
Cannabidiol Modulates the Expression of Alzheimer’s Disease-Related Genes in Mesenchymal Stem Cells

 , , and
, , and
Abstract
:1. Introduction
2. Results
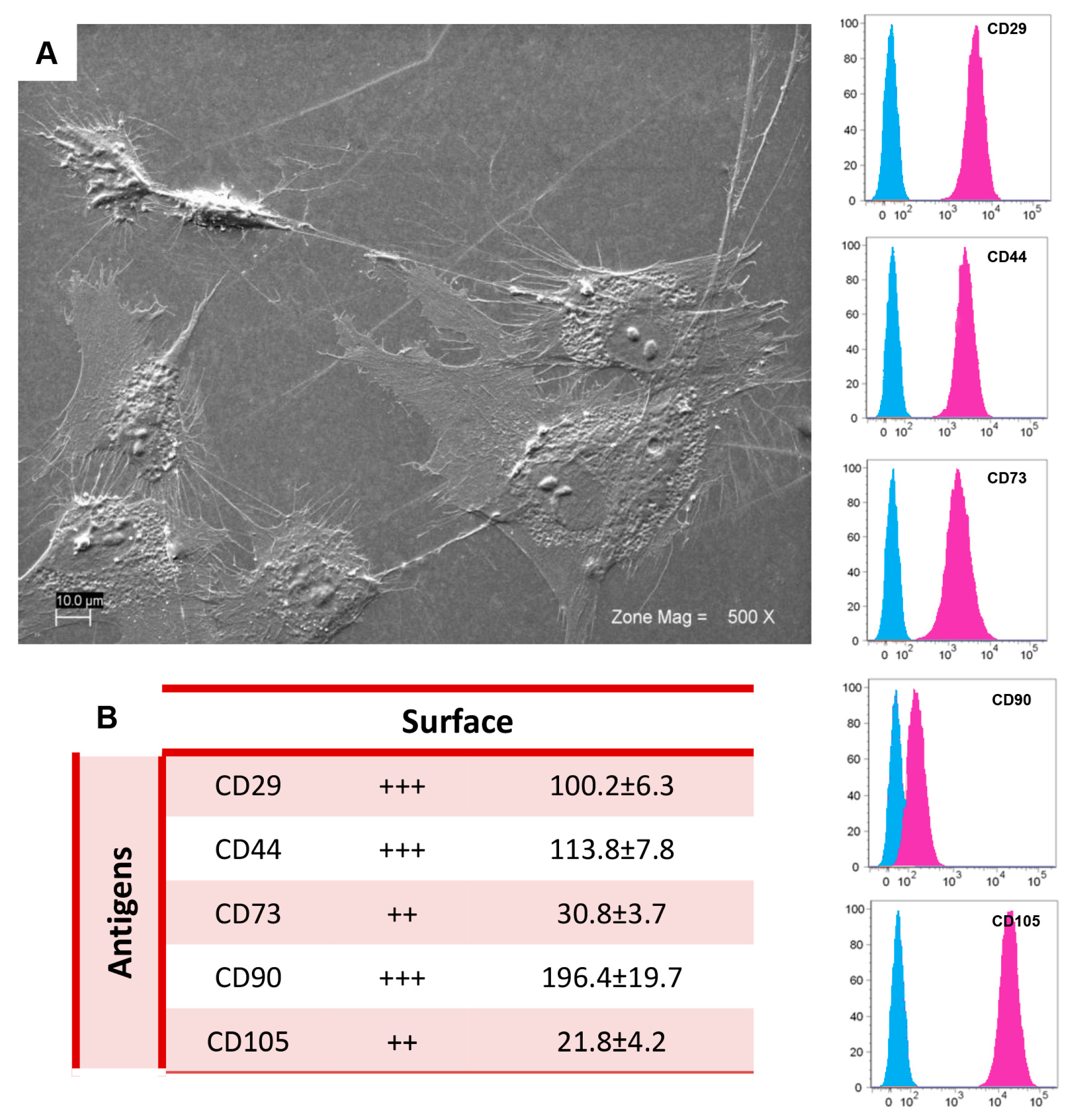
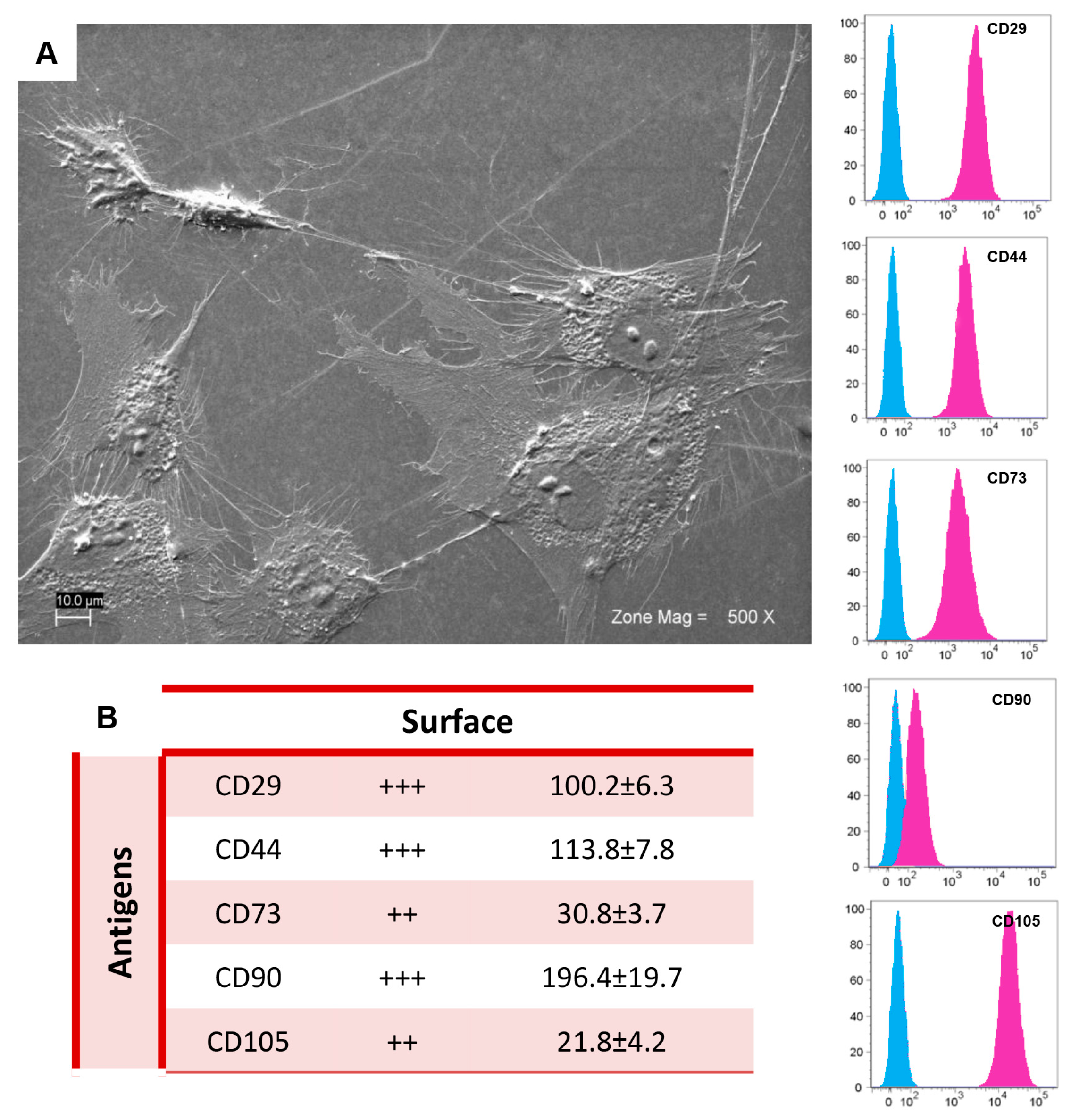
2.1. Morphological and Cytofluorimetric Characterization of GMSCs
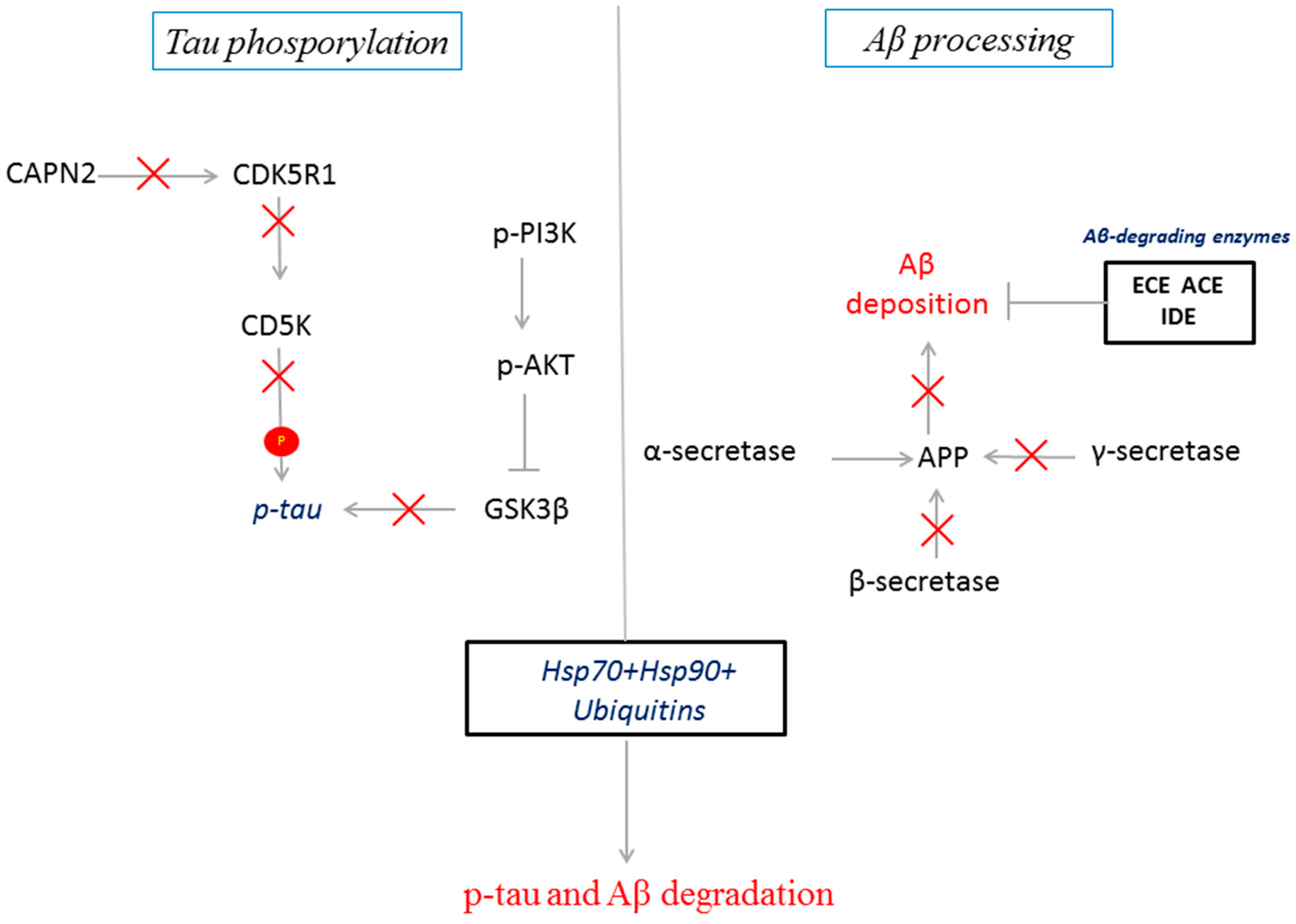
2.2. CBD Treatment in GMSCs Modulated the Transcription of Genes Involved in AD Pathogenesis
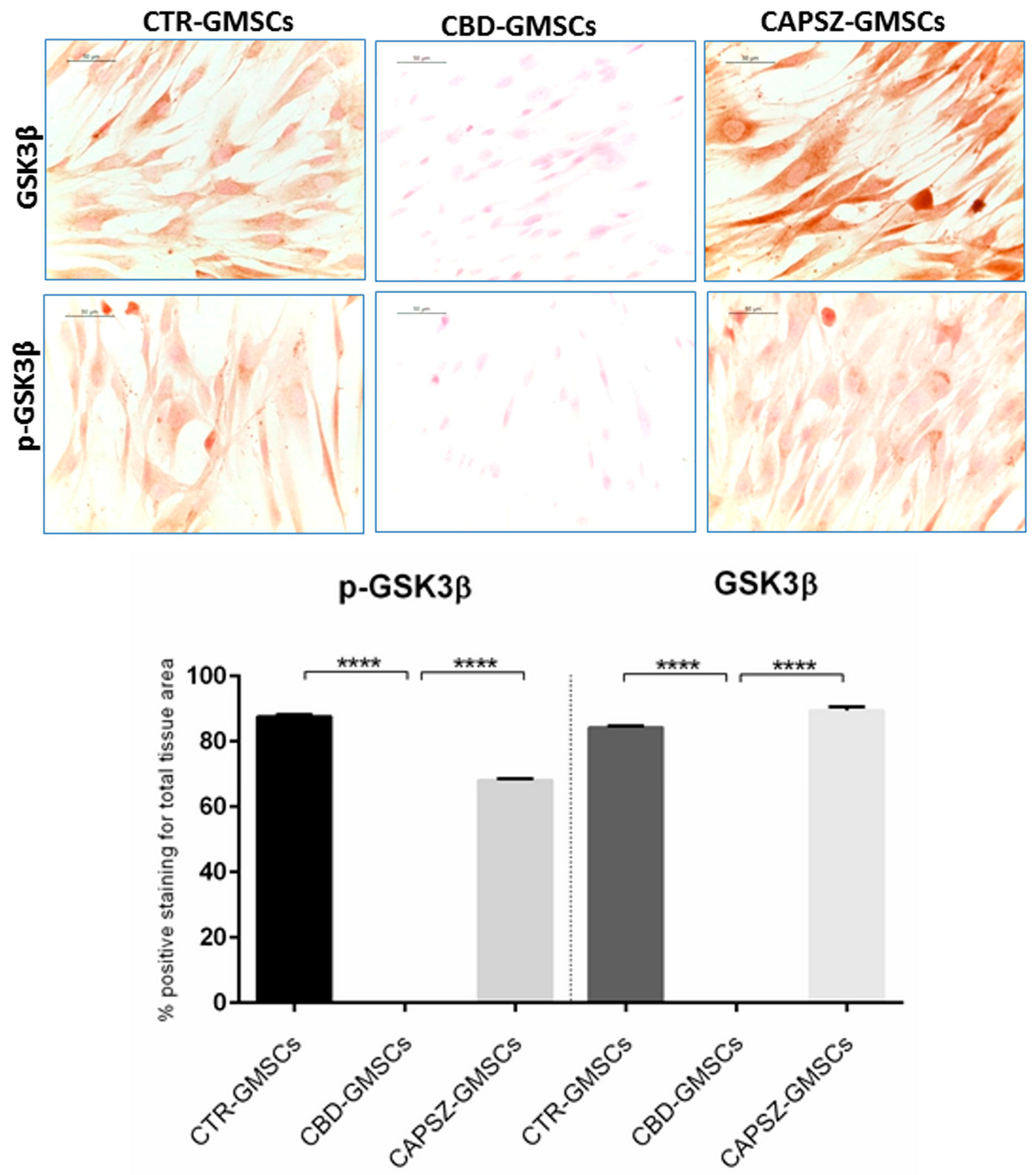
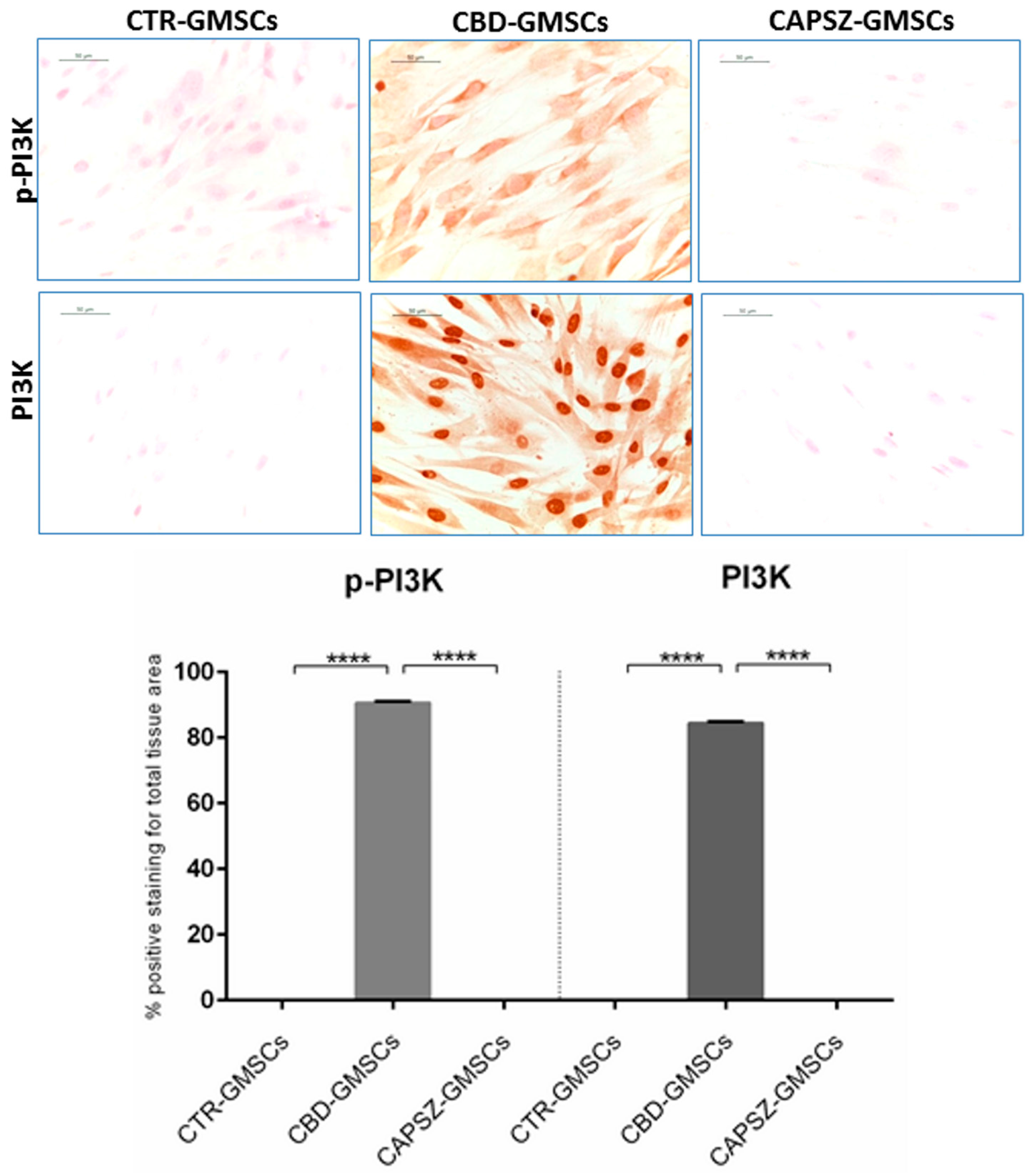
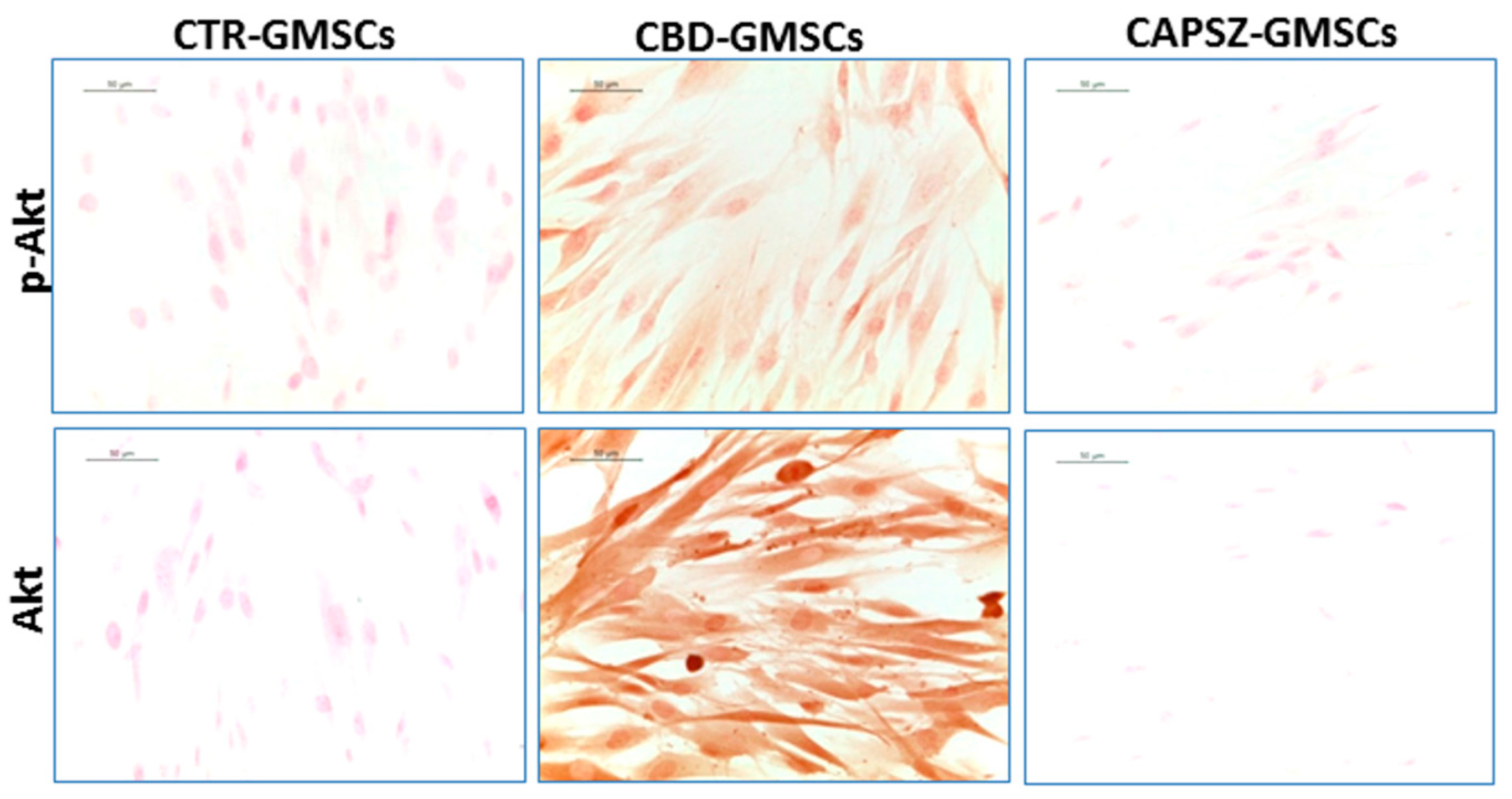
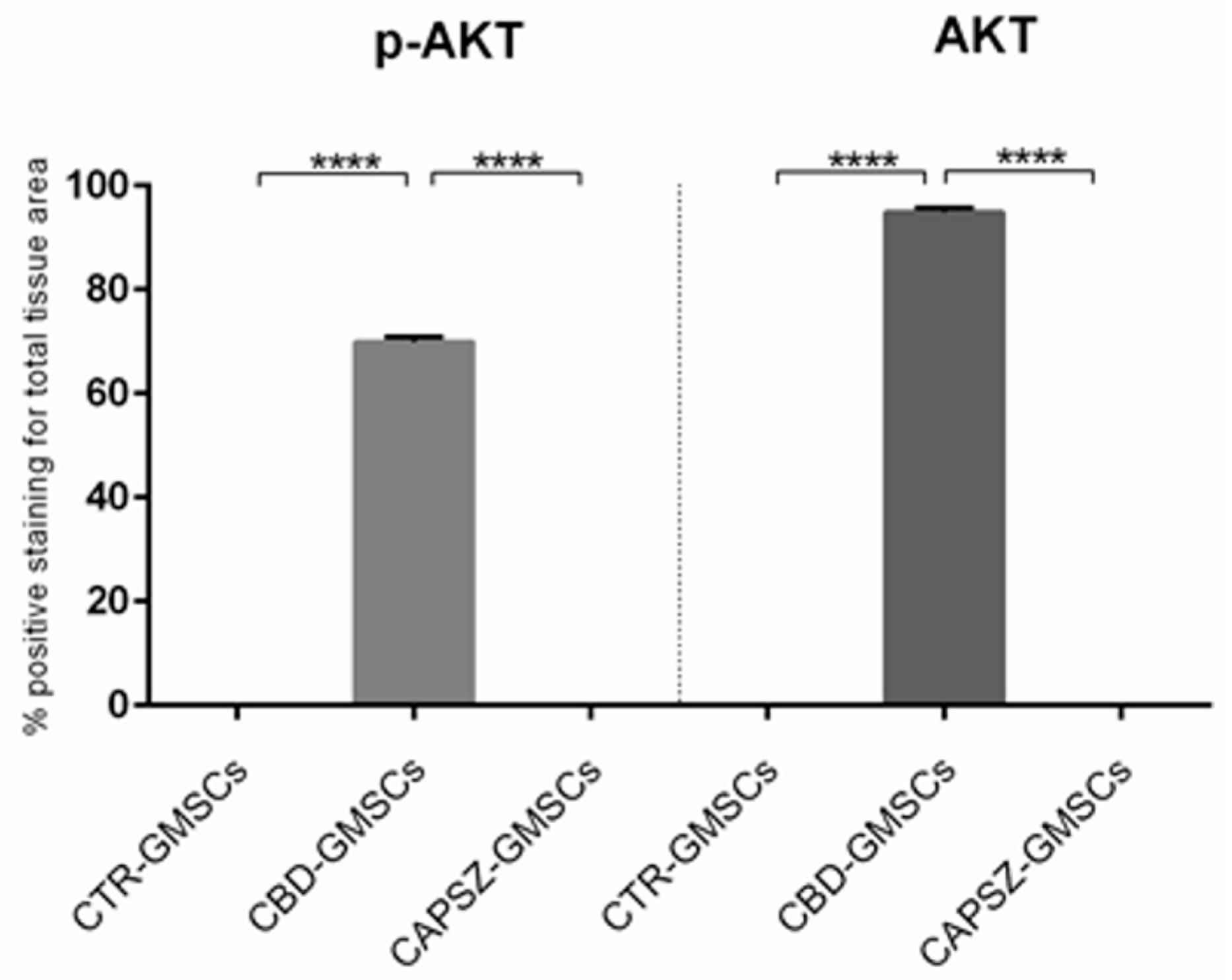
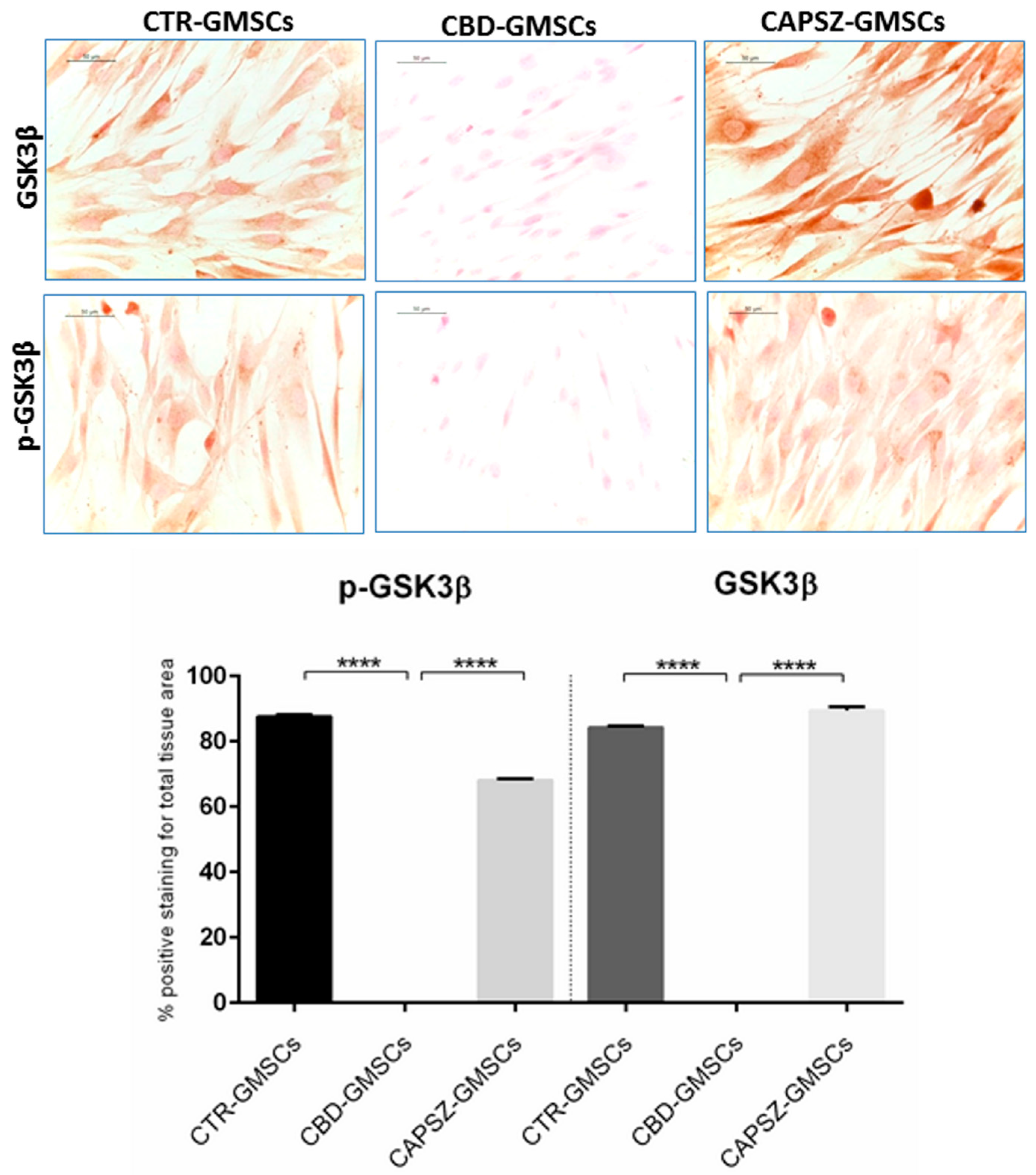
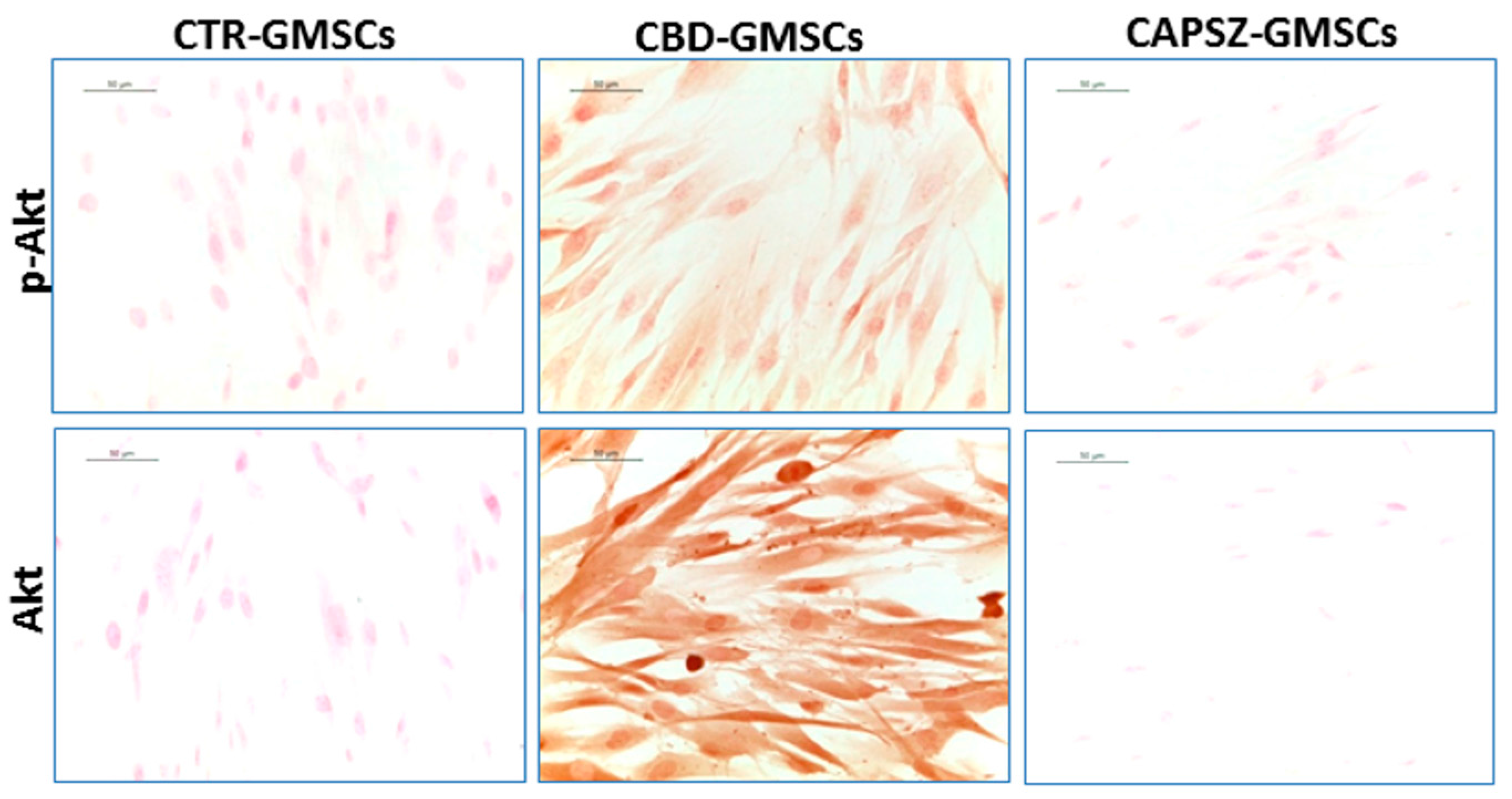
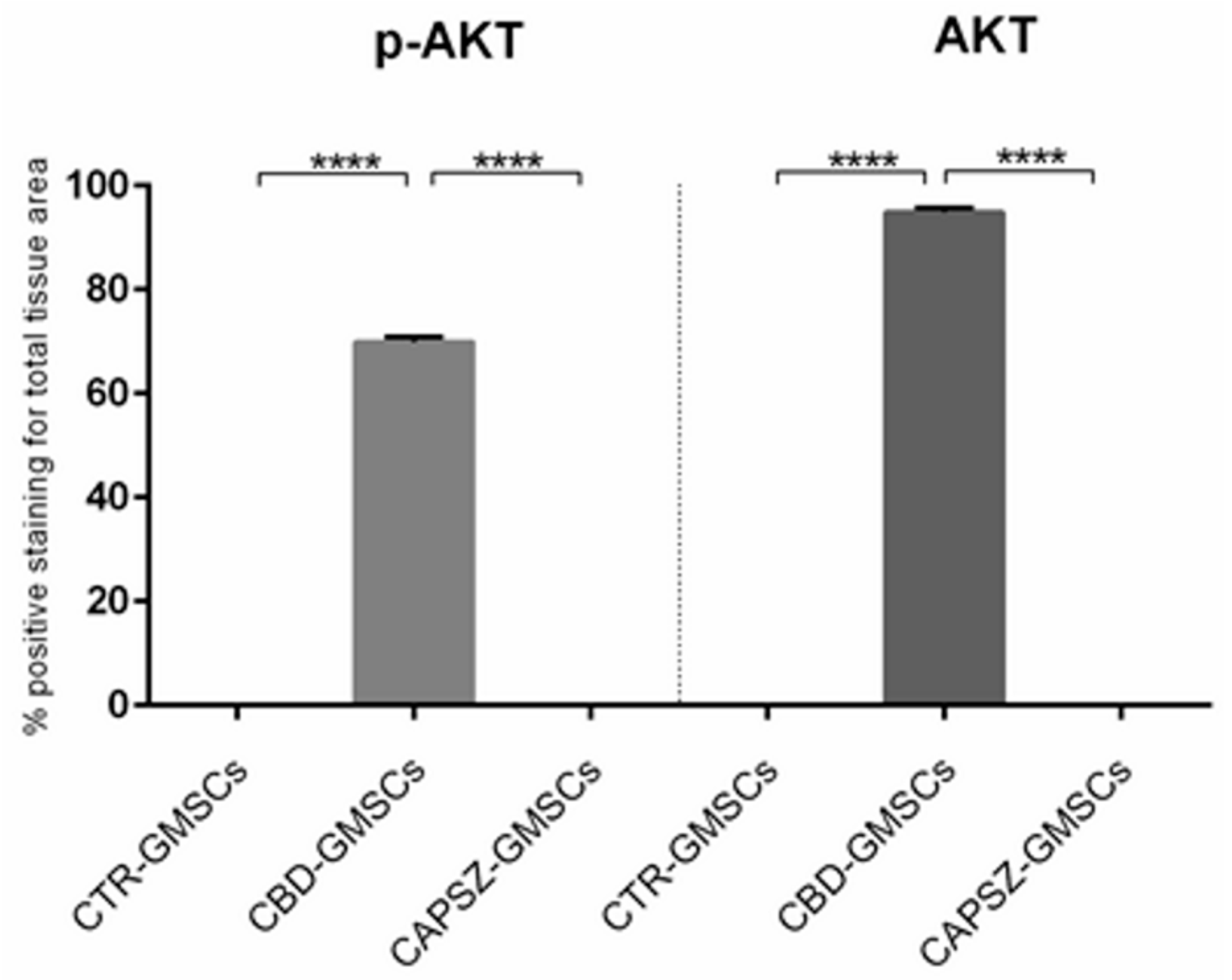
2.3. CBD Inhibited GSK3β Activity by Promoting the PI3K/AKT Signalling Pathway
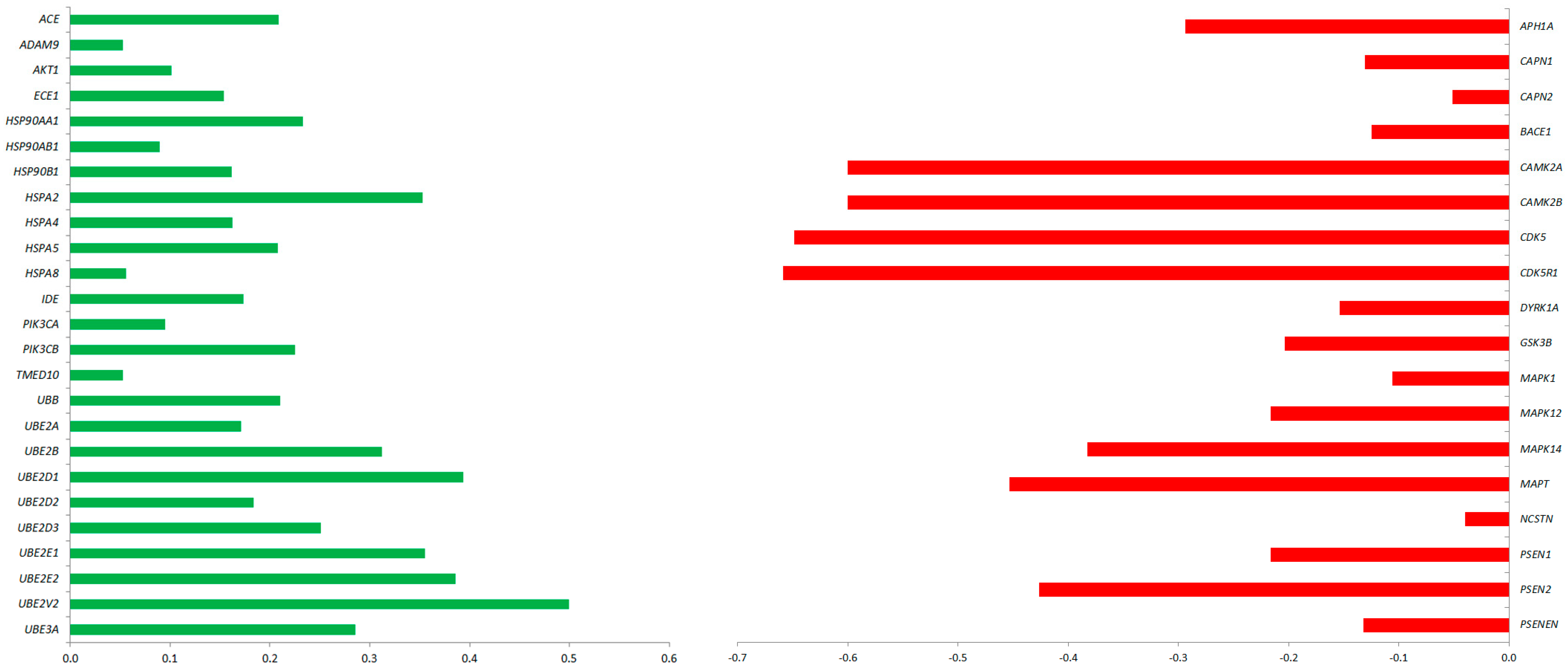
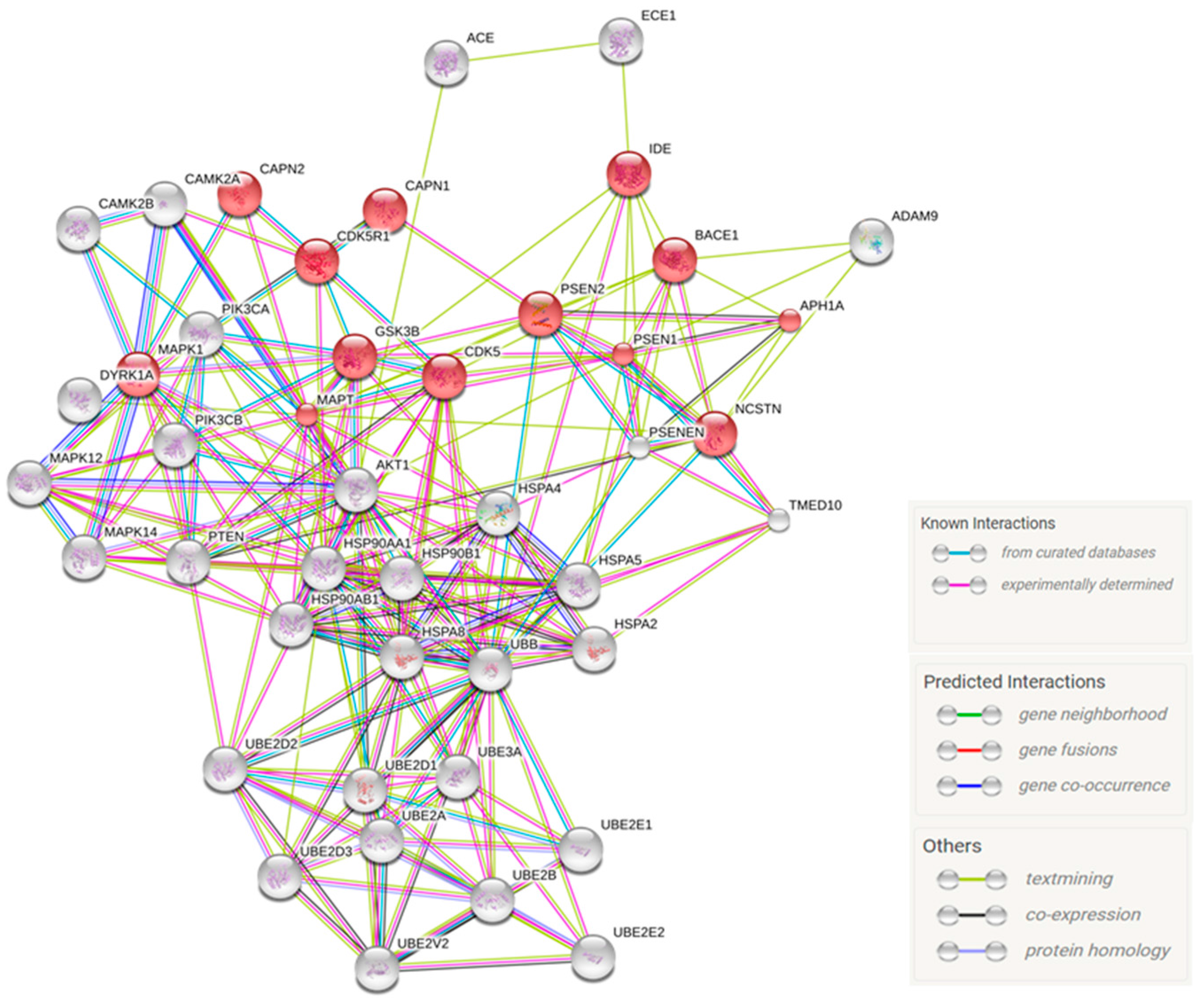
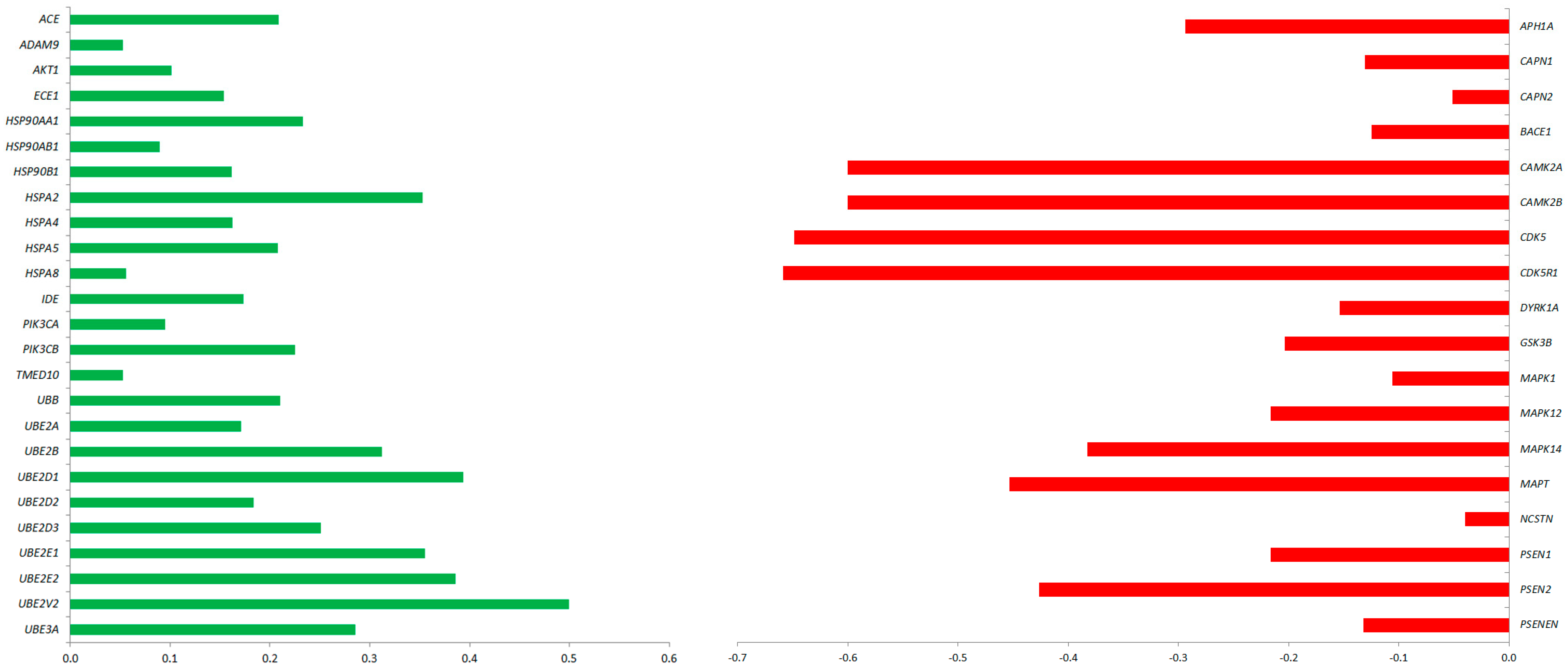
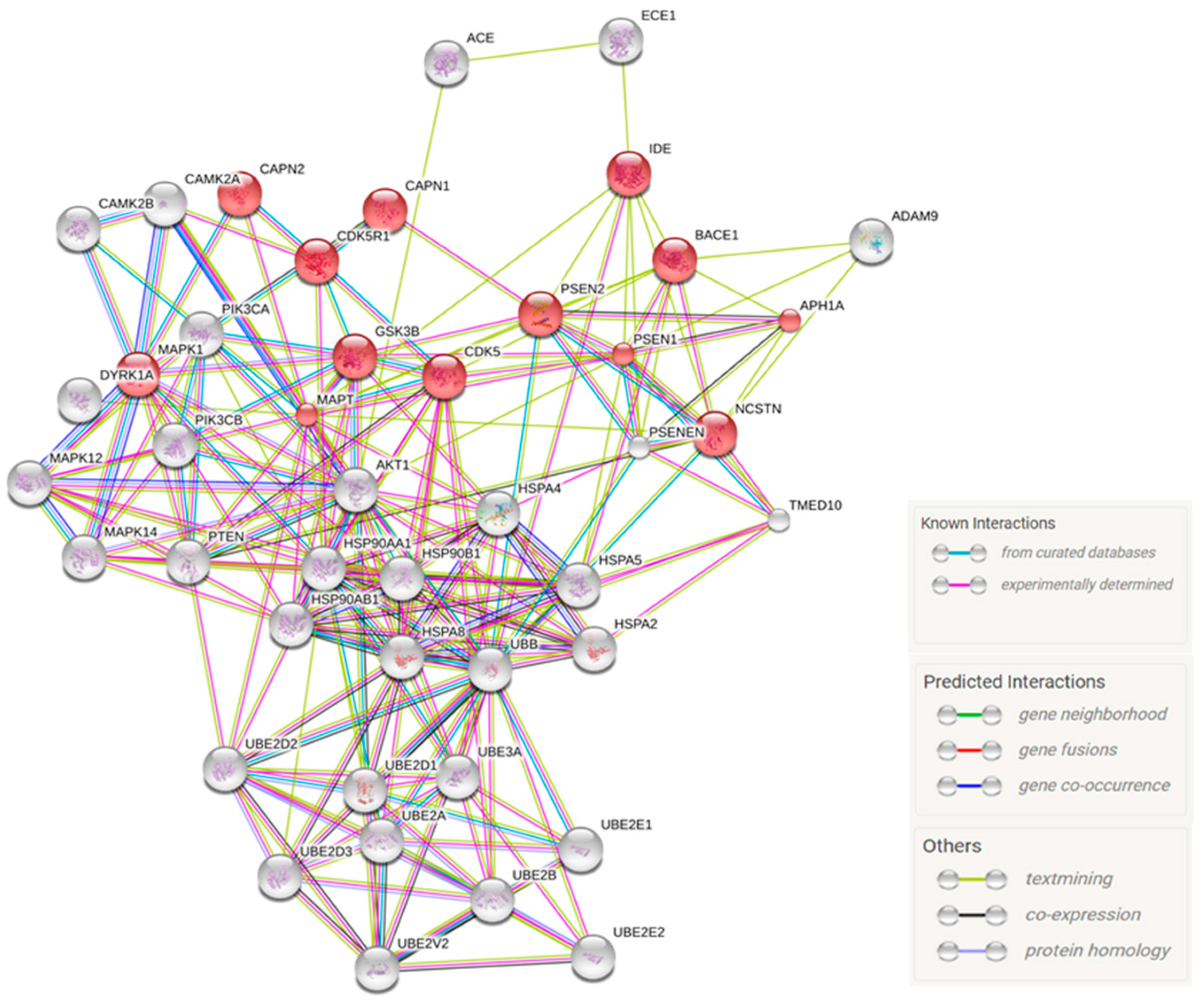
2.4. Predicted Protein Interaction Network in CBD-GMSCs
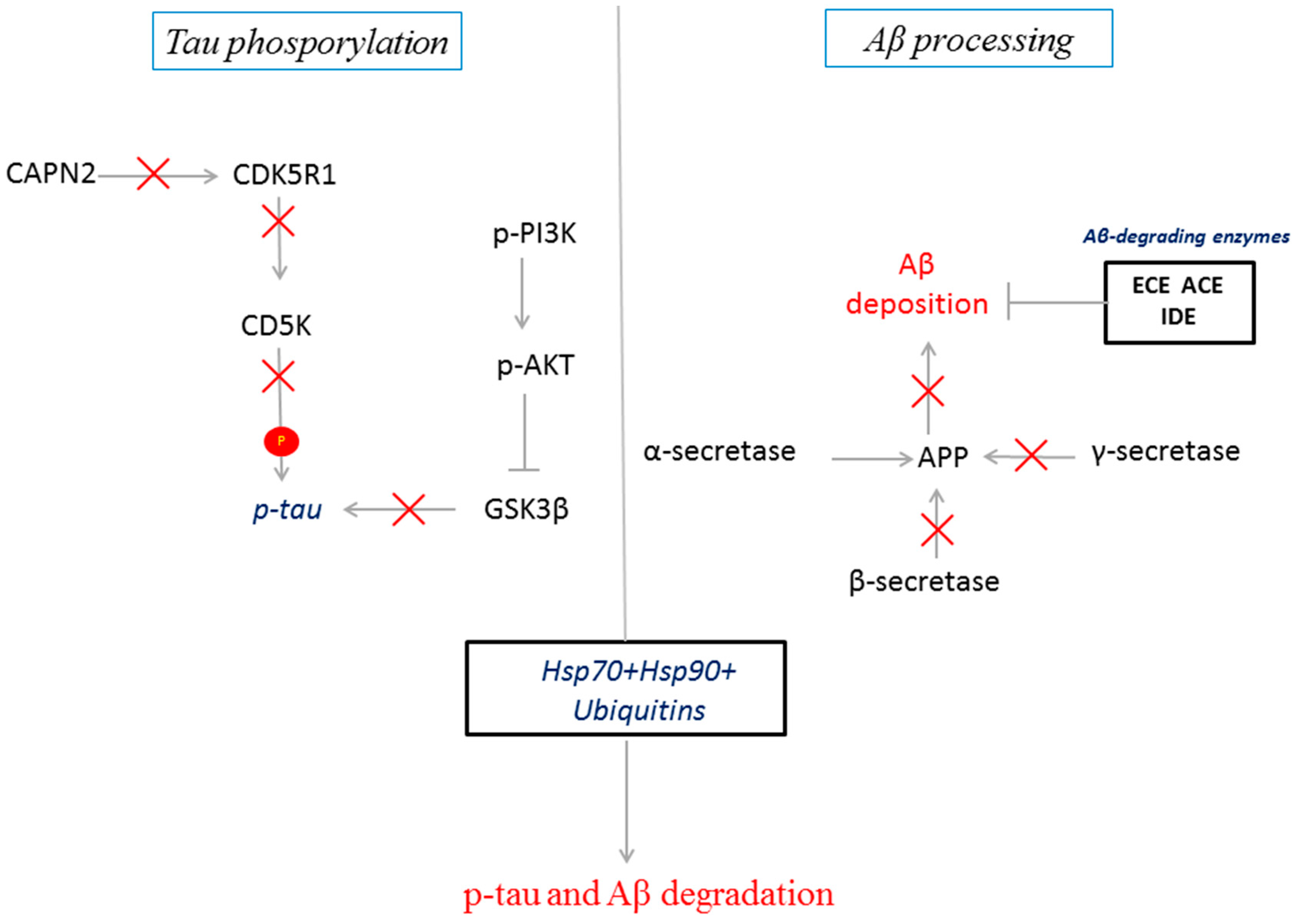
3. Discussion
4. Materials and Methods
4.1. Extraction and Isolation of CBD
4.2. Cell Culture Isolation and Characterization
4.3. Scanning Electron Microscopy Analysis
4.4. Cell Cultures and Drug Treatment
4.5. Preparation of RNA Libraries for Deep Sequencing
4.6. NGS Data Processing
4.7. Bioinformatics Analysis
4.8. Immunocytochemistry
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s Disease |
| Aβ | Amyloid Beta |
| CBD | Cannabidiol |
| CD | Cluster of Differentiation |
| FPKM | Fragments per Kilobase of Exon per Million Fragments Mapped |
| GMSCs | Gingiva Mesenchymal Stem Cells |
| GO | Gene Ontology |
| GSK3β | Glycogen Synthase Kinase 3β |
| HLA | Human Leukocyte Antigen-DR |
| HSPs | Heat Shock Proteins |
| MSCs | Mesenchymal Stem Cells |
References
- Du, L.; Yang, P.; Ge, S. Isolation and characterization of human gingiva-derived mesenchymal stem cells using limiting dilution method. J. Dent. Sci. 2016, 11, 304–314. [Google Scholar] [CrossRef]
- Gao, Y.; Zhao, G.; Li, D.; Chen, X.; Pang, J.; Ke, J. Isolation and multiple differentiation potential assessment of human gingival mesenchymal stem cells. Int. J. Mol. Sci. 2014, 15, 20982–20996. [Google Scholar] [CrossRef] [PubMed]
- Mitrano, T.I.; Grob, M.S.; Carrion, F.; Nova-Lamperti, E.; Luz, P.A.; Fierro, F.S.; Quintero, A.; Chaparro, A.; Sanz, A. Culture and characterization of mesenchymal stem cells from human gingival tissue. J. Periodontol. 2010, 81, 917–925. [Google Scholar] [CrossRef] [PubMed]
- Hass, R.; Kasper, C.; Bohm, S.; Jacobs, R. Different populations and sources of human mesenchymal stem cells (MSC): A comparison of adult and neonatal tissue-derived MSC. Cell Commun. Signal. 2011, 9, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santamaria, S.; Sanchez, N.; Sanz, M.; Garcia-Sanz, J.A. Comparison of periodontal ligament and gingiva-derived mesenchymal stem cells for regenerative therapies. Clin. Oral Investig. 2016. [Google Scholar] [CrossRef] [PubMed]
