Fast Detection of a BRCA2 Large Genomic Duplication by Next Generation Sequencing as a Single Procedure: A Case Report

 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
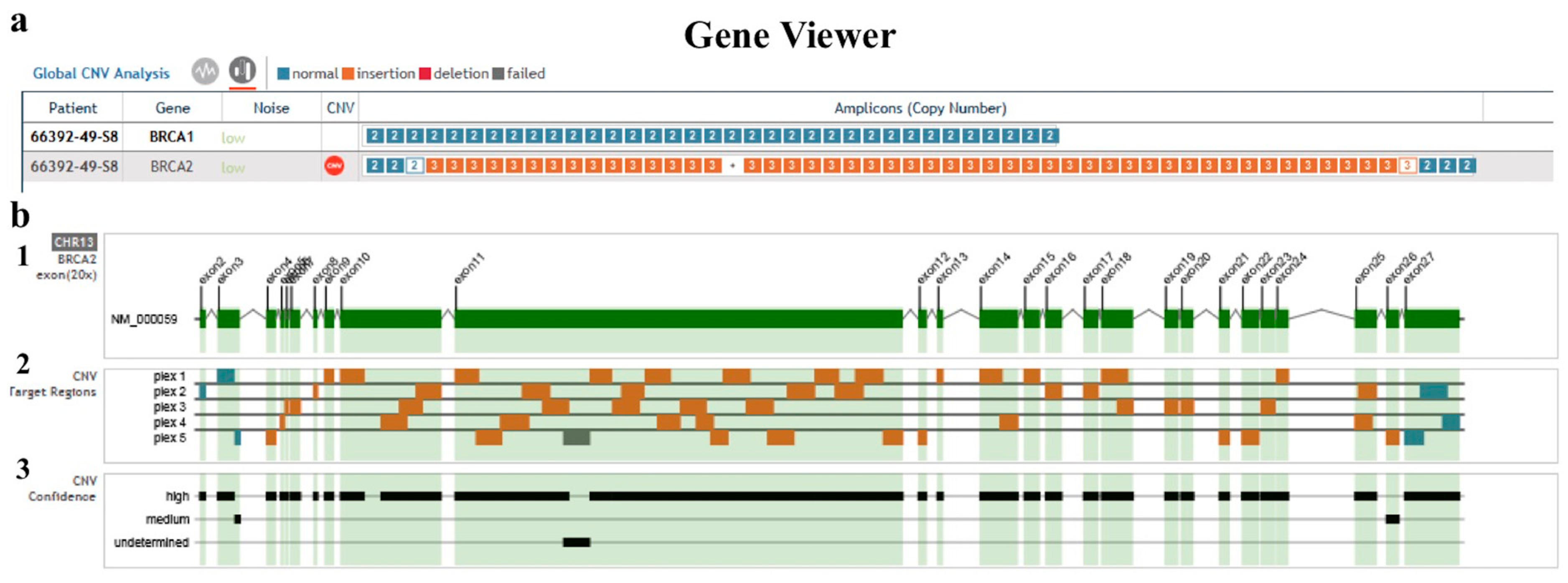
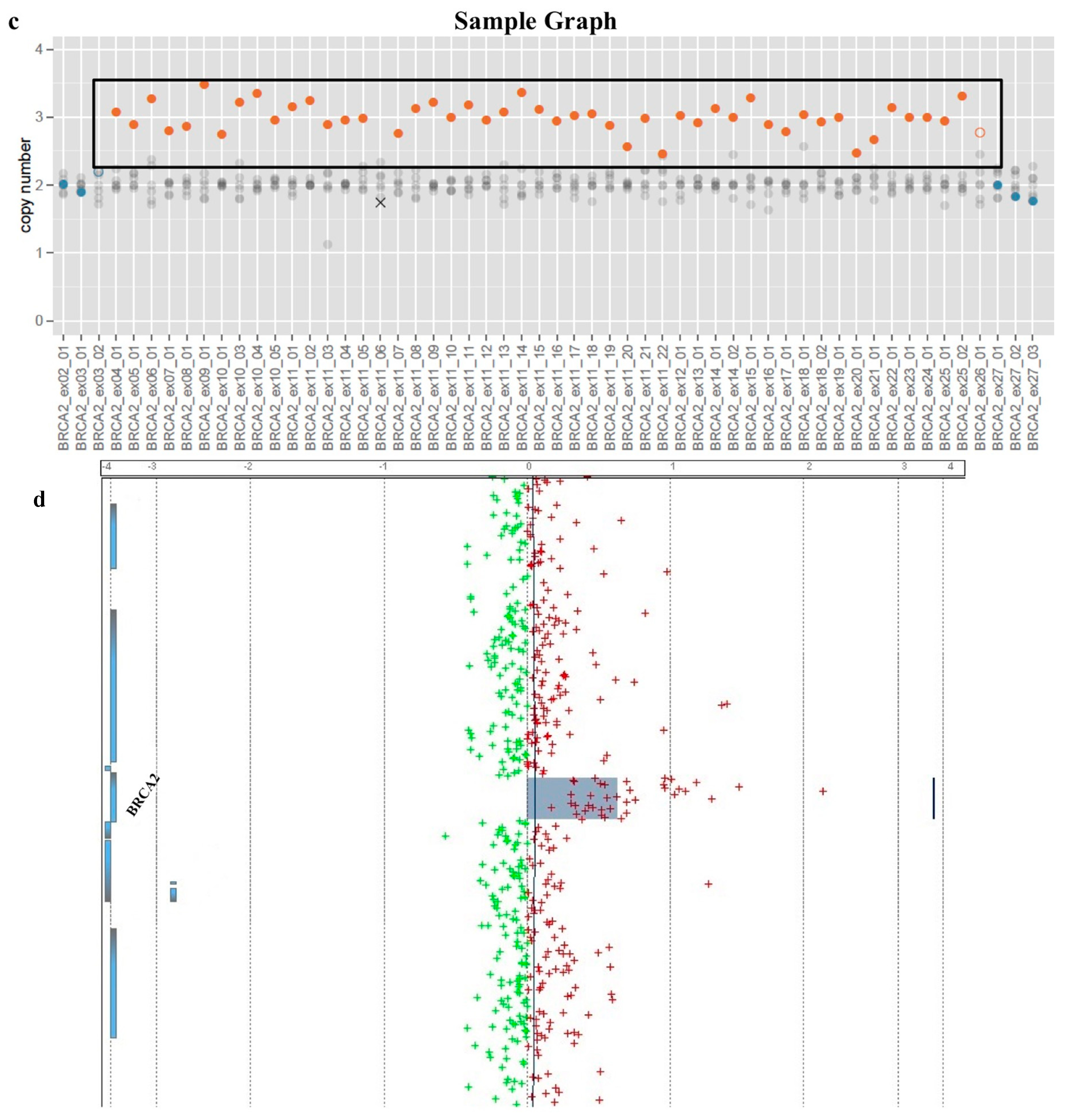
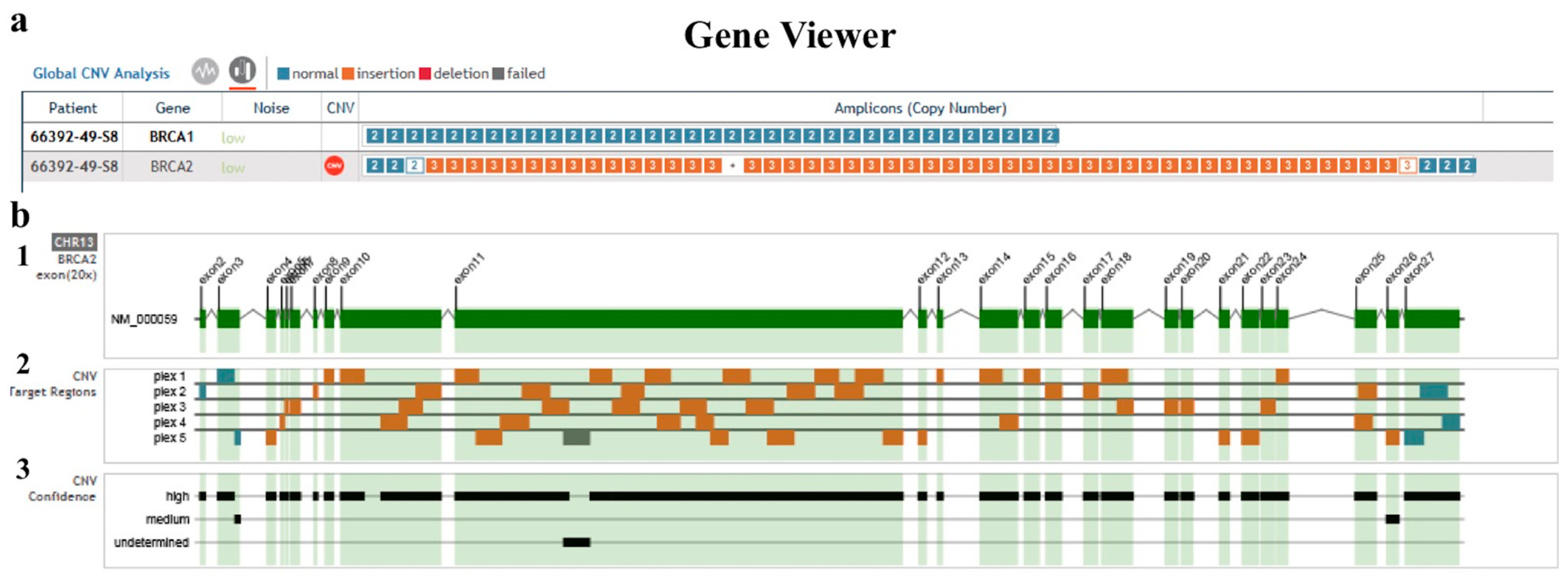
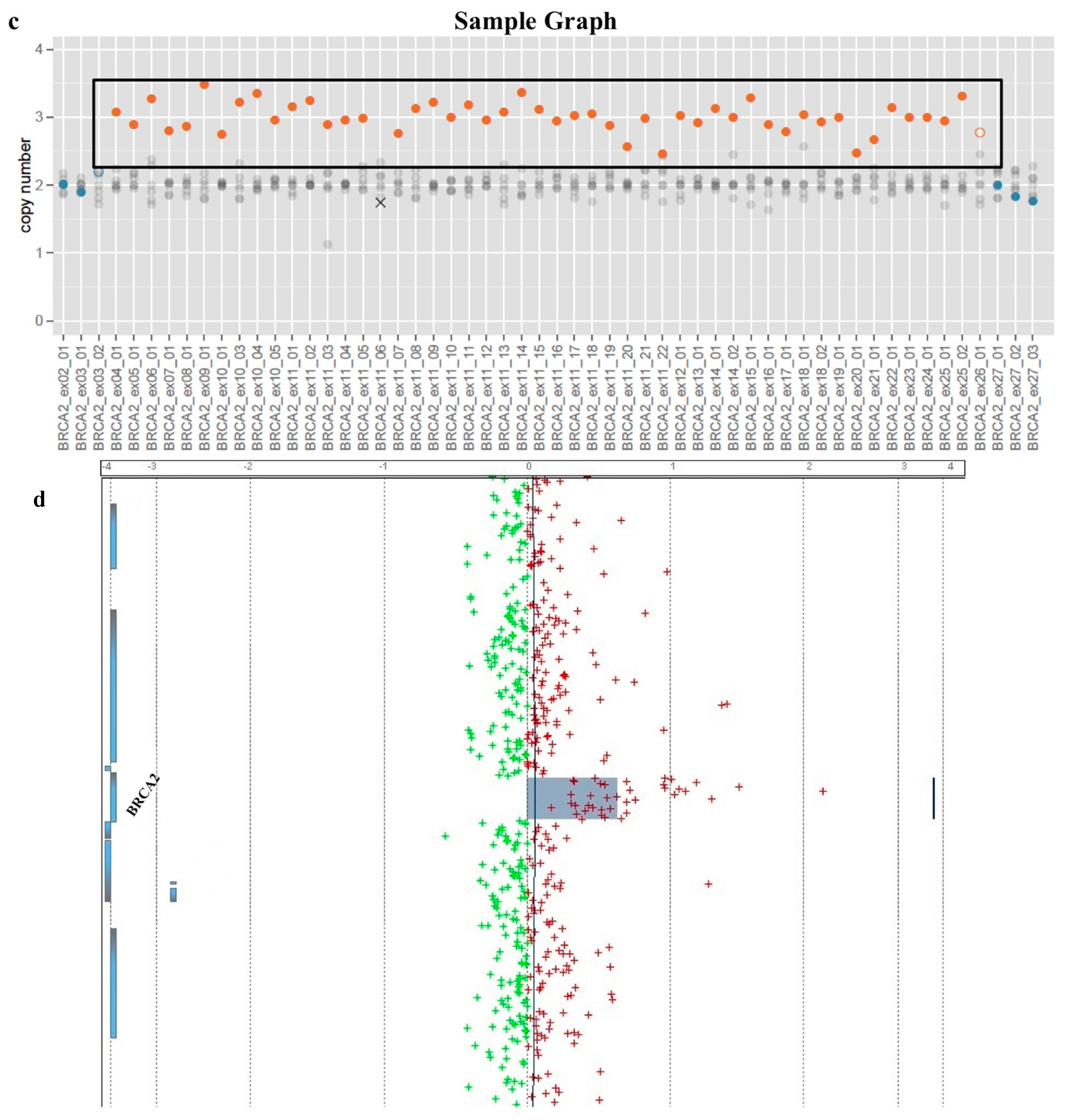
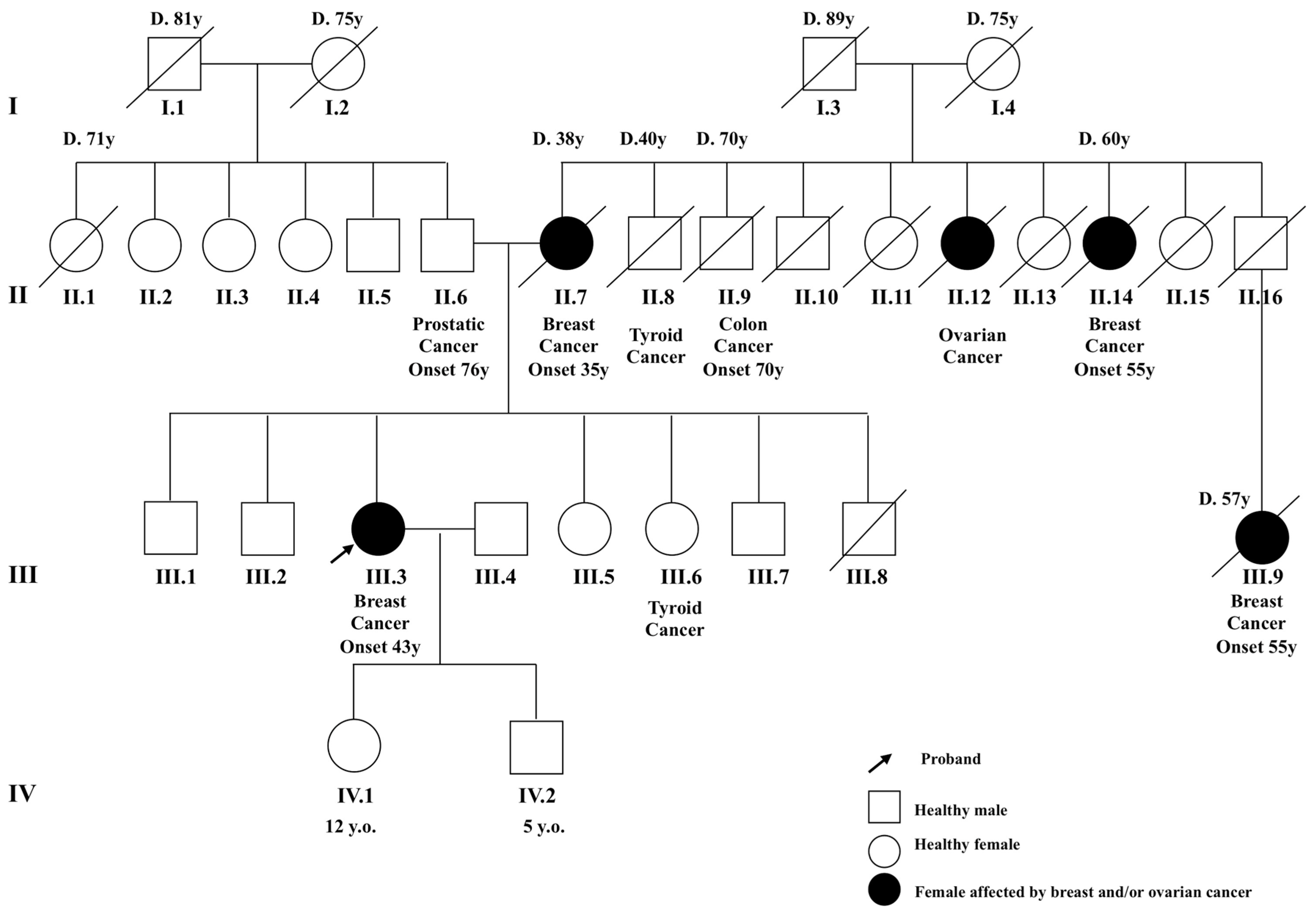
2. Results
3. Discussion
Note after Discussion
4. Materials and Methods
4.1. Enrollment of Patients and Collection of Samples
4.2. Next Generation Sequencing
4.3. Bioinformatic Analysis
4.4. Comparative Genomic Hybridization Array
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| HBOC | hereditary breast and ovarian cancer |
| LGR | large genomic rearrangement |
| NGS | next generation sequencing |
| aCGH | array-based comparative genomic hybridization |
| CNV | copy number variant |
References
- Lord, C.J.; Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Engel, C.; Fischer, C. Breast cancer risks and risk prediction models. Breast Care 2015, 10, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Hoogerbrugge, N.; Jongmans, M.C. Finding all BRCA pathogenic mutation carriers: Best practice models. Eur. J. Hum. Genet. 2016, 24, S19–S26. