
Pharmacogenetic Foundations of Therapeutic Efficacy and Adverse Events of Statins

 and
and
Abstract
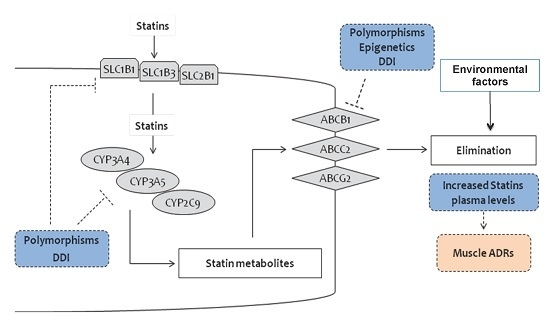
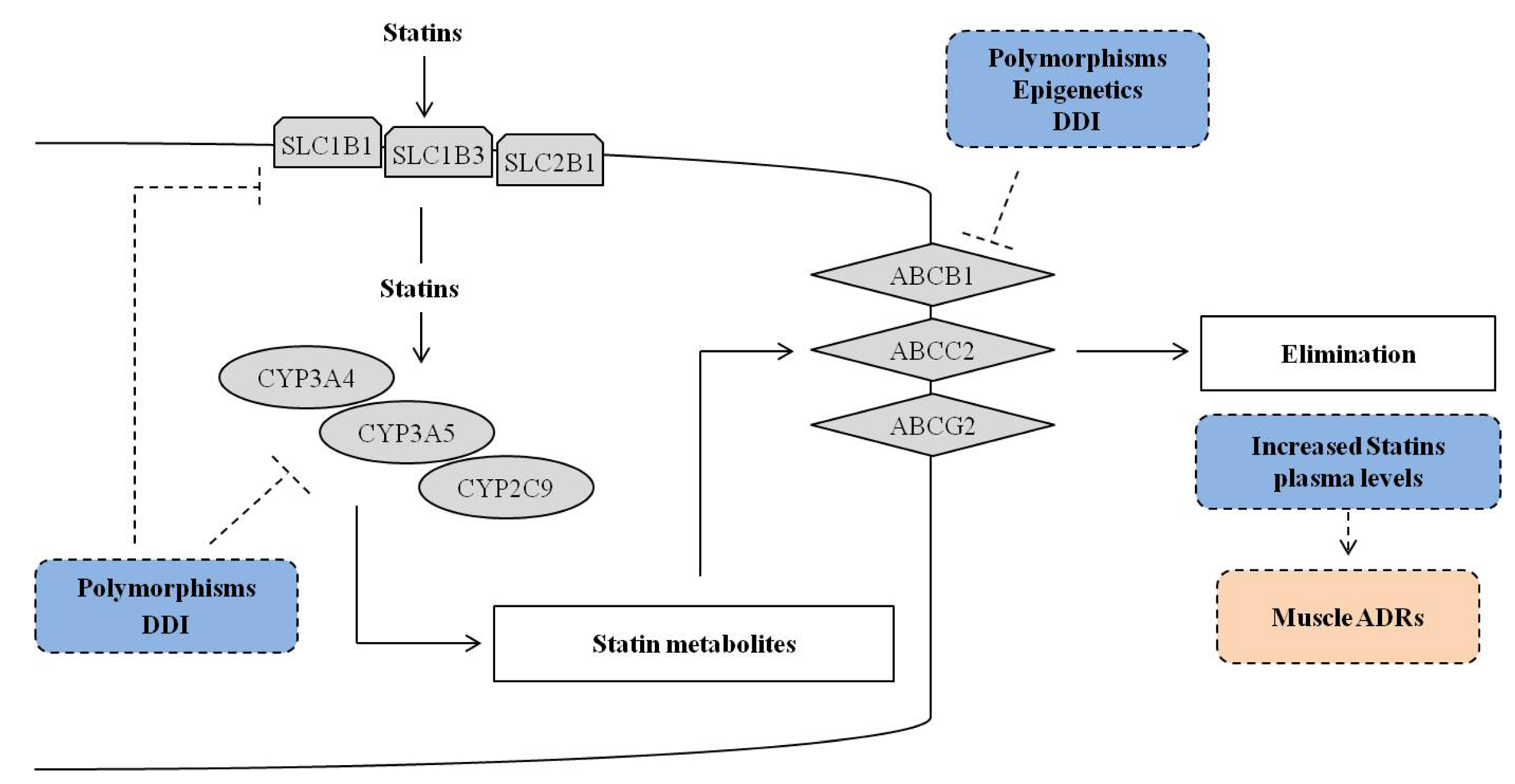
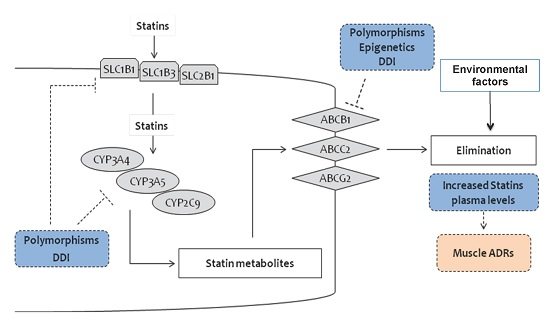
:
1. Introduction
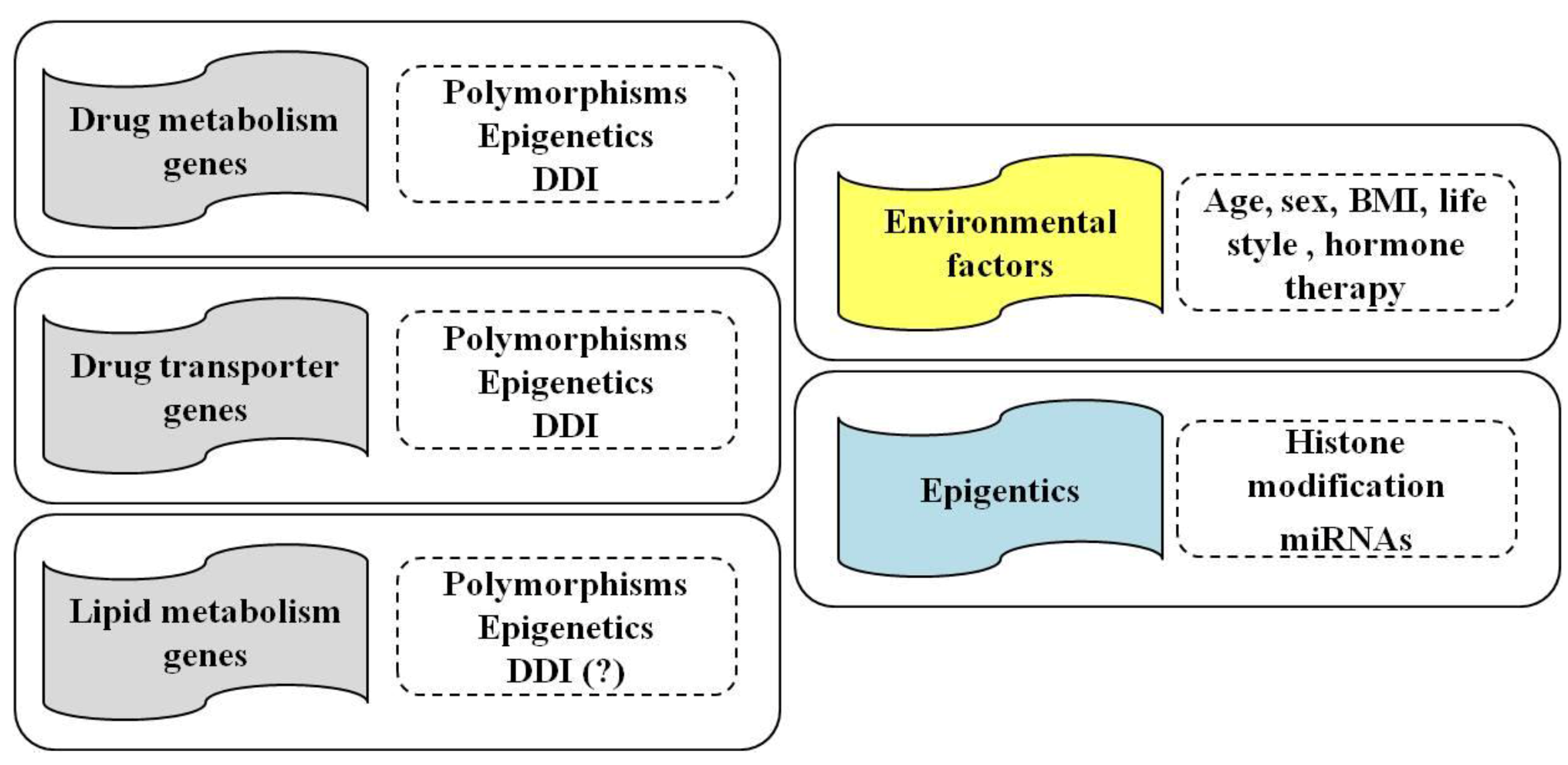
2. Pharmacogenetics of Statins
2.1. Genes and SNPs Involved in Statin Metabolism
2.2. Genes and SNPs of Transmembrane Transport
2.2.1. Efflux Transporters
2.2.2. ABCB1
2.2.3. ABCG2
2.2.4. ABCC2
2.2.5. Uptake Transporters
2.2.6. Polymorphisms in Other Genes
3. Statins and Epigenetics
4. Statins, Individual and Environmental Factors
5. Clinically Relevant Drug–Drug Interaction with Statins
5.1. Statins and Cardiovascular-Anti-Platelet/Anti-Coagulant Drugs
5.2. Statins and Immunosuppressant Drugs
5.3. Statins and Anti-Microbial/Anti-Viral Drugs
5.4. Other Interactions
6. Conclusions
Author Contributions
Conflicts of Interest
References
- Leusink, M.; Onland-Moret, N.C.; de Bakker, P.I.; de Boer, A.; Maitland-van der Zee, A.H. Seventeen years of statin pharmacogenetics: A systematic review. Pharmacogenomics 2016, 17, 163–180. [Google Scholar] [CrossRef] [PubMed]
- Mihaylova, B.; Emberson, J.; Blackwell, L.; Keech, A.; Simes, J.; Barnes, E.H.; Voysey, M.; Gray, A.; Collins, R.; Baigent, C. The effects of lowering LDL cholesterol with statin therapy in people at low risk of vascular disease: Meta-analysis of individual data from 27 randomised trials. Lancet 2012, 380, 581–590. [Google Scholar] [PubMed]
- Zambrano, T.; Hirata, R.D.C.; Hirata, M.H.; Cerda, A.; Salazar, L.A. Altered microRNome profiling in statin-induced HepG2 cells: A pilot study identifying potential new biomarkers involved in lipid-lowering treatment. Cardiovasc. Drugs Ther. 2015, 29, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Soko, N.D.; Masimirembwa, C.; Dandara, C. Pharmacogenomics of rosuvastatin: A glocal (global + local) african perspective and expert review on a statin drug. OMICS 2016, 20, 498–509. [Google Scholar] [CrossRef] [PubMed]
- Paoletti, R.; Fahmy, M.; Mahla, G.; Mizan, J.; Southworth, H. Rosuvastatin demonstrates greater reduction of low-density lipoprotein cholesterol compared with pravastatin and simvastatin in hypercholesterolaemic patients: A randomized, double-blind study. J. Cardiovasc. Risk 2001, 8, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Zemankova, L.; Varejckova, M.; Dolezalova, E.; Fikrova, P.; Jezkova, K.; Rathouska, J.; Cerveny, L.; Botella, L.M.; Bernabeu, C.; Nemeckova, I.; et al. Atorvastatin-induced endothelial nitric oxide synthase expression in endothelial cells is mediated by endoglin. J. Physiol. Pharmacol. 2015, 66, 403–413. [Google Scholar] [PubMed]
- Lefer, D.J. Statins as potent antiinflammatory drugs. Circulation 2002, 106, 2041–2042. [Google Scholar] [CrossRef] [PubMed]
- Miyaki, K.; Matsubara, A.; Nishiwaki, A.; Tomida, K.; Morita, H.; Yoshida, M.; Ogura, Y. Pitavastatin attenuates leukocyte-endothelial interactions induced by ischemia-reperfusion injury in the rat retina. Curr. Eye Res. 2009, 34, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Pruefer, D.; Scalia, R.; Lefer, A.M. Simvastatin inhibits leukocyte-endothelial cell interactions and protects against inflammatory processes in normocholesterolemic rats. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 2894–2900. [Google Scholar] [CrossRef] [PubMed]
- Alfonsi, J.E.; Hegele, R.A.; Gryn, S.E. Pharmacogenetics of lipid-lowering agents: Precision or indecision medicine? Curr. Atheroscler. Rep. 2016. [Google Scholar] [CrossRef] [PubMed]
- Postmus, I.; Warren, H.R.; Trompet, S.; Arsenault, B.J.; Avery, C.L.; Bis, J.C.; Chasman, D.I.; de Keyser, C.E.; Deshmukh, H.A.; Evans, D.S.; et al. Meta-analysis of genome-wide association studies of HDL cholesterol response to statins. J. Med. Genet. 2016, 53, 835–845. [Google Scholar] [CrossRef] [PubMed]
- Kei, A.A.; Filippatos, T.D.; Elisaf, M.S. The safety of ezetimibe and simvastatin combination for the treatment of hypercholesterolemia. Exp. Opin. Drug Saf. 2016, 15, 559–569. [Google Scholar] [CrossRef] [PubMed]
- Gazzerro, P.; Proto, M.C.; Gangemi, G.; Malfitano, A.M.; Ciaglia, E.; Pisanti, S.; Santoro, A.; Laezza, C.; Bifulco, M. Pharmacological actions of statins: A critical appraisal in the management of cancer. Pharmacol. Rev. 2012, 64, 102–146. [Google Scholar] [CrossRef] [PubMed]
