Apoptosis Induced by the Curcumin Analogue EF-24 Is Neither Mediated by Oxidative Stress-Related Mechanisms nor Affected by Expression of Main Drug Transporters ABCB1 and ABCG2 in Human Leukemia Cells



Abstract
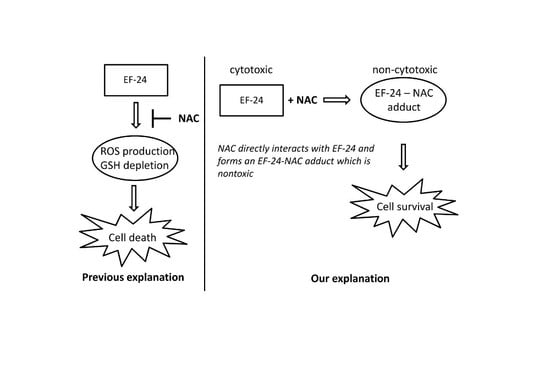
:
1. Introduction
2. Results
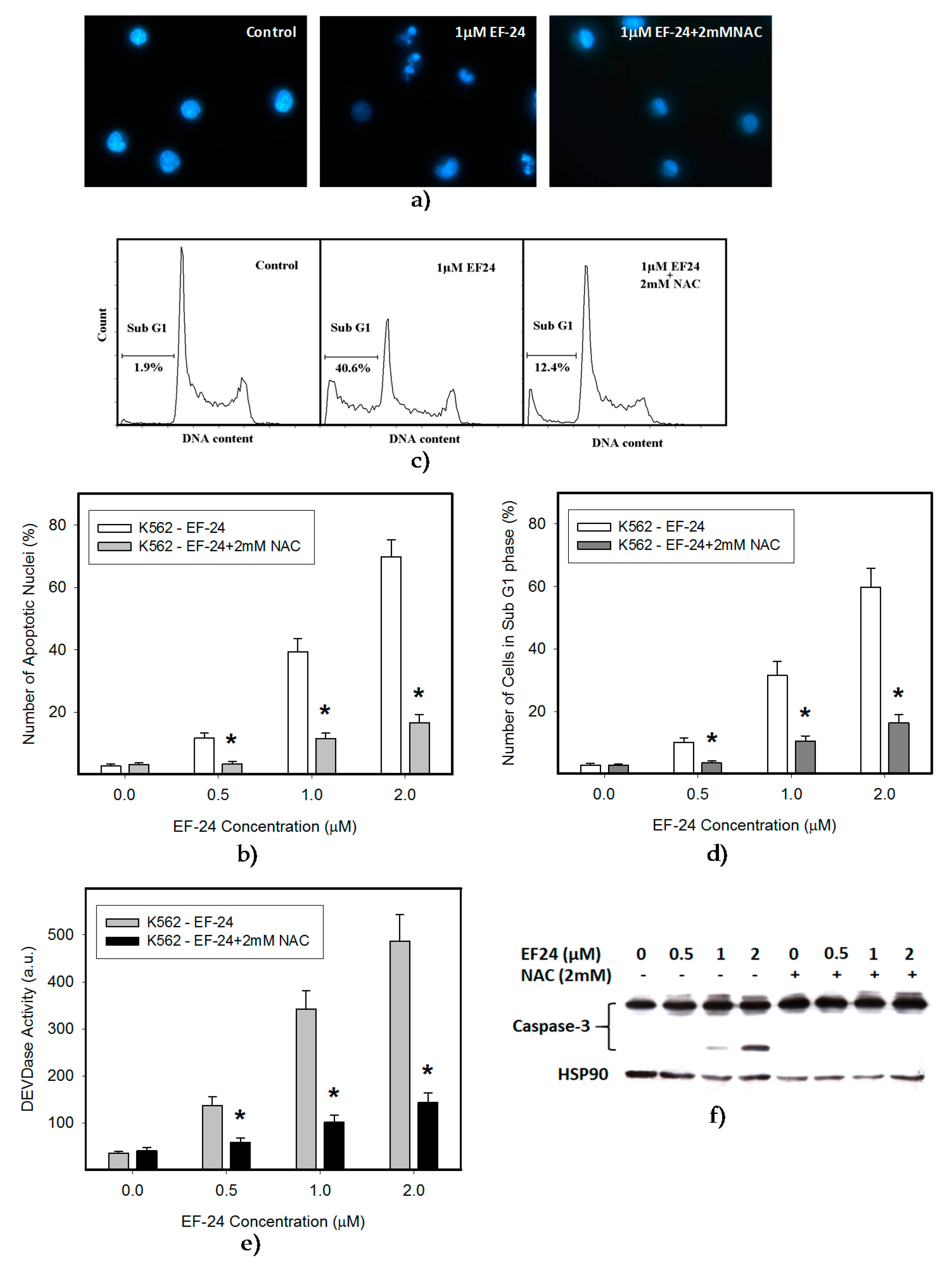
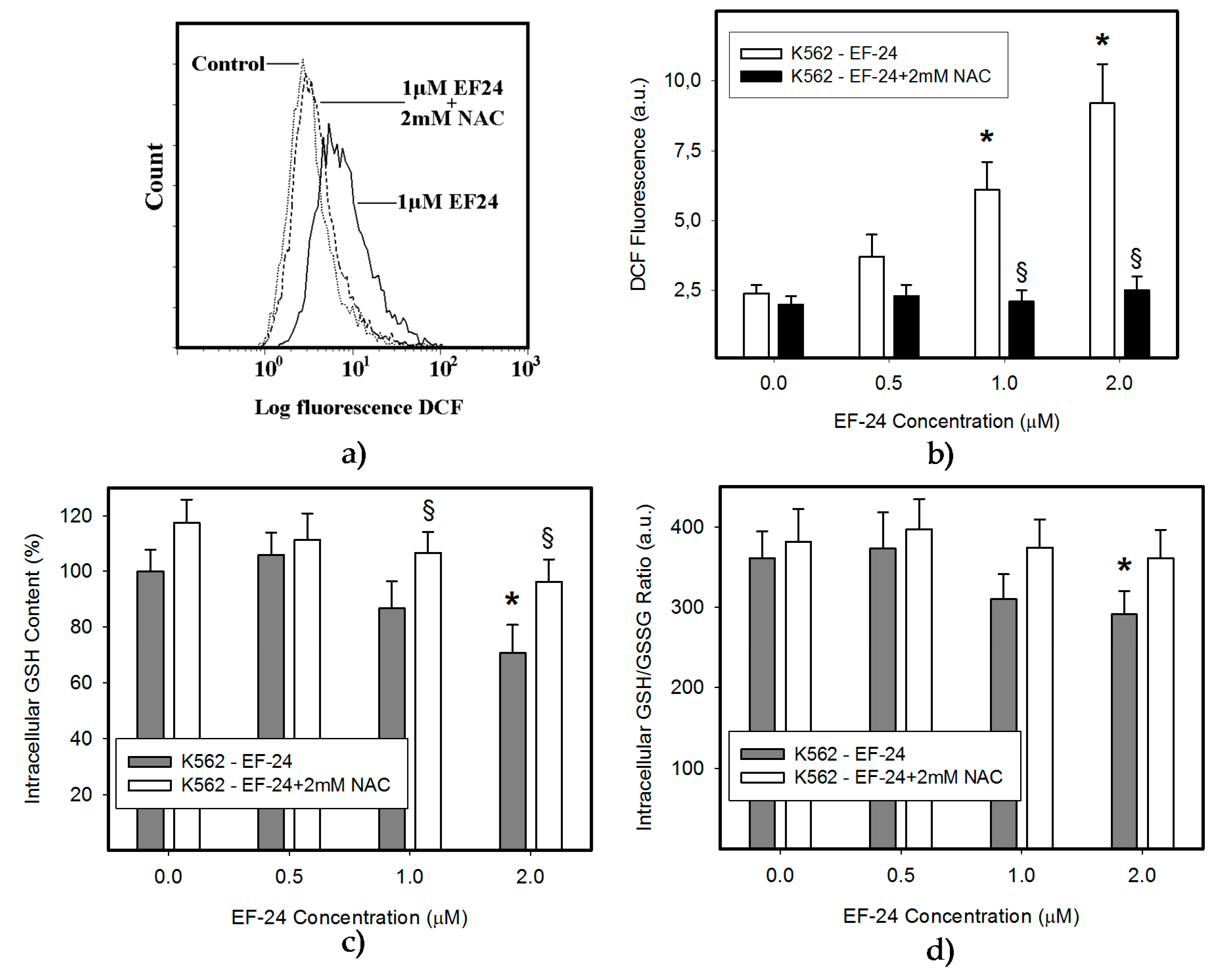
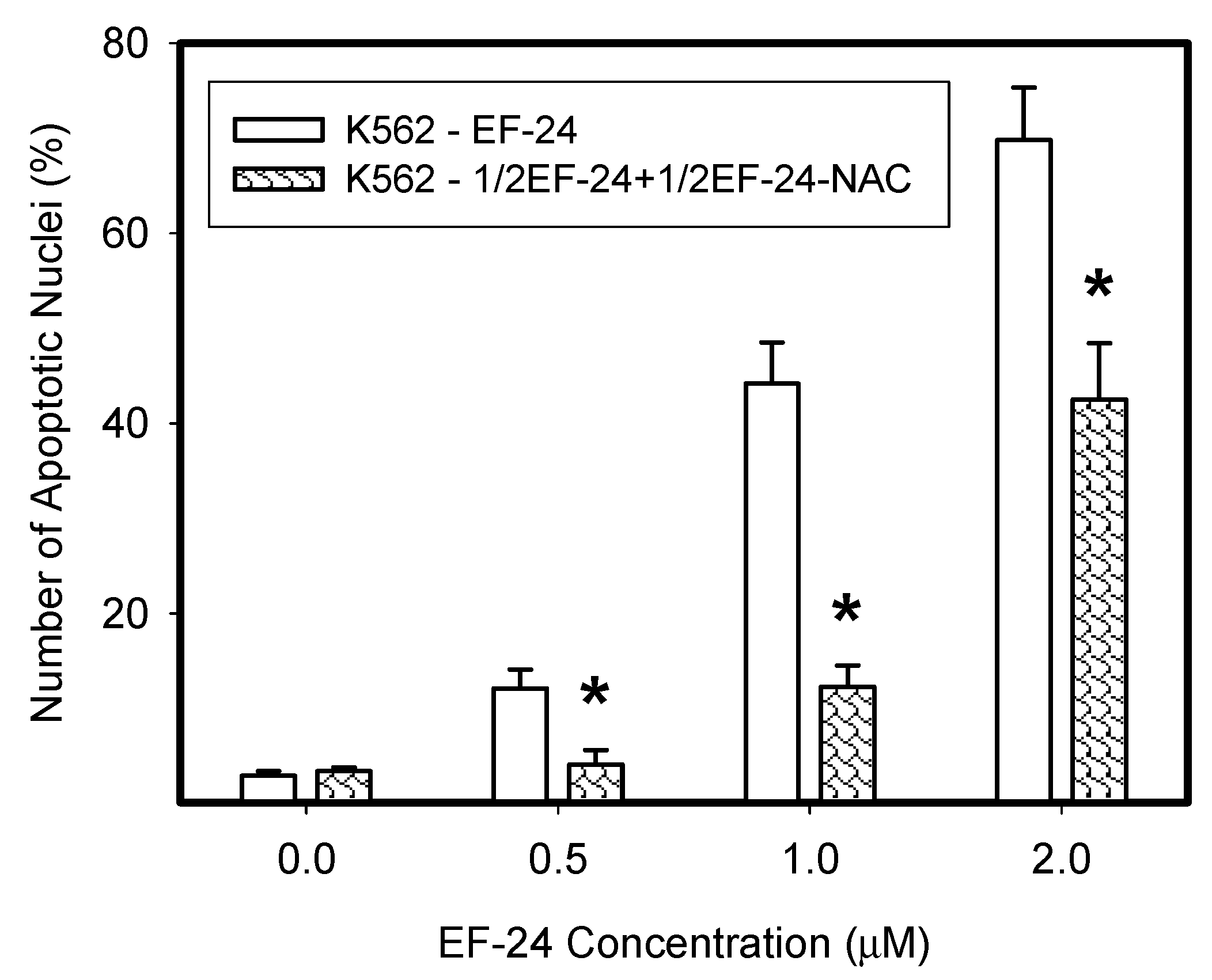
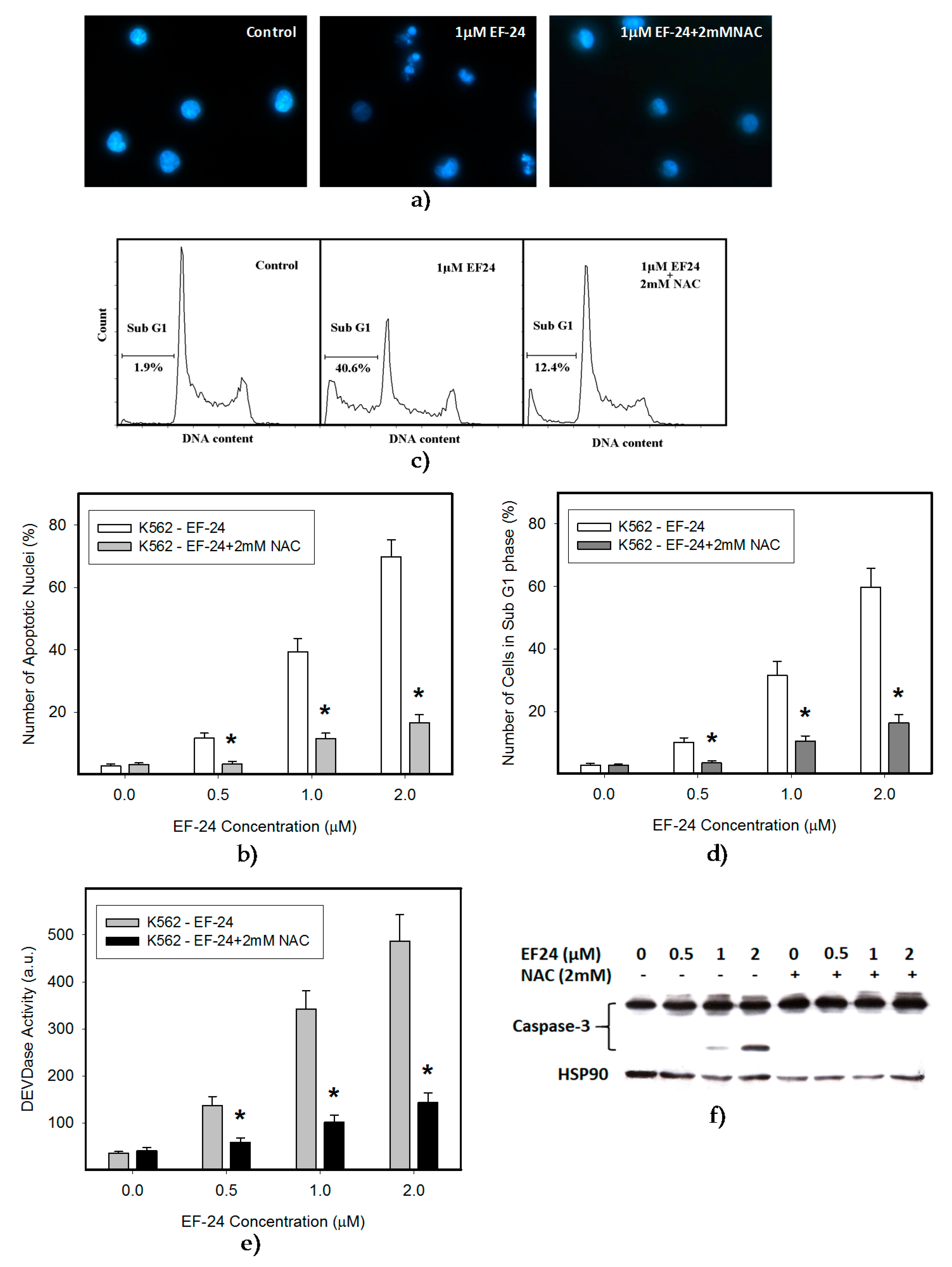
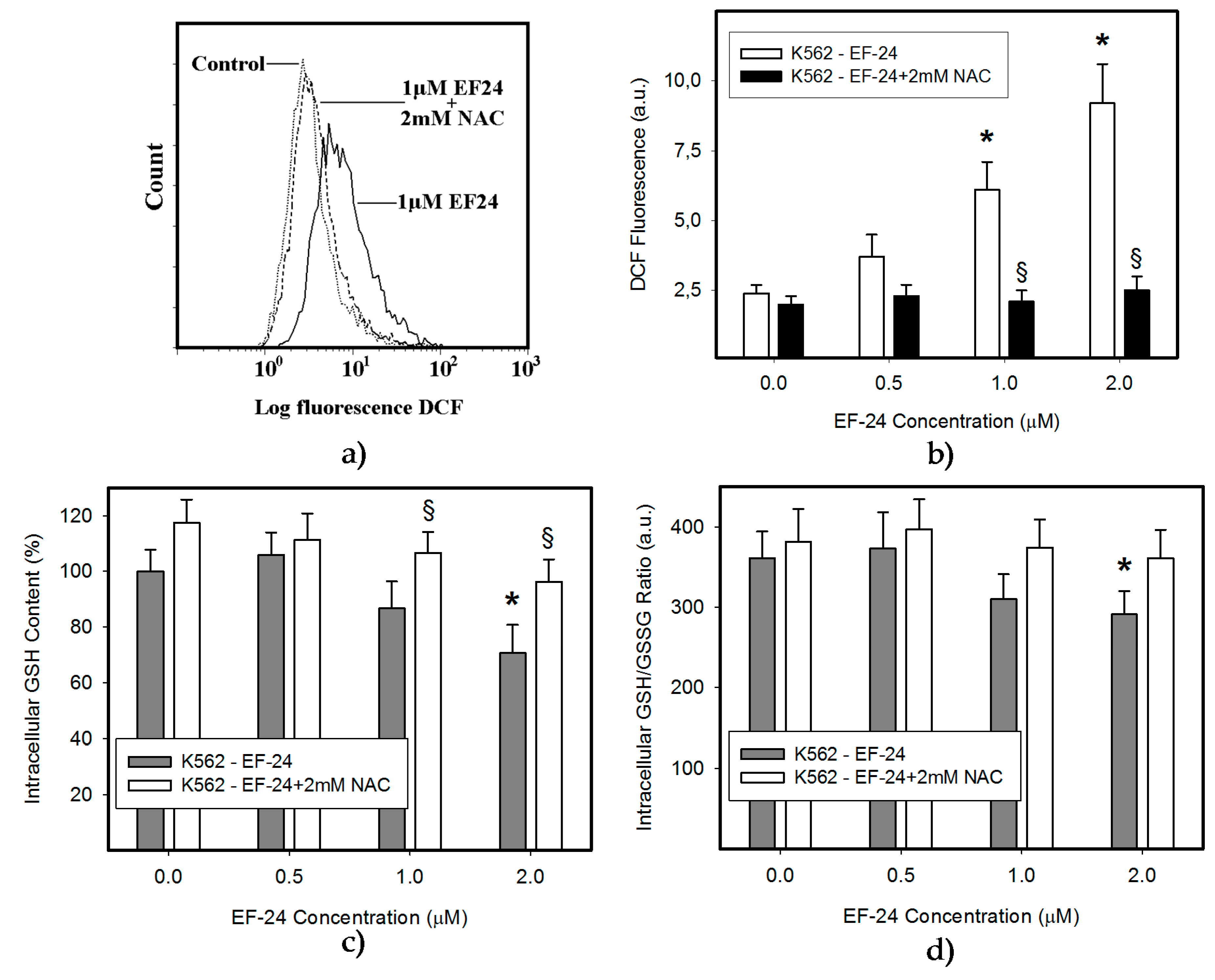
2.1. N-Acetylcysteine (NAC) Prevented 3,5-bis[(2-Fluorophenyl)Methylene]-4-Piperidinone (EF-24)-Induced Cell Death with Apoptotic Hallmarks in K562 Cells
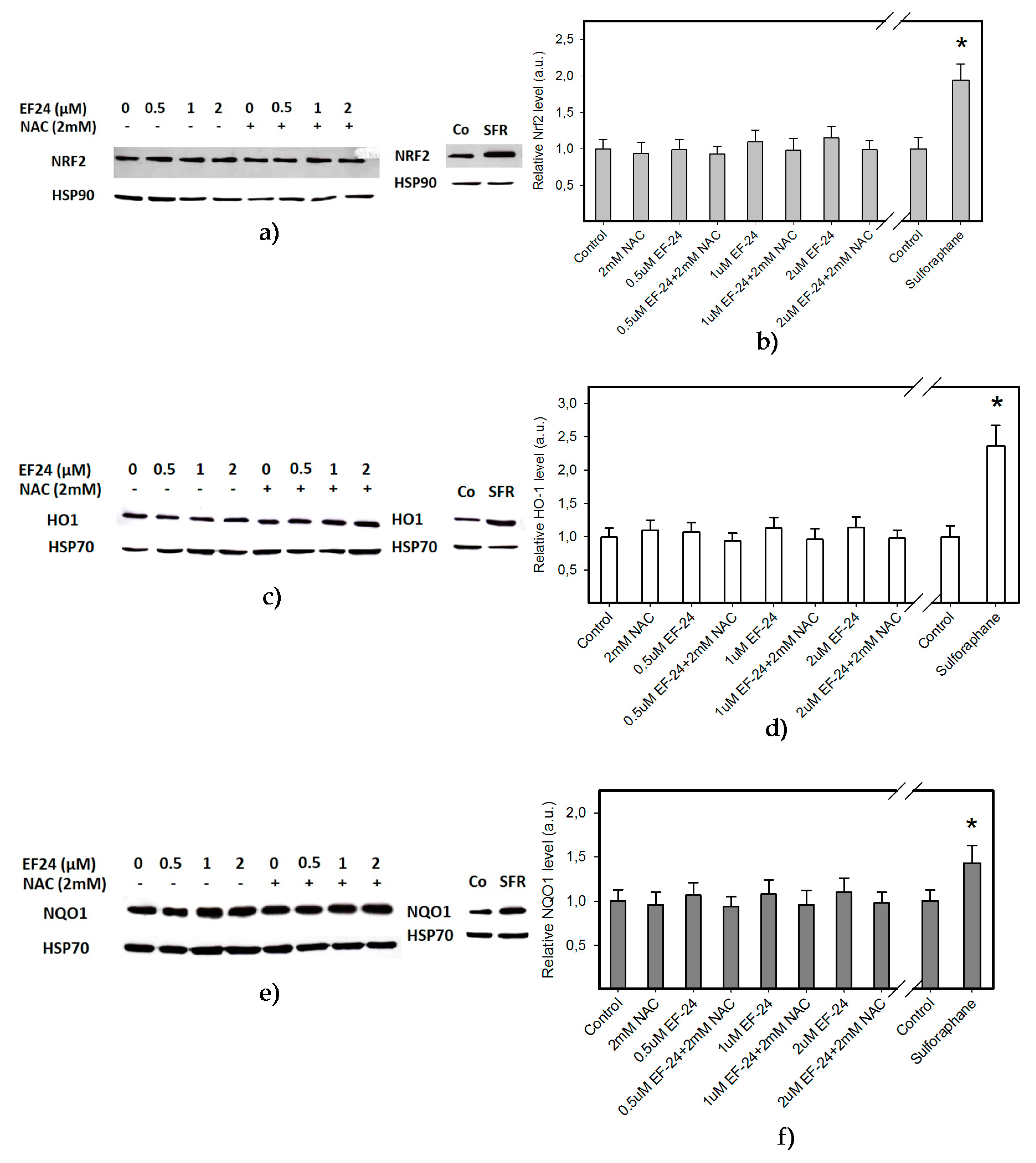
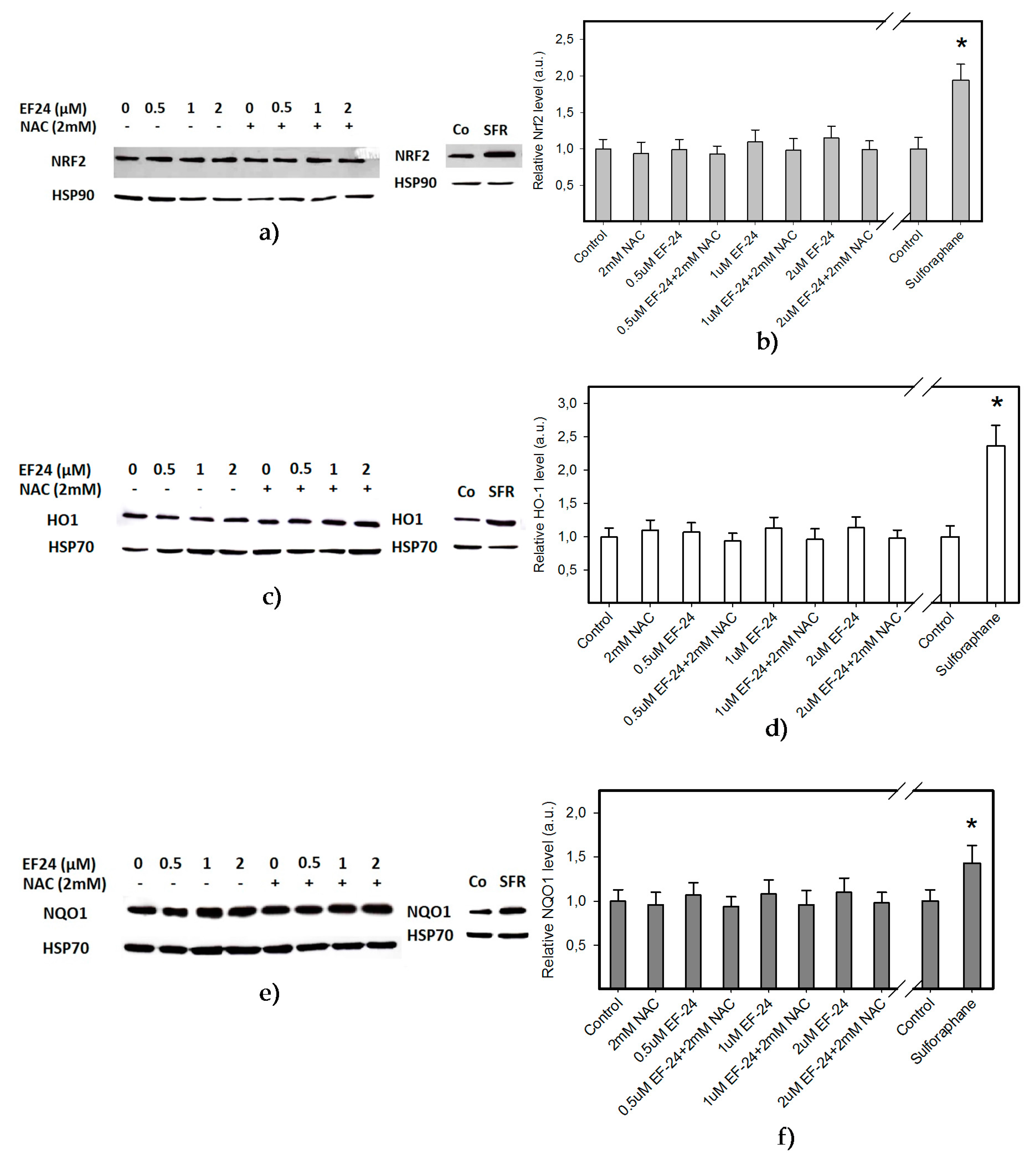
2.2. EF-24 Did Not Activate Nuclear Factor-Erythroid 2 Related Factor 2 (Nrf2) in K562 Cells
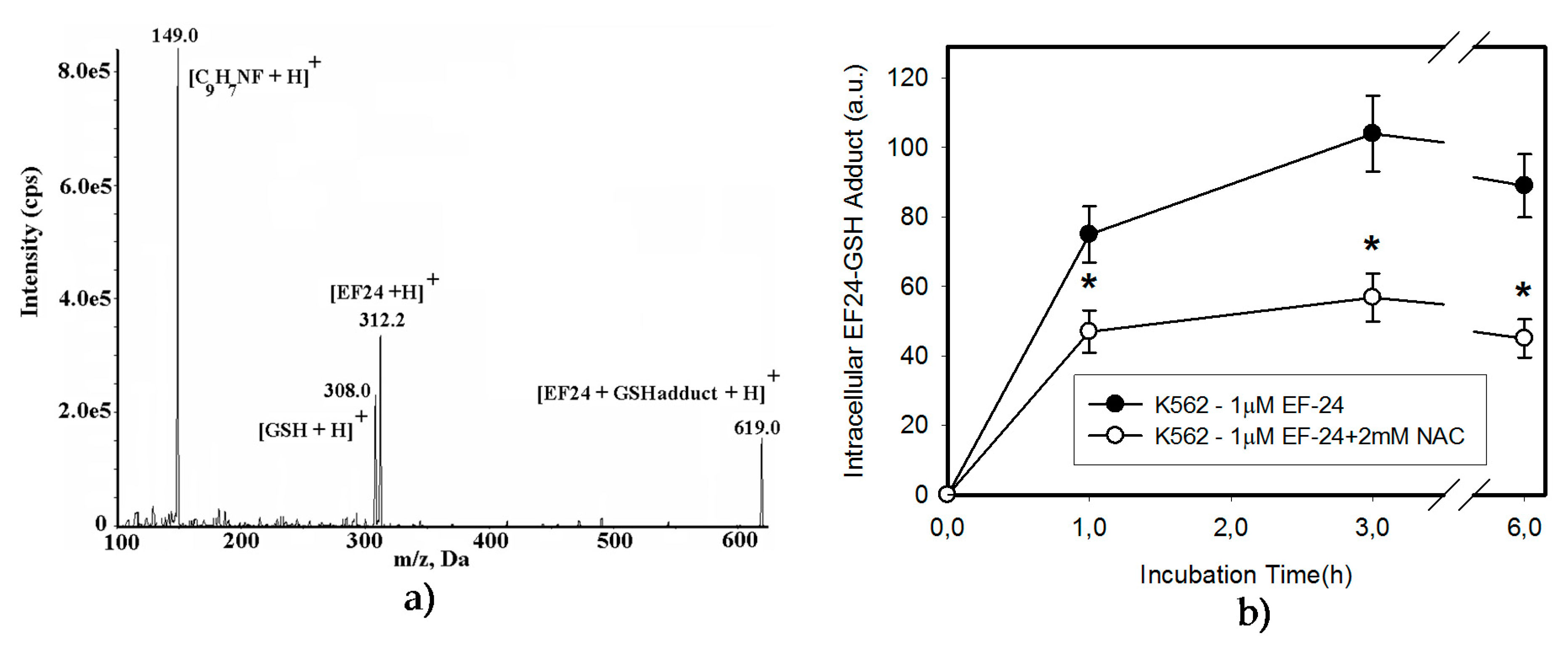
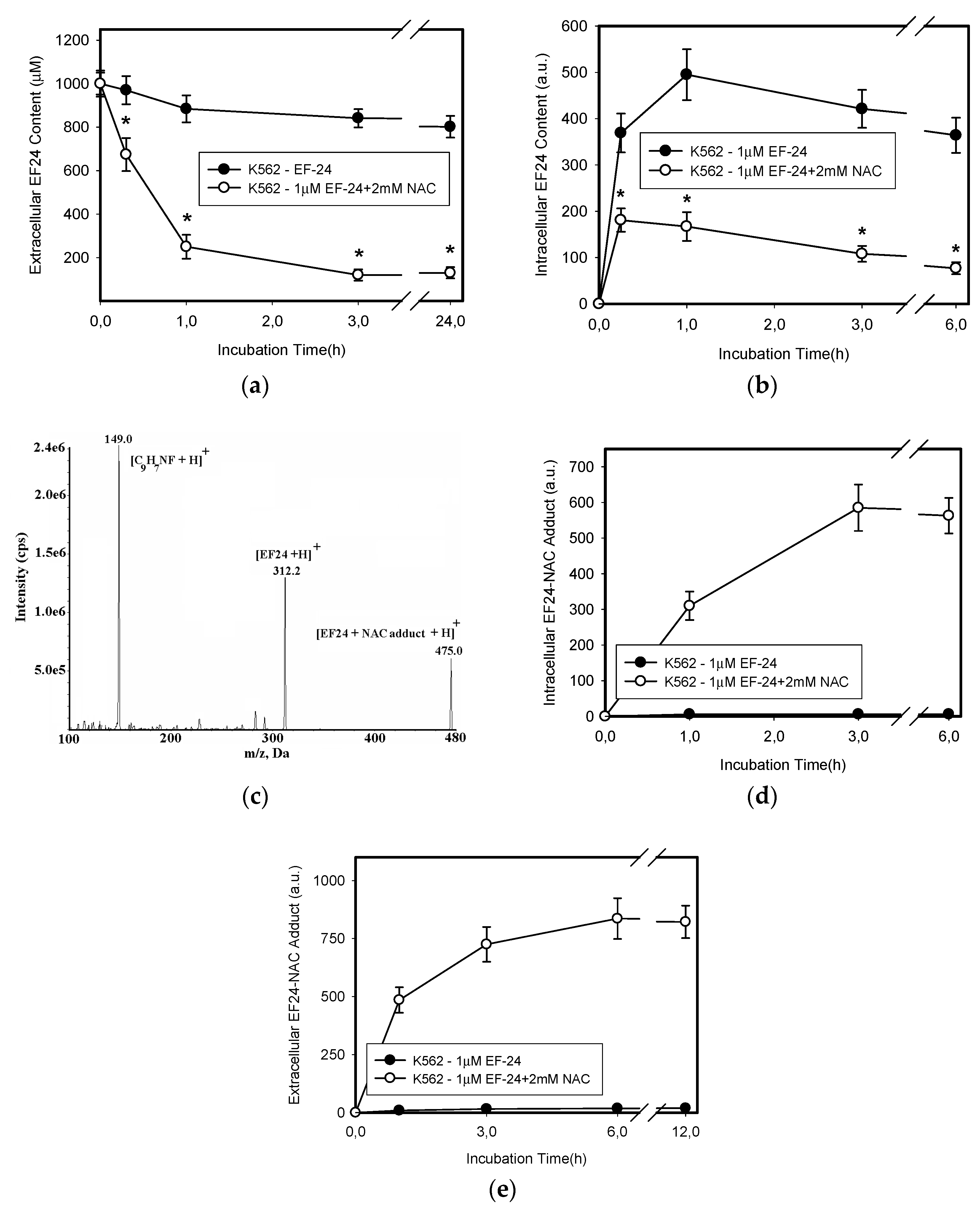
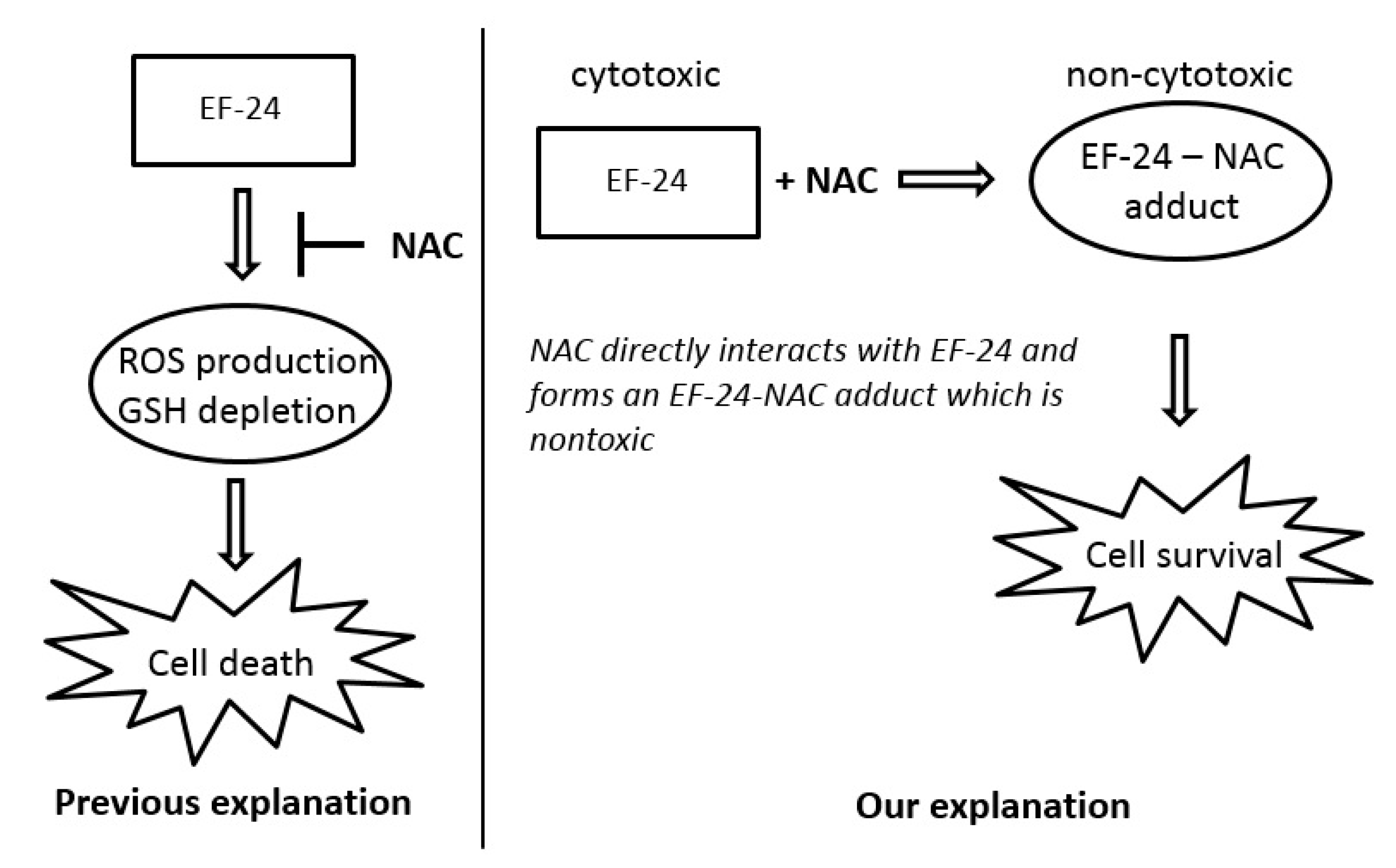
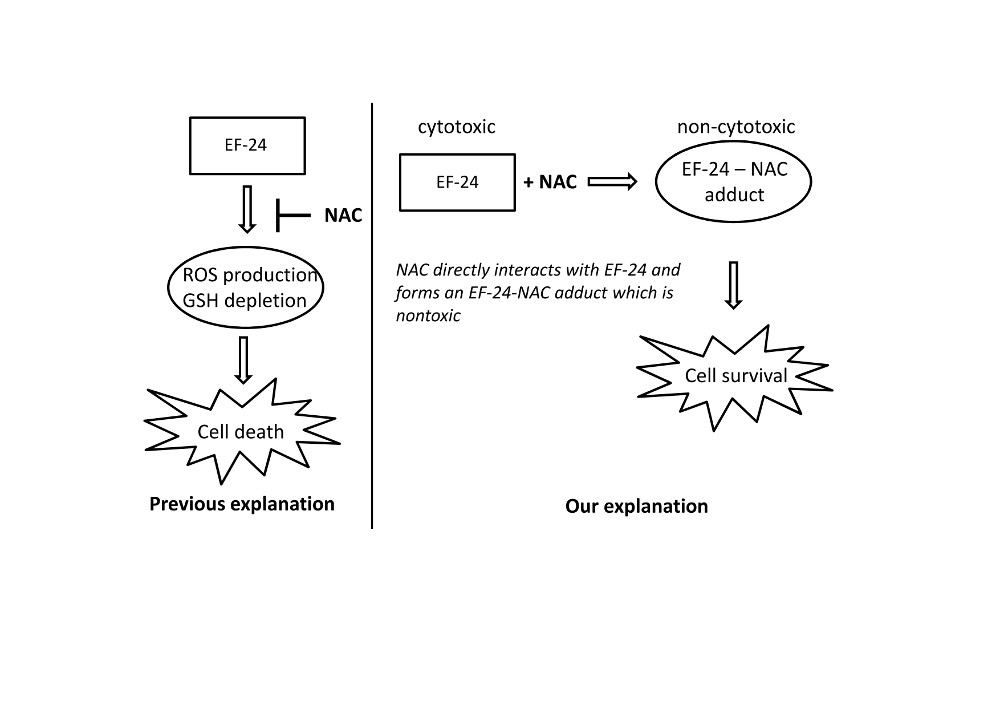
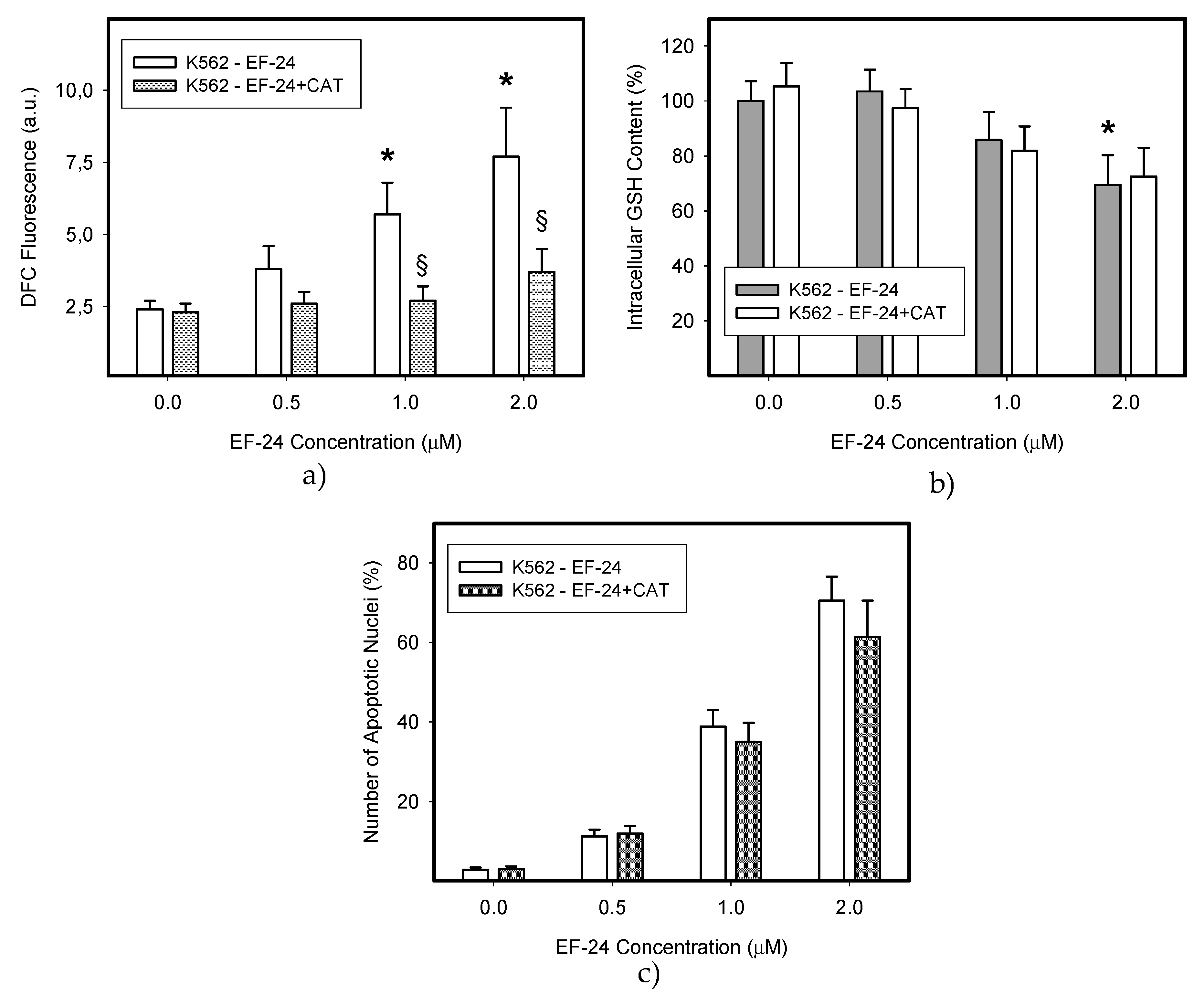
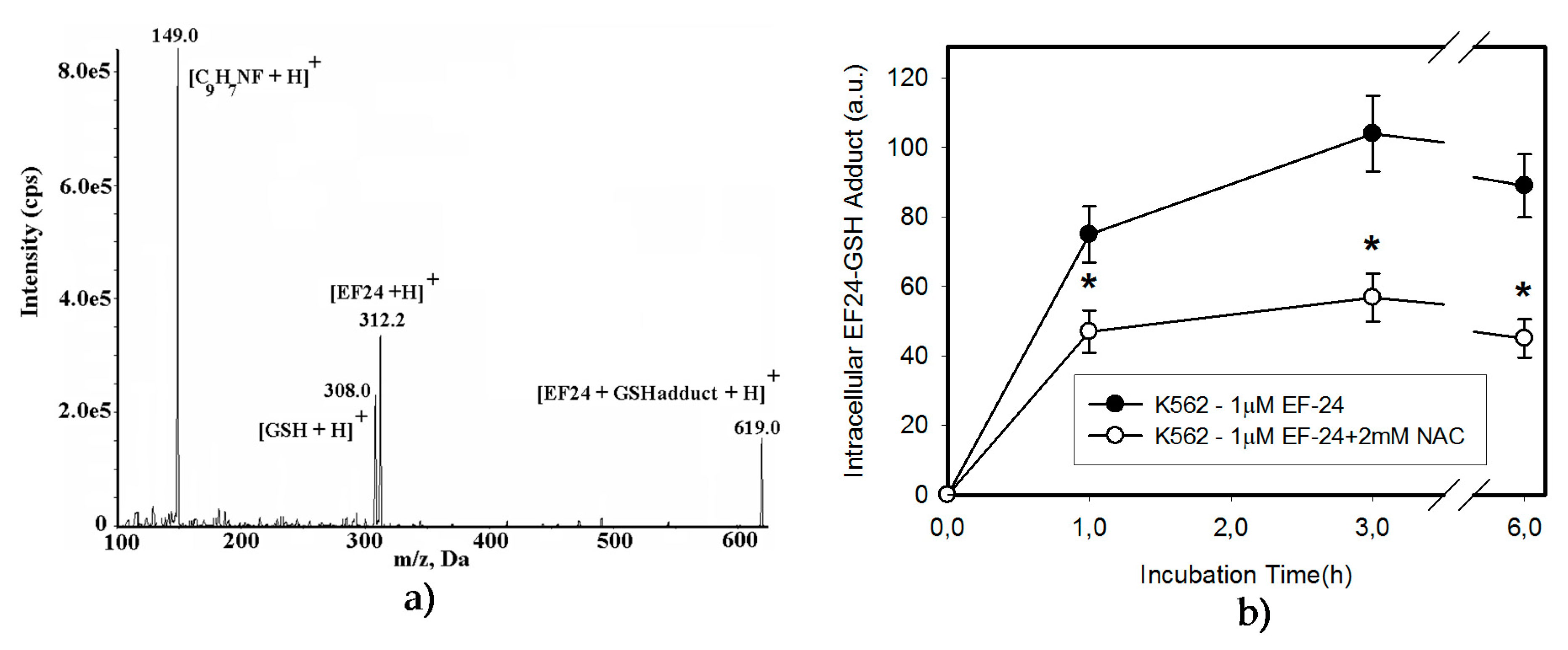
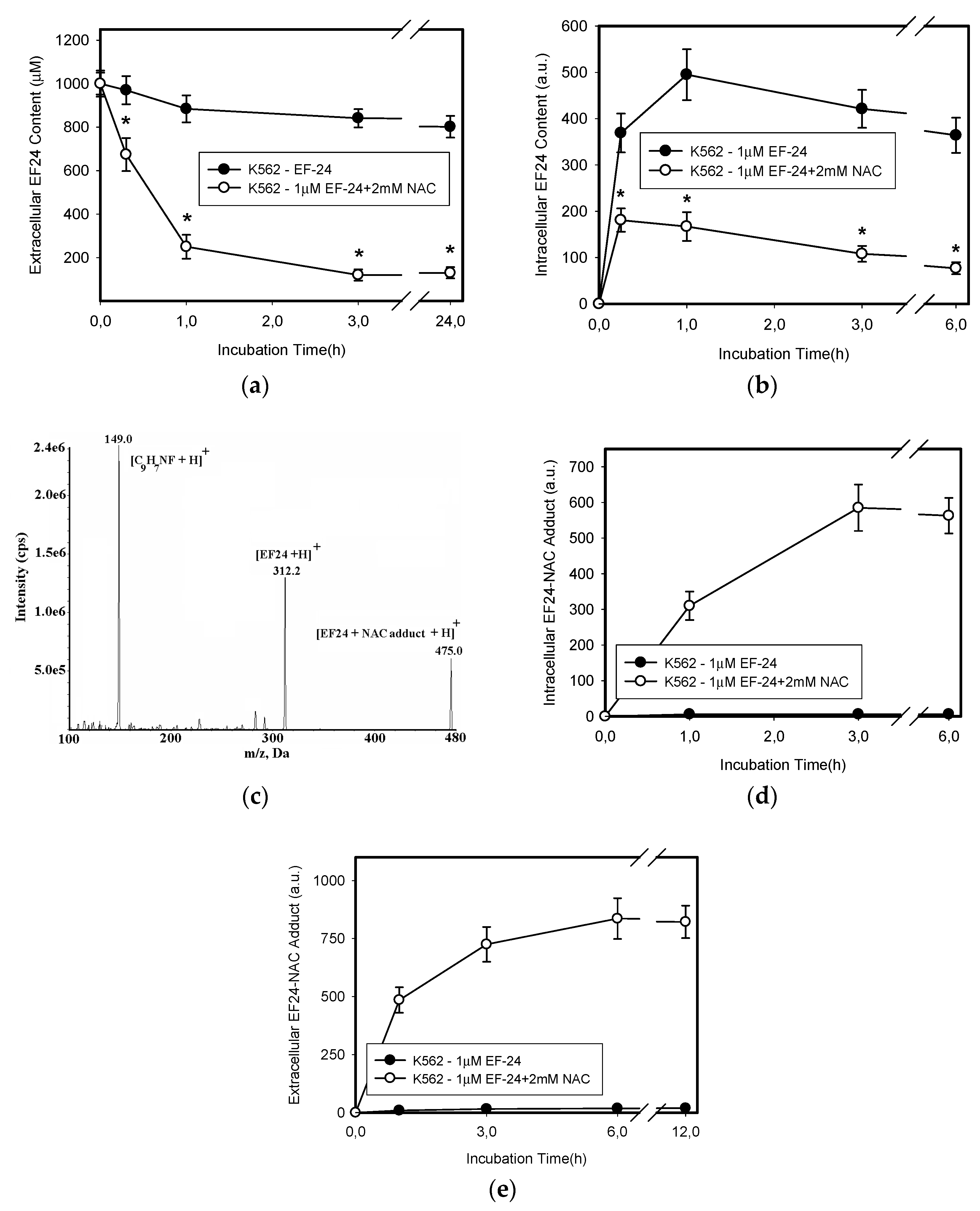
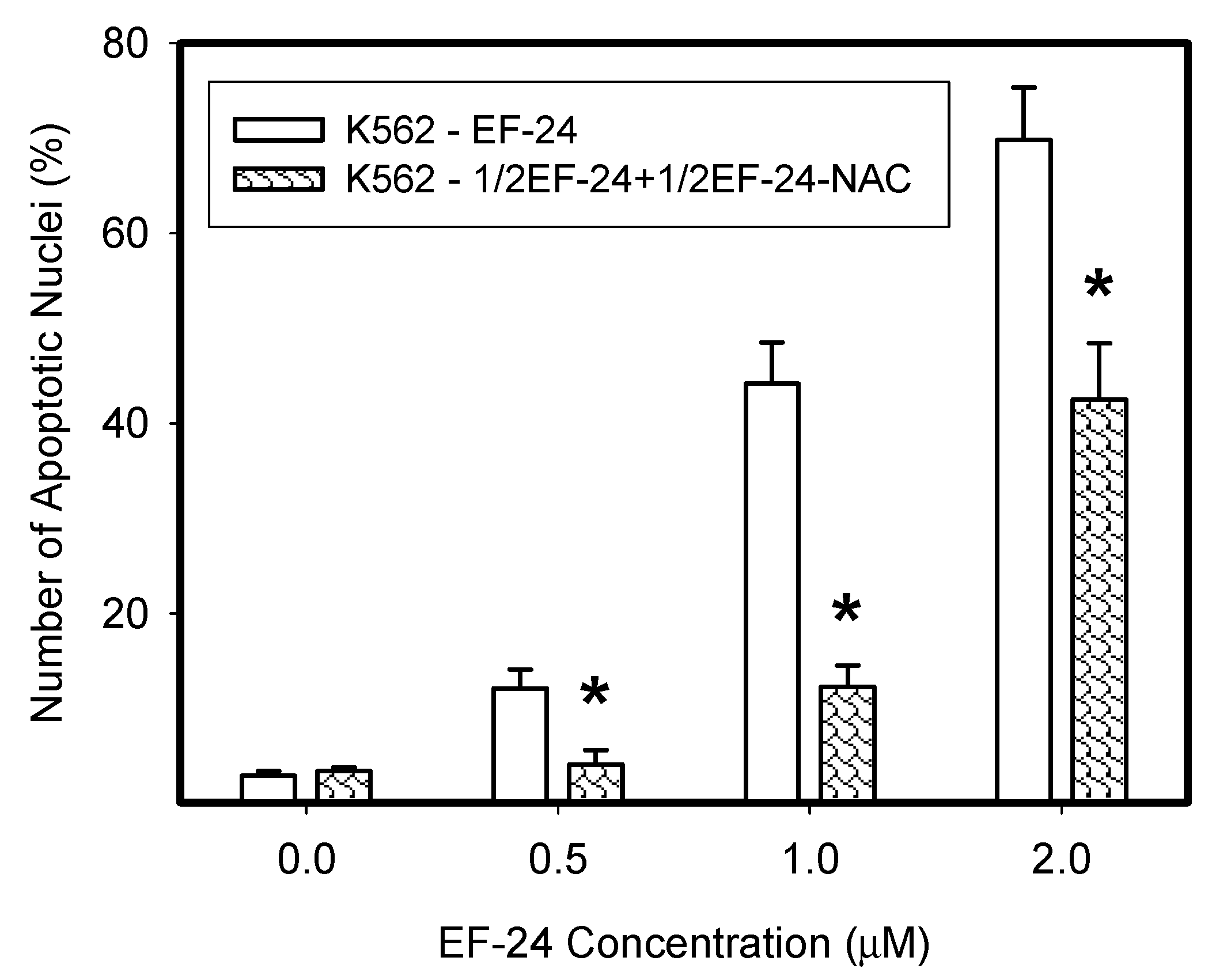
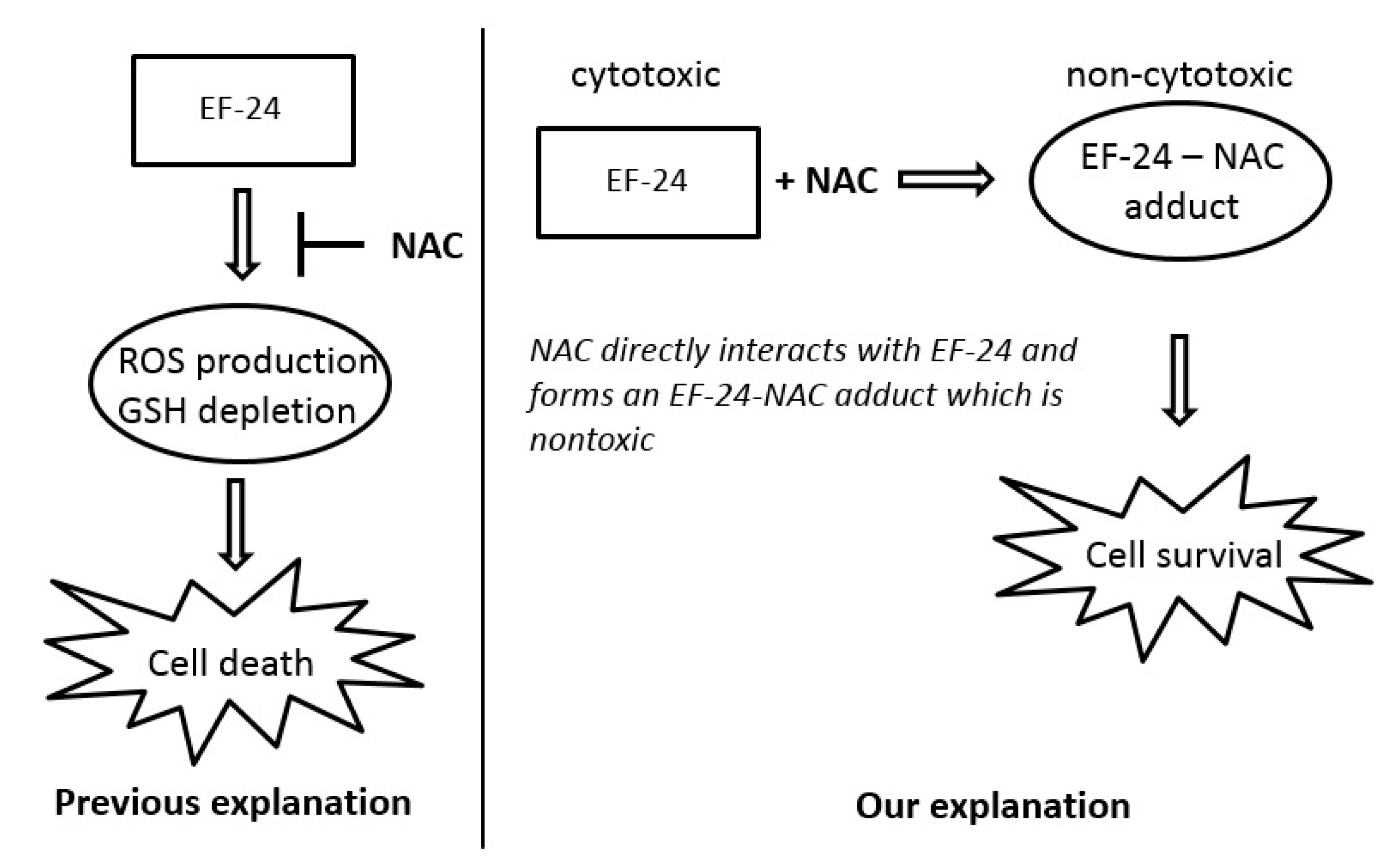
2.3. Conversion of Cytotoxic EF-24 Into the Non-Cytotoxic EF-24-NAC Adduct is the Main Mechanism of the NAC Protection Against the Cytotoxic Effects of EF-24
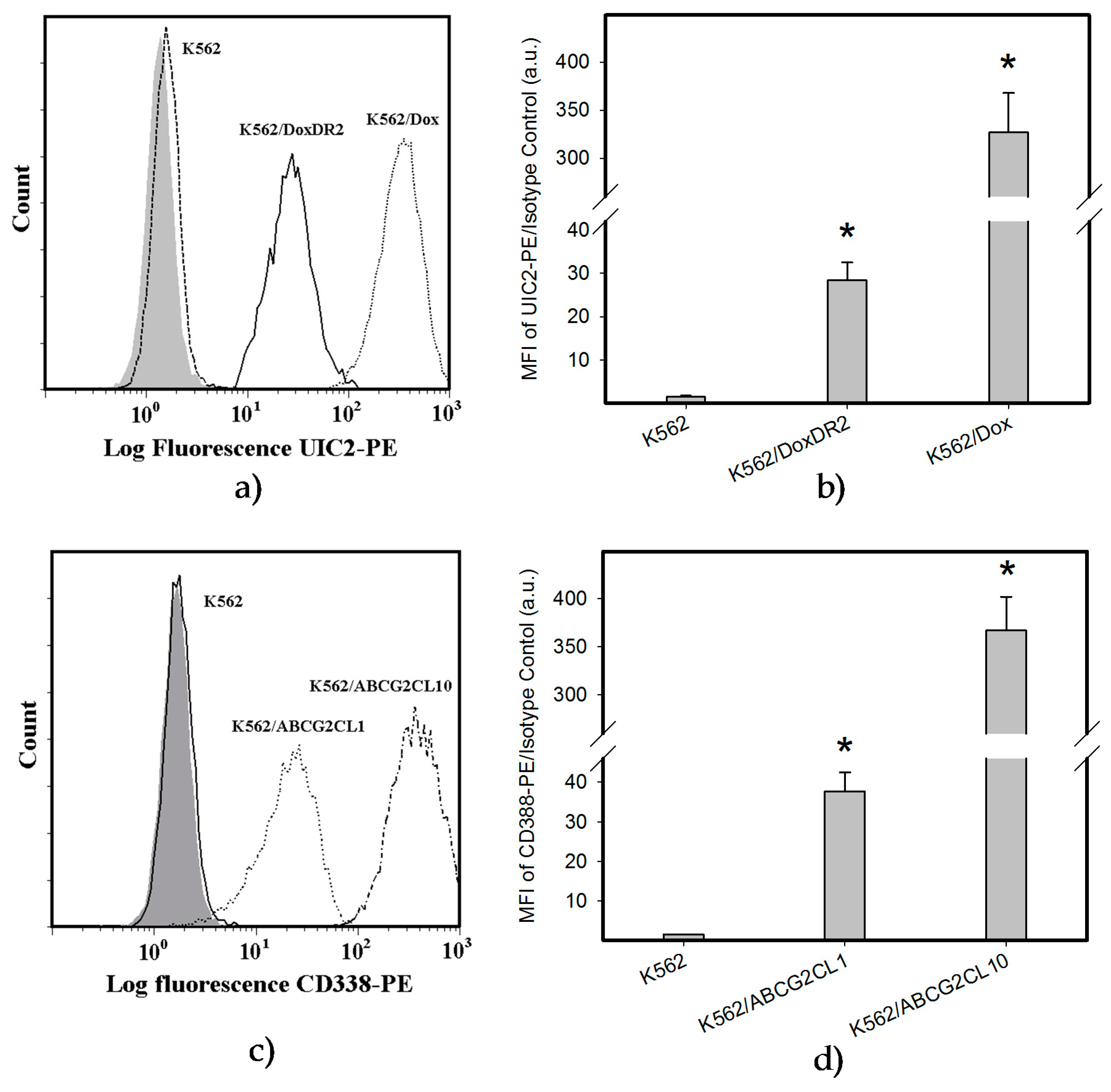
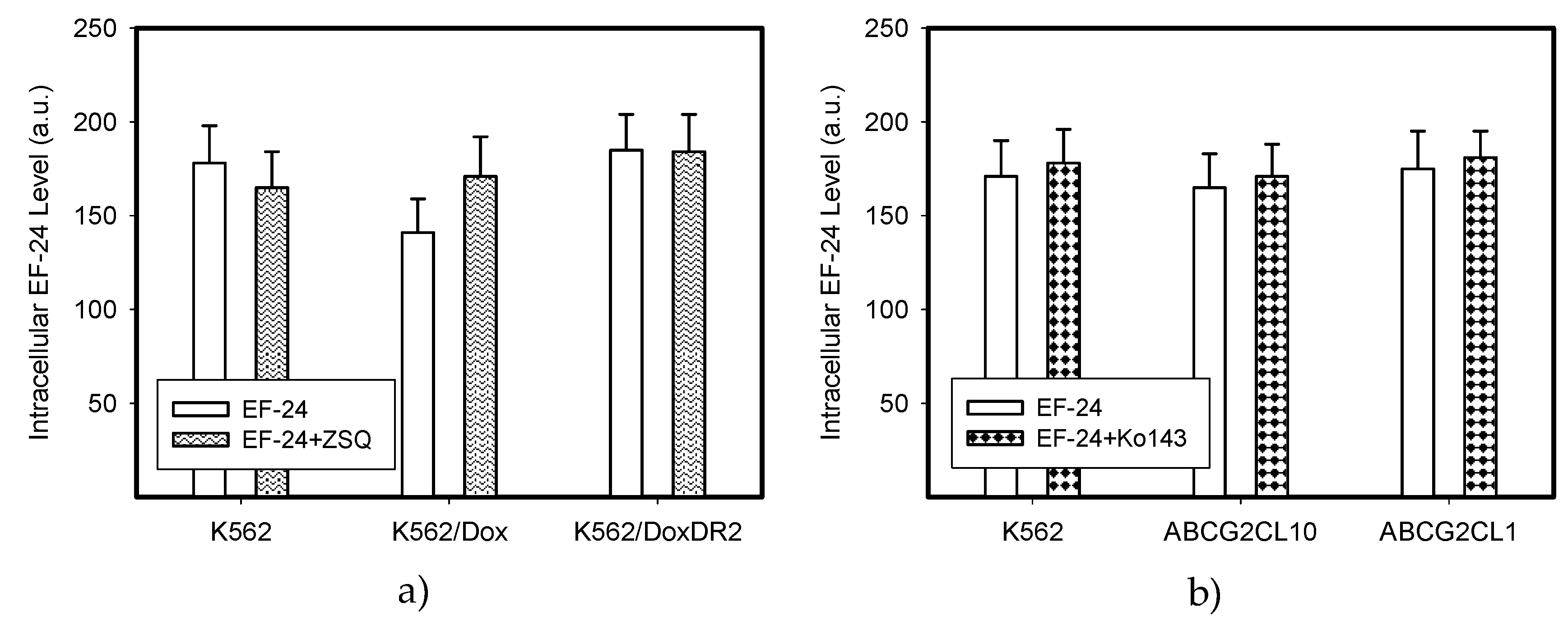
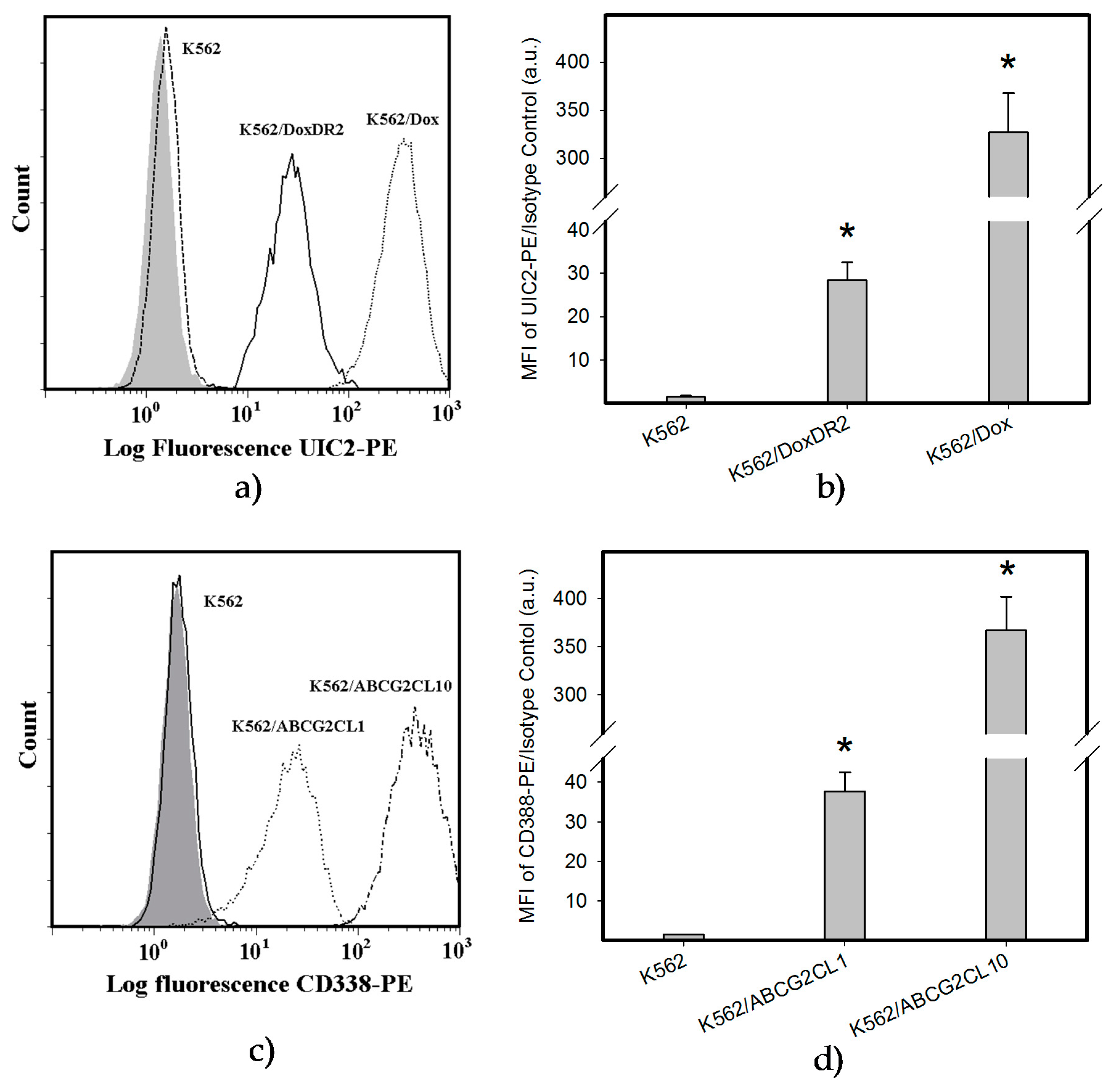
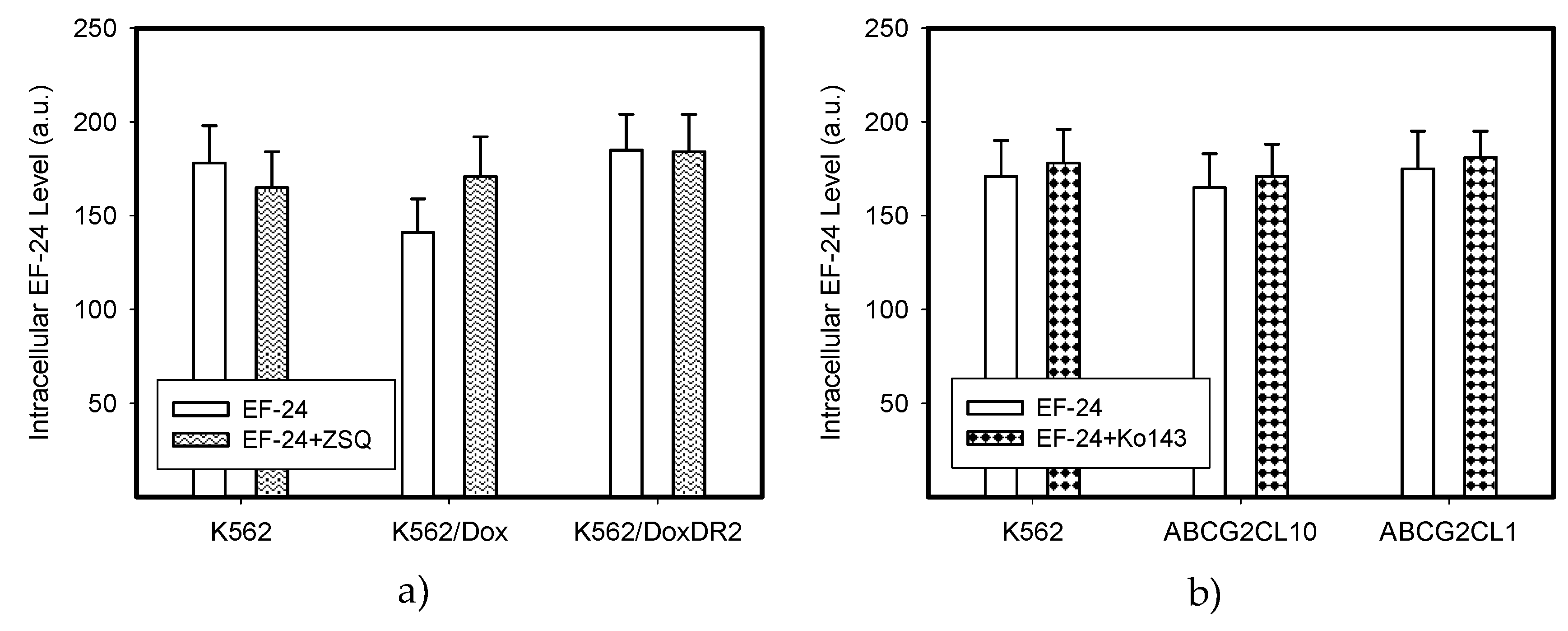
2.4. Overexpression of ABCB1 and ABCG2 Did Not Compromise the Antiproliferative Effects of EF-24 in K562 Cells
3. Discussion
4. Material and Methods
4.1. Chemicals and Cell Treatment
4.2. Cell Culture
4.3. Determination of Cell Survival and Proliferation
4.4. Analysis of Cell Cycle and Apoptotic Cells
4.5. Measurement of ROS Production
4.6. Morphological Analysis of Apoptosis
4.7. Measurement of Caspase-3 Enzymatic Activity
4.8. Measurement of Caspase-3 Processing