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, L.C.; Lindor, N.M. The Role of Risk-Reducing Surgery in Hereditary Breast and Ovarian Cancer. N. Engl. J. Med. 2016, 374, 454–468. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q. Cancer predisposition genes: Molecular mechanisms and clinical impact on personalized cancer care: Examples of Lynch and HBOC syndromes. Acta Pharmacol. Sin. 2016, 37, 143–149. [Google Scholar] [CrossRef] [PubMed]
- D’Argenio, V.; Esposito, M.V.; Telese, A.; Precone, V.; Starnone, F.; Nunziato, M.; Cantiello, P.; Iorio, M.; Evangelista, E.; D’Aiuto, M.; et al. The molecular analysis of BRCA1 and BRCA2: Next-generation sequencing supersedes conventional approaches. Clin. Chim. Acta 2015, 446, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Kwong, A.; Chen, J.; Shin, V.Y.; Ho, J.C.; Law, F.B.; Au, C.H.; Chan, T.L.; Ma, E.S.; Ford, J.M. The importance of analysis of long-range rearrangement of BRCA1 and BRCA2 in genetic diagnosis of familial breast cancer. Cancer Genet. 2015, 208, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Sluiter, M.D.; van Rensburg, E.J. Large genomic rearrangements of the BRCA1 and BRCA2 genes: Review of the literature and report of a novel BRCA1 mutation. Breast Cancer Res. Treat. 2011, 125, 325–349. [Google Scholar] [CrossRef] [PubMed]
