
Predicting Zoonotic Risk of Influenza A Viruses from Host Tropism Protein Signature Using Random Forest

Abstract
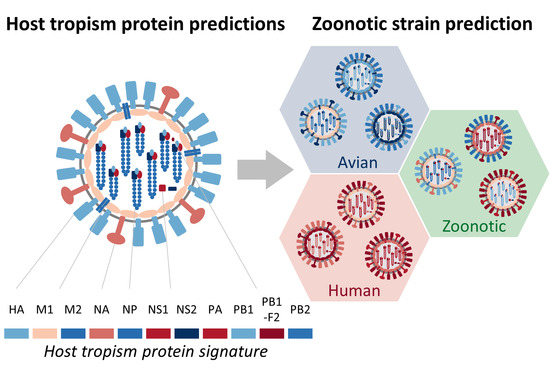
:
1. Introduction
2. Results
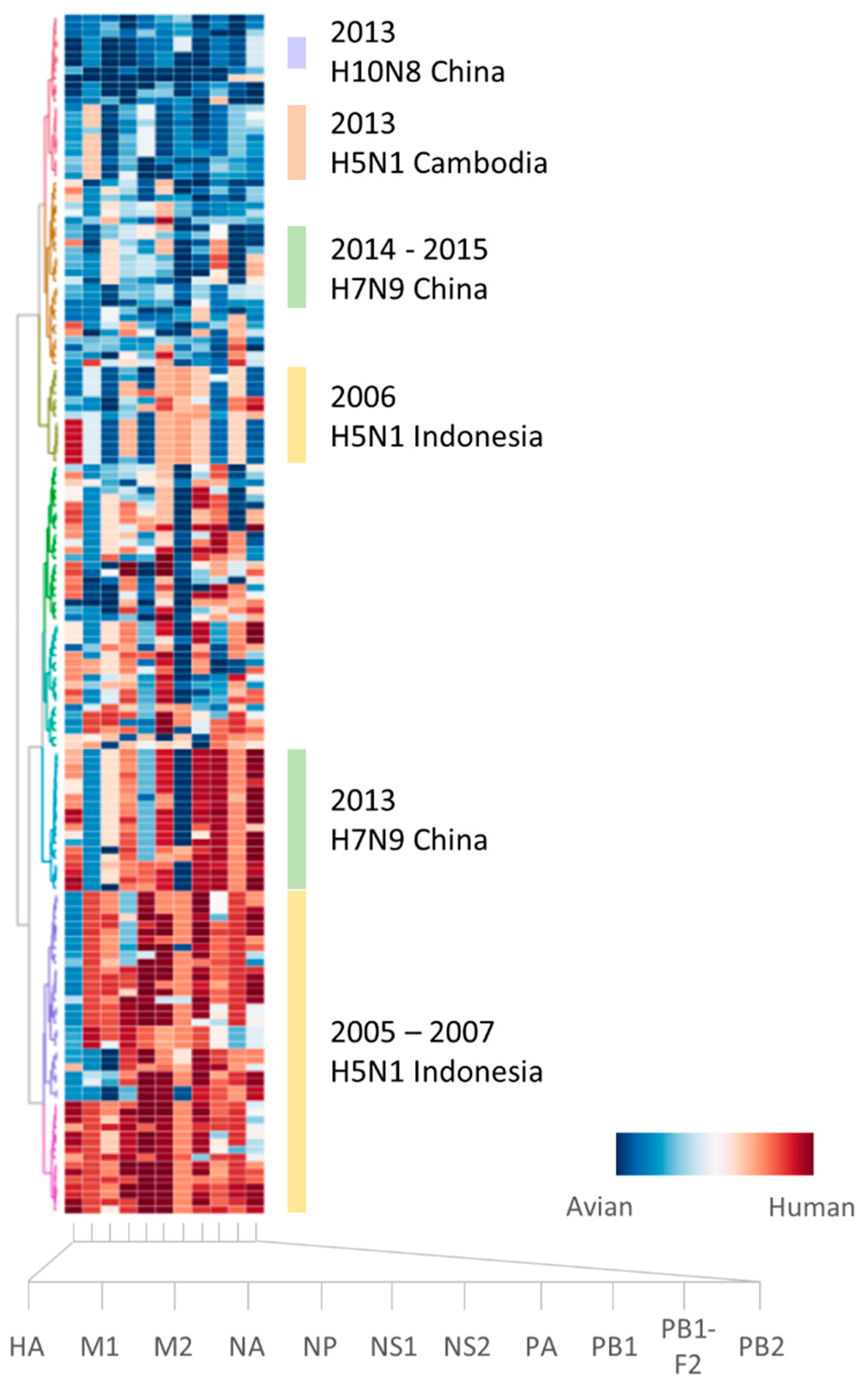
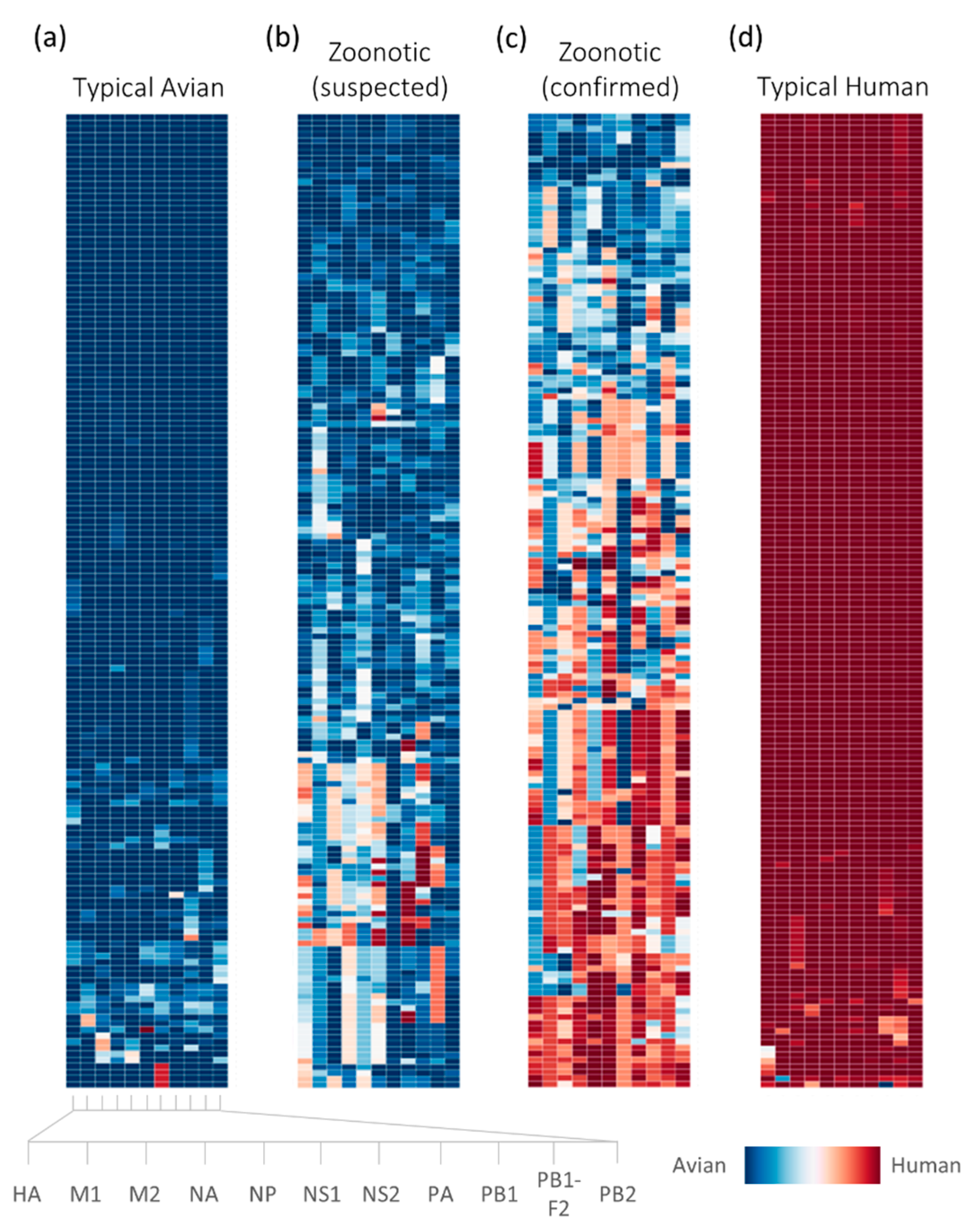
2.1. Sufficient Distinction in Host Tropism Protein Signatures to Characterize Zoonotic Strains
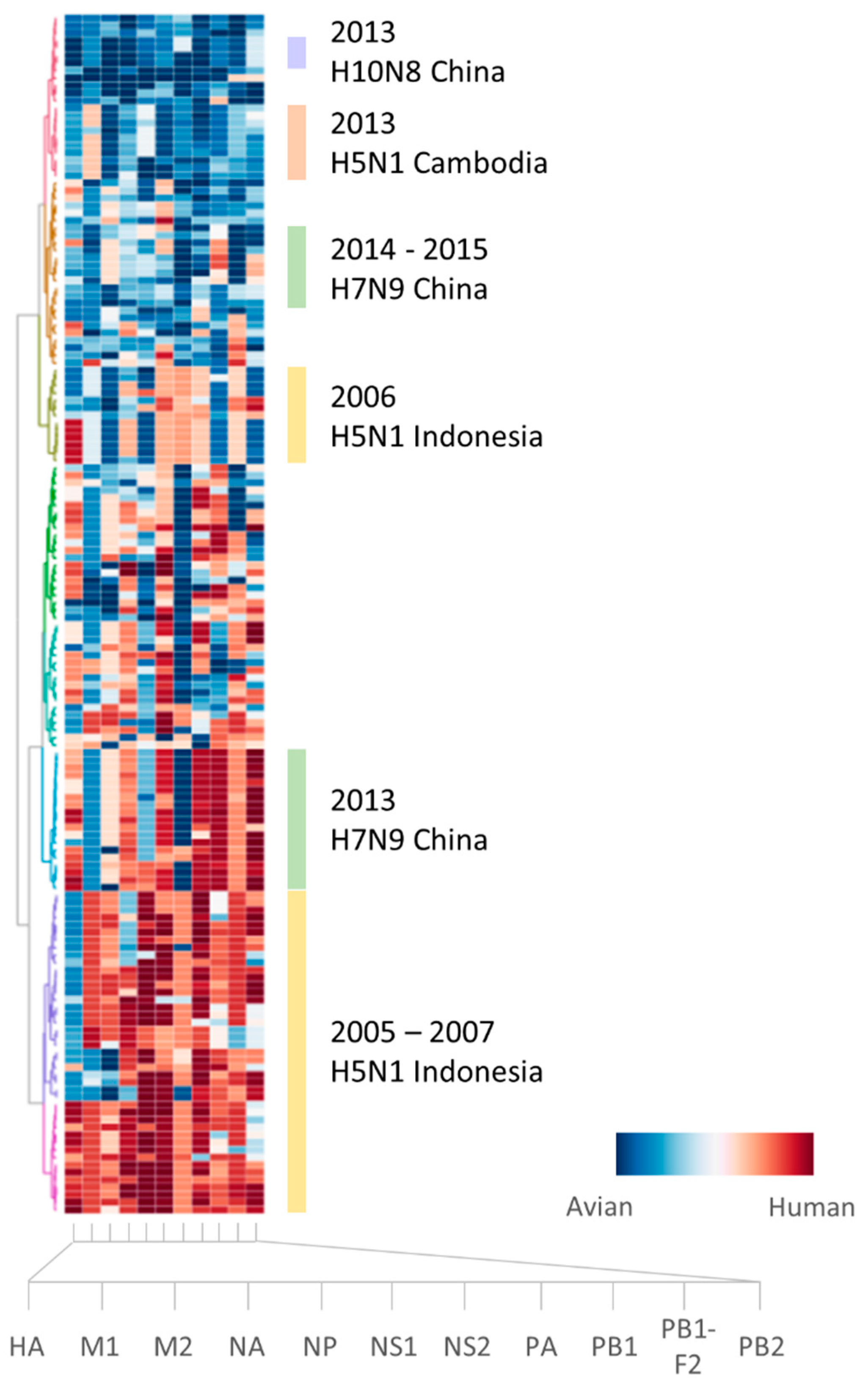
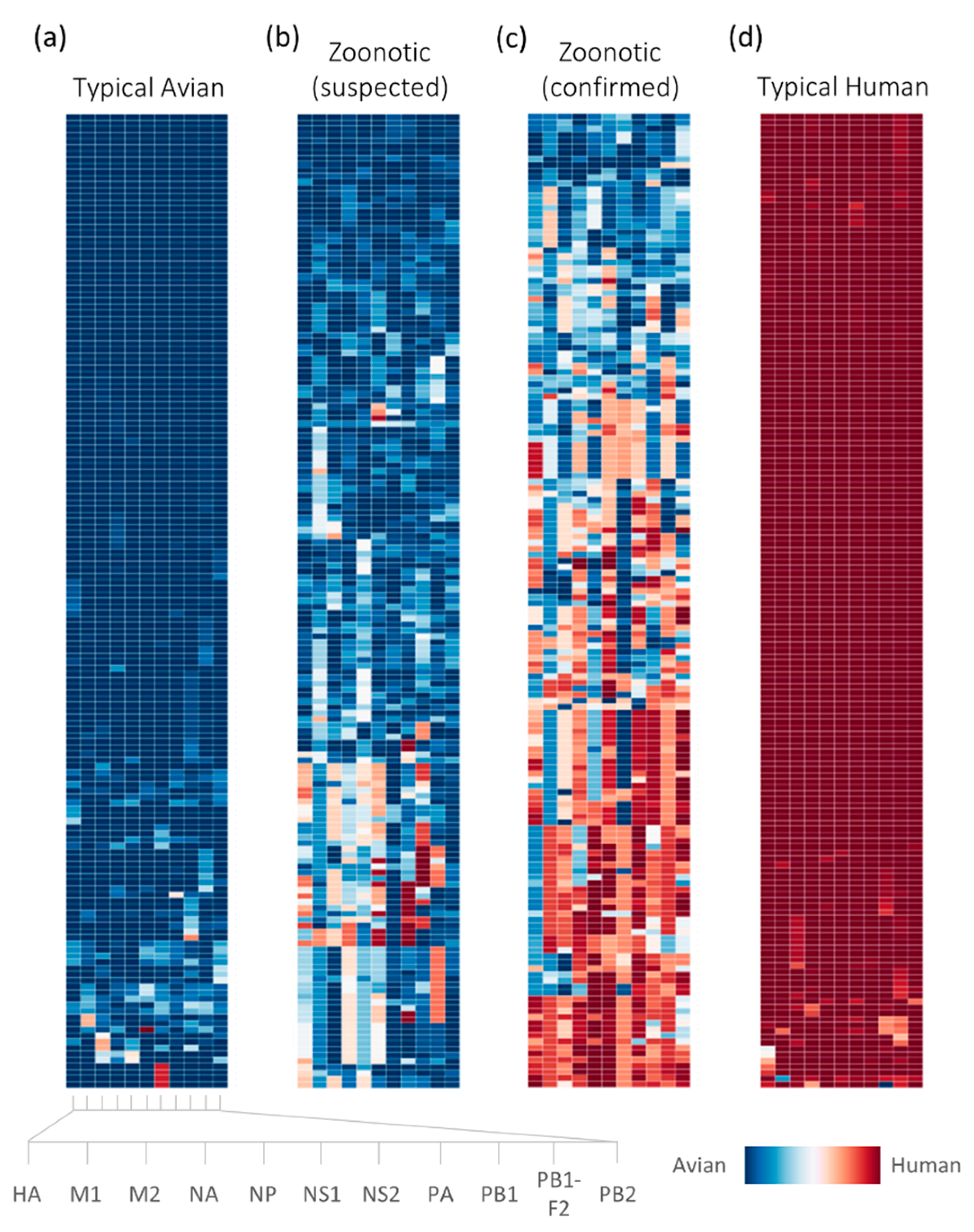
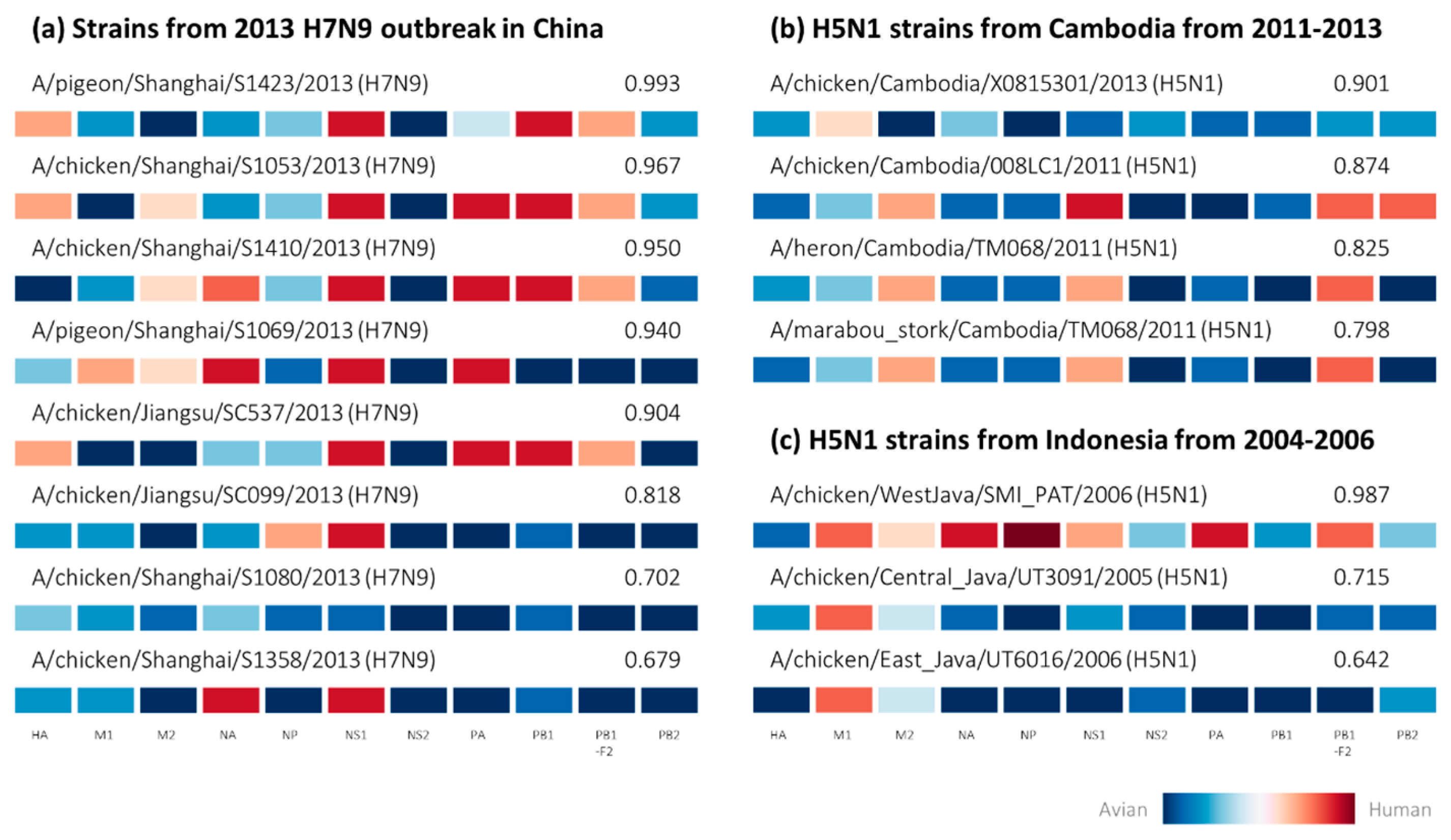
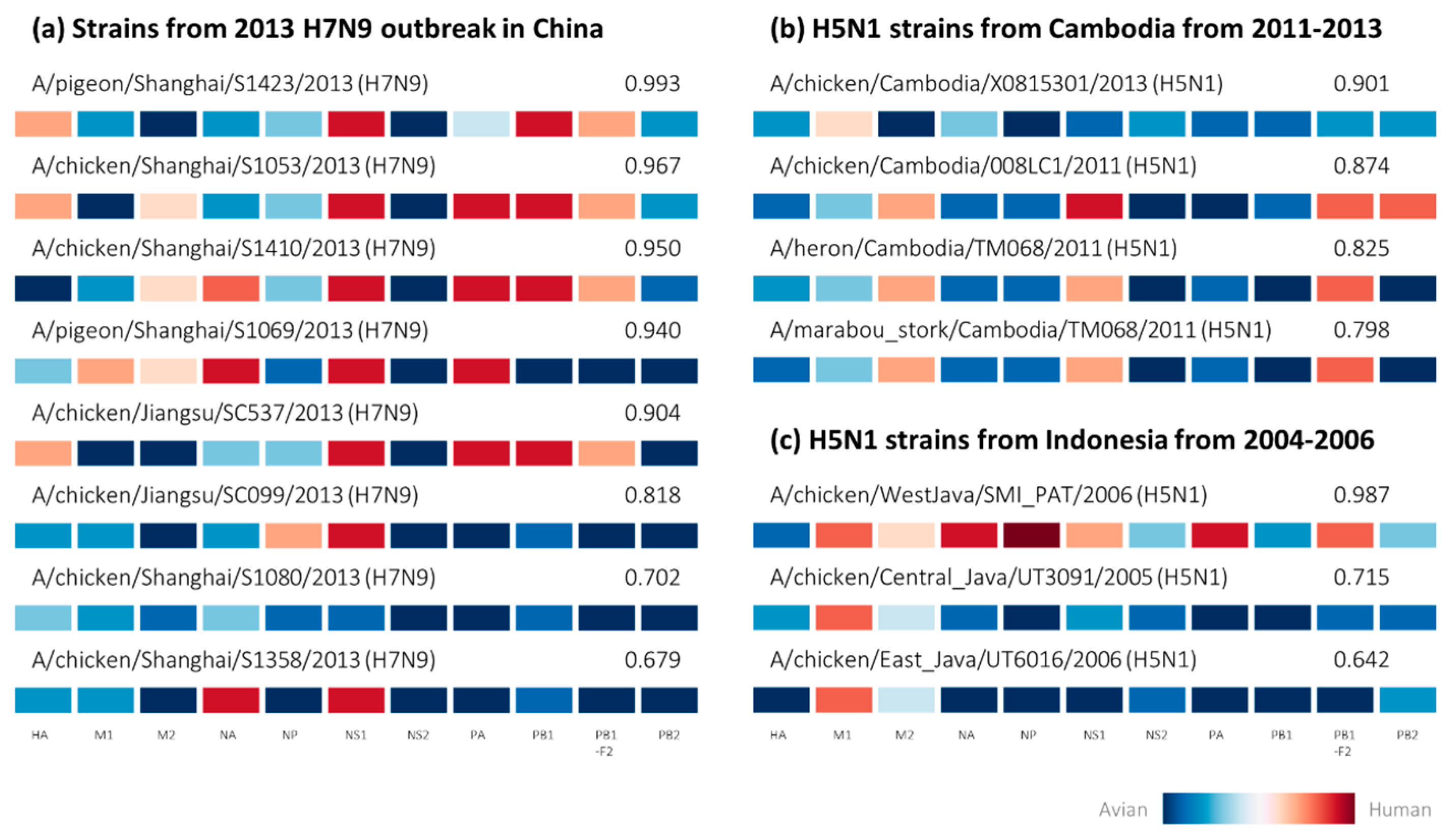
2.2. Retrospective Analysis of Avian Strains from Outbreaks Demonstrate Capability of Zoonotic Strain Prediction Model
3. Discussion
4. Materials and Methods
4.1. Data Collection and Preparation
4.2. Host Tropism Protein Signature Feature Transformation
4.3. Construction of Zoonotic Strain Prediction Model
4.4. Analysis of Avian-Isolated Suspected Zoonotic Strains
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ANN | Artificial neural network |
| AUC | Area under the receiver operating characteristic curve |
| CDC | Center for Control, Disease and Prevention |
| HA | Hemagglutinin |
| M1 | Matrix protein 1 |
| M2 | Matrix protein 2 |
| NA | Neuraminidase |
| NP | Nucleoprotein |
| NS1 | Non-structural protein 1 |
| NS2 | Non-structural protein 2 |
| PA | Polymerase acidic protein |
| PB1 | Polymerase basic protein 1 |
| PB1-F2 | Accessory protein F2 translated from PB1 segment |