- Golomb, B.A.; Evans, M.A. Statin adverse effects: A review of the literature and evidence for a mitochondrial mechanism. Am. J. Cardiovasc. Drugs 2008, 8, 373–418. [Google Scholar] [CrossRef] [PubMed]
- Finegold, J.A.; Manisty, C.H.; Goldacre, B.; Barron, A.J.; Francis, D.P. What proportion of symptomatic side effects in patients taking statins are genuinely caused by the drug? Systematic review of randomized placebo-controlled trials to aid individual patient choice. Eur. J. Prev. Cardiol. 2014, 21, 464–474. [Google Scholar] [CrossRef] [PubMed]
- Taylor, F.; Huffman, M.D.; Macedo, A.F.; Moore, T.H.M.; Burke, M.; Davey Smith, G.; Ward, K.; Ebrahim, S. Statins for the primary prevention of cardiovascular disease. Cochrane Database Syst. Rev. 2013, 1, CD004816. [Google Scholar]
- Silva, M.A.; Swanson, A.C.; Gandhi, P.J.; Tataronis, G.R. Statin-related adverse events: A meta-analysis. Clin. Ther. 2006, 28, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Joy, T.R.; Hegele, R.A. Narrative review: Statin-related myopathy. Ann. Intern. Med. 2009, 150, 858–868. [Google Scholar] [CrossRef] [PubMed]
- Rahal, A.J.; ElMallah, A.I.; Poushuju, R.J.; Itani, R. Do statins really cause diabetes? A meta-analysis of major randomized controlled clinical trials. Saudi Med. J. 2016, 37, 1051–1060. [Google Scholar] [CrossRef] [PubMed]
- Naci, H.; Brugts, J.; Ades, T. Comparative tolerability and harms of individual statins: A study-level network meta-analysis of 246,955 participants from 135 randomized, controlled trials. Circ. Cardiovasc. Qual. Outcomes 2013, 6, 390–399. [Google Scholar] [CrossRef] [PubMed]
- Mancini, G.B.J.; Tashakkor, A.Y.; Baker, S.; Bergeron, J.; Fitchett, D.; Frohlich, J.; Genest, J.; Gupta, M.; Hegele, R.A.; Ng, D.S.; et al. Diagnosis, prevention, and management of statin adverse effects and intolerance: Canadian working group consensus update. Can. J. Cardiol. 2013, 29, 1553–1568. [Google Scholar] [CrossRef] [PubMed]
- Jukema, J.W.; Cannon, C.P.; de Craen, A.J.M.; Westendorp, R.G.J.; Trompet, S. The controversies of statin therapy: Weighing the evidence. J. Am. Coll. Cardiol. 2012, 60, 875–881. [Google Scholar] [CrossRef] [PubMed]
- Dale, K.M.; Coleman, C.I.; Henyan, N.N.; Kluger, J.; White, C.M. Statins and cancer risk: A meta-analysis. JAMA 2006, 295, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.; Song, X.; Zhang, G.; Peng, A.; Li, X.; Li, M.; Liu, Y.; Wang, C. Statins and the risk of lung cancer: A meta-analysis. PLoS ONE 2013, 8, e57349. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, M.; Qian, J.; Zheng, J.; Zhang, X.; Guo, C.; Geng, J.; Peng, B.; Che, J.; Wu, Y. Statin use and risk of kidney cancer: A meta-analysis of observational studies and randomized trials. Br. J. Clin. Pharmacol. 2014, 77, 458–465. [Google Scholar] [CrossRef] [PubMed]
- Undela, K.; Srikanth, V.; Bansal, D. Statin use and risk of breast cancer: A meta-analysis of observational studies. Breast Cancer Res. Treat. 2012, 135, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Xie, Y.; Chen, M.; Li, J.; Liao, X.; Shen, J.; Shi, M.; Li, W.; Zheng, H.; Jiang, B. Statin use and risk of pancreatic cancer: A meta-analysis. Cancer Causes Control 2012, 23, 1099–1111. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Geng, J.; Zhang, X.; Peng, B.; Che, J.; Yan, Y.; Wang, G.; Xia, S.; Wu, Y.; Zheng, J. Statin use and risk of bladder cancer: A meta-analysis. Cancer Causes Control 2013, 24, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Singh, A.G.; Singh, P.P.; Murad, M.H.; Iyer, P.G. Statins are associated with reduced risk of esophageal cancer, particularly in patients with Barrett’s esophagus: A systematic review and meta-analysis. Clin. Gastroenterol. Hepatol. 2013, 11, 620–629. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tang, W.; Wang, J.; Xie, L.; Li, T.; He, Y.; Deng, Y.; Peng, Q.; Li, S.; Qin, X. Association between statin use and colorectal cancer risk: A meta-analysis of 42 studies. Cancer Causes Control 2014, 25, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.D.; Zeng, K.; Xue, F.Q.; Chen, J.H.; Chen, Y.Q. Statins are associated with reduced risk of gastric cancer: A meta-analysis. Eur. J. Clin. Pharmacol. 2013, 69, 1855–1860. [Google Scholar] [CrossRef] [PubMed]
