
Genetic Variation Controlling Wrinkled Seed Phenotypes in Pisum: How Lucky Was Mendel?

Abstract
:
1. Introduction
2. Results
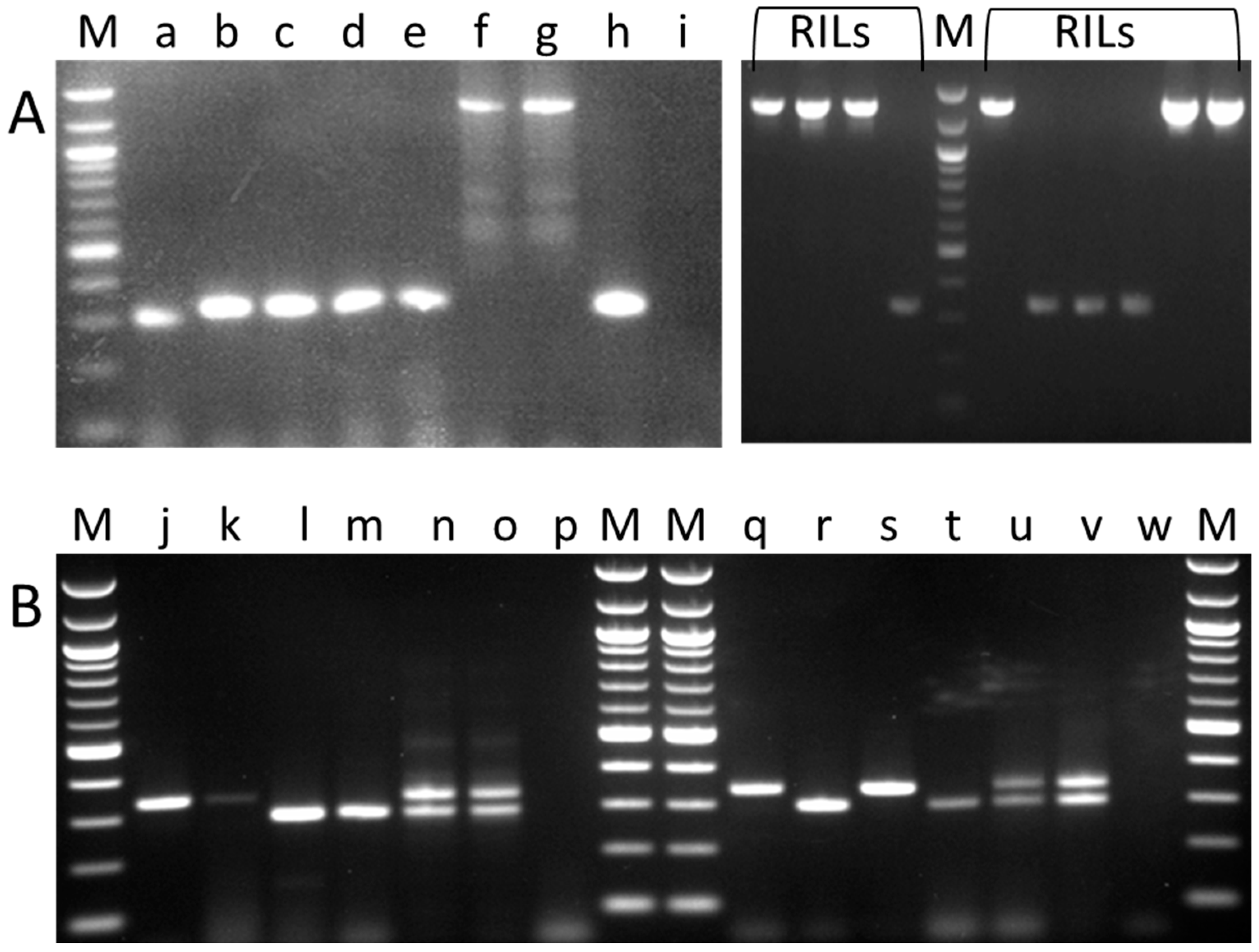
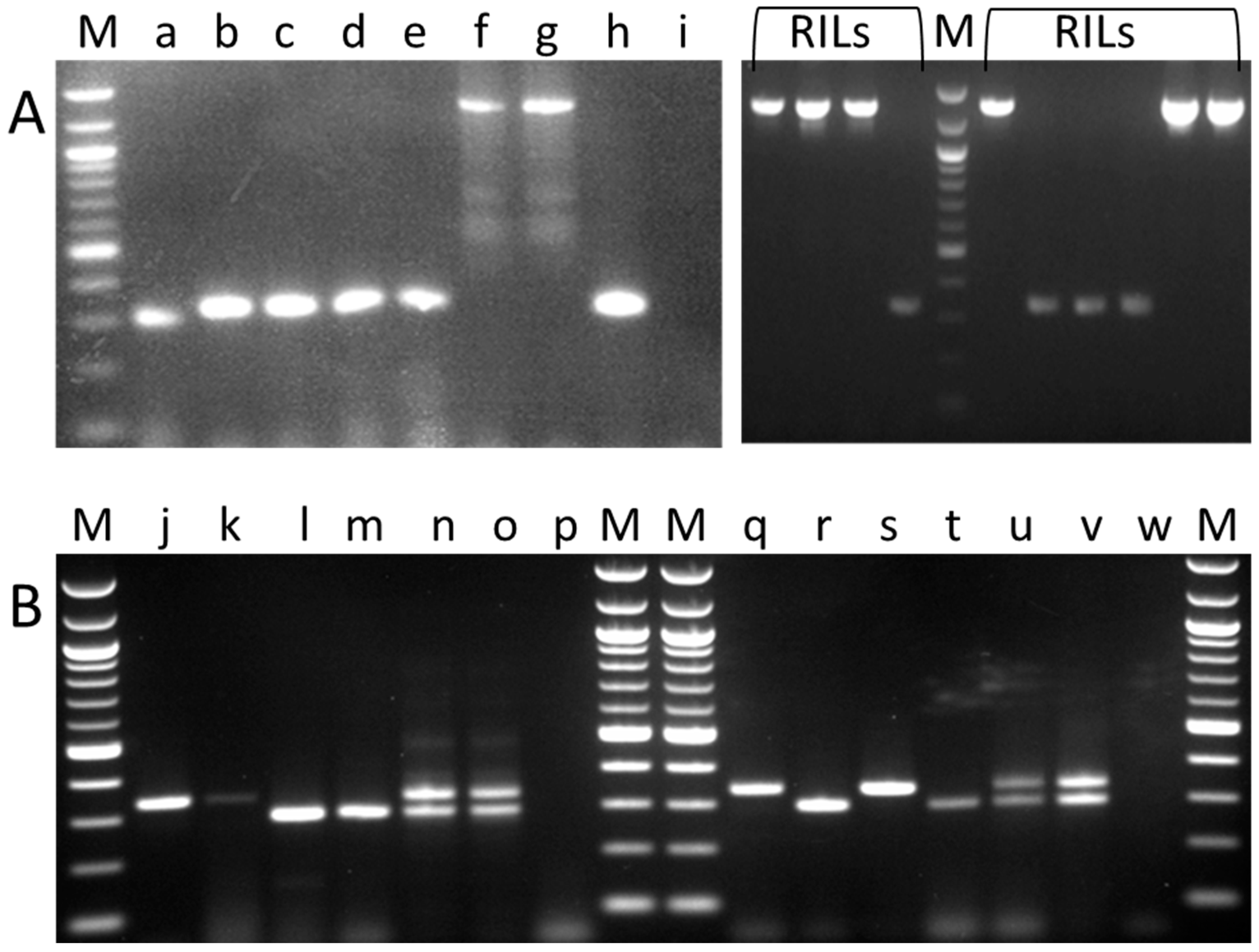
2.1. Germplasm Screen for sbeI Variants
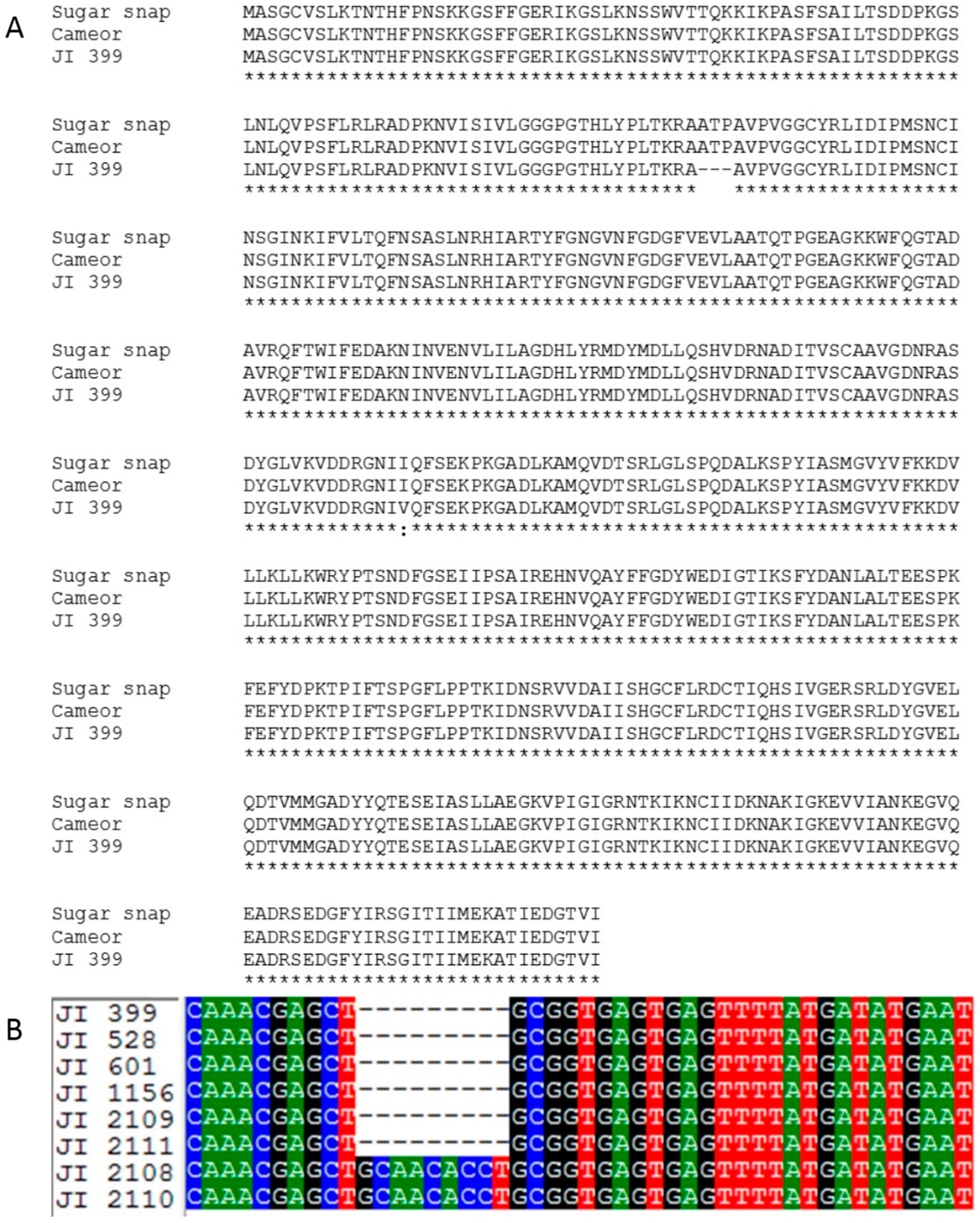
2.2. Screening for the AgpL Mutation
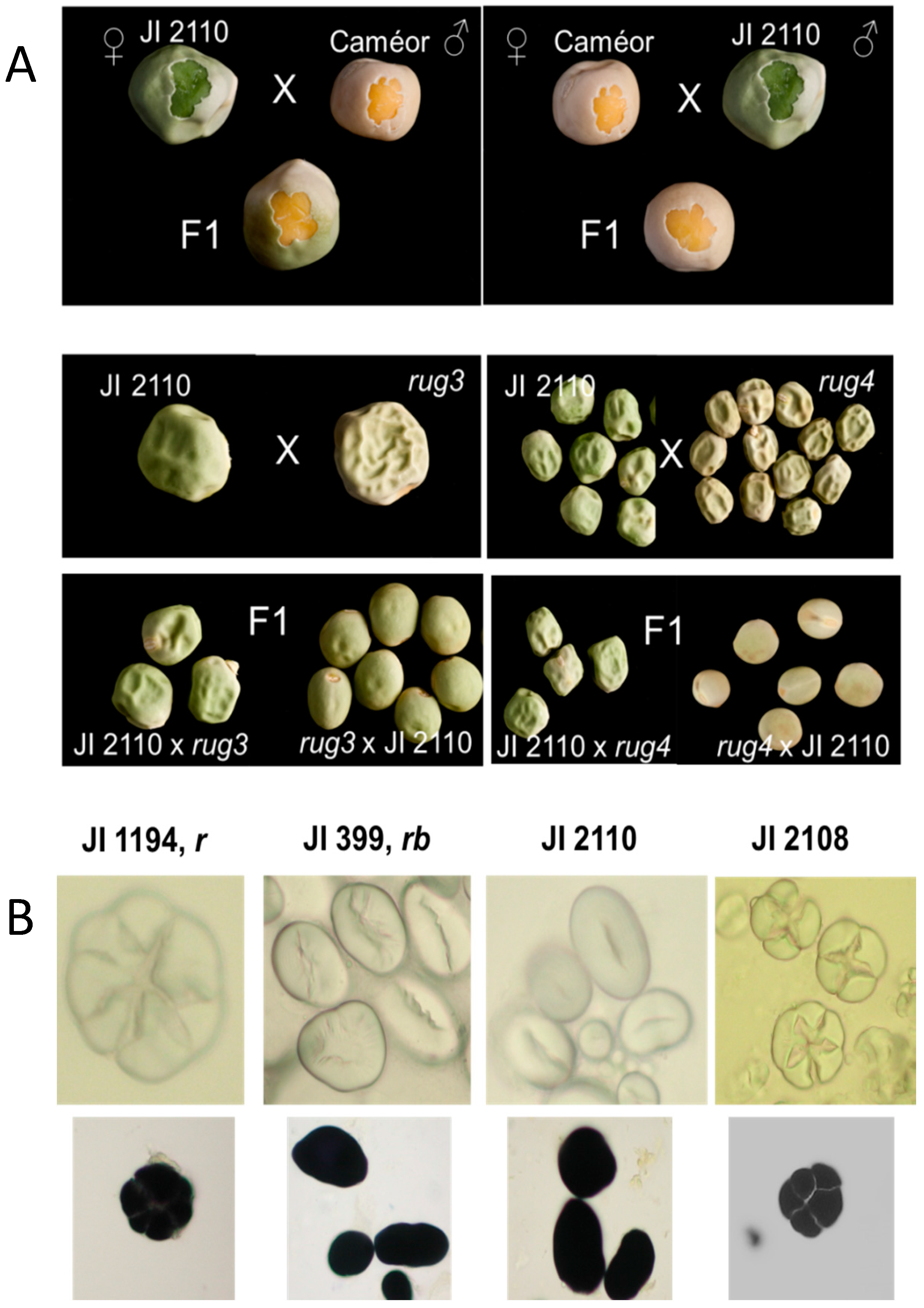
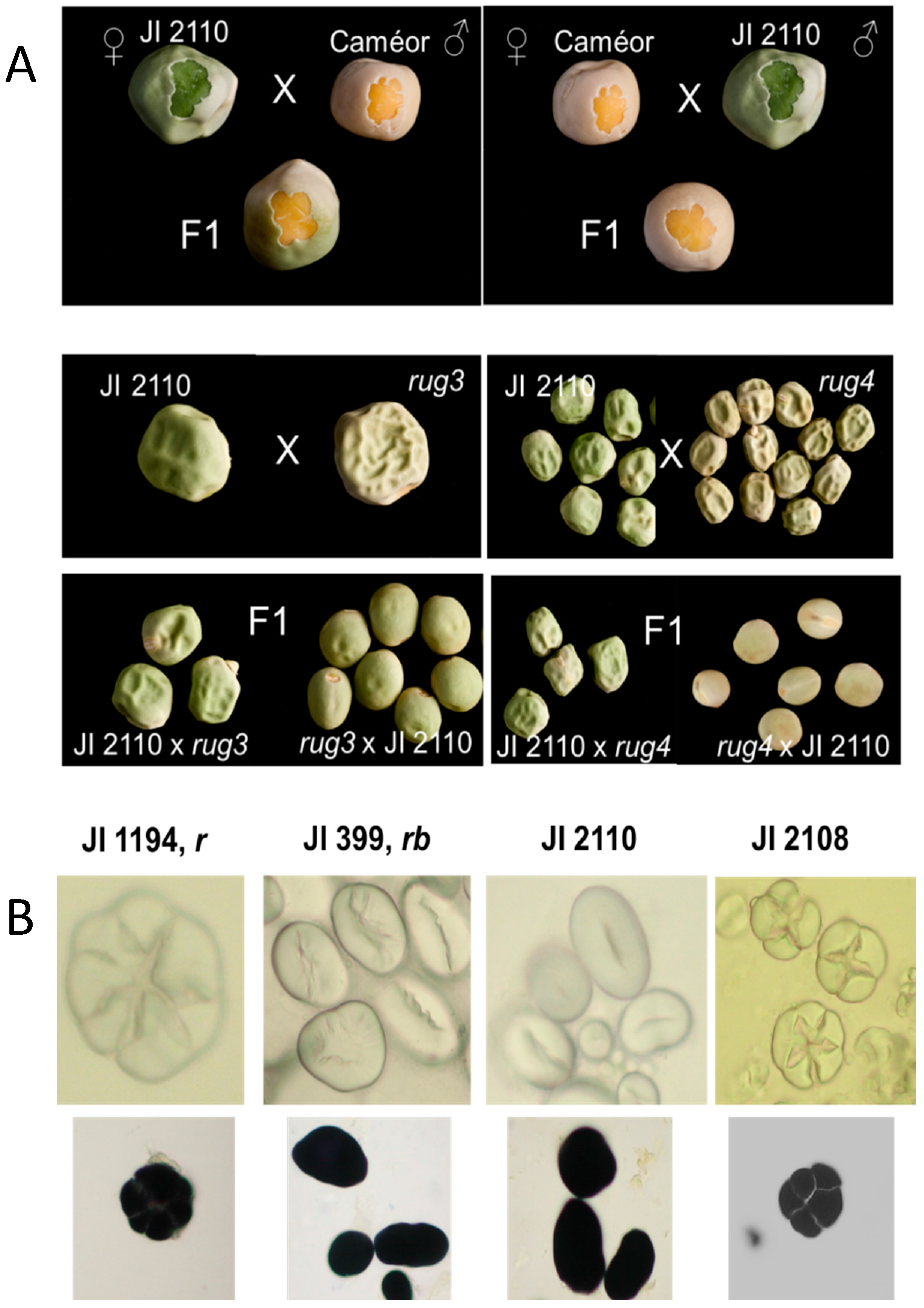
2.3. Allelism Tests of JI 2110, A Novel Wrinkled-Seeded Mutant
2.4. Genetic Mapping of the JI 2110 Seed Phenotype
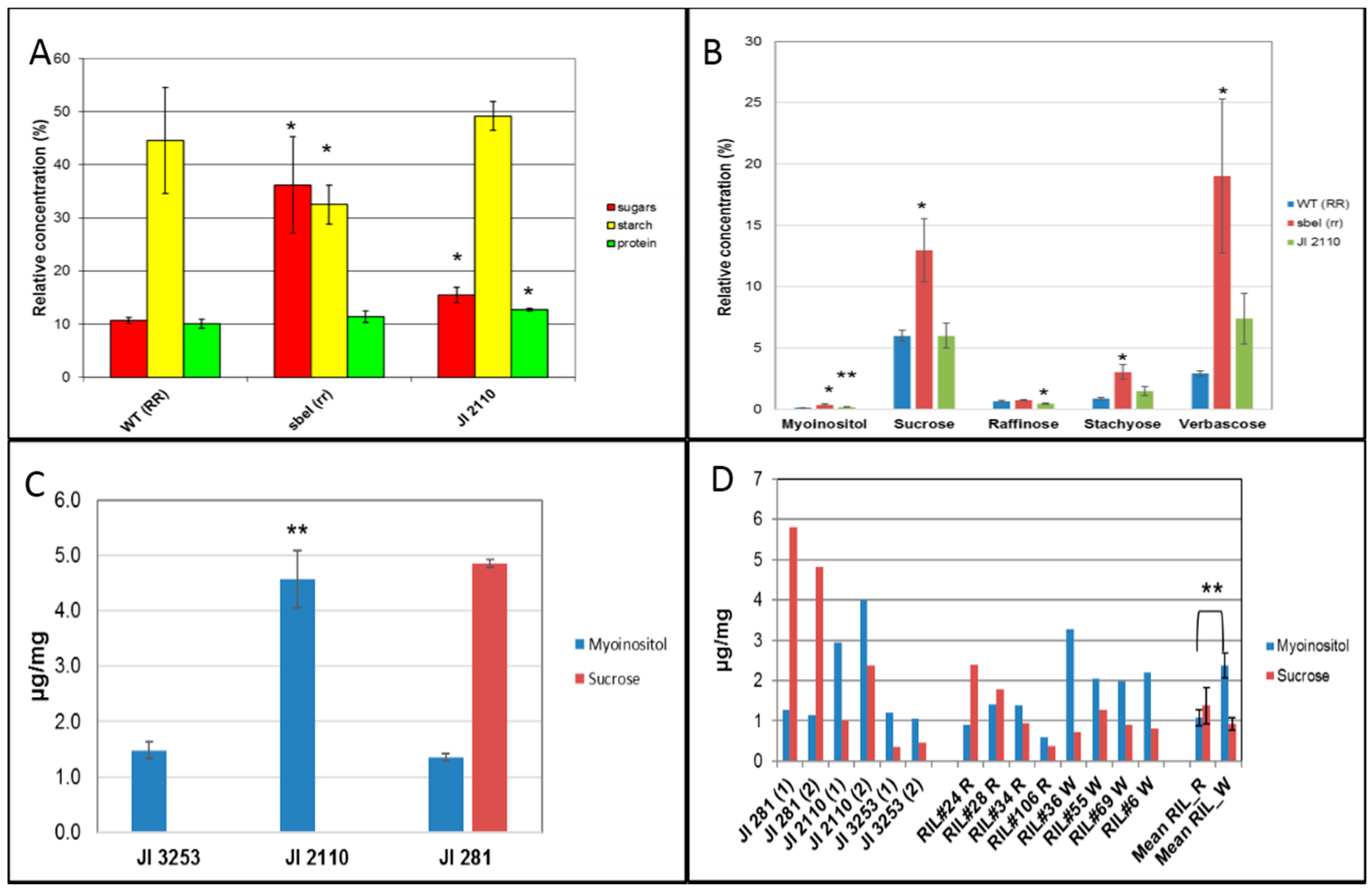
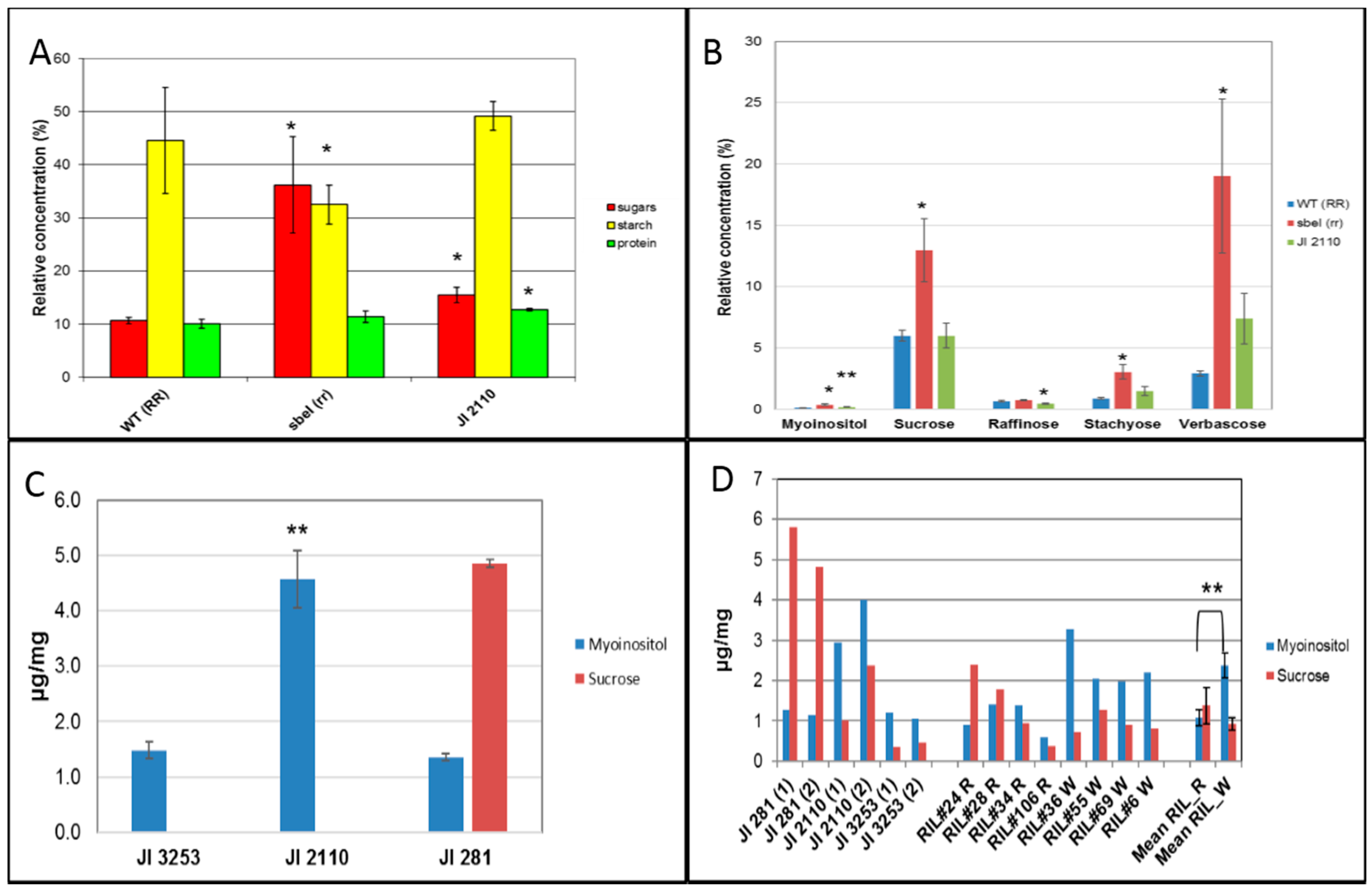
2.5. Compositional and Metabolite Analysis of Cotyledons and Testas in JI 2110
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Germplasm Genotype Screens
4.2. Allelism Tests of Seed Phenotype
4.3. Genetic Mapping of Novel Variant Seed Phenotype
4.4. Metabolite Analysis of Cotyledon and Testa Samples
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bhattacharyya, M.K.; Smith, A.M.; Ellis, T.H.N.; Hedley, C.; Martin, C. The wrinkled-seed character of pea described by Mendel is caused by a transposon-like insertion in a gene encoding starch-branching enzyme. Cell 1990, 60, 115–122. [Google Scholar] [CrossRef]
- Wang, T.L.; Bogracheva, T.Y.; Hedley, C.L. Starch: As simple as A, B, C? J. Exp. Bot. 1998, 49, 481–502. [Google Scholar] [CrossRef]
- Tieman, D.; Zhu, G.; Resende, M.F.R., Jr.; Lin, T.; Nguyen, C.; Bies, D.; Rambla, J.L.; Beltran, K.S.O.; Taylor, M.; Zhang, B.; et al. A chemical genetic roadmap to improved tomato flavour. Science 2017, 355, 391–394. [Google Scholar] [CrossRef] [PubMed]
- Mendel, G. Versuche über pflanzen-hybriden. Verhandlungen des Naturforschenden Vereines in Brünn 1866, 4, 3–47. [Google Scholar]