4.9. Western Blot Analysis of Activation of Nrf2 and Stress-Response Pathway
4.10. Preparation of Cell Extracts
4.11. High-Performance Liquid Chromatography (HPLC) Analysis of Glutathione (GSH) and Oxidized Glutathione (GSSG)
4.12. Determination of EF-24
4.13. Determination of EF-24-GSH and EF-24-NAC Adducts
4.14. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cheng, A.L.; Hsu, C.H.; Lin, J.K.; Hsu, M.M.; Ho, Y.F.; Shen, T.S.; Ko, J.Y.; Lin, J.T.; Lin, B.R.; Ming-Shiang, W.; et al. Phase I clinical trial of curcumin, a chemopreventive agent, in patients with high-risk or pre-malignant lesions. Anticancer Res. 2001, 21, 2895–2900. [Google Scholar] [PubMed]
- Sharma, R.A.; Euden, S.A.; Platton, S.L.; Cooke, D.N.; Shafayat, A.; Hewitt, H.R.; Marczylo, T.H.; Morgan, B.; Hemingway, D.; Plummer, S.M.; et al. Phase I clinical trial of oral curcumin: Biomarkers of systemic activity and compliance. Clin. Cancer Res. 2004, 10, 6847–6854. [Google Scholar] [CrossRef] [PubMed]
- Adams, B.K.; Ferstl, E.M.; Davis, M.C.; Herold, M.; Kurtkaya, S.; Camalier, R.F.; Hollingshead, M.G.; Kaur, G.; Sausville, E.A.; Rickles, F.R.; et al. Synthesis and biological evaluation of novel curcumin analogs as anti-cancer and anti-angiogenesis agents. Bioorg. Med. Chem. 2004, 12, 3871–3883. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Sidell, N.; Mancini, A.; Huang, R.P.; Shenming, W.; Horowitz, I.R.; Liotta, D.C.; Taylor, R.N.; Wieser, F. Multiple anticancer activities of EF24, a novel curcumin analog, on human ovarian carcinoma cells. Reprod. Sci. 2010, 17, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Kasinski, A.L.; Du, Y.; Thomas, S.L.; Zhao, J.; Sun, S.Y.; Khuri, F.R.; Wang, C.Y.; Shoji, M.; Sun, A.; Snyder, J.P.; et al. Inhibition of IkappaB kinase-nuclear factor-kappaB signaling pathway by 3,5-bis(2-flurobenzylidene)piperidin-4-one (EF24), a novel monoketone analog of curcumin. Mol. Pharmacol. 2008, 74, 654–661. [Google Scholar] [CrossRef] [PubMed]