| PB2 | Polymerase basic protein 2 |
| SVM | Support vector machine |
| WHO | World Health Organization |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year | Subtype | Country | Avian-isolated Suspected Zoonotic Samples | Human-Isolated Confirmed Zoonotic Samples |
|---|---|---|---|---|
| 1997 | H5N1 | Hong Kong | ||
| H9N2 | Hong Kong | 2 | ||
| 1998 | H9N2 | China | 1 | |
| 1999 | H9N2 | China | 2 | |
| 2003 | H5N1 | China | 32 | |
| Vietnam | ||||
| H7N7 | Netherlands | 1 | ||
| H9N2 | Hong Kong | 2 | ||
| 2004 | H5N1 | Thailand | 10 | |
| Vietnam | 14 | |||
| H7N3 | Canada | 1 | 1 | |
| 2005 | H5N1 | Cambodia | ||
| China | 2 | |||
| Indonesia | 11 | 8 | ||
| Thailand | 22 | |||
| Vietnam | 46 | |||
| 2006 | H5N1 | Azerbaijan | ||
| Cambodia | ||||
| China | 19 | |||
| Djibouti | ||||
| Egypt | 4 | 2 | ||
| Indonesia | 11 | 46 | ||
| Iraq | 1 | |||
| Thailand | 7 | 1 | ||
| Turkey | 3 | |||
| 2007 | H3N8 | Laos | 1 | |
| H5N1 | Cambodia | |||
| China | 7 | |||
| Egypt | 2 | |||
| Indonesia | 4 | 10 | ||
| Laos | 16 | |||
| Myanmar | ||||
| Nigeria | 20 | |||
| Pakistan | ||||
| Vietnam | 54 | |||
| H7N2 | United Kingdom | |||
| 2008 | H3N8 | Vietnam | 1 | |
| H5N1 | Bangladesh | 1 | ||
| Cambodia | ||||
| China | 5 | |||
| Egypt | 18 | |||
| Indonesia | 1 | |||
| Vietnam | 4 | |||
| H9N2 | Hong Kong | 9 | ||
| H11N9 | Vietnam | 2 | ||
| 2009 | H5N1 | Cambodia | 1 | |