- Wang, T.L.; Hedley, C.L. Seed development in peas: Knowing your three “r’s” (or four, or five). Seed Sci. Res. 1991, 1, 3–14. [Google Scholar] [CrossRef]
- Casey, R.; Domoney, C.; Forster, C.; Hedley, C.; Hitchin, E.; Wang, T. The effect of modifying carbohydrate metabolism on seed protein gene expression in peas. J. Plant Physiol. 1998, 152, 636–640. [Google Scholar] [CrossRef]
- Lyall, T.W.; Ellis, R.H.; John, P.; Hedley, C.L.; Wang, T.L. Mutant alleles at the rugosus loci in pea affect seed moisture sorption isotherms and the relations between seed longevity and moisture content. J. Exp. Bot. 2003, 54, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Sestili, F.; Janni, M.; Doherty, A.; Botticella, E.; D’Ovidio, R.; Masci, S.; Jones, H.D.; Lafiandra, D. Increasing the amylose content of durum wheat through silencing of the SBEIIa genes. BMC Plant Biol. 2010, 10, 144. [Google Scholar] [CrossRef] [PubMed]
- Petropoulou, K.; Chambers, E.S.; Morrison, D.J.; Preston, T.; Godsland, I.F.; Wilde, P.; Narbad, A.; Parker, R.; Salt, L.; Morris, V.J.; et al. Identifying crop variants with high resistant starch content to maintain healthy glucose homeostasis. Nutrit. Bull. 2016, 41, 372–377. [Google Scholar] [CrossRef]
- Hylton, C.; Smith, A.M. The rb mutation of peas causes structural and regulatory changes in ADP glucose pyrophosphorylase from developing embryos. Plant Physiol. 1992, 99, 1626–1634. [Google Scholar] [CrossRef] [PubMed]
- Kooistra, E. On the differences between smooth and three types of wrinkled peas. Euphytica 1962, 11, 357–373. [Google Scholar]
- Reid, J.B.; Ross, J.J. Mendel’s genes: Toward a full molecular characterization. Genetic 2011, 189, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Jing, R.C.; Vershinin, A.; Grzebyta, J.; Shaw, P.; Smỳkal, P.; Marshall, D.; Ambrose, M.J.; Ellis, T.H.N.; Flavell, A.J. The genetic diversity and evolution of field pea (Pisum) studied by high throughput retrotransposon based insertion polymorphism (RBIP) marker analysis. BMC Evol. Biol. 2010, 10, 44. [Google Scholar] [CrossRef] [PubMed]
