
Aquaporins in Brain Edema and Neuropathological Conditions


Abstract
1. Introduction
2. Structure and Function of Aquaporins
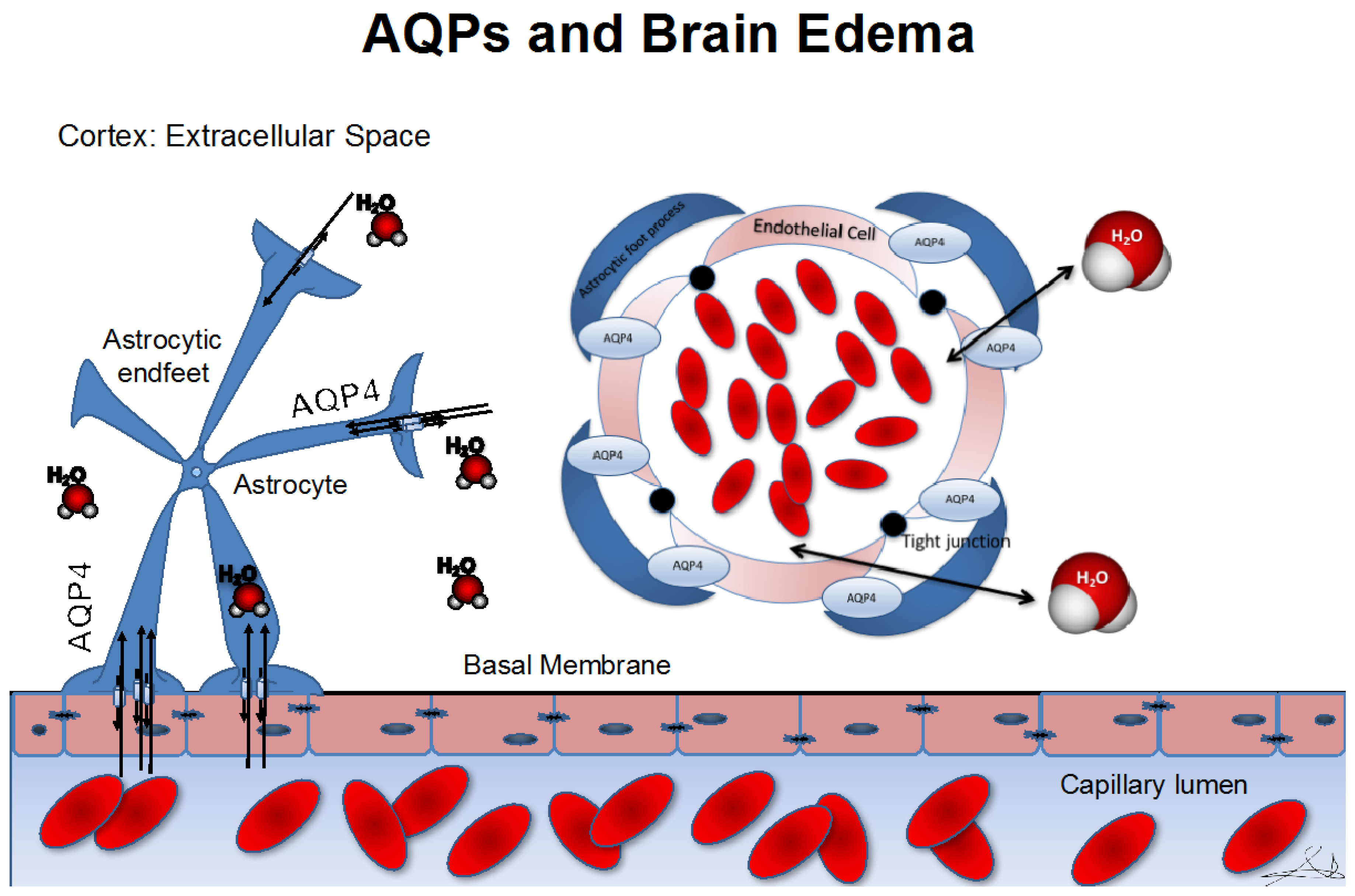
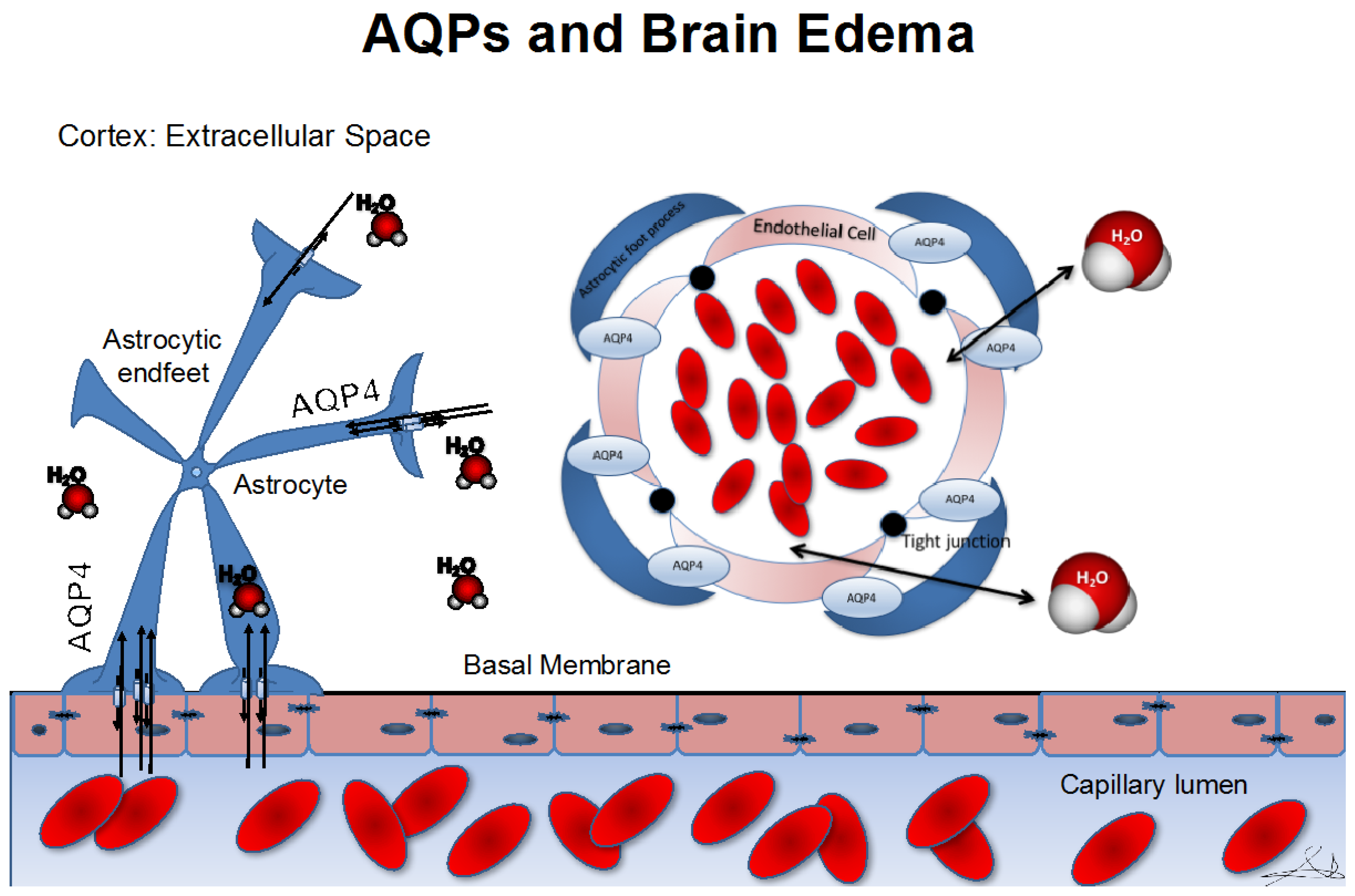
Aquaporin Distribution in the Central Nervous System (CNS)
3. Role of Aquaporins in Cerebral Edema
3.1. Hypoxia-Induced Changes in Aquaporin Expression
3.2. Traumatic Brain Injury
3.3. Edematous Brain Tumors
4. Hydrocephalus
5. Role of Aquaporins in Cellular Migration
6. Epilepsy, Memory Consolidation, and Aquaporins
Glutamate Metabolism and Synaptic Plasticity
7. Aquaporins and the Glymphatic System
8. Looking Forward
Author Contributions
Conflicts of Interest
References
- Saadoun, S.; Papadopoulos, M.C.; Davies, D.C.; Bell, B.A.; Krishna, S. Increased aquaporin 1 water channel expression in human brain tumours. Br. J. Cancer 2002, 87, 621–623. [Google Scholar] [CrossRef] [PubMed]
- Manley, G.T.; Fujimura, M.; Ma, T.; Noshita, N.; Filiz, F.; Bollen, A.W.; Chan, P.; Verkman, A.S. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat. Med. 2000, 6, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, M.C.; Manley, G.T.; Krishna, S.; Verkman, A.S. Aquaporin-4 facilitates reabsorption of excess fluid in vasogenic brain edema. FASEB J. 2004, 18, 1291–1293. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, J.A.; Szu, J.I.; Yonan, J.M.; Binder, D.K. Regulation of astrocyte glutamate transporter-1 (GLT1) and aquaporin-4 (AQP4) expression in a model of epilepsy. Exp. Neurol. 2016, 283, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.; Huang, C.; Ding, H.; Dong, J.; Gao, Z.; Yang, X.; Tang, Y.; Dong, Q. Aquaporin-4 and Cerebrovascular Diseases. Int. J. Mol. Sci. 2016, 17, 1249. [Google Scholar] [CrossRef] [PubMed]
- Previch, L.E.; Ma, L.; Wright, J.C.; Singh, S.; Geng, X.; Ding, Y. Progress in AQP research and new developments in therapeutic approaches to ischemic and hemorrhagic stroke. Int. J. Mol. Sci. 2016, 17, 1146. [Google Scholar] [CrossRef] [PubMed]
- Ikeshima-Kataoka, H. Neuroimmunological Implications of AQP4 in Astrocytes. Int. J. Mol. Sci. 2016, 17, 1306. [Google Scholar] [CrossRef] [PubMed]
- Sánchez Gomar, I.; Díaz Sánchez, M.; Uclés Sánchez, A.J.; Casado Chocán, J.L.; Suárez-Luna, N.; Ramírez-Lorca, R.; Villadiego, J.; Toledo-Aral, J.J.; Echevarría, M. Comparative analysis for the presence of IgG anti-Aquaporin-1 in patients with NMO-spectrum disorders. Int. J. Mol. Sci. 2016, 17, 1195. [Google Scholar] [CrossRef] [PubMed]
- Gleiser, C.; Wagner, A.; Fallier-Becker, P.; Wolburg, H.; Hirt, B.; Mack, A.F. Aquaporin-4 in astroglial cells in the CNS and supporting cells of sensory organs-A comparative perspective. Int. J. Mol. Sci. 2016, 17, 1411. [Google Scholar] [CrossRef] [PubMed]
- Agre, P.; King, L.S.; Yasui, M.; Guggino, W.B.; Ottersen, O.P.; Fujiyoshi, Y.; Engel, A.; Nielsen, S. Aquaporin water channels—From atomic structure to clinical medicine. J. Physiol. 2002, 542, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Benga, G.; Popescu, O.; Borza, V.; Pop, V.I.; Muresan, A.; Mocsy, I.; Brain, A.; Wrigglesworth, J.M. Water permeability in human erythrocytes: Identification of membrane proteins involved in water transport. Eur. J. Cell Biol. 1986, 41, 252–262. [Google Scholar] [PubMed]
