadav, G.; Hammond, M. A defined methodology for reliable quantification of Western blot data. Mol. Biotechnol. 2013, 55, 217–226. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Waller, K.A.; Zhang, L.X.; Jay, G.D. Friction-Induced Mitochondrial Dysregulation Contributes to Joint Deterioration in Prg4 Knockout Mice. Int. J. Mol. Sci. 2017, 18, 1252. https://doi.org/10.3390/ijms18061252
Waller KA, Zhang LX, Jay GD. Friction-Induced Mitochondrial Dysregulation Contributes to Joint Deterioration in Prg4 Knockout Mice. International Journal of Molecular Sciences. 2017; 18(6):1252. https://doi.org/10.3390/ijms18061252
Chicago/Turabian StyleWaller, Kimberly A., Ling X. Zhang, and Gregory D. Jay. 2017. "Friction-Induced Mitochondrial Dysregulation Contributes to Joint Deterioration in Prg4 Knockout Mice" International Journal of Molecular Sciences 18, no. 6: 1252. https://doi.org/10.3390/ijms18061252
APA StyleWaller, K. A., Zhang, L. X., & Jay, G. D. (2017). Friction-Induced Mitochondrial Dysregulation Contributes to Joint Deterioration in Prg4 Knockout Mice. International Journal of Molecular Sciences, 18(6), 1252. https://doi.org/10.3390/ijms18061252