| China | 6 | |||
| Egypt | 6 | |||
| Indonesia | 1 | |||
| Vietnam | ||||
| H9N2 | Hong Kong | 4 | ||
| 2010 | H5N1 | Cambodia | 5 | 1 |
| China | 3 | 1 | ||
| Egypt | 25 | |||
| Indonesia | ||||
| Vietnam | 3 | |||
| 2011 | H5N1 | Bangladesh | 1 | 1 |
| Cambodia | 6 | 4 | ||
| China | 11 | |||
| Egypt | 13 | |||
| Indonesia | ||||
| 2012 | H5N1 | Bangladesh | 18 | |
| Cambodia | 1 | 3 | ||
| China | 3 | |||
| Egypt | 6 | |||
| Indonesia | ||||
| H5N1 | Vietnam | 30 | ||
| H7N3 | Mexico | 1 | ||
| 2013 | H5N1 | Bangladesh | 5 | |
| Cambodia | 8 | 10 | ||
| Canada | ||||
| China | 3 | |||
| Egypt | 3 | |||
| Indonesia | ||||
| Vietnam | 19 | |||
| H7N9 | China | 114 | 35 | |
| Hong Kong | 1 | |||
| Taiwan | 2 | |||
| H9N2 | China | 135 | 1 | |
| H10N8 | China | 26 | 4 | |
| 2014 | H5N1 | Cambodia | ||
| China | 7 | |||
| Egypt | 3 | 1 | ||
| Indonesia | ||||
| Vietnam | 24 | |||
| H7N9 | China | 226 | 19 | |
| 2015 | H5N1 | China | ||
| Egypt | ||||
| Indonesia | ||||
| H5N6 | China | 1 | ||
| H7N9 | Canada | |||
| China | 2 | |||
| Total | 1047 | 160 |
References
- Centers for Disease Control and Prevention. Isolation of avian influenza A (H5N1) viruses from humans—Hong Kong, May–December 1997. Morb. Mortal. Wkly. Rep. 1997, 46, 1204–1207. [Google Scholar]