- Tomar, G.B.; Srivastava, R.K.; Gupta, N.; Barhanpurkar, A.P.; Pote, S.T.; Jhaveri, H.M.; Mishra, G.C.; Wani, M.R. Human gingiva-derived mesenchymal stem cells are superior to bone marrow-derived mesenchymal stem cells for cell therapy in regenerative medicine. Biochem. Biophys. Res. Commun. 2010, 393, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Chen, C.; Akiyama, K.; Chai, Y.; Le, A.D.; Wang, Z.; Shi, S. Gingivae contain neural-crest- and mesoderm-derived mesenchymal stem cells. J. Dent. Res. 2013, 92, 825–832. [Google Scholar] [CrossRef] [PubMed]
- Gouras, G.K.; Almeida, C.G.; Takahashi, R.H. Intraneuronal Aβ accumulation and origin of plaques in Alzheimer’s disease. Neurobiol. Aging 2005, 26, 1235–1244. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Alzheimer’s disease results from the cerebral accumulation and cytotoxicity of amyloid β-protein. J. Alzheimers Dis. 2001, 3, 75–80. [Google Scholar] [PubMed]
- Chow, V.W.; Mattson, M.P.; Wong, P.C.; Gleichmann, M. An overview of APP processing enzymes and products. Neuromol. Med. 2010, 12, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Cruchaga, C.; Haller, G.; Chakraverty, S.; Mayo, K.; Vallania, F.L.M.; Mitra, R.D.; Faber, K.; Williamson, J.; Bird, T.; Diaz-Arrastia, R.; et al. NIA-LOAD/NCRAD family study consortium rare variants in APP, PSEN1 and PSEN2 increase risk for AD in late-onset Alzheimer’s disease families. PLoS ONE 2012, 7, e31039. [Google Scholar] [CrossRef]
- Dolan, P.J.; Johnson, G.V.W. The role of tau kinases in Alzheimer’s disease. Curr. Opin. Drug Discov. Dev. 2010, 13, 595–603. [Google Scholar]
- Ferrer, I.; Barrachina, M.; Puig, B.; Martinez de Lagran, M.; Marti, E.; Avila, J.; Dierssen, M. Constitutive Dyrk1A is abnormally expressed in Alzheimer disease, Down syndrome, Pick disease, and related transgenic models. Neurobiol. Dis. 2005, 20, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, Y.; Ichinose, T.; Yamauchi, T. Phosphorylation of tau protein to sites found in Alzheimer’s disease brain is catalyzed by Ca2+/calmodulin-dependent protein kinase II as demonstrated tandem mass spectrometry. Neurosci. Lett. 2003, 353, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Esposito, G.; de Filippis, D.; Carnuccio, R.; Izzo, A.A.; Iuvone, T. The marijuana component cannabidiol inhibits β-amyloid-induced tau protein hyperphosphorylation through Wnt/β-catenin pathway rescue in PC12 cells. J. Mol. Med. (Berl.) 2006, 84, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Scuderi, C.; Steardo, L.; Esposito, G. Cannabidiol promotes amyloid precursor protein ubiquitination and reduction of β amyloid expression in SHSY5YAPP+ cells through PPARγ involvement. Phytother. Res. 2014, 28, 1007–1013. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Low, J.K.; Logge, W.; Garner, B.; Karl, T. Chronic cannabidiol treatment improves social and object recognition in double transgenic APPswe/PS1∆E9 mice. Psychopharmacology (Berl.) 2014, 231, 3009–3017. [Google Scholar] [CrossRef] [PubMed]