- Pradelli, D.; Soranna, D.; Scotti, L.; Zambon, A.; Catapano, A.; Mancia, G.; La Vecchia, C.; Corrao, G. Statins and primary liver cancer: A meta-analysis of observational studies. Eur. J. Cancer Prev. 2013, 22, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Bansal, D.; Undela, K.; D’Cruz, S.; Schifano, F. Statin use and risk of prostate cancer: A meta-analysis of observational studies. PLoS ONE 2012, 7, e46691. [Google Scholar] [CrossRef] [PubMed]
- Mangravite, L.M.; Thorn, C.F.; Krauss, R.M. Clinical implications of pharmacogenomics of statin treatment. Pharmacogenom. J. 2006, 6, 360–374. [Google Scholar] [CrossRef] [PubMed]
- Sirtori, C.R.; Mombelli, G.; Triolo, M.; Laaksonen, R. Clinical response to statins: Mechanism(s) of variable activity and adverse effects. Ann. Med. 2012, 44, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Verschuren, J.J.W.; Trompet, S.; Wessels, J.A.M.; Guchelaar, H.J.; de Maat, M.P.M.; Simoons, M.L.; Jukema, J.W. A systematic review on pharmacogenetics in cardiovascular disease: Is it ready for clinical application? Eur. Heart J. 2012, 33, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Arrigoni, E.; Galimberti, S.; Petrini, M.; Danesi, R.; di Paolo, A. ATP-binding cassette transmembrane transporters and their epigenetic control in cancer: An overview. Exp. Opin. Drug Metab. Toxicol. 2016, 5255, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Frudakis, T.N.; Thomas, M.J.; Ginjupalli, S.N.; Handelin, B.; Gabriel, R.; Gomez, H.J. CYP2D6*4 polymorphism is associated with statin-induced muscle effects. Pharmacogenet. Genom. 2007, 17, 695–707. [Google Scholar] [CrossRef] [PubMed]
- Zuccaro, P.; Mombelli, G.; Calabresi, L.; Baldassarre, D.; Palmi, I.; Sirtori, C.R. Tolerability of statins is not linked to CYP450 polymorphisms, but reduced CYP2D6 metabolism improves cholesteraemic response to simvastatin and fluvastatin. Pharmacol. Res. 2007, 55, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, X.; Zhang, Z.; Zou, J.; Chen, Y.; Wang, X.; Wu, J. Statin therapy correlated CYP2D6 gene polymorphism and hyperlipidemia. Curr. Med. Res. Opin. 2014, 30, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Mirosevic Skvrce, N.; Bozina, N.; Zibar, L.; Barisic, I.; Pejnovic, L.; Macolic Sarinic, V. CYP2C9 and ABCG2 polymorphisms as risk factors for developing adverse drug reactions in renal transplant patients taking fluvastatin: A case-control study. Pharmacogenomics 2013, 14, 1419–1431. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Zhang, Y.; Zhou, H.; Wang, X.; Wang, W. CYP2C9 genetic polymorphism is a potential predictive marker for the efficacy of rosuvastatin therapy. Clin. Lab. 2015, 61, 1317–1324. [Google Scholar] [PubMed]
- Buzková, H.; Pechandová, K.; Danzig, V.; Vareka, T.; Perlik, F.; Zak, A.; Slanar, O. Lipid-lowering effect of fluvastatin in relation to cytochrome P450 2C9 variant alleles frequently distributed in the Czech population. Med. Sci. Monit. 2012, 18, 512–517. [Google Scholar] [CrossRef]
- Kajinami, K.; Brousseau, M.E.; Ordovas, J.M.; Schaefer, E.J. CYP3A4 genotypes and plasma lipoprotein levels before and after treatment with atorvastatin in primary hypercholesterolemia. Am. J. Cardiol. 2004, 93, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Becker, M.L.; Visser, L.E.; van Schaik, R.H.N.; Hofman, A.; Uitterlinden, A.G.; Stricker, B.H.C. Influence of genetic variation in CYP3A4 and ABCB1 on dose decrease or switching during simvastatin and atorvastatin therapy. Pharmacoepidemiol. Drug Saf. 2010, 19, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Leusink, M.; de Keyser, C.E.; Onland-Moret, N.C.; Hofman, A.; Visser, L.E.; Stricker, B.H.; de Bakker, P.I.W.; de Boer, A.; van Schaik, R.H.N.; Maitland-van der Zee, A.H. No association between CYP3A4*22 and statin effectiveness in reducing the risk for myocardial infarction. Pharmacogenomics 2014, 15, 1471–1477. [Google Scholar] [CrossRef] [PubMed]