- Peiris, J.S.; de Jong, M.D.; Guan, Y. Avian influenza virus (H5N1): A threat to human health. Clin. Microbiol. Rev. 2007, 20, 243–267. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Cumulative Number of Confirmed Human Cases for Avian Influenza A (H5N1) Reported to WHO, 2003–2016. Available online: http://www.who.int/influenza/human_animal_interface/EN_GIP_20160404cumulativenumberH5N1cases.pdf (accessed on 18 December 2016).
- Fouchier, R.A.; Schneeberger, P.M.; Rozendaal, F.W.; Broekman, J.M.; Kemink, S.A.; Munster, V.; Kuiken, T.; Rimmelzwaan, G.F.; Schutten, M.; van Doornum, G.J.; et al. Avian influenza A virus (H7N7) associated with human conjunctivitis and a fatal case of acute respiratory distress syndrome. Proc. Natl. Acad. Sci. USA 2004, 101, 1356–1361. [Google Scholar] [CrossRef] [PubMed]
- Koopmans, M.; Wilbrink, B.; Conyn, M.; Natrop, G.; van der Nat, H.; Vennema, H.; Meijer, A.; van Steenbergen, J.; Fouchier, R.; Osterhaus, A.; et al. Transmission of H7N7 avian influenza A virus to human beings during a large outbreak in commercial poultry farms in the netherlands. Lancet 2004, 363, 587–593. [Google Scholar] [CrossRef]
- Kurtz, J.; Manvell, R.J.; Banks, J. Avian influenza virus isolated from a woman with conjunctivitis. Lancet 1996, 348, 901–902. [Google Scholar] [CrossRef]
- Butt, K.M.; Smith, G.J.; Chen, H.; Zhang, L.J.; Leung, Y.H.; Xu, K.M.; Lim, W.; Webster, R.G.; Yuen, K.Y.; Peiris, J.S.; et al. Human infection with an avian H9N2 influenza A virus in Hong Kong in 2003. J. Clin. Microbiol. 2005, 43, 5760–5767. [Google Scholar] [CrossRef] [PubMed]