- Agre, P.; Preston, G.M.; Smith, B.L.; Jung, J.S.; Raina, S.; Moon, C.; Guggino, W.B.; Nielsen, S. Aquaporin CHIP: The archetypal molecular water channel. Am. J. Physiol. 1993, 265, F463–F476. [Google Scholar] [PubMed]
- Gonen, T.; Walz, T. The structure of aquaporins. Q. Rev. Biophys. 2006, 39, 361–396. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.S.; Preston, G.M.; Smith, B.L.; Guggino, W.B.; Agre, P. Molecular structure of the water channel through aquaporin CHIP. The hourglass model. J. Biol. Chem. 1994, 269, 14648–14654. [Google Scholar] [PubMed]
- Maunsbach, A.B.; Marples, D.; Chin, E.; Ning, G.; Bondy, C.; Agre, P.; Nielsen, S. Aquaporin-1 water channel expression in human kidney. J. Am. Soc. Nephrol. 1997, 8, 1–14. [Google Scholar] [PubMed]
- Pallone, T.L.; Kishore, B.K.; Nielsen, S.; Agre, P.; Knepper, M.A. Evidence that aquaporin-1 mediates NaCl-induced water flux across descending vasa recta. Am. J. Physiol. 1997, 272, F587–F596. [Google Scholar] [PubMed]
- Nielsen, S.; Chou, C.L.; Marples, D.; Christensen, E.I.; Kishore, B.K.; Knepper, M.A. Vasopressin increases water permeability of kidney collecting duct by inducing translocation of aquaporin-CD water channels to plasma membrane. Proc. Natl. Acad. Sci. USA 1995, 92, 1013–1017. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.; Smith, B.L.; Christensen, E.I.; Agre, P. Distribution of the aquaporin CHIP in secretory and resorptive epithelia and capillary endothelia. Proc. Natl. Acad. Sci. USA 1993, 90, 7275–7279. [Google Scholar] [CrossRef] [PubMed]
- Verkman, A.S. Aquaporins: Translating bench research to human disease. J. Exp. Biol. 2009, 212, 1707–1715. [Google Scholar] [CrossRef] [PubMed]
- Badaut, J.; Regli, L. Distribution and possible roles of aquaporin 9 in the brain. Neuroscience 2003, 129, 971–981. [Google Scholar] [CrossRef] [PubMed]
- Gorelick, D.A.; Praetorius, J.; Tsunenari, T.; Nielsen, S.; Agre, P. Aquaporin-11: A channel protein lacking apparent transport function expressed in brain. BMC Biochem. 2006, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Shin, I.; Kim, H.J.; Lee, J.E.; Gye, M.C. Aquaporin 7 expression during perinatal development of mouse brain. Neurosci. Lett. 2006, 409, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, H.; Ma, T.; Skach, W.; Matthay, M.A.; Verkman, A.S. Molecular cloning of a mercurial-insensitive water channel expressed in selected water-transporting tissues. J. Biol. Chem. 1994, 269, 5497–5500. [Google Scholar] [PubMed]
- Preston, G.M.; Jung, J.S.; Guggino, W.B.; Agre, P. The mercury-sensitive residue at cysteine 189 in the CHIP28 water channel. J. Biol. Chem. 1993, 268, 17–20. [Google Scholar] [PubMed]
