
Engineering Exosomes for Cancer Therapy



Abstract
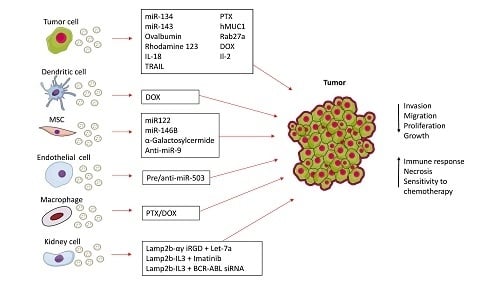
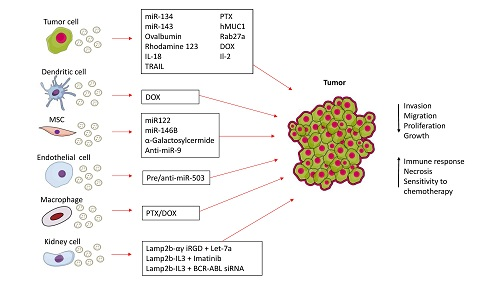
:
1. Introduction
2. Direct Modification of Isolated Exosomes for Cancer Therapy
3. Genetic Engineering of Exosomes
3.1. Modifying the Surface of Exosomes
3.2. Genetic Engineering of Exosome Content
3.3. Genetically Modified Exosomes for Immune Modulation
4. Discussion
Acknowledgments
Conflicts of Interest
References
- American Cancer Society. Global Cancer Facts & Figures, 3rd ed.; American Cancer Society: Atlanta, GA, USA, 2015. [Google Scholar]
- Thomas, C.E.; Ehrhardt, A.; Kay, M.A. Progress and problems with the use of viral vectors for gene therapy. Nat. Rev. Genet. 2003, 4, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Mizrak, A.; Bolukbasi, M.F.; Ozdener, G.B.; Brenner, G.J.; Madlener, S.; Erkan, E.P.; Strobel, T.; Breakefield, X.O.; Saydam, O. Genetically engineered microvesicles carrying suicide mRNA/protein inhibit schwannoma tumor growth. Mol. Ther. 2013, 21, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Johnsen, K.B.; Gudbergsson, J.M.; Skov, M.N.; Pilgaard, L.; Moos, T.; Duroux, M. A comprehensive overview of exosomes as drug delivery vehicles—Endogenous nanocarriers for targeted cancer therapy. Biochim. Biophys. Acta 2014, 1846, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Valadi, H.; Ekstrom, K.; Bossios, A.; Sjostrand, M.; Lee, J.J.; Lotvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Pegtel, D.M.; Cosmopoulos, K.; Thorley-Lawson, D.A.; van Eijndhoven, M.A.; Hopmans, E.S.; Lindenberg, J.L.; de Gruijl, T.D.; Wurdinger, T.; Middeldorp, J.M. Functional delivery of viral miRNAs via exosomes. Proc. Natl. Acad. Sci. USA 2010, 107, 6328–6333. [Google Scholar] [CrossRef] [PubMed]
- Kosaka, N.; Iguchi, H.; Yoshioka, Y.; Takeshita, F.; Matsuki, Y.; Ochiya, T. Secretory mechanisms and intercellular transfer of microRNAs in living cells. J. Biol. Chem. 2010, 285, 17442–17452. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, D.; Chen, X.; Li, J.; Li, L.; Bian, Z.; Sun, F.; Lu, J.; Yin, Y.; Cai, X.; et al. Secreted monocytic miR-150 enhances targeted endothelial cell migration. Mol. Cell 2010, 39, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Haney, M.J.; Zhao, Y.; Mahajan, V.; Deygen, I.; Klyachko, N.L.; Inskoe, E.; Piroyan, A.; Sokolsky, M.; Okolie, O.; et al. Development of exosome-encapsulated paclitaxel to overcome MDR in cancer cells. Nanomedicine 2016, 12, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Lamichhane, T.N.; Raiker, R.S.; Jay, S.M. Exogenous DNA Loading into Extracellular Vesicles via Electroporation is Size-Dependent and Enables Limited Gene Delivery. Mol. Pharm. 2015, 12, 3650–3657. [Google Scholar] [CrossRef] [PubMed]