- Peiris, M.; Yuen, K.Y.; Leung, C.W.; Chan, K.H.; Ip, P.L.; Lai, R.W.; Orr, W.K.; Shortridge, K.F. Human infection with influenza H9N2. Lancet 1999, 354, 916–917. [Google Scholar] [CrossRef]
- Chen, Y.; Liang, W.; Yang, S.; Wu, N.; Gao, H.; Sheng, J.; Yao, H.; Wo, J.; Fang, Q.; Cui, D.; et al. Human infections with the emerging avian influenza A H7N9 virus from wet market poultry: Clinical analysis and characterisation of viral genome. Lancet 2013, 381, 1916–1925. [Google Scholar] [CrossRef]
- Gao, R.; Cao, B.; Hu, Y.; Feng, Z.; Wang, D.; Hu, W.; Chen, J.; Jie, Z.; Qiu, H.; Xu, K.; et al. Human infection with a novel avian-origin influenza A (H7N9) virus. N. Engl. J. Med. 2013, 368, 1888–1897. [Google Scholar] [CrossRef] [PubMed]
- Richard, M.; de Graaf, M.; Herfst, S. Avian influenza A viruses: From zoonosis to pandemic. Future Virol. 2014, 9, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Wu, J.T.; Cowling, B.J.; Liao, Q.; Fang, V.J.; Zhou, S.; Wu, P.; Zhou, H.; Lau, E.H.; Guo, D.; et al. Effect of closure of live poultry markets on poultry-to-person transmission of avian influenza A H7N9 virus: An ecological study. Lancet 2014, 383, 541–548. [Google Scholar] [CrossRef]
- World Health Organization. H5N1 Research Issues. Available online: http://www.who.int/influenza/human_animal_interface/avian_influenza/h5n1_research/en/ (accessed on 14 October 2014).