- Mobasheri, A.; Marples, D. Expression of the AQP-1 water channel in normal human tissues: A semiquantitative study using tissue microarray technology. Am. J. Physiol. Cell Physiol. 2004, 286, C529–C537. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Gao, F.; Liu, H.; Yu, W.H.; Sun, S.Q. Temporal changes in expression of aquaporin-3, -4, -5 and -8 in rat brains after permanent focal cerebral ischemia. Brain Res. 2009, 1290, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Badaut, J.; Lasbennes, F.; Magistretti, P.J.; Regli, L. Aquaporins in brain: Distribution, physiology, and pathophysiology. J. Cereb. Blood Flow Metab. 2002, 22, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Lambertz, N.; Hindy, N.E.; Adler, C.; Rump, K.; Adamzik, M.; Keyvani, K.; Bankfalvi, A.; Siffert, W.; Erol Sandalcioglu, I.; Bachmann, H.S. Expression of aquaporin 5 and the AQP5 polymorphism A(-1364)C in association with peritumoral brain edema in meningioma patients. J. Neurooncol. 2013, 112, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Chai, R.C.; Jiang, J.H.; Wong, A.Y.K.; Jiang, F.; Gao, K.; Vatcher, G.; Hoi Yu, A.C. AQP5 is differentially regulated in astrocytes during metabolic and traumatic injuries. Glia 2013, 61, 1748–1765. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.-J.; Wang, K.-J.; Gan, S.-W.; Xu, J.; Xu, S.-Y.; Sun, S.Q. Expression of aquaporin8 in human astrocytomas: Correlation with pathologic grade. Biochem. Biophys. Res. Commun. 2013, 440, 168–172. [Google Scholar] [CrossRef] [PubMed]
- Koike, S.; Tanaka, Y.; Matsuzaki, T.; Morishita, Y.; Ishibashi, K. Aquaporin-11 (AQP11) Expression in the Mouse Brain. Int. J. Mol. Sci. 2016, 17, 861. [Google Scholar] [CrossRef] [PubMed]
- Fishman, R.A. Brain edema. N. Engl. J. Med. 1975, 293, 706–711. [Google Scholar] [CrossRef] [PubMed]
- Solenov, E.; Watanabe, H.; Manley, G.T.; Verkman, A.S. Sevenfold-reduced osmotic water permeability in primary astrocyte cultures from AQP-4-deficient mice, measured by a fluorescence quenching method. Am. J. Physiol. Cell Physiol. 2004, 286, C426–C432. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, M.C.; Verkman, A.S. Aquaporin-4 gene disruption in mice reduces brain swelling and mortality in pneumococcal meningitis. J. Biol. Chem. 2005, 280, 13906–13912. [Google Scholar] [CrossRef] [PubMed]
- Barzó, P.; Marmarou, A.; Fatouros, P.; Hayasaki, K.; Corwin, F. Biphasic pathophysiological response of vasogenic and cellular edema in traumatic brain swelling. Acta Neurochir. Suppl. 1997, 70, 119–122. [Google Scholar] [PubMed]
- Saadoun, S.; Papadopoulos, M.C.; Davies, D.C.; Krishna, S.; Bell, B.A. Aquaporin-4 expression is increased in oedematous human brain tumours. J. Neurol. Neurosurg. Psychiatr. 2002, 72, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Smith, Z.A.; Moftakhar, P.; Malkasian, D.; Xiong, Z.; Vinters, H.V.; Lazareff, J.A. Choroid plexus hyperplasia: Surgical treatment and immunohistochemical results. Case report. J. Neurosurg. 2007, 107, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Tourdias, T.; Dragonu, I.; Fushimi, Y.; Deloire, M.S.A.; Boiziau, C.; Brochet, B.; Moonen, C.; Petry, K.G.; Dousset, V. Aquaporin 4 correlates with apparent diffusion coefficient and hydrocephalus severity in the rat brain: A combined MRI-histological study. Neuroimage 2009, 47, 659–666. [Google Scholar] [CrossRef] [PubMed]
- Saadoun, S.; Papadopoulos, M.C.; Hara-Chikuma, M.; Verkman, A.S. Impairment of angiogenesis and cell migration by targeted aquaporin-1 gene disruption. Nature 2005, 434, 786–792. [Google Scholar] [CrossRef] [PubMed]