- Ragia, G.; Kolovou, V.; Tavridou, A.; Elens, L.; Tselepis, A.D.; Elisaf, M.; van Schaik, R.H.N.; Kolovou, G.; Manolopoulos, V.G. No effect of CYP3A4 intron 6 C>T polymorphism (CYP3A4*22) on lipid-lowering response to statins in Greek patients with primary hypercholesterolemia. Drug Metab. Pers. Ther. 2015, 30, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Kivistö, K.T.; Niemi, M.; Schaeffeler, E.; Pitkälä, K.; Tilvis, R.; Fromm, M.F.; Schwab, M.; Eichelbaum, M.; Strandberg, T. Lipid-lowering response to statins is affected by CYP3A5 polymorphism. Pharmacogenetics 2004, 14, 523–525. [Google Scholar] [CrossRef] [PubMed]
- Fiegenbaum, M.; da Silveira, F.R.; van der Sand, C.R.; van der Sand, L.C.; Ferreira, M.E.W.; Pires, R.C.; Hutz, M.H. The role of common variants of ABCB1, CYP3A4, and CYP3A5 genes in lipid-lowering efficacy and safety of simvastatin treatment. Clin. Pharmacol. Ther. 2005, 78, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Rebecchi, I.M.M.; Rodrigues, A.C.; Arazi, S.S.; Genvigir, F.D.V.; Willrich, M.A.V.; Hirata, M.H.; Soares, S.A.; Bertolami, M.C.; Faludi, A.A.; Bernik, M.M.S.; et al. ABCB1 and ABCC1 expression in peripheral mononuclear cells is influenced by gene polymorphisms and atorvastatin treatment. Biochem. Pharmacol. 2009, 77, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Mega, J.L.; Morrow, D.A.; Brown, A.; Cannon, C.P.; Sabatine, M.S. Identification of genetic variants associated with response to statin therapy. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1310–1315. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Ruan, Z.R.; Yuan, H.; Xu, D.H.; Zeng, S. ABCB1 gene polymorphisms, ABCB1 haplotypes and ABCG2 c.421C>A are determinants of inter-subject variability in rosuvastatin pharmacokinetics. Die Pharm. 2013, 68, 129–134. [Google Scholar]
- Sałacka, A.; Bińczak-kuleta, A.; Kaczmarczyk, M.; Hornowska, I.; Safranow, K.; Clark, J.S.C. Possible association of ABCB1: c.3435T>C polymorphism with high-density-lipoprotein-cholesterol response to statin treatment—A pilot study. Bosn. J. Basic Med. Sci. 2014, 14, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Xu, H.; Yang, J.; Yu, Q.; Yang, S.; Zhang, J.; Yao, Q.; Zhu, Y.; Luo, Y.; Ji, L.; et al. ABCB1 C3435T polymorphism and the lipid-lowering response in hypercholesterolemic patients on statins: A meta-analysis. Lipids Health Dis. 2015, 14, 122. [Google Scholar] [CrossRef] [PubMed]
- Hoenig, M.R.; Walker, P.J.; Gurnsey, C.; Beadle, K.; Johnson, L. The C3435T polymorphism in ABCB1 influences atorvastatin efficacy and muscle symptoms in a high-risk vascular cohort. J. Clin. Lipidol. 2011, 5, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Becker, M.L.; Visser, L.E.; van Schaik, R.H.N.; Hofman, A.; Uitterlinden, A.G.; Stricker, B.H.C. Common genetic variation in the ABCB1 gene is associated with the cholesterol-lowering effect of simvastatin in males. Pharmacogenomics 2009, 10, 1743–1751. [Google Scholar] [CrossRef] [PubMed]
- Kadam, P.; Ashavaid, T.F.; Ponde, C.K.; Rajani, R.M. Genetic determinants of lipid-lowering response to atorvastatin therapy in an Indian population. J. Clin. Pharm. Ther. 2016, 41, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Niemi, M.; Arnold, K.A.; Backman, J.T.; Pasanen, M.K.; Godtel-Armbrust, U.; Wojnowski, L.; Zanger, U.M.; Neuvonen, P.J.; Eichelbaum, M.; Kivisto, K.T.; et al. Association of genetic polymorphism in ABCC2 with hepatic multidrug resistance-associated protein 2 expression and pravastatin pharmacokinetics. Pharmacogenet. Genom. 2006, 16, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Becker, M.L.; Elens, L.L.F.S.; Visser, L.E.; Hofman, A.; Uitterlinden, A.G.; van Schaik, R.H.N.; Stricker, B.H. Genetic variation in the ABCC2 gene is associated with dose decreases or switches to other cholesterol-lowering drugs during simvastatin and atorvastatin therapy. Pharmacogenom. J. 2013, 13, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Oh, E.S.; Kim, C.O.; Cho, S.K.; Park, M.S.; Chung, J.Y. Impact of ABCC2, ABCG2 and SLCO1B1 polymorphisms on the pharmacokinetics of pitavastatin in humans. Drug Metab. Pharmacokinet. 2013, 28, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Keskitalo, J.E.; Zolk, O.; Fromm, M.F.; Kurkinen, K.J.; Neuvonen, P.J.; Niemi, M. ABCG2 polymorphism markedly affects the pharmacokinetics of atorvastatin and rosuvastatin. Clin. Pharmacol. Ther. 2009, 86, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Keskitalo, J.E.; Pasanen, M.K.; Neuvonen, P.J.; Niemi, M. Different effects of the ABCG2 c.421C>A SNP on the pharmacokinetics of fluvastatin, pravastatin and simvastatin. Pharmacogenomics 2009, 10, 1617–1624. [Google Scholar] [CrossRef] [PubMed]