- Fawzy El-Sayed, K.M.; Dorfer, C.E. Gingival Mesenchymal Stem/Progenitor Cells: A Unique Tissue Engineering Gem. Stem Cells Int. 2016, 2016, 7154327. [Google Scholar] [CrossRef] [PubMed]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Schuchman, E.H.; Jin, H.K.; Bae, J. Soluble CCL5 derived from bone marrow-derived mesenchymal stem cells and activated by amyloid β ameliorates Alzheimer’s disease in mice by recruiting bone marrow-induced microglia immune responses. Stem Cells 2012, 30, 1544–1555. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Lee, J.K.; Lee, H.; Carter, J.E.; Chang, J.W.; Oh, W.; Yang, Y.S.; Suh, J.-G.; Lee, B.-H.; Jin, H.K.; et al. Human umbilical cord blood-derived mesenchymal stem cells improve neuropathology and cognitive impairment in an Alzheimer’s disease mouse model through modulation of neuroinflammation. Neurobiol. Aging 2012, 33, 588–602. [Google Scholar] [CrossRef] [PubMed]
- Juknat, A.; Rimmerman, N.; Levy, R.; Vogel, Z.; Kozela, E. Cannabidiol affects the expression of genes involved in zinc homeostasis in BV-2 microglial cells. Neurochem. Int. 2012, 61, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 2008, 104, 1433–1439. [Google Scholar] [CrossRef] [PubMed]
- Noble, W.; Olm, V.; Takata, K.; Casey, E.; Mary, O.; Meyerson, J.; Gaynor, K.; LaFrancois, J.; Wang, L.; Kondo, T.; et al. CDK5 is a key factor in tau aggregation and tangle formation in vivo. Neuron 2003, 38, 555–565. [Google Scholar] [CrossRef]
- Ryoo, S.-R.; Jeong, H.K.; Radnaabazar, C.; Yoo, J.-J.; Cho, H.-J.; Lee, H.-W.; Kim, I.-S.; Cheon, Y.-H.; Ahn, Y.S.; Chung, S.-H.; et al. DYRK1A-mediated hyperphosphorylation of Tau. A functional link between Down syndrome and Alzheimer disease. J. Biol. Chem. 2007, 282, 34850–34857. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Hiragami, Y.; Murayama, M.; Ishizuka, K.; Kawahara, M.; Takashima, A. Phosphorylation of tau at serine 416 by Ca2+/calmodulin-dependent protein kinase II in neuronal soma in brain. J. Neurochem. 2005, 94, 1438–1447. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Hasegawa, M.; Jakes, R.; Lawler, S.; Cuenda, A.; Cohen, P. Phosphorylation of microtubule-associated protein tau by stress-activated protein kinases. FEBS Lett. 1997, 409, 57–62. [Google Scholar] [CrossRef]
- Tsai, L.-H.; Patrick, G.N.; Zukerberg, L.; Nikolic, M.; de la Monte, S.; Dikkes, P. Conversion of p35 to p25 deregulates CDK5 activity and promotes neurodegeneration. Nature 1999, 402, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, H.; Feng, Y.; Wang, J. The effect of CDK-5 overexpression and overactivation on tau hyperphosphorylation in cultured N2a cells. Wuhan Univ. J. Nat. Sci. 2005, 10, 472–476. [Google Scholar]