- Ruiz de Garibay, G.; Gutiérrez-Enríquez, S.; Garre, P.; Bonache, S.; Romero, A.; Palomo, L.; Sánchez de Abajo, A.; Benítez, J.; Balmaña, J.; Pérez-Segura, P.; et al. Characterization of four novel BRCA2 large genomic rearrangements in Spanish breast/ovarian cancer families: Review of the literature, and reevaluation of the genetic mechanisms involved in their origin. Breast Cancer Res. Treat. 2012, 133, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Judkins, T.; Rosenthal, E.; Arnell, C.; Burbidge, L.A.; Geary, W.; Barrus, T.; Schoenberger, J.; Trost, J.; Wenstrup, R.J.; Roa, B.B. Clinical significance of large rearrangements in BRCA1 and BRCA2. Cancer 2012, 118, 5210–5216. [Google Scholar] [CrossRef] [PubMed]
- Riahi, A.; Chabouni-Bouhamed, H.; Kharrat, M. Prevalence of BRCA1 and BRCA2 large genomic rearrangements in Tunisian high risk breast/ovarian cancer families: Implications for genetic testing. Cancer Genet. 2017, 210, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Timoteo, A.R.; Albuquerque, B.M.; Moura, P.C.; Ramos, C.C.; Agnez-Lima, L.F.; Walsh, T.; King, M.C.; Lajus, T.B. Identification of a new BRCA2 large genomic deletion associated with high risk male breast cancer. Hered. Cancer Clin. Pract. 2015, 13, 2. [Google Scholar] [CrossRef] [PubMed]