- Selvendiran, K.; Tong, L.; Vishwanath, S.; Bratasz, A.; Trigg, N.J.; Kutala, V.K.; Hideg, K.; Kuppusamy, P. EF24 induces G2/M arrest and apoptosis in cisplatin-resistant human ovarian cancer cells by increasing PTEN expression. J. Biol. Chem. 2007, 282, 28609–28618. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.L.; Zhong, D.; Zhou, W.; Malik, S.; Liotta, D.; Snyder, J.P.; Hamel, E.; Giannakakou, P. EF24, a novel curcumin analog, disrupts the microtubule cytoskeleton and inhibits HIF-1. Cell Cycle 2008, 7, 2409–2417. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Zheng, T.; Song, R.; Wang, J.; Yin, D.; Wang, L.; Liu, H.; Tian, L.; Fang, X.; Meng, X.; et al. Hypoxia-mediated sorafenib resistance can be overcome by EF24 through Von Hippel-Lindau tumor suppressor-dependent HIF-1α inhibition in hepatocellular carcinoma. Hepatology 2013, 57, 1847–1857. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zou, P.; Zhao, Z.; Chen, X.; Fan, X.; Vinothkumar, R.; Cui, R.; Wu, F.; Zhang, Q.; Liang, G.; et al. Synergistic antitumor activity of rapamycin and EF24 via increasing ROS for the treatment of gastric cancer. Redox Biol. 2016, 10, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, D.; May, R.; Sureban, S.M.; Lee, K.B.; George, R.; Kuppusamy, P.; Ramanujam, R.P.; Hideg, K.; Dieckgraefe, B.K.; Houchen, C.W.; et al. Diphenyl difluoroketone: A curcumin derivative with potent in vivo anticancer activity. Cancer Res. 2008, 68, 1962–1969. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Liang, Y.; Wang, L.; Tian, L.; Song, R.; Han, T.; Pan, S.; Liu, L. In vivo and in vitro suppression of hepatocellular carcinoma by EF24, a curcumin analog. PLoS ONE 2012, 7, e48075. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.H.; Yue, J.; Sims, M.; Pfeffer, L.M. The curcumin analog EF24 targets NF-κB and miRNA-21, and has potent anticancer activity in vitro and in vivo. PLoS ONE 2013, 8, e71130. [Google Scholar] [CrossRef] [PubMed]