- Bukau, B.; Weissman, J.; Horwich, A. Molecular chaperones and protein quality control. Cell 2006, 125, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Hong, L.; Huang, H.-C.; Jiang, Z.-F. Relationship between amyloid-β and the ubiquitin-proteasome system in Alzheimer’s disease. Neurol. Res. 2014, 36, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Gadhave, K.; Bolshette, N.; Ahire, A.; Pardeshi, R.; Thakur, K.; Trandafir, C.; Istrate, A.; Ahmed, S.; Lahkar, M.; Muresanu, D.F.; et al. The ubiquitin proteasomal system: A potential target for the management of Alzheimer’s disease. J. Cell. Mol. Med. 2016, 20, 1392–1407. [Google Scholar] [CrossRef] [PubMed]
- Patterson, K.R.; Ward, S.M.; Combs, B.; Voss, K.; Kanaan, N.M.; Morfini, G.; Brady, S.T.; Gamblin, T.C.; Binder, L.I. Heat shock protein 70 prevents both tau aggregation and the inhibitory effects of preexisting tau aggregates on fast axonal transport. Biochemistry 2011, 50, 10300–10310. [Google Scholar] [CrossRef] [PubMed]
- Ou, J.-R.; Tan, M.-S.; Xie, A.-M.; Yu, J.-T.; Tan, L. Heat shock protein 90 in Alzheimer’s Disease. BioMed Res. Int. 2014, 2014, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, F.; Gomez de Barreda, E.; Fuster-Matanzo, A.; Lucas, J.J.; Avila, J. GSK3: A possible link between β amyloid peptide and tau protein. Exp. Neurol. 2010, 223, 322–325. [Google Scholar] [CrossRef] [PubMed]
- Trazzi, S.; Steger, M.; Mitrugno, V.M.; Bartesaghi, R.; Ciani, E. CB1 cannabinoid receptors increase neuronal precursor proliferation through AKT/glycogen synthase kinase-3β/β-catenin signaling. J. Biol. Chem. 2010, 285, 10098–10109. [Google Scholar] [CrossRef] [PubMed]
- Ozaita, A.; Puighermanal, E.; Maldonado, R. Regulation of PI3K/Akt/GSK-3 pathway by cannabinoids in the brain. J. Neurochem. 2007, 102, 1105–1114. [Google Scholar] [CrossRef] [PubMed]
- Iannotti, F.A.; Hill, C.L.; Leo, A.; Alhusaini, A.; Soubrane, C.; Mazzarella, E.; Russo, E.; Whalley, B.J.; di Marzo, V.; Stephens, G.J. Nonpsychotropic plant cannabinoids, cannabidivarin (CBDV) and cannabidiol (CBD), activate and desensitize transient receptor potential vanilloid 1 (TRPV1) channels in vitro: Potential for the treatment of neuronal hyperexcitability. ACS Chem. Neurosci. 2014, 5, 1131–1141. [Google Scholar] [CrossRef] [PubMed]
- Trubiani, O.; Guarnieri, S.; Diomede, F.; Mariggio, M.A.; Merciaro, I.; Morabito, C.; Cavalcanti, M.F.X.B.; Cocco, L.; Ramazzotti, G. Nuclear translocation of PKCα isoenzyme is involved in neurogenic commitment of human neural crest-derived periodontal ligament stem cells. Cell Signal. 2016, 28, 1631–1641. [Google Scholar] [CrossRef] [PubMed]
- Bisogno, T.; Hanus, L.; de Petrocellis, L.; Tchilibon, S.; Ponde, D.E.; Brandi, I.; Moriello, A.S.; Davis, J.B.; Mechoulam, R.; di Marzo, V. Molecular targets for cannabidiol and its synthetic analogues: Effect on vanilloid VR1 receptors and on the cellular uptake and enzymatic hydrolysis of anandamide. Br. J. Pharmacol. 2001, 134, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Hassan, S.; Eldeeb, K.; Millns, P.J.; Bennett, A.J.; Alexander, S.P.H.; Kendall, D.A. Cannabidiol enhances microglial phagocytosis via transient receptor potential (TRP) channel activation. Br. J. Pharmacol. 2014, 171, 2426–2439. [Google Scholar] [CrossRef] [PubMed]