- Kooijmans, S.A.; Stremersch, S.; Braeckmans, K.; de Smedt, S.C.; Hendrix, A.; Wood, M.J.; Schiffelers, R.M.; Raemdonck, K.; Vader, P. Electroporation-Induced siRNA precipitation obscures the efficiency of siRNA loading into extracellular vesicles. J. Control. Release 2013, 172, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Felgner, P.L.; Gadek, T.R.; Holm, M.; Roman, R.; Chan, H.W.; Wenz, M.; Northrop, J.P.; Ringold, G.M.; Danielsen, M. Lipofection: A highly efficient, lipid-mediated DNA-transfection procedure. Proc. Natl. Acad. Sci. USA 1987, 84, 7413–7417. [Google Scholar] [CrossRef] [PubMed]
- Gresch, O.; Engel, F.B.; Nesic, D.; Tran, T.T.; England, H.M.; Hickman, E.S.; Korner, I.; Gan, L.; Chen, S.; Castro-Obregon, S.; et al. New non-viral method for gene transfer into primary cells. Methods 2004, 33, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.; Wei, D.; Wu, Z.; Zhou, X.; Wei, X.; Huang, H.; Li, G. Phase I clinical trial of autologous ascites-derived exosomes combined with GM-CSF for colorectal cancer. Mol. Ther. 2008, 16, 782–790. [Google Scholar] [CrossRef] [PubMed]
- Escudier, B.; Dorval, T.; Chaput, N.; Andre, F.; Caby, M.P.; Novault, S.; Flament, C.; Leboulaire, C.; Borg, C.; Amigorena, S.; et al. Vaccination of metastatic melanoma patients with autologous dendritic cell (DC) derived-exosomes: Results of thefirst phase I clinical trial. J. Transl. Med. 2005, 3, 10. [Google Scholar] [CrossRef] [PubMed]
- Morse, M.A.; Garst, J.; Osada, T.; Khan, S.; Hobeika, A.; Clay, T.M.; Valente, N.; Shreeniwas, R.; Sutton, M.A.; Delcayre, A.; et al. A phase I study of dexosome immunotherapy in patients with advanced non-small cell lung cancer. J. Transl. Med. 2005, 3, 9. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Regnault, A.; Lozier, A.; Wolfers, J.; Flament, C.; Tenza, D.; Ricciardi-Castagnoli, P.; Raposo, G.; Amigorena, S. Eradication of established murine tumors using a novel cell-free vaccine: Dendritic cell-derived exosomes. Nat. Med. 1998, 4, 594–600. [Google Scholar] [CrossRef] [PubMed]
- Andre, F.; Chaput, N.; Schartz, N.E.; Flament, C.; Aubert, N.; Bernard, J.; Lemonnier, F.; Raposo, G.; Escudier, B.; Hsu, D.H.; et al. Exosomes as potent cell-free peptide-based vaccine. I. Dendritic cell-derived exosomes transfer functional MHC class I/peptide complexes to dendritic cells. J. Immunol. 2004, 172, 2126–2136. [Google Scholar] [CrossRef] [PubMed]
- Chaput, N.; Schartz, N.E.C.; Andre, F.; Taieb, J.; Novault, S.; Bonnaventure, P.; Aubert, N.; Bernard, J.; Lemonnier, F.; Merad, M.; et al. Exosomes as potent cell-free peptide-based vaccine. II. Exosomes in CpG adjuvants efficiently prime naive Tc1 lymphocytes leading to tumor rejection. J. Immunol. 2004, 172, 2137–2146. [Google Scholar] [CrossRef] [PubMed]
- Viaud, S.; Terme, M.; Flament, C.; Taieb, J.; Andre, F.; Novault, S.; Escudier, B.; Robert, C.; Caillat-Zucman, S.; Tursz, T.; et al. Dendritic cell-derived exosomes promote natural killer cell activation and proliferation: A role for NKG2D ligands and IL-15Ralpha. PLoS ONE 2009, 4, e4942. [Google Scholar] [CrossRef] [PubMed]