- Saadoun, S.; Papadopoulos, M.C.; Watanabe, H.; Yan, D.; Manley, G.T.; Verkman, A.S. Involvement of aquaporin-4 in astroglial cell migration and glial scar formation. J. Cell Sci. 2005, 118, 5691–5698. [Google Scholar] [CrossRef] [PubMed]
- Nakahama, K.; Nagano, M.; Fujioka, A.; Shinoda, K.; Sasaki, H. Effect of TPA on aquaporin 4 mRNA expression in cultured rat astrocytes. Glia 1999, 25, 240–246. [Google Scholar] [CrossRef]
- Fazzina, G.; Amorini, A.M.; Marmarou, C.R.; Fukui, S.; Okuno, K.; Dunbar, J.G.; Glisson, R.; Marmarou, A.; Kleindienst, A. The protein kinase C activator phorbol myristate acetate decreases brain edema by aquaporin 4 downregulation after middle cerebral artery occlusion in the rat. J. Neurotrauma 2010, 27, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Amiry-Moghaddam, M.; Otsuka, T.; Hurn, P.D.; Traystman, R.J.; Haug, F.-M.; Froehner, S.C.; Adams, M.E.; Neely, J.D.; Agre, P.; Ottersen, O.P.; et al. An α-syntrophin-dependent pool of AQP4 in astroglial end-feet confers bidirectional water flow between blood and brain. Proc. Natl. Acad. Sci. USA 2003, 100, 2106–2111. [Google Scholar] [CrossRef] [PubMed]
- Eng, L.F.; Ghirnikar, R.S.; Lee, Y.L. Glial fibrillary acidic protein: GFAP-thirty-one years (1969–2000). Neurochem. Res. 2000, 25, 1439–1451. [Google Scholar] [CrossRef] [PubMed]
- Bullock, R.; Maxwell, W.L.; Graham, D.I.; Teasdale, G.M.; Adams, J.H. Glial swelling following human cerebral contusion: An ultrastructural study. J. Neurol. Neurosurg. Psychiatr. 1991, 54, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Hernando, F.; Schoots, O.; Lolait, S.J.; Burbach, J.P. Immunohistochemical localization of the vasopressin V1b receptor in the rat brain and pituitary gland: Anatomical support for its involvement in the central effects of vasopressin. Endocrinology 2001, 142, 1659–1668. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.D.; Pan, J.; Xu, M.; Su, W.; Lu, Y.Q.; Chen, Z.J.; Jiang, T.Y.; Yang, Y.M. Changes and effects of plasma arginine vasopressin in traumatic brain injury. J. Endocrinol. Investig. 2008, 31, 996–1000. [Google Scholar] [CrossRef] [PubMed]
- Bemana, I.; Nagao, S. Treatment of brain edema with a nonpeptide arginine vasopressin V1 receptor antagonist OPC-21268 in rats. Neurosurgery 1999, 44, 148–154, discussion 154–155. [Google Scholar] [CrossRef] [PubMed]
- Serradeil-Le Gal, C.; Wagnon, J.; Garcia, C.; Lacour, C.; Guiraudou, P.; Christophe, B.; Villanova, G.; Nisato, D.; Maffrand, J.P.; Le Fur, G. Biochemical and pharmacological properties of SR 49059, a new, potent, nonpeptide antagonist of rat and human vasopressin V1a receptors. J. Clin. Investig. 1993, 92, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Marmarou, C.R.; Liang, X.; Abidi, N.H.; Parveen, S.; Taya, K.; Henderson, S.C.; Young, H.F.; Filippidis, A.S.; Baumgarten, C.M. Selective vasopressin-1a receptor antagonist prevents brain edema, reduces astrocytic cell swelling and GFAP, V1aR and AQP4 expression after focal traumatic brain injury. Brain Res. 2014, 1581, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Filippidis, A.S.; Liang, X.; Wang, W.; Parveen, S.; Baumgarten, C.M.; Marmarou, C.R. Real-time monitoring of changes in brain extracellular sodium and potassium concentrations and intracranial pressure after selective vasopressin-1a receptor inhibition following focal traumatic brain injury in rats. J. Neurotrauma 2014, 31, 1258–1267. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Arrastia, R.; Kochanek, P.M.; Bergold, P.; Kenney, K.; Marx, C.E.; Grimes, C.J.; Loh, L.T.; Adam, L.T.; Oskvig, D.; Curley, K.C.; et al. Pharmacotherapy of traumatic brain injury: State of the science and the road forward: Report of the Department of Defense Neurotrauma Pharmacology Workgroup. J. Neurotrauma 2014, 31, 135–158. [Google Scholar] [CrossRef] [PubMed]
- Marmarou, A. A review of progress in understanding the pathophysiology and treatment of brain edema. Neurosurg. Focus 2007, 22, E1. [Google Scholar] [CrossRef] [PubMed]
- Staub, F.; Baethmann, A.; Peters, J.; Weigt, H.; Kempski, O. Effects of lactacidosis on glial cell volume and viability. J. Cereb. Blood Flow Metab. 1990, 10, 866–876. [Google Scholar] [CrossRef] [PubMed]
- Bouvier, M.; Szatkowski, M.; Amato, A.; Attwell, D. The glial cell glutamate uptake carrier countertransports pH-changing anions. Nature 1992, 360, 471–474. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Umenishi, F.; Inamasu, G.; Sato, M.; Ishikawa, M.; Nishizawa, M.; Oizumi, T. Expression of water channel mRNA following cerebral ischemia. Acta Neurochir. Suppl. 2000, 76, 239–241. [Google Scholar] [PubMed]
- Yamamoto, N.; Yoneda, K.; Asai, K.; Sobue, K.; Tada, T.; Fujita, Y.; Katsuya, H.; Fujita, M.; Aihara, N.; Mase, M.; et al. Alterations in the expression of the AQP family in cultured rat astrocytes during hypoxia and reoxygenation. Brain Res. Mol. Brain Res. 2001, 90, 26–38. [Google Scholar] [CrossRef]
- Taniguchi, M.; Yamashita, T.; Kumura, E.; Tamatani, M.; Kobayashi, A.; Yokawa, T.; Maruno, M.; Kato, A.; Ohnishi, T.; Kohmura, E.; et al. Induction of aquaporin-4 water channel mRNA after focal cerebral ischemia in rat. Brain Res. Mol. Brain Res. 2000, 78, 131–137. [Google Scholar] [CrossRef]
- Kaur, C.; Sivakumar, V.; Zhang, Y.; Ling, E.A. Hypoxia-induced astrocytic reaction and increased vascular permeability in the rat cerebellum. Glia 2006, 54, 826–839. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, T.; Katayama, Y.; Aoyama, N.; Mori, T. Heterogeneous mechanisms of early edema formation in cerebral contusion: Diffusion MRI and ADC mapping study. Acta Neurochir. Suppl. 2000, 76, 9–12. [Google Scholar] [PubMed]
- Marmarou, A.; Signoretti, S.; Fatouros, P.P.; Portella, G.; Aygok, G.A.; Bullock, M.R. Predominance of cellular edema in traumatic brain swelling in patients with severe head injuries. J. Neurosurg. 2006, 104, 720–730. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.-C.; Honey, C.R.; Berk, C.; Wong, N.L.M.; Tsui, J.K.C. Regulation of aquaporin-4 in a traumatic brain injury model in rats. J. Neurosurg. 2003, 98, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Vizuete, M.L.; Venero, J.L.; Vargas, C.; Ilundáin, A.A.; Echevarría, M.; Machado, A.; Cano, J. Differential upregulation of aquaporin-4 mRNA expression in reactive astrocytes after brain injury: Potential role in brain edema. Neurobiol. Dis. 1999, 6, 245–258. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, M.C.; Saadoun, S.; Binder, D.K.; Manley, G.T.; Krishna, S.; Verkman, A.S. Molecular mechanisms of brain tumor edema. Neuroscience 2003, 129, 1011–1020. [Google Scholar] [CrossRef] [PubMed]