- Stein, A.T.; Ufret-Vincenty, C.A.; Hua, L.; Santana, L.F.; Gordon, S.E. Phosphoinositide 3-kinase binds to TRPV1 and mediates NGF-stimulated TRPV1 trafficking to the plasma membrane. J. Gen. Physiol. 2006, 128, 509–522. [Google Scholar] [CrossRef] [PubMed]
- Taglialatela-Scafati, O.; Pagani, A.; Scala, F.; De Petrocellis, L.; Di Marzo, V.; Grassi, G.; Appendino, G. Cannabimovone, a cannabinoid with a rearranged terpenoid skeleton from hemp. Eur. J. Org. Chem. 2010, 2010, 2067–2072. [Google Scholar] [CrossRef]
- Diomede, F.; Caputi, S.; Merciaro, I.; Frisone, S.; D’Arcangelo, C.; Piattelli, A.; Trubiani, O. Pro-inflammatory cytokine release and cell growth inhibition in primary human oral cells after exposure to endodontic sealer. Int. Endod. J. 2014, 47, 864–872. [Google Scholar] [CrossRef] [PubMed]
- Diomede, F.; Zini, N.; Gatta, V.; Fulle, S.; Merciaro, I.; D’Aurora, M.; La Rovere, R.M.; Traini, T.; Pizzicannella, J.; Ballerini, P.; et al. Human periodontal ligament stem cells cultured onto cortico-cancellous scaffold drive bone regenerative process. Eur. Cell Mater. 2016, 32, 181–201. [Google Scholar] [CrossRef] [PubMed]
- Rajan, T.S.; Giacoppo, S.; Scionti, D.; Diomede, F.; Grassi, G.; Pollastro, F.; Piattelli, A.; Bramanti, P.; Mazzon, E.; Trubiani, O. Cannabidiol activates neuronal precursor genes in human gingival mesenchymal stromal cells. J. Cell. Biochem. 2016. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Won, S.J.; Mao, X.O.; Jin, K.; Greenberg, D.A. Involvement of protein kinase A in Cannabinoid receptor-mediated protection from oxidative neuronal injury. J. Pharmacol. Exp. Ther. 2005, 313, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Whalley, B.J.; Wilkinson, J.D.; Williamson, E.M.; Constanti, A. A novel component of cannabis extract potentiates excitatory synaptic transmission in rat olfactory cortex in vitro. Neurosci. Lett. 2004, 365, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Ambrosino, P.; Soldovieri, M.V.; Russo, C.; Taglialatela, M. Activation and desensitization of TRPV1 channels in sensory neurons by the PPARα agonist palmitoylethanolamide. Br. J. Pharmacol. 2013, 168, 1430–1444. [Google Scholar] [CrossRef] [PubMed]
- Kwak, J. Capsaicin blocks the hyperpolarization-activated inward currents via TRPV1 in the rat dorsal root ganglion neurons. Exp. Neurobiol. 2012, 21, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Geng, D.C.; Xu, Y.Z.; Yang, H.L.; Zhu, X.S.; Zhu, G.M.; Wang, X.B. Inhibition of titanium particle-induced inflammatory osteolysis through inactivation of cannabinoid receptor 2 by AM630. J. Biomed. Mater. Res. Part A 2010, 95A, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Cui, N.; Du, Y.; Ma, H.; Zhang, Y. Anandamide reduces intracellular Ca2+ concentration through suppression of Na+/Ca2+ exchanger current in rat cardiac myocytes. PLoS ONE 2013, 8, e63386. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Description | GO Processes | CTR-GMSCs Exp_Value | CBD-GMSCs Exp_Value | FC Log10 | FDR q-Values |