- Heymann, D.L.; Dixon, M. Infections at the animal/human interface: Shifting the paradigm from emergency response to prevention at source. Curr. Top. Microbiol. Immunol. 2013, 366, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Reid, A.H.; Taubenberger, J.K. The origin of the 1918 pandemic influenza virus: A continuing enigma. J. Gen. Virol. 2003, 84, 2285–2292. [Google Scholar] [CrossRef] [PubMed]
- Peiris, M.; Yen, H.L. Animal and human influenzas. Rev. Sci. Tech. 2014, 33, 539–553. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Antigenic and Genetic Characteristics of Influenza A(H5N1) and Influenza A(H9N2) Viruses and Candidate Vaccine Viruses Developed for Potential Use in Human Vaccines. Available online: http://www.who.int/influenza/resources/documents/201009_H5_H9_VaccineVirusUpdate.pdf (accessed on 8 April 2014).
- Chien, Y.J. How did international agencies perceive the avian influenza problem? The adoption and manufacture of the “one world, one health” framework. Soc. Health Illn. 2013, 35, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Hartaningsih, N.; Wibawa, H.; Pudjiatmoko; Rasa, F.S.; Irianingsih, S.H.; Dharmawan, R.; Azhar, M.; Siregar, E.S.; McGrane, J.; Wong, F.; et al. Surveillance at the molecular level: Developing an integrated network for detecting variation in avian influenza viruses in indonesia. Prev. Vet. Med. 2015, 120, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Russell, C.A.; Kasson, P.M.; Donis, R.O.; Riley, S.; Dunbar, J.; Rambaut, A.; Asher, J.; Burke, S.; Davis, C.T.; Garten, R.J.; et al. Improving pandemic influenza risk assessment. eLife 2014, 3, e03883. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.W.; Chang, S.C.; Mok, C.K.; Lo, Y.L.; Kung, Y.N.; Huang, J.H.; Shih, Y.H.; Wang, J.Y.; Chiang, C.; Chen, C.J.; et al. Genomic signatures of human versus avian influenza a viruses. Emerg. Infect. Dis. 2006, 12, 1353–1360. [Google Scholar] [CrossRef] [PubMed]
- Finkelstein, D.B.; Mukatira, S.; Mehta, P.K.; Obenauer, J.C.; Su, X.; Webster, R.G.; Naeve, C.W. Persistent host markers in pandemic and H5N1 influenza viruses. J. Virol. 2007, 81, 10292–10299. [Google Scholar] [CrossRef] [PubMed]
- Steel, J.; Lowen, A.C.; Mubareka, S.; Palese, P. Transmission of influenza virus in a mammalian host is increased by PB2 amino acids 627K or 627E/701N. PLoS Pathog. 2009, 5, e1000252. [Google Scholar] [CrossRef] [PubMed]
- Subbarao, E.K.; London, W.; Murphy, B.R. A single amino acid in the PB2 gene of influenza A virus is a determinant of host range. J. Virol. 1993, 67, 1761–1764. [Google Scholar] [PubMed]
- Khaliq, Z.; Leijon, M.; Belak, S.; Komorowski, J. Identification of combinatorial host-specific signatures with a potential to affect host adaptation in influenza A H1N1 and H3N2 subtypes. BMC Genom. 2016, 17, 529. [Google Scholar] [CrossRef] [PubMed]
- Sjaugi, M.F.; Tan, S.; Abd Raman, H.S.; Lim, W.C.; Nik Mohamed, N.E.; August, J.; Khan, A.M. g-FLUA2H: A web-based application to study the dynamics of animal-to-human mutation transmission for influenza viruses. BMC Med. Genom. 2015, 8, S5. [Google Scholar] [CrossRef] [PubMed]
- Taubenberger, J.K.; Kash, J.C. Influenza virus evolution, host adaptation, and pandemic formation. Cell Host Microbe 2010, 7, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Global Influenza Programme. Tool for Influenza Pandemic Risk Assessment (TIPRA). Available online: http://www.who.int/influenza/publications/TIPRA_manual_v1/en/ (accessed on 14 March 2017).
- Trock, S.C.; Burke, S.A.; Cox, N.J. Development of framework for assessing influenza virus pandemic risk. Emerg. Infect. Dis. 2015, 21, 1372–1378. [Google Scholar] [CrossRef] [PubMed]
- Qiang, X.; Kou, Z. Prediction of interspecies transmission for avian influenza A virus based on a back-propagation neural network. Math. Comput. Model. 2010, 52, 2060–2065. [Google Scholar] [CrossRef]
- Wang, J.; Ma, C.; Kou, Z.; Zhou, Y.; Liu, H. Predicting transmission of avian influenza A viruses from avian to human by using informative physicochemical properties. Int. J. Data Min. Bioinform. 2013, 7, 166–179. [Google Scholar] [CrossRef] [PubMed]
- Eng, C.L.; Tong, J.C.; Tan, T.W. Predicting host tropism of influenza A virus proteins using random forest. BMC Med. Genom. 2014, 7, S1. [Google Scholar] [CrossRef] [PubMed]
- Eng, C.L.; Tong, J.C.; Tan, T.W. Distinct host tropism protein signatures to identify possible zoonotic influenza A viruses. PLoS ONE 2016, 11, e0150173. [Google Scholar] [CrossRef] [PubMed]
- Kageyama, T.; Fujisaki, S.; Takashita, E.; Xu, H.; Yamada, S.; Uchida, Y.; Neumann, G.; Saito, T.; Kawaoka, Y.; Tashiro, M. Genetic analysis of novel avian A(H7N9) influenza viruses isolated from patients in China, February to April 2013. Euro Surveill. 2013, 18, 20453. [Google Scholar] [PubMed]
- World Health Organization. Overview of the Emergence and Characteristics of the Avian Influenza A(H7N9) Virus. Available online: Http://www.who.int/influenza/human_animal_interface/influenza_h7n9/WHO_H7N9_review_31May13.pdf?ua=1 (accessed on 8 April 2014).