- Precone, V.; Del Monaco, V.; Esposito, M.V.; De Palma, F.D.; Ruocco, A.; Salvatore, F.; D’Argenio, V. Cracking the Code of Human Diseases Using Next-Generation Sequencing: Applications, Challenges, and Perspectives. Biomed. Res. Int. 2015, 2015, 1–15. [Google Scholar] [CrossRef] [PubMed]
- D’Argenio, V.; Esposito, M.V.; Gilder, J.A.; Frisso, G.; Salvatore, F. Should a BRCA2 stop codon human variant, usually considered a polymorphism, be classified as a predisposing mutation? Cancer 2014, 120, 1594–1595. [Google Scholar] [CrossRef] [PubMed]
- Wallace, A.J. New challenges for BRCA testing: A view from the diagnostic laboratory. Eur. J. Hum. Genet. 2016, 24, S10–S18. [Google Scholar] [CrossRef] [PubMed]
- Esposito, M.V.; Nunziato, M.; Starnone, F.; Telese, A.; Calabrese, A.; D’Aiuto, G.; Pucci, P.; D’Aiuto, M.; Baralle, F.; D’Argenio, V.; et al. A Novel Pathogenic BRCA1 Splicing Variant Produces Partial Intron Retention in the Mature Messenger RNA. Int. J. Mol. Sci. 2016, 17, 2145. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.Y.; Hansen, T.V.O.; Ahlborn, L.B.; Jønson, L.; Yde, C.W.; Nielsen, F.C. Next-Generation Sequencing-Based Detection of Germline Copy Number Variations in BRCA1/BRCA2: Validation of a One-Step Diagnostic Workflow. J. Mol. Diagn. 2017, 19, 809–816. [Google Scholar] [CrossRef] [PubMed]