- Mosley, C.A.; Liotta, D.C.; Snyder, J.P. Highly active anticancer curcumin analogues. Adv. Exp. Med. Biol. 2007, 595, 77–103. [Google Scholar] [CrossRef] [PubMed]
- Adams, B.K.; Cai, J.; Armstrong, J.; Herold, M.; Lu, Y.J.; Sun, A.; Snyder, J.P.; Liotta, D.C.; Jones, D.P.; Shoji, M. EF-24, a novel synthetic curcumin analog, induces apoptosis in cancer cells via a redox-dependent mechanism. Anticancer Drugs 2005, 16, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Sun, A.; Lu, Y.J.; Hu, H.; Shoji, M.; Liotta, D.C.; Snyder, J.P. Curcumin analog cytotoxicity against breast cancer cells: Exploitation of a redox-dependent mechanism. Bioorg. Med. Chem. Lett. 2009, 19, 6627–6631. [Google Scholar] [CrossRef] [PubMed]
- Zou, P.; Xia, Y.; Chen, W.; Chen, X.; Ying, S.; Feng, Z.; Chen, T.; Ye, Q.; Wang, Z.; Qiu, C.; et al. EF24 induces ROS-mediated apoptosis via targeting thioredoxin reductase 1 in gastric cancer cells. Oncotarget 2016, 7, 18050–18064. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Feng, C.; Vinothkumar, R.; Chen, W.; Dai, X.; Chen, X.; Ye, Q.; Qiu, C.; Zhou, H.; Wang, Y.; et al. Curcumin analog EF24 induces apoptosis via ROS-dependent mitochondrial dysfunction in human colorectal cancer cells. Cancer Chemother. Pharmacol. 2016, 78, 1151–1161. [Google Scholar] [CrossRef] [PubMed]
- Braun, T.; Carvalho, G.; Fabre, C.; Grosjean, J.; Fenaux, P.; Kroemer, G. Targeting NF-kappaB in hematologic malignancies. Cell Death Differ. 2006, 13, 748–758. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Yamamoto, M. Molecular basis of the Keap1-Nrf2 system. Free Radic. Biol. Med. 2015, 88, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef] [PubMed]