- Romagnoli, G.G.; Zelante, B.B.; Toniolo, P.A.; Migliori, I.K.; Barbuto, J.A. Dendritic Cell-Derived Exosomes may be a Tool for Cancer Immunotherapy by Converting Tumor Cells into Immunogenic Targets. Front. Immunol. 2014, 5, 692. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Martin, P.; Fogarty, B.; Brown, A.; Schurman, K.; Phipps, R.; Yin, V.P.; Lockman, P.; Bai, S. Exosome delivered anticancer drugs across the blood-brain barrier for brain cancer therapy in Danio rerio. Pharm. Res. 2015, 32, 2003–2014. [Google Scholar] [CrossRef] [PubMed]
- Koppers-Lalic, D.; Hogenboom, M.M.; Middeldorp, J.M.; Pegtel, D.M. Virus-modified exosomes for targeted RNA delivery; a new approach in nanomedicine. Adv. Drug Deliv. Rev. 2013, 65, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Vlassov, A.V.; Magdaleno, S.; Setterquist, R.; Conrad, R. Exosomes: Current knowledge of their composition, biological functions, and diagnostic and therapeutic potentials. Biochim. Biophys. Acta 2012, 1820, 940–948. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, A.; Costa-Silva, B.; Shen, T.L.; Rodrigues, G.; Hashimoto, A.; Tesic, M.; Molina, H.; Kohsaka, S.; di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tew, S.R.; Russell, A.M.; Gonzalez, K.R.; Hardingham, T.E.; Hawkins, R.E. Transduction of passaged human articular chondrocytes with adenoviral, retroviral, and lentiviral vectors and the effects of enhanced expression of SOX9. Tissue Eng. 2004, 10, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Ohno, S.I.; Takanashi, M.; Sudo, K.; Ueda, S.; Ishikawa, A.; Matsuyama, N.; Fujita, K.; Mizutani, T.; Ohgi, T.; Ochiya, T.; et al. Systemically Injected Exosomes Targeted to EGFR Deliver Antitumor MicroRNA to Breast Cancer Cells. Mol. Ther. 2013, 21, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Li, S.; Song, J.; Ji, T.; Zhu, M.; Anderson, G.J.; Wei, J.; Nie, G. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials 2014, 35, 2383–2390. [Google Scholar] [CrossRef] [PubMed]
- Bellavia, D.; Raimondo, S.; Calabrese, G.; Forte, S.; Cristaldi, M.; Patinella, A.; Memeo, L.; Manno, M.; Raccosta, S.; Diana, P.; et al. Interleukin 3-receptor targeted exosomes inhibit in vitro and in vivo Chronic Myelogenous Leukemia cell growth. Theranostics 2017, 7, 1333–1345. [Google Scholar] [CrossRef] [PubMed]
- Rivoltini, L.; Chiodoni, C.; Squarcina, P.; Tortoreto, M.; Villa, A.; Vergani, B.; Burdek, M.; Botti, L.; Arioli, I.; Cova, A.; et al. TNF-Related Apoptosis-Inducing Ligand (TRAIL)-Armed Exosomes Deliver Proapoptotic Signals to Tumor Site. Clin. Cancer Res. 2016, 22, 3499–3512. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Kolluri, K.K.; Gowers, K.H.; Janes, S.M. TRAIL delivery by MSC-derived extracellular vesicles is an effective anticancer therapy. J. Extracell. Vesicles 2017, 6, 1265291. [Google Scholar] [CrossRef] [PubMed]
- O'Brien, K.; Lowry, M.C.; Corcoran, C.; Martinez, V.G.; Daly, M.; Rani, S.; Gallagher, W.M.; Radomski, M.W.; MacLeod, R.A.; O'Driscoll, L. miR-134 in extracellular vesicles reduces triple-negative breast cancer aggression and increases drug sensitivity. Oncotarget 2015, 6, 32774–32789. [Google Scholar] [PubMed]