- Lim, B.C.; Lee, S.; Shin, J.Y.; Kim, J.I.; Hwang, H.; Kim, K.J.; Hwang, Y.S.; Seo, J.S.; Chae, J.H. Genetic diagnosis of Duchenne and Becker muscular dystrophy using next-generation sequencing technology: Comprehensive mutational search in a single platform. J. Med. Genet. 2011, 48, 731–736. [Google Scholar] [CrossRef] [PubMed]
- Concolino, P.; Rizza, R.; Hackmann, K.; Minucci, A.; Scaglione, G.L.; De Bonis, M.; Costella, A.; Zuppi, C.; Schrock, E.; Capoluongo, E. Identification and Characterization of a New BRCA2 Rearrangement in an Italian Family with Hereditary Breast and Ovarian Cancer Syndrome. Mol. Diagn. Ther. 2017. [Google Scholar] [CrossRef] [PubMed]
- Daly, M.B.; Pilarski, R.; Berry, M.; Buys, S.S.; Farmer, M.; Friedman, S.; Garber, J.E.; Kauff, N.D.; Khan, S.; Klein, C.; et al. NCCN Guidelines Insights: Genetic/Familial High-Risk Assessment: Breast and Ovarian, Version 2.2017. J. Natl. Compr. Cancer Netw. 2017, 15, 9–20. [Google Scholar] [CrossRef]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nunziato, M.; Starnone, F.; Lombardo, B.; Pensabene, M.; Condello, C.; Verdesca, F.; Carlomagno, C.; De Placido, S.; Pastore, L.; Salvatore, F.; et al. Fast Detection of a BRCA2 Large Genomic Duplication by Next Generation Sequencing as a Single Procedure: A Case Report. Int. J. Mol. Sci. 2017, 18, 2487. https://doi.org/10.3390/ijms18112487