- Fallier-Becker, P.; Nieser, M.; Wenzel, U.; Ritz, R.; Noell, S. Is upregulation of Aquaporin 4-M1 isoform responsible for the loss of typical orthogonal arrays of particles in astrocytomas? Int. J. Mol. Sci. 2016, 17, 1230. [Google Scholar] [CrossRef] [PubMed]
- Dubois, L.G.; Campanati, L.; Righy, C.; D’Andrea-Meira, I.; Spohr, T.C.; Porto-Carreiro, I.; Pereira, C.M.; Balça-Silva, J.; Kahn, S.A.; DosSantos, M.F.; et al. Gliomas and the vascular fragility of the blood brain barrier. Front. Cell. Neurosci. 2014, 8, 418. [Google Scholar] [CrossRef] [PubMed]
- Maugeri, R.; Schiera, G.; di Liegro, C.M.; Fricano, A.; Iacopino, D.G.; di Liegro, I. Aquaporins and brain tumors. Int. J. Mol. Sci. 2016, 17, 1029. [Google Scholar] [CrossRef] [PubMed]
- Rekate, H.L. The definition and classification of hydrocephalus: A personal recommendation to stimulate debate. Cerebrospinal Fluid Res. 2008, 5, 2. [Google Scholar] [CrossRef] [PubMed]
- Oshio, K.; Song, Y.; Verkman, A.S.; Manley, G.T. Aquaporin-1 deletion reduces osmotic water permeability and cerebrospinal fluid production. Acta Neurochir. Suppl. 2003, 86, 525–528. [Google Scholar] [PubMed]
- Oshio, K.; Watanabe, H.; Song, Y.; Verkman, A.S.; Manley, G.T. Reduced cerebrospinal fluid production and intracranial pressure in mice lacking choroid plexus water channel Aquaporin-1. FASEB J. 2005, 19, 76–78. [Google Scholar] [CrossRef] [PubMed]
- Longatti, P.; Basaldella, L.; Orvieto, E.; Dei Tos, A.; Martinuzzi, A. Aquaporin(s) expression in choroid plexus tumours. Pediatr. Neurosurg. 2005, 42, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Leena, P. The altered expression of aquaporin 1 and 4 in choroid plexus of congenital hydrocephalus. Cerebrospinal Fluid Res. 2009, 6, S7. [Google Scholar]
- Bloch, O.; Auguste, K.I.; Manley, G.T.; Verkman, A.S. Accelerated progression of kaolin-induced hydrocephalus in aquaporin-4-deficient mice. J. Cereb. Blood Flow Metab. 2006, 26, 1527–1537. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Enno, T.L.; del Bigio, M.R. Aquaporin 4 changes in rat brain with severe hydrocephalus. Eur. J. Neurosci. 2006, 23, 2929–2936. [Google Scholar] [CrossRef] [PubMed]
- Auguste, K.I.; Jin, S.; Uchida, K.; Yan, D.; Manley, G.T.; Papadopoulos, M.C.; Verkman, A.S. Greatly impaired migration of implanted aquaporin-4-deficient astroglial cells in mouse brain toward a site of injury. FASEB J. 2007, 21, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Vacca, A.; Frigeri, A.; Ribatti, D.; Nicchia, G.P.; Nico, B.; Ria, R.; Svelto, M.; Dammacco, F. Microvessel overexpression of aquaporin 1 parallels bone marrow angiogenesis in patients with active multiple myeloma. Br. J. Haematol. 