- Myzak, M.C.; Dashwood, R.H. Chemoprotection by sulforaphane: Keep one eye beyond Keap1. Cancer Lett. 2006, 233, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Ketterer, B.; Coles, B.; Meyer, D.J. The role of glutathione in detoxication. Environ. Health Perspect. 1983, 49, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Cidlowski, J.A. Apoptosis and glutathione: Beyond an antioxidant. Cell Death Differ. 2009, 16, 1303–1314. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M. How cancer cells evade chemotherapy: Sixteenth Richard and Hinda Rosenthal Foundation Award Lecture. Cancer Res. 1993, 53, 747–754. [Google Scholar] [PubMed]
- Ambudkar, S.V.; Kimchi-Sarfaty, C.; Sauna, Z.E.; Gottesman, M.M. P-glycoprotein: From genomics to mechanism. Oncogene 2003, 22, 7468–7485. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Sherratt, P.J.; Huang, H.C.; Yang, C.S.; Pickett, C.B. Increased protein stability as a mechanism that enhances Nrf2-mediated transcriptional activation of the antioxidant response element. Degradation of Nrf2 by the 26 S proteasome. J. Biol. Chem. 2003, 278, 4536–4541. [Google Scholar] [CrossRef] [PubMed]
- Mlejnek, P.; Dolezel, P. N-acetylcysteine prevents the geldanamycin cytotoxicity by forming geldanamycin-N-acetylcysteine adduct. Chem. Biol. Interact. 2014, 220, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Mlejnek, P.; Dolezel, P. Loss of mitochondrial transmembrane potential and glutathione depletion are not sufficient to account for induction of apoptosis by carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone in human leukemia K562 cells. Chem. Biol. Interact. 2015, 239, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Türk, D.; Szakács, G. Relevance of multidrug resistance in the age of targeted therapy. Curr. Opin. Drug Discov. Dev. 2009, 12, 246–252. [Google Scholar]
- Kosztyu, P.; Dolezel, P.; Mlejnek, P. Can P-glycoprotein mediate resistance to nilotinib in human leukaemia cells? Pharmacol. Res. 2013, 67, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Kosztyu, P.; Bukvova, R.; Dolezel, P.; Mlejnek, P. Resistance to daunorubicin, imatinib, or nilotinib depends on expression levels of ABCB1 and ABCG2 in human leukemia cells. Chem. Biol. Interact. 