- Bovy, N.; Blomme, B.; Freres, P.; Dederen, S.; Nivelles, O.; Lion, M.; Carnet, O.; Martial, J.A.; Noel, A.; Thiry, M.; et al. Endothelial exosomes contribute to the antitumor response during breast cancer neoadjuvant chemotherapy via microRNA transfer. Oncotarget 2015, 6, 10253–10266. [Google Scholar] [CrossRef] [PubMed]
- Akao, Y.; Nakagawa, Y.; Hirata, I.; Iio, A.; Itoh, T.; Kojima, K.; Nakashima, R.; Kitade, Y.; Naoe, T. Role of anti-oncomirs miR-143 and -145 in human colorectal tumors. Cancer Gene Ther. 2010, 17, 398–408. [Google Scholar] [CrossRef] [PubMed]
- Lou, G.; Song, X.; Yang, F.; Wu, S.; Wang, J.; Chen, Z.; Liu, Y. Exosomes derived from miR-122-modified adipose tissue-derived MSCs increase chemosensitivity of hepatocellular carcinoma. J. Hematol. Oncol. 2015, 8, 122. [Google Scholar] [CrossRef] [PubMed]
- Katakowski, M.; Buller, B.; Zheng, X.; Lu, Y.; Rogers, T.; Osobamiro, O.; Shu, W.; Jiang, F.; Chopp, M. Exosomes from marrow stromal cells expressing miR-146b inhibit glioma growth. Cancer Lett. 2013, 335, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.A.; Yeo, D.J.; Son, H.Y.; Kim, H.W.; Jung, D.S.; Ko, J.K.; Koh, J.S.; Kim, Y.N.; Kim, C.W. Exosomes: A new delivery system for tumor antigens in cancer immunotherapy. Int. J. Cancer 2005, 114, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Mu, D.; Tian, F.; Hu, Y.; Jiang, T.; Han, Y.; Chen, J.; Han, G.; Li, X. Exosomes derived from Rab27aoverexpressing tumor cells elicit efficient induction of antitumor immunity. Mol. Med. Rep. 2013, 8, 1876–1882. [Google Scholar] [PubMed]
- Gehrmann, U.; Hiltbrunner, S.; Georgoudaki, A.M.; Karlsson, M.C.; Naslund, T.I.; Gabrielsson, S. Synergistic induction of adaptive antitumor immunity by codelivery of antigen with alpha-galactosylceramide on exosomes. Cancer Res. 2013, 73, 3865–3876. [Google Scholar] [CrossRef] [PubMed]
- Munoz, J.L.; Bliss, S.A.; Greco, S.J.; Ramkissoon, S.H.; Ligon, K.L.; Rameshwar, P. Delivery of Functional Anti-miR-9 by Mesenchymal Stem Cell-derived Exosomes to Glioblastoma Multiforme Cells Conferred Chemosensitivity. Mol. Ther. Nucleic Acids 2013, 2, e126. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.; Zhou, X.; Wang, B.; Wang, Q.; Fu, Y.; Chen, T.; Wan, T.; Yu, Y.; Cao, X. Enhanced induction of dendritic cell maturation and HLA-A*0201-restricted CEA-specific CD8(+) CTL response by exosomes derived from IL-18 gene-modified CEA-positive tumor cells. J. Mol. Med. 2006, 84, 1067–1076. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Xiu, F.; Cai, Z.; Wang, J.; Wang, Q.; Fu, Y.; Cao, X. Increased induction of antitumor response by exosomes derived from interleukin-2 gene-modified tumor cells. J. Cancer Res. Clin. Oncol. 2007, 133, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Drescher, K.M.; Chen, X.M. Exosomal miRNAs: Biological Properties and Therapeutic Potential. Front. Genet. 2012, 3, 56. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K.; et al. Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524–15529. [Google Scholar] [CrossRef] [PubMed]