- Hellens, R.P.; Moreau, C.; Lin-Wang, K.; Schwinn, K.E.; Thomson, S.J.; Fiers, M.W.; Frew, T.J.; Murray, S.R.; Hofer, J.M.I.; Jacobs, J.M.E.; et al. Identification of Mendel’s white flower character. PLoS ONE 2010, 5, e13230. [Google Scholar] [CrossRef] [PubMed]
- Clemente, A.; Arques, M.C.; Dalmais, M.; le Signor, C.; Chinoy, C.; Olias, R.; Rayner, T.; Isaac, P.G.; Lawson, D.M.; Bendahmane, A.; et al. Eliminating anti-nutritional plant food proteins: The case of seed protease inhibitors in pea. PLoS ONE 2015, 10, e0134634. [Google Scholar]
- Hedley, C.L.; Bogracheva, T.Y.; Wang, T.L. A genetic approach to studying the morphology, structure and function of starch granules using pea as a model. Starke 2002, 54, 235–242. [Google Scholar] [CrossRef]
- Coxon, D.T.; Davies, D.R. The effect of the ra and rb loci on the lipid content of the seed of Pisum sativum. Theor. Appl. Genet. 1982, 64, 47–50. [Google Scholar] [CrossRef] [PubMed]
- Harrison, C.J.; Mould, R.M.; Leech, M.J.; Johnson, S.A.; Turner, L.; Schreck, S.L.; Baird, K.M.; Jack, P.L.; Rawsthorne, S.; Hedley, C.L.; et al. The rug3 locus of pea encodes plastidial phosphoglucomutase. Plant Physiol. 2000, 122, 1187–1192. [Google Scholar] [CrossRef] [PubMed]
- Barratt, D.H.; Barber, L.; Kruger, N.J.; Smith, A.M.; Wang, T.L.; Martin, C. Multiple, distinct isoforms of sucrose synthase in pea. Plant Physiol. 