2001, 113, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Verkman, A.S. More than just water channels: Unexpected cellular roles of aquaporins. J. Cell Sci. 2005, 118, 3225–3232. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, M.T.; Dehghani, G.A. Nitric oxide as a regulatory factor for aquaporin-1 and 4 gene expression following brain ischemia/reperfusion injury in rat. Pathol. Res. Pract. 2015, 211, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Hesdorffer, D.C.; Logroscino, G.; Benn, E.K.T.; Katri, N.; Cascino, G.; Hauser, W.A. Estimating risk for developing epilepsy: A population-based study in Rochester, Minnesota. Neurology 2011, 76, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Ransom, B.; Behar, T.; Nedergaard, M. New roles for astrocytes (stars at last). Trends Neurosci. 2003, 26, 520–522. [Google Scholar] [CrossRef] [PubMed]
- Chebabo, S.R.; Hester, M.A.; Aitken, P.G.; Somjen, G.G. Hypotonic exposure enhances synaptic transmission and triggers spreading depression in rat hippocampal tissue slices. Brain Res. 1995, 695, 203–216. [Google Scholar] [CrossRef]
- Binder, D.K.; Oshio, K.; Ma, T.; Verkman, A.S.; Manley, G.T. Increased seizure threshold in mice lacking aquaporin-4 water channels. Neuroreport 2004, 15, 259–262. [Google Scholar] [CrossRef] [PubMed]
- Binder, D.K.; Yao, X.; Zador, Z.; Sick, T.J.; Verkman, A.S.; Manley, G.T. Increased seizure duration and slowed potassium kinetics in mice lacking aquaporin-4 water channels. Glia 2006, 53, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Nagelhus, E.A.; Mathiisen, T.M.; Ottersen, O.P. Aquaporin-4 in the central nervous system: Cellular and subcellular distribution and coexpression with KIR4.1. Neuroscience 2004, 129, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Vajda, Z.; Pedersen, M.; Füchtbauer, E.-M.; Wertz, K.; Stødkilde-Jørgensen, H.; Sulyok, E.; Dóczi, T.; Neely, J.D.; Agre, P.; Frøkiaer, J.; et al. Delayed onset of brain edema and mislocalization of aquaporin-4 in dystrophin-null transgenic mice. Proc. Natl. Acad. Sci. USA 2002, 99, 13131–13136. [Google Scholar] [CrossRef] [PubMed]
- Binder, D.K.; Nagelhus, E.A.; Ottersen, O.P. Aquaporin-4 and epilepsy. Glia 2012, 60, 1203–1214. [Google Scholar] [CrossRef] [PubMed]
- Danbolt, N.C. Glutamate uptake. Prog. Neurobiol. 2001, 65, 1–105. [Google Scholar] [CrossRef]
- Tanaka, K.; Watase, K.; Manabe, T.; Yamada, K.; Watanabe, M.; Takahashi, K.; Iwama, H.; Nishikawa, T.; Ichihara, N.; Kikuchi, T.; et al. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science 1997, 276, 1699–1702. [Google Scholar] [CrossRef] [PubMed]
- Kong, Q.; Takahashi, K.; Schulte, D.; Stouffer, N.; Lin, Y.; Lin, C.-L.G. Increased glial glutamate transporter EAAT2 expression reduces epileptogenic processes following pilocarpine-induced status epilepticus. Neurobiol. Dis. 2012, 47, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, J.A.; Hsu, M.S.; Seldin, M.M.; Binder, D.K. Expression of the astrocyte water channel Aquaporin-4 in the mouse brain. ASN Neuro 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Skucas, V.A.; Mathews, I.B.; Yang, J.; Cheng, Q.; Treister, A.; Duffy, A.M.; Verkman, A.S.; Hempstead, B.L.; Wood, M.A.; Binder, D.K.; et al. Impairment of select forms of spatial memory and neurotrophin-dependent synaptic plasticity by deletion of glial aquaporin-4. J. Neurosci. 