- Wan, Z.; Wang, G.; Li, T.; Xu, B.; Pei, Q.; Peng, Y.; Sun, H.; Cheng, L.; Zeng, Y.; Yang, G.; et al. Marked alteration of rosuvastatin pharmacokinetics in healthy Chinese with ABCG2 34G>A and 421C>A homozygote or compound heterozygote. J. Pharmacol. Exp. Ther. 2015, 354, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.Y.; Bae, K.S.; Cho, S.H.; Ghim, J.L.; Choe, S.; Jung, J.A.; Jin, S.J.; Kim, H.S.; Lim, H.S. Impact of CYP2D6, CYP3A5, CYP2C19, CYP2A6, SLCO1B1, ABCB1, and ABCG2 gene polymorphisms on the pharmacokinetics of simvastatin and simvastatin acid. Pharmacogenet. Genom. 2015, 25, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Mirosevic Skvrce, N.; Macolic Sarinic, V.; Simic, I.; Ganoci, L.; Muacevic Katanec, D.; Bozina, N. ABCG2 gene polymorphisms as risk factors for atorvastatin adverse reactions: A case-control study. Pharmacogenomics 2015, 16, 803–815. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; To, K.K.W.; Mak, V.W.L.; Tomlinson, B. The ABCG2 transporter and its relations with the pharmacokinetics, drug interaction and lipid-lowering effects of statins. Exp. Opin. Drug Metab. Toxicol. 2011, 7, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Lui, S.S.H.; Mak, V.W.L.; Chu, T.T.W.; Lee, V.W.Y.; Poon, E.W.M.; Tsui, T.K.C.; Ko, G.T.C.; Baum, L.; Tam, L.S.; et al. Pharmacogenetic analysis of lipid responses to rosuvastatin in Chinese patients. Pharmacogenet. Genom. 2010, 20, 634–637. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, B.; Hu, M.; Lee, V.W.Y.; Lui, S.S.H.; Chu, T.T.W.; Poon, E.W.M.; Ko, G.T.C.; Baum, L.; Tam, L.S.; Li, E.K. ABCG2 polymorphism is associated with the low-density lipoprotein cholesterol response to rosuvastatin. Clin. Pharmacol. Ther. 2010, 87, 558–562. [Google Scholar] [CrossRef] [PubMed]
- Bailey, K.M.; Romaine, S.P.R.; Jackson, B.M.; Farrin, A.J.; Efthymiou, M.; Barth, J.H.; Copeland, J.; McCormack, T.; Whitehead, A.; Flather, M.D.; et al. Hepatic metabolism and transporter gene variants enhance response to rosuvastatin in patients with acute myocardial infarction: The GEOSTAT-1 Study. Circ. Cardiovasc. Genet. 2010, 3, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Chasman, D.I.; Giulianini, F.; MacFadyen, J.; Barratt, B.J.; Nyberg, F.; Ridker, P.M. Genetic determinants of statin-induced low-density lipoprotein cholesterol reduction: The justification for the use of statins in prevention: An intervention trial evaluating rosuvastatin (JUPITER) trial. Circ. Cardiovasc. Genet. 2012, 5, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Gryn, S.E.; Hegele, R.A. Pharmacogenomics, lipid disorders, and treatment options. Clin. Pharmacol. Ther. 2014, 96, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Mwinyi, J.; Johne, A.; Bauer, S.; Roots, I.; Gerloff, T. Evidence for inverse effects of OATP-C (SLC21A6) 5 and 1B haplotypes on pravastatin kinetics. Clin. Pharmacol. Ther. 2004, 75, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Maeda, K.; Ieiri, I.; Yasuda, K.; Fujino, A.; Fujiwara, H.; Otsubo, K.; Hirano, M.; Watanabe, T.; Kitamura, Y.; Kusuhara, H.; et al. Effects of organic anion transporting polypeptide 1B1 haplotype on pharmacokinetics of pravastatin, valsartan, and temocapril. Clin. Pharmacol. Ther. 2006, 79, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Postmus, I.; Trompet, S.; Deshmukh, H.A.; Barnes, M.R.; Li, X.; Warren, H.R.; Chasman, D.I.; Zhou, K.; Arsenault, B.J.; Donnelly, L.A.; et al. Pharmacogenetic meta-analysis of genome-wide association studies of LDL cholesterol response to statins. Nat. Commun. 2014, 5, 5068–5078. [Google Scholar] [CrossRef] [PubMed]
- Pasanen, M.K.; Neuvonen, M.; Neuvonen, P.J.; Niemi, M. SLCO1B1 polymorphism markedly affects the pharmacokinetics of simvastatin acid. Pharmacogenet. Genom. 2006, 16, 873–879. [Google Scholar] [CrossRef] [PubMed]
- Pasanen, M.K.; Fredrikson, H.; Neuvonen, P.J.; Niemi, M. Different effects of SLCO1B1 polymorphism on the pharmacokinetics of atorvastatin and rosuvastatin. Clin. Pharmacol. Ther. 2007, 82, 726–733. [Google Scholar] [CrossRef] [PubMed]