2001, 127, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Craig, J.; Lloyd, J.R.; Tomlinson, K.; Barber, L.; Edwards, A.; Wang, T.L.; Martin, C.; Hedley, C.L.; Smith, A.M. Mutations in the gene encoding starch synthase II profoundly alter amylopectin structure in pea embryos. Plant Cell 1998, 10, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Knox, M.R.; Moreau, C.; Lipscombe, J.; Ellis, T.H.N. High-throughput retrotransposon-based fluorescent markers: Improved information content and allele discrimination. Plant Methods 2009, 5, 10. [Google Scholar] [CrossRef] [PubMed]
- Ellis, T.H.N.; Hofer, J.I.; Timmerman-Vaughan, G.M.; Coyne, C.J.; Hellens, R.P. Mendel, 150 years on. Trends Plant Sci. 2011, 16, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Kaló, P.; Seres, A.; Taylor, S.A.; Jakab, J.; Kevei, Z.; Kereszt, A.; Endre, G.; Ellis, T.H.N.; Kiss, G.B. Comparative mapping between Medicago sativa and Pisum sativum. Mol. Gen. Genom. 2004, 272, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Aubert, G.; Morin, J.; Jacquin, F.; Loridon, K.; Quillet, M.C.; Petit, A.; Rameau, C.; Lejeune-Hénaut, I.; Huguet, T.; Burstin, J. Functional mapping in pea, as an aid to the candidate gene selection and for investigating synteny with the model legume Medicago truncatula. Theor. Appl. Genet. 2006, 112, 1024–1041. [Google Scholar] [CrossRef] [PubMed]
- Sindhu, A.; Ramsay, L.; Sanderson, L.A.; Stonehouse, R.; Li, R.; Condie, J.; Shunmugam, A.S.K.; Liu, Y.; Jha, A.B.; Diapari, M.; et al. Gene-based SNP discovery and genetic mapping in pea. Theor. Appl. Genet. 2014, 127, 2225–2241. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Ballicora, M.A.; Preiss, J.; Geiger, J.H. Crystal structure of potato tuber ADP-glucose pyrophosphorylase. EMBO J. 2005, 24, 694–704. [Google Scholar] [CrossRef] [PubMed]
- Dalmais, M.; Schmidt, J.; Le Signor, C.; Moussy, F.; Burstin, J.; Savois, V.; Aubert, G.; Brunaud, V.; de Oliveira, Y.; Guichard, C.; et al. UTILLdb, a Pisum sativum in silico forward and reverse genetics tool. Genome Biol. 2008, 9, R43. [Google Scholar] [CrossRef] [PubMed]