2014, 219, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Mlejnek, P.; Dolezel, P.; Ruzickova, E. Drug resistance of cancer cells is crucially affected by expression levels of ABC-transporters. BioDiscovery 2017, 20, e11211. [Google Scholar] [CrossRef]
- Ruzickova, E.; Janska, R.; Dolezel, P.; Mlejnek, P. A clinically relevant study of interactions of inhibitors of anti-apoptotic Bcl-2 proteins with the ABC transporters. Pharmazie 2017, in press. [Google Scholar]
- Fojo, A.T.; Ueda, K.; Slamon, D.J.; Poplack, D.G.; Gottesman, M.M.; Pastan, I. Expression of a multidrug-resistance gene in human tumors and tissues. Proc. Natl. Acad. Sci. USA 1987, 84, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, L.J.; Galski, H.; Fojo, A.; Willingham, M.; Lai, S.L.; Gazdar, A.; Pirker, R.; Green, A.; Crist, W.; Brodeur, G.M.; et al. Expression of a multidrug resistance gene in human cancers. J. Natl. Cancer Inst. 1989, 81, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Mlejnek, P.; Kosztyu, P.; Dolezel, P.; Bates, S.E.; Ruzickova, E. Reversal of ABCB1 mediated efflux by imatinib and nilotinib in cells expressing various transporter levels. Chem. Biol. Interact. 2017, 273, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Chearwae, W.; Anuchapreeda, S.; Nandigama, K.; Ambudkar, S.V.; Limtrakul, P. Biochemical mechanism of modulation of human P-glycoprotein (ABCB1) by curcumin I, II, and III purified from Turmeric powder. Biochem. Pharmacol. 2004, 68, 2043–2052. [Google Scholar] [CrossRef] [PubMed]
- Chearwae, W.; Shukla, S.; Limtrakul, P.; Ambudkar, S.V. Modulation of the function of the multidrug resistance-linked ATP-binding cassette transporter ABCG2 by the cancer chemopreventive agent curcumin. Mol. Cancer Ther. 2006, 5, 1995–2006. [Google Scholar] [CrossRef] [PubMed]