Nunziato M, Starnone F, Lombardo B, Pensabene M, Condello C, Verdesca F, Carlomagno C, De Placido S, Pastore L, Salvatore F, et al. Fast Detection of a BRCA2 Large Genomic Duplication by Next Generation Sequencing as a Single Procedure: A Case Report. International Journal of Molecular Sciences. 2017; 18(11):2487. https://doi.org/10.3390/ijms18112487
Chicago/Turabian StyleNunziato, Marcella, Flavio Starnone, Barbara Lombardo, Matilde Pensabene, Caterina Condello, Francesco Verdesca, Chiara Carlomagno, Sabino De Placido, Lucio Pastore, Francesco Salvatore, and et al. 2017. "Fast Detection of a BRCA2 Large Genomic Duplication by Next Generation Sequencing as a Single Procedure: A Case Report" International Journal of Molecular Sciences 18, no. 11: 2487. https://doi.org/10.3390/ijms18112487
APA StyleNunziato, M., Starnone, F., Lombardo, B., Pensabene, M., Condello, C., Verdesca, F., Carlomagno, C., De Placido, S., Pastore, L., Salvatore, F., & D’Argenio, V. (2017). Fast Detection of a BRCA2 Large Genomic Duplication by Next Generation Sequencing as a Single Procedure: A Case Report. International Journal of Molecular Sciences, 18(11), 2487. https://doi.org/10.3390/ijms18112487