- Cernac, A.; Benning, C. WRINKLED1 encodes an AP2/EREB domain protein involved in the control of storage compound biosynthesis in Arabidopsis. Plant J. 2004, 40, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Baud, S.; Wuillème, S.; To, A.; Rochat, C.; Lepiniec, L. Role of WRINKLED1 in the transcriptional regulation of glycolytic and fatty acid biosynthetic genes in Arabidopsis. Plant J. 2009, 60, 933–947. [Google Scholar] [CrossRef] [PubMed]
- To, A.; Joubes, J.; Barthole, G.; Lecureuil, A.; Scagnelli, A.; Jasinski, S.; Lepiniec, L.; Baud, S. WRINKLED transcription factors orchestrate tissue-specific regulation of fatty acid biosynthesis in Arabidopsis. Plant Cell 2012, 24, 5007–5023. [Google Scholar] [CrossRef] [PubMed]
- Weigelt, K.; Küster, H.; Rutten, T.; Fait, A.; Fernie, A.R.; Miersch, O.; Wasternack, C.; Emery, R.J.N.; Desel, C.; Hosein, F.; et al. ADP-glucose pyrophosphorylase-deficient pea embryos reveal specific transcriptional and metabolic changes of carbon-nitrogen metabolism and stress responses. Plant Physiol. 2009, 149, 395–411. [Google Scholar] [CrossRef] [PubMed]
- Shunmugam, A.S.K.; Bock, C.; Arganosa, G.C.; Georges, F.; Gray, G.R.; Warkentin, T.D. Accumulation of phosphorus-containing compounds in developing seeds of low-phytate pea (Pisum sativum L.) mutants. Plants 2015, 4, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Alves-Carvalho, S.; Aubert, G.; Carrère, S.; Cruaud, C.; Brochot, A.L.; Jacquin, F.; Klein, A.; Martin, C.; Boucherot, K.; Kreplak, J.; et al. Full-length de novo assembly of RNA-seq data in pea (Pisum sativum L.) provides a gene expression atlas and gives insights into root nodulation in this species. Plant J. 2015, 84, 1–19. [Google Scholar] [PubMed]