|---|---|---|---|---|---|---|
| CAMK2A | Calcium/Calmodulin Dependent Protein Kinase II α | peptidyl-serine phosphorylation, cell cycle, phosphorylation | 0.02 | 0.00 | −0.60 | 0.002 |
| CAMK2B | Calcium/Calmodulin Dependent Protein Kinase II β | peptidyl-serine phosphorylation, protein autophosphorylation, cell cycle | 0.02 | 0.00 | −0.60 | 0.0082 |
| CAPN1 | Calpain 1 | receptor catabolic process | 76.18 | 56.41 | −0.13 | 9.79 × 105 |
| CAPN2 | Calpain 2 | receptor catabolic process | 521.29 | 463.30 | −0.05 | 9.79 × 105 |
| CDK5 | Cyclin Dependent Kinase 5 | protein autophosphorylation, cell cycle, phosphorylation | 12.15 | 2.73 | −0.65 | 9.79 × 105 |
| CDK5R1 | Cyclin Dependent Kinase 5 Regulatory Subunit 1 | protein autophosphorylation | 0.46 | 0.00 | −0.66 | 9.79 × 105 |
| DYRK1A | Dual Specificity Tyrosine Phosphorylation Regulated Kinase 1A | peptidyl-serine phosphorylation, protein autophosphorylation, cell cycle, phosphorylation | 7.34 | 5.15 | −0.15 | 1.9 × 103 |
| GSK3B | Glycogen Synthase Kinase 3β | peptidyl-serine phosphorylation, protein autophosphorylation, cell cycle, phosphorylation | 6.31 | 4.97 | −0.20 | 0.005 |
| MAPK1 | Mitogen-Activated Protein Kinase 1 | peptidyl-serine phosphorylation, cell cycle, phosphorylation | 35.19 | 27.59 | −0.11 | 0.002 |
| MAPK12 | Mitogen-Activated Protein Kinase 12 | peptidyl-serine phosphorylation, cell cycle, phosphorylation | 7.17 | 4.36 | −0.72 | 0.0082 |
| MAPK14 | Mitogen-Activated Protein Kinase 14 | peptidyl-serine phosphorylation, cell cycle, phosphorylation | 14.07 | 5.83 | −1.27 | 9.79 × 105 |
| MAPT | Microtubule Associated Protein Tau | cell cycle | 0.34 | 0.00 | −0.45 | 0.005 |
| APH1A | Aph-1 Homolog A | – | 17.8 | 9.04 | −0.30 | 9.79 × 105 |
| BACE1 | β-Secretase 1 | β-amyloid metabolic process | 7.65 | 5.74 | −0.12 | 0.0011 |
| NCSTN | Nicastrin | amyloid precursor protein catabolic process, apoptotic process | 93.23 | 92.34 | −0.04 | 0.05 |
| PSENEN | Presenilin Enhancer γ-Secretase Subunit | amyloid precursor protein catabolic process, apoptotic process | 69.70 | 51.45 | −0.13 | 0.001 |
| PSEN1 | Presenilin 1 | amyloid precursor protein catabolic process, apoptotic process | 14.06 | 8.55 | −0.22 | 9.79 × 105 |
| PSEN2 | Presenilin 2 | amyloid precursor protein catabolic process, apoptotic process | 11.89 | 4.45 | −0.43 | 9.79 × 105 |
| Gene | Description | GO Processes | CTR-GMSCs Exp_Value | CBD-GMSCs Exp_Value | FC Log10 | FDR q-Values |
|---|---|---|---|---|---|---|