- Hou, Q.; Li, S.; Li, L.; Li, Y.; Sun, X.; Tian, H. Association between SLCO1B1 gene T521C polymorphism and statin-related myopathy risk: A meta-analysis of case-control studies. Medicine 2015, 94, e1268. [Google Scholar] [CrossRef] [PubMed]
- Link, E.; Parish, S.; Armitage, J.; Bowman, L.; Heath, S.; Matsuda, F.; Gut, I.; Lathrop, M.; Collins, R. SLCO1B1 variants and statin-induced myopathy—A genomewide study. N. Engl. J. Med. 2008, 359, 789–799. [Google Scholar] [PubMed]
- Wilke, R.A.; Ramsey, L.B.; Johnson, S.G.; Maxwell, W.D.; McLeod, H.L.; Voora, D.; Krauss, R.M.; Roden, D.M.; Feng, Q.; Cooper-Dehoff, R.M.; et al. The clinical pharmacogenomics implementation consortium: CPIC guideline for SLCO1B1 and simvastatin-induced myopathy. Clin. Pharmacol. Ther. 2012, 92, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Brunham, L.R.; Lansberg, P.J.; Zhang, L.; Miao, F.; Carter, C.; Hovingh, G.K.; Visscher, H.; Jukema, J.W.; Stalenhoef, A.F.; Ross, C.J.D.; et al. Differential effect of the rs4149056 variant in SLCO1B1 on myopathy associated with simvastatin and atorvastatin. Pharmacogenom. J. 2012, 12, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Niemi, M.; Schaeffeler, E.; Lang, T.; Fromm, M.F.; Neuvonen, M.; Kyrklund, C.; Backman, J.T.; Kerb, R.; Schwab, M.; Neuvonen, P.J.; et al. High plasma pravastatin concentrations are associated with single nucleotide polymorphisms and haplotypes of organic anion transporting polypeptide-C (OATP-C, SLCO1B1). Pharmacogenetics 2004, 14, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Carr, D.F.; O’Meara, H.; Jorgensen, A.L.; Campbell, J.; Hobbs, M.; McCann, G.; van Staa, T.; Pirmohamed, M. SLCO1B1 genetic variant associated with statin-induced myopathy: A proof-of-concept study using the clinical practice research datalink. Clin. Pharmacol. Ther. 2013, 94, 695–701. [Google Scholar] [CrossRef] [PubMed]
- Voora, D.; Shah, S.H.; Spasojevic, I.; Ali, S.; Reed, C.R.; Salisbury, B.A.; Ginsburg, G.S. The SLCO1B1*5 genetic variant is associated with statin-induced side effects. J. Am. Coll. Cardiol. 2009, 54, 1609–1616. [Google Scholar] [CrossRef] [PubMed]
- Marciante, K.D.; Durda, J.P.; Heckbert, S.R.; Lumley, T.; Rice, K.; McKnight, B.; Totah, R.A.; Tamraz, B.; Kroetz, D.L.; Fukushima, H.; et al. Cerivastatin, genetic variants, and the risk of rhabdomyolysis. Pharmacogenet. Genom. 2011, 21, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Tornio, A.; Vakkilainen, J.; Neuvonen, M.; Backman, J.T.; Neuvonen, P.J.; Niemi, M. SLCO1B1 polymorphism markedly affects the pharmacokinetics of lovastatin acid. Pharmacogenet. Genom. 2015, 25, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Nishizato, Y.; Ieiri, I.; Suzuki, H.; Kimura, M.; Kawabata, K.; Hirota, T.; Takane, H.; Irie, S.; Kusuhara, H.; Urasaki, Y.; et al. Polymorphisms of OATP-C (SLC21A6) and OAT3 (SLC22A8) genes: Consequences for pravastatin pharmacokinetics. Clin. Pharmacol. Ther. 2003, 73, 554–565. [Google Scholar] [CrossRef]
- Deng, J.W.; Song, I.S.; Shin, H.J.; Yeo, C.W.; Cho, D.Y.; Shon, J.H.; Shin, J.G. The effect of SLCO1B1*15 on the disposition of pravastatin and pitavastatin is substrate dependent: The contribution of transporting activity changes by SLCO1B1*15. Pharmacogenet. Genom. 2008, 18, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, A.C. Efflux and uptake transporters as determinants of statin response. Exp. Opin. Drug Metab. Toxicol. 2010, 6, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Grube, M.; Kock, K.; Oswald, S.; Draber, K.; Meissner, K.; Eckel, L.; Bohm, M.; Felix, S.B.; Vogelgesang, S.; Jedlitschky, G.; et al. Organic anion transporting polypeptide 2B1 is a high-affinity transporter for atorvastatin and is expressed in the human heart. Clin. Pharmacol. Ther. 2006, 80, 607–620. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.F.; Man, M.; Johnson, K.J.; Wood, L.S.; Lira, M.E.; Lloyd, D.B.; Banerjee, P.; Milos, P.M.; Myrand, S.P.; Paulauskis, J.; et al. An association study of 43 SNPs in 16 candidate genes with atorvastatin response. Pharmacogenom. J. 2005, 5, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Zintzaras, E.; Kitsios, G.D.; Triposkiadis, F.; Lau, J.; Raman, G. APOE gene polymorphisms and response to statin therapy. Pharmacogenom. J. 2009, 9, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, L.A.; Doney, A.S.F.; Dannfald, J.; Whitley, A.L.; Lang, C.C.; Morris, A.D.; Donnan, P.T.; Palmer, C.N.A. A paucimorphic variant in the HMG-CoA reductase gene is associated with lipid-lowering response to statin treatment in diabetes: A GoDARTS study. Pharmacogenet. Genom. 2008, 18, 1021–1026. [Google Scholar] [CrossRef] [PubMed]