- Tang, R.; Faussat, A.M.; Perrot, J.Y.; Marjanovic, Z.; Cohen, S.; Storme, T.; Morjani, H.; Legrand, O.; Marie, J.P. Zosuquidar restores drug sensitivity in P-glycoprotein expressing acute myeloid leukemia (AML). BMC Cancer 2008, 8, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Mlejnek, P.; Dolezel, P.; Kosztyu, P. P-glycoprotein mediates resistance to A3 adenosine receptor agonist 2-chloro-N6-(3-iodobenzyl)-adenosine-5′-N-methyluronamide in human leukemia cells. J. Cell. Physiol. 2012, 227, 676–685. [Google Scholar] [CrossRef] [PubMed]
- Elkind, N.B.; Szentpétery, Z.; Apáti, Á.; Özvegy-Laczka, C.; Várady, G.; Ujhelly, O.; Szabó, K.; Homolya, L.; Váradi, A.; Buday, L.; et al. Multidrug transporter ABCG2 prevents tumor cell death induced by the epidermal growth factor receptor inhibitor Iressa (ZD1839, Gefitinib). Cancer Res. 2005, 65, 1770–1777. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Nicoletti, I.; Migliorati, G.; Pagliacci, M.C.; Grignani, F.; Riccardi, C. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J. Immunol. Methods 1991, 139, 271–279. [Google Scholar] [CrossRef]
- Mlejnek, P.; Kuglik, P. Induction of apoptosis in HL-60 cells by N6-Benzyladenosine. J. Cell. Biochem. 2000, 77, 6–17. [Google Scholar] [CrossRef]
- Frydrych, I.; Mlejnek, P. Serine protease inhibitors N-alpha-tosyl-L-lysinyl-chloromethylketone (TLCK) and N-tosyl-L-phenylalaninyl-chloromethylketone (TPCK) are potent inhibitors of activated caspase proteases. J. Cell. Biochem. 2008, 103, 1646–1656. [Google Scholar] [CrossRef] [PubMed]
- Frydrych, I.; Mlejnek, P.; Dolezel, P.; Zoumpourlis, V.; Krumpochova, P. The broad-spectrum caspase inhibitor Boc-Asp-CMK induces cell death in human leukaemia cells. Toxicol. In Vitro 2008, 22, 1356–1360. [Google Scholar] [CrossRef] [PubMed]
- Mlejnek, P.; Novak, O.; Dolezel, P. A non-radioactive assay for precise determination of intracellular levels of imatinib and its main metabolite in Bcr-Abl positive cells. Talanta 2011, 283, 1466–1471. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Treatment | IC50 |