| HSPA2 | Heat Shock Protein Family A (Hsp70) Member 2 | positive regulation of cellular protein metabolic process, response to unfolded protein, regulation of protein modification process | 4.81 | 10.84 | 0.35 | 9.79 × 105 |
| HSPA4 | Heat Shock Protein Family A (Hsp70) Member 4 | response to unfolded protein | 67.8627 | 98.572 | 0.16 | 9.79 × 105 |
| HSPA5 | Heat Shock Protein Family A (Hsp70) Member 5 | response to unfolded protein, regulation of protein metabolic process, proteasome-mediated ubiquitin-dependent protein catabolic process | 52.14 | 84.21 | 0.21 | 9.79 × 105 |
| HSPA8 | Heat Shock Protein Family A (Hsp70) Member 8 | response to unfolded protein, regulation of protein metabolic process | 593.285 | 674.165 | 0.06 | 9.79 × 105 |
| HSP90AA1 | Heat Shock Protein 90α Family Class A Member 1 | response to unfolded protein | 423.83 | 724.10 | 0.23 | 9.79 × 105 |
| HSP90AB1 | Heat Shock Protein 90α Family Class B Member 1 | response to unfolded protein, positive regulation of cellular protein metabolic process, regulation of protein modification process | 572.907 | 704.588 | 0.08 | 9.79 × 105 |
| HSP90B1 | Heat Shock Protein 90β Family Member 1 | response to unfolded protein, proteolysis, proteasome-mediated ubiquitin-dependent protein catabolic process | 262.43 | 380.62 | 0.16 | 9.79 × 105 |
| UBB | Ubiquitin B | proteolysis, protein polyubiquitination, protein catabolic process, proteasome-mediated ubiquitin-dependent protein catabolic process | 2458.27 | 3992.14 | 0.21 | 9.79 × 105 |
| UBE2A | Ubiquitin Conjugating Enzyme E2 A | proteolysis, protein polyubiquitination, protein catabolic process, proteasome-mediated ubiquitin-dependent protein catabolic process | 21.79 | 32.28 | 0.17 | 9.79 × 105 |
| UBE2B | Ubiquitin Conjugating Enzyme E2 B | proteolysis, protein polyubiquitination, protein catabolic process, proteasome-mediated ubiquitin-dependent protein catabolic process | 15.83 | 32.47 | 0.31 | 9.79 × 105 |
| UBE2D1 | Ubiquitin Conjugating Enzyme E2 D1 | proteolysis, protein polyubiquitination, protein catabolic process, proteasome-mediated ubiquitin-dependent protein catabolic process | 2.71 | 6.7154 | 0.39 | 9.79 × 105 |
| UBE2D2 | Ubiquitin Conjugating Enzyme E2 D2 | proteolysis, protein polyubiquitination, protein catabolic process, proteasome-mediated ubiquitin-dependent protein catabolic process | 11.55 | 17.63 | 0.18 | 9.79 × 105 |
| UBE2D3 | Ubiquitin Conjugating Enzyme E2 D3 | proteolysis, protein polyubiquitination, protein catabolic process, proteasome-mediated ubiquitin-dependent protein catabolic process | 21.63 | 38.56 | 0.25 | 9.79 × 105 |
| UBE2E1 | Ubiquitin Conjugating Enzyme E2 E1 | proteolysis, protein polyubiquitination, protein catabolic process | 26.54 | 60.20 | 0.36 | 9.79 × 105 |
| UBE2E2 | Ubiquitin Conjugating Enzyme E2 E2 | proteolysis, protein polyubiquitination, protein catabolic process | 8.10 | 19.69 | 0.39 | 9.79 × 105 |
| UBE2V2 | Ubiquitin Conjugating Enzyme E2 V2 | proteolysis, protein polyubiquitination, protein catabolic process | 8.10 | 19.70 | 0.39 | 9.79 × 105 |