- Cimmino, A.; Calin, G.A.; Fabbri, M.; Iorio, M.V.; Ferracin, M.; Shimizu, M.; Wojcik, S.E.; Aqeilan, R.I.; Zupo, S.; Dono, M.; et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc. Natl. Acad. Sci. USA 2005, 102, 13944–13949. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.W.; Ferland-McCollough, D.; Jackson, T.J.; Bushell, M. microRNAs in cancer management. Lancet Oncol. 2012, 13, e249–e258. [Google Scholar] [CrossRef]
- Consortium, E.-T.; van Deun, J.; Mestdagh, P.; Agostinis, P.; Akay, O.; Anand, S.; Anckaert, J.; Martinez, Z.A.; Baetens, T.; Beghein, E.; et al. EV-TRACK: Transparent reporting and centralizing knowledge in extracellular vesicle research. Nat. Methods 2017, 14, 228–232. [Google Scholar]

{kind=link}
| Exosome Source | Setting | Therapy | Tumour | Study Outcome | Reference |
|---|---|---|---|---|---|
| Macrophages (RAW 264.7) | In Vivo | PTX/DOX | Lung Mets | Exosomal PTX preferentially accumulated in cancer cells | [9] |
| Ascites-derived | Clinical trial | AEX alone or AEX + GM-CSF | Colorectal | AEX + GM-CSF was safe, nontoxic, tolerable, and induced a beneficial tumour-specific anti-tumour CTL response | [14] |
| Dendritic cells | Clinical trial | MHC Class II peptides | Melanoma | Large scale exosome production was feasible and exosome administration was safe and well tolerated | [15] |
| Dendritic cells | Clinical trial | MAGE (tumour antigens) | Lung | Therapy well tolerated with some experiencing long term stable disease and activation of immune effectors | [16] |
| Dendritic cells | In Vivo | IL-4 + GM-CSF | Breast | Eradication/suppression of growth of pre-established tumours in a T-cell dependant manner | [17] |
| Dendritic cells | In Vivo | MHC Class I | Melanoma | MHC Class I restricted CD8+ T-cell expansion and differentiation | [18] |
| Dendritic cells | In Vivo | CpG Adjuvant | Melanoma | Combination of exosomes and TLR 3 + 9 triggered efficient MHC-restricted CD8+ T-cell responses | [19] |
| Dendritic cells | In Vivo | DC-Exo alone | Melanoma | DC-Exo promoted IL-15Rα- and NKG2D-dependent NK cell proliferation and activation which resulted in anti-metastatic effects | [20] |
| Dendritic cells | In Vitro | DC-Exo alone | Breast | Incorporation of DC-Exo by tumour cells increased ability to activate T-cells for a more effective response | [21] |
| Brain endothelial cells | In Vivo | rhodamine 123, PTX, DOX | Brain | Exosome delivery allowed DOX and PTX to cross the BBB which resulted in cytotoxicity against U-87 MG cells | [22] |
| Exosome Source | Setting | Therapeutic Agent | Tumour Model | Study Outcome | Reference |
|---|---|---|---|---|---|
| Kidney cells (HEK293) | In Vivo | GE11 peptide + Let-7a | Breast | Tumor targeted delivery of Let-7a suppressed tumour growth | [27] |
| Dendritic cells | In Vivo | Lamp2b fused to αγ iRGD peptide + DOX | Breast | Significant inhibition of tumour growth, with no overt toxicity | [28] |
| Kidney cells (HEK293T) | In Vivo | Lamp2b IL3 + Imatinib or BCR-ABL siRNA | Chronic Myeloid Leukemia | IL3L surface improved tumor targeting. IL3L-Imatinib: reduced tumor size; IL3L BCR-ABL siRNA: slower tumor growth | [29] |