- Pantin-Jackwood, M.J.; Miller, P.J.; Spackman, E.; Swayne, D.E.; Susta, L.; Costa-Hurtado, M.; Suarez, D.L. Role of poultry in the spread of novel H7N9 influenza virus in china. J. Virol. 2014, 88, 5381–5390. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.R.; Kuo, C.Y.; Huang, H.Y.; Wu, F.T.; Huang, Y.L.; Cheng, C.Y.; Su, Y.T.; Wu, H.S.; Liu, M.T. Characterization of influenza A (H7N9) viruses isolated from human cases imported into Taiwan. PLoS ONE 2015, 10, e0119792. [Google Scholar] [CrossRef] [PubMed]
- Rith, S.; Davis, C.T.; Duong, V.; Sar, B.; Horm, S.V.; Chin, S.; Ly, S.; Laurent, D.; Richner, B.; Oboho, I.; et al. Identification of molecular markers associated with alteration of receptor-binding specificity in a novel genotype of highly pathogenic avian influenza A(H5N1) viruses detected in Cambodia in 2013. J. Virol. 2014, 88, 13897–13909. [Google Scholar] [CrossRef] [PubMed]
- Setiawaty, V.; Dharmayanti, N.L.; Misriyah; Pawestri, H.A.; Azhar, M.; Tallis, G.; Schoonman, L.; Samaan, G. Avian influenza A(H5N1) virus outbreak investigation: Application of the FAO-OIE-WHO four-way linking framework in Indonesia. Zoonoses Public Health 2015, 62, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Wibawa, H.; Henning, J.; Wong, F.; Selleck, P.; Junaidi, A.; Bingham, J.; Daniels, P.; Meers, J. A molecular and antigenic survey of H5N1 highly pathogenic avian influenza virus isolates from smallholder duck farms in Central Java, Indonesia during 2007–2008. J. Virol. 2011, 8, 425. [Google Scholar] [CrossRef] [PubMed]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Squires, R.B.; Noronha, J.; Hunt, V.; Garcia-Sastre, A.; Macken, C.; Baumgarth, N.; Suarez, D.; Pickett, B.E.; Zhang, Y.; Larsen, C.N.; et al. Influenza research database: An integrated bioinformatics resource for influenza research and surveillance. Influenza Respir. Viruses 2012, 6, 404–416. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.S.; Themudo, G.E.; Christensen, J.P.; Biswas, P.K.; Giasuddin, M.; Samad, M.A.; Toft, N.; Ersboll, A.K. Molecular epidemiology of circulating highly pathogenic avian influenza (H5N1) virus in chickens, in Bangladesh, 2007–2010. Vaccine 2012, 30, 7381–7390. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Update: Influenza activity—United States and worldwide, 2005–06 season, and composition of the 2006–07 influenza vaccine. Morb. Mortal. Wkly. Rep. 2006, 55, 648–653. [Google Scholar]
- Pabbaraju, K.; Tellier, R.; Wong, S.; Li, Y.; Bastien, N.; Tang, J.W.; Drews, S.J.; Jang, Y.; Davis, C.T.; Fonseca, K.; et al. Full-genome analysis of avian influenza A(H5N1) virus from a human, North America, 2013. Emerg. Infect. Dis. 2014, 20, 887–891. [Google Scholar] [CrossRef] [PubMed]
- Rabinowitz, P.M.; Galusha, D.; Vegso, S.; Michalove, J.; Rinne, S.; Scotch, M.; Kane, M. Comparison of human and animal surveillance data for H5N1 influenza A in Egypt 2006–2011. PLoS ONE 2012, 7, e43851. [Google Scholar] [CrossRef] [PubMed]
- Claas, E.C.; de Jong, J.C.; van Beek, R.; Rimmelzwaan, G.F.; Osterhaus, A.D. Human influenza virus A/HongKong/156/97 (H5N1) infection. Vaccine 1998, 16, 977–978. [Google Scholar] [CrossRef]
- Kandun, I.N.; Wibisono, H.; Sedyaningsih, E.R.; Yusharmen; Hadisoedarsuno, W.; Purba, W.; Santoso, H.; Septiawati, C.; Tresnaningsih, E.; Heriyanto, B.; et al. Three Indonesian clusters of H5N1 virus infection in 2005. N. Engl. J. Med. 2006, 355, 2186–2194. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Update: Influenza activity—United States and worldwide, 2006–07 season, and composition of the 2007–08 influenza vaccine. Morb. Mortal. Wkly. Rep. 2007, 56, 789–794. [Google Scholar]
- Zaman, M.; Ashraf, S.; Dreyer, N.A.; Toovey, S. Human infection with avian influenza virus, Pakistan, 2007. Emerg. Infect. Dis. 2011, 17, 1056–1059. [Google Scholar] [CrossRef] [PubMed]
- Tiensin, T.; Chaitaweesub, P.; Songserm, T.; Chaisingh, A.; Hoonsuwan, W.; Buranathai, C.; Parakamawongsa, T.; Premashthira, S.; Amonsin, A.; Gilbert, M.; et al. Highly pathogenic avian influenza H5N1, Thailand, 2004. Emerg. Infect. Dis. 2005, 11, 1664–1672. [Google Scholar] [CrossRef] [PubMed]
- Oner, A.F.; Bay, A.; Arslan, S.; Akdeniz, H.; Sahin, H.A.; Cesur, Y.; Epcacan, S.; Yilmaz, N.; Deger, I.; Kizilyildiz, B.; et al. Avian influenza A (H5N1) infection in Eastern Turkey in 2006. N. Engl. J. Med. 2006, 355, 2179–2185. [Google Scholar] [CrossRef] [PubMed]
- Manabe, T.; Yamaoka, K.; Tango, T.; Binh, N.G.; Co, D.X.; Tuan, N.D.; Izumi, S.; Takasaki, J.; Chau, N.Q.; Kudo, K. Chronological, geographical, and seasonal trends of human cases of avian influenza A (H5N1) in Vietnam, 2003–2014: A spatial analysis. BMC Infect. Dis. 2016, 16, 64. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Cui, L.; Yu, H.; Ge, Y.; Tang, F. Whole-genome sequence of a reassortant H5N6 avian influenza virus isolated from a live poultry market in China, 2013. Genome Announc. 2014, 2. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Zhang, L.; Kan, X.; Jiang, L.; Yang, J.; Guo, Z.; Ren, Q. Origin and molecular characteristics of a novel 2013 avian influenza A(H6N1) virus causing human infection in Taiwan. Clin. Infect. Dis. 2013, 57, 1367–1368. [Google Scholar] [CrossRef] [PubMed]
- Editorial Team. Avian influenza A/(H7N2) outbreak in the United Kingdom. Euro Surveill. 2007, 12, E070531. [Google Scholar] [PubMed]
- Editorial team. Avian influenza H5N1 detected in poultry in Nigeria, further human cases reported in Iraq, Indonesia and China. Euro Surveill. 2006, 11, E060209 060201. [Google Scholar] [PubMed]