- Bordat, A.; Savois, V.; Nicolas, M.; Salse, J.; Chauveau, A.; Bourgeois, M.; Potier, J.; Houtin, H.; Rond, C.; Murat, F.; et al. Translational genomics in legumes allowed placing in silico 5460 unigenes on the pea functional map and identified candidate genes in Pisum sativum L. Genes Genom. Genet. 2011, 1, 93–103. [Google Scholar]
- Shunmugam, A.S.K.; Liu, X.; Stonehouse, R.; Tar’an, B.; Bett, K.E.; Sharpe, A.G.; Warkentin, T.D. Mapping seed phytic acid concentration and iron bioavailability in a pea recombinant inbred line population. Crop Sci. 2015, 55, 828–836. [Google Scholar] [CrossRef]
- Rehman, A.U.; Shunmugam, A.; Arganosa, G.; Bett, K.E.; Warkentin, T.D. Inheritance of the low-phytate trait in pea. Crop Sci. 2012, 52, 1171–1175. [Google Scholar] [CrossRef]
- Donahue, J.L.; Alford, S.R.; Torabinejad, J.; Kerwin, R.E.; Nourbakhsh, A.; Ray, W.K.; Hernick, M.; Huang, X.; Lyons, B.M.; Hein, P.P.; et al. The Arabidopsis thaliana myo-inositol 1-phosphate synthase1 gene is required for myo-inositol synthesis and suppression of cell death. Plant Cell 2010, 22, 888–903. [Google Scholar] [CrossRef] [PubMed]
- Vandecasteele, C.; Teulat-Merah, B.; Morère-Le Paven, M.C.; Leprince, O.; Vu, B.L.; Viau, L.; Ledroit, L.; Pelletier, S.; Payet, N.; Satour, P.; et al. Quantitative trait loci analysis reveals a correlation between the ratio of sucrose/raffinose family oligosaccharides and seed vigour in Medicago truncatula. Plant Cell Env. 2011, 34, 1473–1487. [Google Scholar] [CrossRef] [PubMed]
- Hartl, D.L. Mendel and Galton: Contrasting approaches to the study of heredity. Mendel Newslett. 1997, 6, 1–3. [Google Scholar]
- Domoney, C.; Knox, M.; Moreau, C.; Ambrose, M.; Palmer, S.; Smith, P.; Christodoulou, V.; Isaac, P.G.; Hegarty, M.; Blackmore, T.; et al. Exploiting a fast neutron mutant genetic resource in Pisum sativum (pea) for functional genomics. Funct. Plant Biol. 2013, 40, 1261–1270. [Google Scholar] [CrossRef]