2011, 31, 6392–6397. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Liu, M.; Wu, X.; Wang, F.; Ding, J.; Chen, J.; Hu, G. Aquaporin-4 promotes memory consolidation in Morris water maze. Brain Struct. Funct. 2013, 218, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-K.; Wang, F.; Wang, W.; Luo, Y.; Wu, P.-F.; Xiao, J.-L.; Hu, Z.-L.; Jin, Y.; Hu, G.; Chen, J.-G. Aquaporin-4 deficiency impairs synaptic plasticity and associative fear memory in the lateral amygdala: Involvement of downregulation of glutamate transporter-1 expression. Neuropsychopharmacology 2012, 37, 1867–1878. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Li, M.-X.; Luo, Y.; Chen, T.; Liu, J.; Fang, P.; Jiang, B.; Hu, Z.-L.; Jin, Y.; Chen, J.-G.; et al. Chronic ceftriaxone treatment rescues hippocampal memory deficit in AQP4 knockout mice via activation of GLT-1. Neuropharmacology 2013, 75, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Katagiri, H.; Tanaka, K.; Manabe, T. Requirement of appropriate glutamate concentrations in the synaptic cleft for hippocampal LTP induction. Eur. J. Neurosci. 2001, 14, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015, 523, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci. Transl. Med. 2012, 4, 147ra111. [Google Scholar] [CrossRef] [PubMed]
- Kress, B.T.; Iliff, J.J.; Xia, M.; Wang, M.; Wei, H.S.; Zeppenfeld, D.; Xie, L.; Kang, H.; Xu, Q.; Liew, J.A.; et al. Impairment of paravascular clearance pathways in the aging brain. Ann. Neurol. 2014, 76, 845–861. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Process | Species | AQPs Studied | Results |
|---|---|---|---|
| Cytotoxic edema [33] | Mice | AQP4 | Upregulated |
| Vasogenic edema [34] | Mice | AQP4 | Upregulated |
| Traumatic brain injury [35] | Rats | AQP4 | Upregulated |
| Edematous brain tumor [36] | Human | AQP4 | Upregulated |
| Hydrocephalus [37,38] | Human | AQP1 | Downregulated |
| Rat | AQP4 | Upregulated | |
| Cellular migration [39,40] | Mice | AQP1 | Upregulated |
| Mice | AQP4 | Upregulated | |
| Prolonged status epilepticus [4] | Mice | AQP4 | Downregulated |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Filippidis, A.S.; Carozza, R.B.; Rekate, H.L. Aquaporins in Brain Edema and Neuropathological Conditions. Int. J. Mol. Sci. 2017, 18, 55. https://doi.org/10.3390/ijms18010055
Filippidis AS, Carozza RB, Rekate HL. Aquaporins in Brain Edema and Neuropathological Conditions. International Journal of Molecular Sciences. 2017; 18(1):55. https://doi.org/10.3390/ijms18010055
Chicago/Turabian StyleFilippidis, Aristotelis S., Richard B. Carozza, and Harold L. Rekate. 2017. "Aquaporins in Brain Edema and Neuropathological Conditions" International Journal of Molecular Sciences 18, no. 1: 55. https://doi.org/10.3390/ijms18010055
APA StyleFilippidis, A. S., Carozza, R. B., & Rekate, H. L. (2017). Aquaporins in Brain Edema and Neuropathological Conditions. International Journal of Molecular Sciences, 18(1), 55. https://doi.org/10.3390/ijms18010055