| Breast cancer (Hs578T) | In Vitro | miR-134 | Breast | Increased miR-134 significantly reduced STAT5B, Hsp90 and Bcl-2 levels resulting in reduced cellular proliferation | [32] |
| HUVECs | In Vitro | Pre/anti-miR-503 | Breast | Increased miR-503 decreased both proliferation and invasion. | [33] |
| Leukemia cells (THP-1) | In Vivo | miR-143 | Colon | Increased miR-143 levels in tumours resulted in suppression of growth. | [34] |
| AMSCs | In Vivo | miR-122 | Hepatocellular carcinoma | Cancer cells were rendered sensitive to chemotherapy through miR-122 expression | [35] |
| MSCs | In Vivo | miR-146b | Glioma | Intra-tumoural exosome injection significantly reduced tumour volume | [36] |
| Mouse colon (CT26) & breast (TA3HA) | In Vivo | hMUC1 | Colon | Tumour size was reduced by MUC-1. CT26-MUC-1 higher dose and TA3HA-MUC-1 lower dose showed best results. | [37] |
| Lung cancer (A549) | In Vivo | Rab27a | Adenocarcinoma | Immunization with Rab27a significantly inhibited tumour growth, with similar results seen in pre-established tumours | [38] |
| Mouse Bone Marrow Cells | In Vivo | α-Galactosylceramide | Melanoma | Induced an early iNKT-cell response, dendritic cell, MZB cell activation as well as NK- and T-cell activation and proliferation | [39] |
| MSCs | In Vitro | Anti-miR-9 | Glioblastoma mutliforme (GBM) | Reverse expression of miR-9 sensitized the GBM cells to TMZ which increased cell death and caspase activity | [40] |
| Colon (LS-174T) | In Vitro | IL-18 | Colon | Exo/IL-18 can chemoattract DCs and T cells which induces IFN-γ augmented release of IL-2 and promoted T-cell proliferation | [41] |
| Mouse thymoma (E.G7-OVA) | In Vivo | Ovalbumin, IL-2 | Thymoma | Induced antigen specific Th1-polarized immune response and CTL more efficiently resulting in tumour regression | [42] |
| Leukemia (K562) | In Vivo | TRAIL | B Lymphoma; Melanoma | Inhibition of tumour growth was seen in both groups, although not significantly in the melanoma group | [30] |
| MSC | In Vitro | TRAIL | Variety of cancer cell lines | Induction of apoptosis in range of cancer cell lines, including some TRAIL resistant cells. Effect enhanced through use of CDK9 inhibitor. | [31] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gilligan, K.E.; Dwyer, R.M. Engineering Exosomes for Cancer Therapy. Int. J. Mol. Sci. 2017, 18, 1122. https://doi.org/10.3390/ijms18061122
Gilligan KE, Dwyer RM. Engineering Exosomes for Cancer Therapy. International Journal of Molecular Sciences. 2017; 18(6):1122. https://doi.org/10.3390/ijms18061122
Chicago/Turabian StyleGilligan, Katie E., and Róisín M. Dwyer. 2017. "Engineering Exosomes for Cancer Therapy" International Journal of Molecular Sciences 18, no. 6: 1122. https://doi.org/10.3390/ijms18061122
APA StyleGilligan, K. E., & Dwyer, R. M. (2017). Engineering Exosomes for Cancer Therapy. International Journal of Molecular Sciences, 18(6), 1122. https://doi.org/10.3390/ijms18061122