- Peterbauer, T.; Lahuta, L.B.; Blöchl, A.; Mucha, J.; Jones, D.A.; Hedley, C.L.; Gòrecki, R.J.; Richter, A. Analysis of the raffinose family oligosaccharide pathway in pea seeds with contrasting carbohydrate composition. Plant Physiol. 2001, 127, 1764–1772. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.M.; Zeeman, S.C. Quantification of starch in plant tissues. Nat. Protocol. 2006, 1, 1342–1345. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. Rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phenotypic Description | Marker Assay Allelic Classes | Total | ||
|---|---|---|---|---|
| SbeI-wt | sbeI-ins | SbeI-wt & sbeI-ins (Presumed Heterozygotes) | ||
| Round/Dimpled | 1766 | 42 | 6 | 1814 |
| Marrowfat | 168 | 29 | 2 | 199 |
| Wrinkled | 59 | 716 | 4 | 779 |
| Total | 1993 | 787 | 12 | 2792 |
| Wrinkled-Seeded Phenotype (Sbei Insertion Minus) | AgpL Genotype Score | |
|---|---|---|
| rb Deletion | WT | |
| JI 399 | + | − |
| JI 492 | + | − |
| JI 528 | + | − |
| JI 601 | + | − |
| JI 1156 | + | − |
| JI 1157 | + | − |
| JI 1408 | + | − |
| JI 2012 | + | − |
| JI 2107 | + | − |
| JI 2109 | + | − |
| JI 2111 | + | − |
| JI 2368 | + | − |
| JI 2369 | + | − |
| JI 1417 | − | + |
| JI 2110 | − | + |
| Observed | Class Group | Chi Square Analysis | |||||
|---|---|---|---|---|---|---|---|
| A | B | total | |||||
| Agreed scores | 78 | 21 | 99 | ||||
| Majority scores | 109 | 38 | 147 | ||||
| Extreme scores | 17 | 13 | 30 | ||||
| A | B | ||||||
| Expected (3:1) | Chi Square | Chi Square | total | p | |||
| Agreed scores | 74.25 | 24.75 | 0.19 | 0.57 | 0.76 | 0.3841 | |
| Majority scores | 110.25 | 36.75 | 0.01 | 0.04 | 0.06 | 0.8118 | |
| Extreme scores | 22.5 | 7.5 | 1.34 | 4.03 | 5.38 | 0.0204 | |