- Ostrowsky, B.; Huang, A.; Terry, W.; Anton, D.; Brunagel, B.; Traynor, L.; Abid, S.; Johnson, G.; Kacica, M.; Katz, J.; et al. Low pathogenic avian influenza A (H7N2) virus infection in immunocompromised adult, New York, USA, 2003. Emerg. Infect. Dis. 2012, 18, 1128–1131. [Google Scholar] [CrossRef] [PubMed]
- Tweed, S.A.; Skowronski, D.M.; Davies, T.M.; Larder, A.; Petric, M.; Lees, W.; Li, Y.; Katz, J.; Krajden, M.; Tellier, R.; et al. Human illness from avian influenza H7N3, British Columbia. Emerg. Infect. Dis. 2004, 10, 2196–2199. [Google Scholar] [CrossRef] [PubMed]
- Puzelli, S.; di Trani, L.; Fabiani, C.; Campitelli, L.; de Marco, M.A.; Capua, I.; Aguilera, J.F.; Zambon, M.; Donatelli, I. Serological analysis of serum samples from humans exposed to avian H7 influenza viruses in Italy between 1999 and 2003. J. Infect. Dis. 2005, 192, 1318–1322. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Martinez, I.; Balish, A.; Barrera-Badillo, G.; Jones, J.; Nunez-Garcia, T.E.; Jang, Y.; Aparicio-Antonio, R.; Azziz-Baumgartner, E.; Belser, J.A.; Ramirez-Gonzalez, J.E.; et al. Highly pathogenic avian influenza A(H7N3) virus in poultry workers, Mexico, 2012. Emerg. Infect. Dis. 2013, 19, 1531–1534. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Van-Tam, J.S.; Nair, P.; Acheson, P.; Baker, A.; Barker, M.; Bracebridge, S.; Croft, J.; Ellis, J.; Gelletlie, R.; Gent, N.; et al. Outbreak of low pathogenicity H7N3 avian influenza in UK, including associated case of human conjunctivitis. Euro Surveill. 2006, 11, E060504 060502. [Google Scholar] [PubMed]
- Puzelli, S.; Rossini, G.; Facchini, M.; Vaccari, G.; di Trani, L.; di Martino, A.; Gaibani, P.; Vocale, C.; Cattoli, G.; Bennett, M.; et al. Human infection with highly pathogenic A(H7N7) avian influenza virus, Italy, 2013. Emerg. Infect. Dis. 2014, 20, 1745–1749. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Background and Summary of Human Infection with Avian Influenza A(H7N9) Virus—As of 31 January 2014. Available online: http://www.who.int/influenza/human_animal_interface/20140131_background_and_summary_H7N9_v1.pdf?ua=1 (accessed on 21 December 2016).
- Huang, Y.; Li, X.; Zhang, H.; Chen, B.; Jiang, Y.; Yang, L.; Zhu, W.; Hu, S.; Zhou, S.; Tang, Y.; et al. Human infection with an avian influenza A (H9N2) virus in the middle region of China. J. Med. Virol. 2015, 87, 1641–1648. [Google Scholar] [CrossRef] [PubMed]
- Pan American Health Organization. Avian Influenza Virus A(H10N7) Circulating among Humans in Egypt. Available online: http://www1.paho.org/hq/dmdocuments/2010/Avian_Influenza_Egypt_070503.pdf (accessed on 17 December 2016).
- Arzey, G.G.; Kirkland, P.D.; Arzey, K.E.; Frost, M.; Maywood, P.; Conaty, S.; Hurt, A.C.; Deng, Y.M.; Iannello, P.; Barr, I.; et al. Influenza virus A (H10N7) in chickens and poultry abattoir workers, Australia. Emerg. Infect. Dis. 2012, 18, 814–816. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Yuan, H.; Gao, R.; Zhang, J.; Wang, D.; Xiong, Y.; Fan, G.; Yang, F.; Li, X.; Zhou, J.; et al. Clinical and epidemiological characteristics of a fatal case of avian influenza A H10N8 virus infection: A descriptive study. Lancet 2014, 383, 714–721. [Google Scholar] [CrossRef]
- Hall, M.; Frank, E.; Holmes, G.; Pfahringer, B.; Reutemann, P.; Witten, I.H. The WEKA data mining software: An update. SIGKDD Explor. 2009, 11, 10–18. [Google Scholar] [CrossRef]
- Fawcett, T. An introduction to ROC analysis. Pattern Recogn. Lett. 2006, 27, 861–874. [Google Scholar] [CrossRef]
- Linden, A. Measuring diagnostic and predictive accuracy in disease management: An introduction to receiver operating characteristic (ROC) analysis. J. Eval. Clin. Pract. 2006, 12, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Herrick, K.A.; Huettmann, F.; Lindgren, M.A. A global model of avian influenza prediction in wild birds: The importance of northern regions. BMC Vet. Res. 2013, 44, 42. [Google Scholar] [CrossRef] [PubMed]

| Stage | Overall Accuracy | Weighted AUC | Zoonotic Accuracy | Zoonotic Sensitivity | Zoonotic Specificity | Zoonotic AUC |
|---|---|---|---|---|---|---|
| Training | 99.20 | 1.000 | 98.40 | 0.984 | 0.996 | 1.000 |
| Testing | 99.06 | 1.000 | 97.14 | 0.971 | 1.000 | 0.999 |
| Zoonotic Probability Estimate | Zoonotic Risk |
|---|---|
| ≥0.7 | High |
| <0.7 | Low to none |
| Category | Training Dataset | Testing Dataset | Total Samples in Group |
|---|---|---|---|
| Avian | 125 | 34 | 159 |
| Human | 127 | 37 | 164 |
| Zoonotic | 125 | 35 | 160 |
| Total samples in dataset | 377 | 106 | 483 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eng, C.L.P.; Tong, J.C.; Tan, T.W. Predicting Zoonotic Risk of Influenza A Viruses from Host Tropism Protein Signature Using Random Forest. Int. J. Mol. Sci. 2017, 18, 1135. https://doi.org/10.3390/ijms18061135
Eng CLP, Tong JC, Tan TW. Predicting Zoonotic Risk of Influenza A Viruses from Host Tropism Protein Signature Using Random Forest. International Journal of Molecular Sciences. 2017; 18(6):1135. https://doi.org/10.3390/ijms18061135
Chicago/Turabian StyleEng, Christine L. P., Joo Chuan Tong, and Tin Wee Tan. 2017. "Predicting Zoonotic Risk of Influenza A Viruses from Host Tropism Protein Signature Using Random Forest" International Journal of Molecular Sciences 18, no. 6: 1135. https://doi.org/10.3390/ijms18061135
APA StyleEng, C. L. P., Tong, J. C., & Tan, T. W. (2017). Predicting Zoonotic Risk of Influenza A Viruses from Host Tropism Protein Signature Using Random Forest. International Journal of Molecular Sciences, 18(6), 1135. https://doi.org/10.3390/ijms18061135