|---|---|
| EF-24 | 0.73 ± 0.11 μM |
| EF-24 + 2 mM NAC | >20 μM |
| EF-24 + CAT (50 U/mL) | 0.84 ± 0.15 μM |
| EF-24 | EF-24 + ZSQ | EF-24 + Ko143 | |
|---|---|---|---|
| K562 | 0.74 ± 0.10 μM | 0.71 ± 0.10 μM | 0.77 ± 0.12 μM |
| K562/Dox | 0.87 ± 0.15 μM | 0.72 ± 0.11 μM | - |
| K562/DoxDR2 | 0.75 ± 0.11 μM | 0.69 ± 0.10 μM | - |
| K562/ABCG2CL10 | 0.71 ± 0.11 μM | - | 0.75 ± 0.11 μM |
| K562/ABCG2CL1 | 0.72 ± 0.10 μM | - | 0.76 ± 0.11 μM |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skoupa, N.; Dolezel, P.; Ruzickova, E.; Mlejnek, P. Apoptosis Induced by the Curcumin Analogue EF-24 Is Neither Mediated by Oxidative Stress-Related Mechanisms nor Affected by Expression of Main Drug Transporters ABCB1 and ABCG2 in Human Leukemia Cells. Int. J. Mol. Sci. 2017, 18, 2289. https://doi.org/10.3390/ijms18112289
Skoupa N, Dolezel P, Ruzickova E, Mlejnek P. Apoptosis Induced by the Curcumin Analogue EF-24 Is Neither Mediated by Oxidative Stress-Related Mechanisms nor Affected by Expression of Main Drug Transporters ABCB1 and ABCG2 in Human Leukemia Cells. International Journal of Molecular Sciences. 2017; 18(11):2289. https://doi.org/10.3390/ijms18112289
Chicago/Turabian StyleSkoupa, Nikola, Petr Dolezel, Eliska Ruzickova, and Petr Mlejnek. 2017. "Apoptosis Induced by the Curcumin Analogue EF-24 Is Neither Mediated by Oxidative Stress-Related Mechanisms nor Affected by Expression of Main Drug Transporters ABCB1 and ABCG2 in Human Leukemia Cells" International Journal of Molecular Sciences 18, no. 11: 2289. https://doi.org/10.3390/ijms18112289
APA StyleSkoupa, N., Dolezel, P., Ruzickova, E., & Mlejnek, P. (2017). Apoptosis Induced by the Curcumin Analogue EF-24 Is Neither Mediated by Oxidative Stress-Related Mechanisms nor Affected by Expression of Main Drug Transporters ABCB1 and ABCG2 in Human Leukemia Cells. International Journal of Molecular Sciences, 18(11), 2289. https://doi.org/10.3390/ijms18112289