- Ordovas, J.M.; Lopez-Miranda, J.; Perez-Jimenez, F.; Rodriguez, C.; Park, J.S.; Cole, T.; Schaefer, E.J. Effect of apolipoprotein E and A-IV phenotypes on the low density lipoprotein response to HMG CoA reductase inhibitor therapy. Atherosclerosis 1995, 113, 157–166. [Google Scholar] [CrossRef]
- Barber, M.J.; Mangravite, L.M.; Hyde, C.L.; Chasman, D.I.; Smith, J.D.; McCarty, C.A.; Li, X.; Wilke, R.A.; Rieder, M.J.; Williams, P.T.; et al. Genome-wide association of lipid-lowering response to statins in combined study populations. PLoS ONE 2010, 5, e9763. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, L.U.; Gerdes, C.; Kervinen, K.; Savolainen, M.; Klausen, I.C.; Hansen, P.S.; Kesaniemi, Y.A.; Faergeman, O. The apolipoprotein epsilon4 allele determines prognosis and the effect on prognosis of simvastatin in survivors of myocardial infarction: A substudy of the Scandinavian simvastatin survival study. Circulation 2000, 101, 1366–1371. [Google Scholar] [CrossRef] [PubMed]
- Chasman, D.I.; Posada, D.; Subrahmanyan, L.; Cook, N.R.; Stanton, V.P.J.; Ridker, P.M. Pharmacogenetic study of statin therapy and cholesterol reduction. JAMA 2004, 291, 2821–2827. [Google Scholar] [CrossRef] [PubMed]
- Krauss, R.M.; Mangravite, L.M.; Smith, J.D.; Medina, M.W.; Wang, D.; Guo, X.; Rieder, M.J.; Simon, J.A.; Hulley, S.B.; Waters, D.; et al. Variation in the 3-hydroxyl-3-methylglutaryl coenzyme a reductase gene is associated with racial differences in low-density lipoprotein cholesterol response to simvastatin treatment. Circulation 2008, 117, 1537–1544. [Google Scholar] [CrossRef] [PubMed]
- Shiffman, D.; Trompet, S.; Louie, J.Z.; Rowland, C.M.; Catanese, J.J.; Iakoubova, O.A.; Kirchgessner, T.G.; Westendorp, R.G.J.; de Craen, A.J.M.; Slagboom, P.E.; et al. Genome-wide study of gene variants associated with differential cardiovascular event reduction by pravastatin therapy. PLoS ONE 2012, 7, e38240. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, D.I.; Preiss, D.; Kuchenbaecker, K.B.; Holmes, M.V.; Engmann, J.E.L.; Shah, T.; Sofat, R.; Stender, S.; Johnson, P.C.D.; Scott, R.A.; et al. HMG-coenzyme A reductase inhibition, type 2 diabetes, and bodyweight: Evidence from genetic analysis and randomised trials. Lancet 2015, 385, 351–361. [Google Scholar] [CrossRef]
- Kuivenhoven, J.A.; Jukema, J.W.; Zwinderman, A.H.; de Knijff, P.; McPherson, R.; Bruschke, A.V.; Lie, K.I.; Kastelein, J.J. The role of a common variant of the cholesteryl ester transfer protein gene in the progression of coronary atherosclerosis. The regression growth evaluation statin study group. N. Engl. J. Med. 1998, 338, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Vrablík, M.; Hubácek, J.A.; Dlouhá, D.; Lánská, V.; Rynekrová, J.; Zlatohlávek, L.; Prusíková, M.; Češka, R.; Adámková, V. Impact of variants within seven candidate genes on statin treatment efficacy. Physiol. Res. 2012, 61, 609–617. [Google Scholar] [PubMed]
- Polisecki, E.; Muallem, H.; Maeda, N.; Peter, I.; Robertson, M.; McMahon, A.D.; Ford, I.; Packard, C.; Shepherd, J.; Jukema, J.W.; et al. Genetic variation at the LDL receptor and HMG-CoA reductase gene loci, lipid levels, statin response, and cardiovascular disease incidence in PROSPER. Atherosclerosis 2008, 200, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Lahoz, C.; Pena, R.; Mostaza, J.M.; Laguna, F.; Garcia-Iglesias, M.F.; Taboada, M.; Pinto, X. Baseline levels of low-density lipoprotein cholesterol and lipoprotein (a) and the AvaII polymorphism of the low-density lipoprotein receptor gene influence the response of low-density lipoprotein cholesterol to pravastatin treatment. Metab. Clin. Exp. 2005, 54, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Iakoubova, O.A.; Sabatine, M.S.; Rowland, C.M.; Tong, C.H.; Catanese, J.J.; Ranade, K.; Simonsen, K.L.; Kirchgessner, T.G.; Cannon, C.P.; Devlin, J.J.; et al. Polymorphism in KIF6 gene and benefit from statins after acute coronary syndromes: Results from the PROVE IT-TIMI 22 study. J. Am. Coll. Cardiol. 