| Expected (1:1) | Chi Square | Chi Square | total | p | |||
| Agreed scores | 49.5 | 49.5 | 16.41 | 16.41 | 32.82 | 0.0000 | |
| Majority scores | 73.5 | 73.5 | 17.15 | 17.15 | 34.29 | 0.0000 | |
| Extreme scores | 15 | 15 | 0.27 | 0.27 | 0.53 | 0.4652 | |
| Primer Name | Sequence (5′–3′) | Direction | Colour |
|---|---|---|---|
| Ps-SBEI-F4 | GAACAGCAGTGGTGTATGCC | F | - |
| Ps-SBEI-R4 | GACTTGATTGCACTGATGCC | R | - |
| r-element-F2 | ATGGATCAAATTGCCACCC | F | - |
| Sbe1F4F | Fam-GATCATTCCTCCACCCGGTA | F | Blue |
| Sbe1R4e | TTTTTGCTGACGGTGAGCTTGC | R | Blue |
| Sbe1rcFV | Vic-CCTTGCGGATGGAGTTGAA | F | Green |
| Sbe1rIR | TCCATTTTGCCACCCCTAATT | R | Green |
| Sbe1rDR | AACTCCAACTGACAGCTCAATTGG | R | Green |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rayner, T.; Moreau, C.; Ambrose, M.; Isaac, P.G.; Ellis, N.; Domoney, C. Genetic Variation Controlling Wrinkled Seed Phenotypes in Pisum: How Lucky Was Mendel? Int. J. Mol. Sci. 2017, 18, 1205. https://doi.org/10.3390/ijms18061205
Rayner T, Moreau C, Ambrose M, Isaac PG, Ellis N, Domoney C. Genetic Variation Controlling Wrinkled Seed Phenotypes in Pisum: How Lucky Was Mendel? International Journal of Molecular Sciences. 2017; 18(6):1205. https://doi.org/10.3390/ijms18061205
Chicago/Turabian StyleRayner, Tracey, Carol Moreau, Mike Ambrose, Peter G. Isaac, Noel Ellis, and Claire Domoney. 2017. "Genetic Variation Controlling Wrinkled Seed Phenotypes in Pisum: How Lucky Was Mendel?" International Journal of Molecular Sciences 18, no. 6: 1205. https://doi.org/10.3390/ijms18061205
APA StyleRayner, T., Moreau, C., Ambrose, M., Isaac, P. G., Ellis, N., & Domoney, C. (2017). Genetic Variation Controlling Wrinkled Seed Phenotypes in Pisum: How Lucky Was Mendel? International Journal of Molecular Sciences, 18(6), 1205. https://doi.org/10.3390/ijms18061205