| UBE3A | Ubiquitin Protein Ligase E3A | proteolysis, protein polyubiquitination, protein catabolic process | 21.35 | 41.18 | 0.29 | 9.79 × 105 |
| ACE1 | angiotensin I converting enzyme | β-amyloid metabolic process | 8.88 | 14.37 | 0.21 | 9.79 × 105 |
| ECE1 | endothelin converting enzyme 1 | β-amyloid metabolic process | 34.80 | 49.57 | 0.15 | 9.79 × 105 |
| IDE | insulin degrading enzyme | β-amyloid metabolic process | 9.41 | 14.047 | 0.17 | 9.79 × 105 |
| ADAM9 | ADAM metallopeptidase domain 9 | regulation of cellular catabolic process | 324.75 | 366.63 | 0.05 | 9.79 × 105 |
| PIK3CA | phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit α | regulation of protein metabolic process | 14.76 | 18.38 | 0.10 | 9.79 × 105 |
| PIK3CB | phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit β | regulation of protein metabolic process | 3.33 | 5.59 | 0.22 | 9.79 × 105 |
| AKT1 | AKT serine/threonine kinase 1 | regulation of protein metabolic process, positive regulation of biological process, protein phosphorylation | 47.16 | 59.5 | 0.10 | 9.79 × 105 |
| TMED10 | transmembrane p24 trafficking protein 10 | – | 40.2452 | 45.459 | 0.05 | 0.002 |
| Antibody | Manufacturer | Concentration |
|---|---|---|
| CD44-FITC | Ancell (Bayport, MN, USA) | 1:50 |
| CD29-PE | ||
| CD105-FITC | ||
| CD14-FITC | Miltenyi Biotec (Bergisch Gladbach, Germany) | |
| HLA-DR-PE | Becton Dickinson (San Jose, CA, USA) | |
| CD90-FITC | ||
| CD73-PE | ||
| CD34-PE | Beckman Coulter (Brea, CA, USA) |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Libro, R.; Diomede, F.; Scionti, D.; Piattelli, A.; Grassi, G.; Pollastro, F.; Bramanti, P.; Mazzon, E.; Trubiani, O. Cannabidiol Modulates the Expression of Alzheimer’s Disease-Related Genes in Mesenchymal Stem Cells. Int. J. Mol. Sci. 2017, 18, 26. https://doi.org/10.3390/ijms18010026
Libro R, Diomede F, Scionti D, Piattelli A, Grassi G, Pollastro F, Bramanti P, Mazzon E, Trubiani O. Cannabidiol Modulates the Expression of Alzheimer’s Disease-Related Genes in Mesenchymal Stem Cells. International Journal of Molecular Sciences. 2017; 18(1):26. https://doi.org/10.3390/ijms18010026
Chicago/Turabian StyleLibro, Rosaliana, Francesca Diomede, Domenico Scionti, Adriano Piattelli, Gianpaolo Grassi, Federica Pollastro, Placido Bramanti, Emanuela Mazzon, and Oriana Trubiani. 2017. "Cannabidiol Modulates the Expression of Alzheimer’s Disease-Related Genes in Mesenchymal Stem Cells" International Journal of Molecular Sciences 18, no. 1: 26. https://doi.org/10.3390/ijms18010026
APA StyleLibro, R., Diomede, F., Scionti, D., Piattelli, A., Grassi, G., Pollastro, F., Bramanti, P., Mazzon, E., & Trubiani, O. (2017). Cannabidiol Modulates the Expression of Alzheimer’s Disease-Related Genes in Mesenchymal Stem Cells. International Journal of Molecular Sciences, 18(1), 26. https://doi.org/10.3390/ijms18010026