2008, 51, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Peng, P.; Lian, J.; Huang, R.S.; Xu, L.; Huang, Y.; Ba, Y.; Yang, X.; Huang, X.; Dong, C.; Zhang, L.; et al. Meta-analyses of KIF6 Trp719Arg in coronary heart disease and statin therapeutic effect. PLoS ONE 2012, 7, e50126. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Tomlinson, B. Evaluation of the pharmacokinetics and drug interactions of the two recently developed statins, rosuvastatin and pitavastatin. Exp. Opin. Drug Metab. Toxicol. 2014, 10, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Wilke, R.A.; Moore, J.H.; Burmester, J.K. Relative impact of CYP3A genotype and concomitant medication on the severity of atorvastatin-induced muscle damage. Pharmacogenet. Genom. 2005, 15, 415–421. [Google Scholar] [CrossRef]
- Willrich, M.A.V.; Hirata, M.H.; Hirata, R.D.C. Statin regulation of CYP3A4 and CYP3A5 expression. Pharmacogenomics 2009, 10, 1017–1024. [Google Scholar] [CrossRef] [PubMed]
- Hustert, E.; Haberl, M.; Burk, O.; Wolbold, R.; He, Y.Q.; Klein, K.; Nuessler, A.C.; Neuhaus, P.; Klattig, J.; Eiselt, R.; et al. The genetic determinants of the CYP3A5 polymorphism. Pharmacogenetics 2001, 11, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Wei, K.K.; Zhang, L.R. Interactions between CYP3A5*3 and POR*28 polymorphisms and lipid lowering response with atorvastatin. Clin. Drug Investig. 2015, 35, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Drogari, E.; Ragia, G.; Mollaki, V.; Elens, L.; van Schaik, R.H.N.; Manolopoulos, V.G. POR*28 SNP is associated with lipid response to atorvastatin in children and adolescents with familial hypercholesterolemia. Pharmacogenomics 2014, 15, 1963–1972. [Google Scholar] [CrossRef] [PubMed]
- Elens, L.; van Gelder, T.; Hesselink, D.A.; Haufroid, V.; van Schaik, R.H.N. CYP3A4*22: Promising newly identified CYP3A4 variant allele for personalizing pharmacotherapy. Pharmacogenomics 2013, 14, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Johnson, A.D.; Papp, A.C.; Kroetz, D.L.; Sadee, W. Multidrug resistance polypeptide 1 (MDR1, ABCB1) variant 3435C>T affects mRNA stability. Pharmacogenet. Genom. 2005, 15, 693–704. [Google Scholar] [CrossRef]
- Mostafa, A.M.; Hamdy, N.M.; Abdel-Rahman, S.Z.; El-Mesallamy, H.O. Effect of vildagliptin and pravastatin combination on cholesterol efflux in adipocytes. IUBMB Life 2016, 68, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Fujino, H.; Saito, T.; Ogawa, S.I.; Kojima, J. Transporter-mediated influx and efflux mechanisms of pitavastatin, a new inhibitor of HMG-CoA reductase. J. Pharm. Pharmacol. 2005, 57, 1305–1311. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, S.; Maeda, K.; Wang, Y.; Sugiyama, Y. Involvement of multiple transporters in the hepatobiliary transport of rosuvastatin. Drug Metab. Dispos. 2008, 36, 2014–2023. [Google Scholar] [CrossRef] [PubMed]
- Niemi, M. Transporter pharmacogenetics and statin toxicity. Clin. Pharmacol. Ther. 2010, 87, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Niemi, M. Role of OATP transporters in the disposition of drugs. Pharmacogenomics 2007, 8, 787–802. [Google Scholar] [CrossRef] [PubMed]
- Niemi, M.; Pasanen, M.K.; Neuvonen, P.J. Organic anion transporting polypeptide 1B1: A genetically polymorphic transporter of major importance for hepatic drug uptake. Pharmacol. Rev. 2011, 63, 157–181. [Google Scholar] [CrossRef] [PubMed]
- Mangravite, L.M.; Krauss, R.M. Pharmacogenomics of statin response. Curr. Opin. Lipidol. 2007, 18, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Ieiri, I.; Higuchi, S.; Sugiyama, Y. Genetic polymorphisms of uptake (OATP1B1, 1B3) and efflux (MRP2, BCRP) transporters: Implications for inter-individual differences in the pharmacokinetics and pharmacodynamics of statins and other clinically relevant drugs. Exp. Opin. Drug Metab. Toxicol. 2009, 5, 703–729. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Lee, M.G.; Cho, J.Y.; Lee, J.E.; Kim, K.H.; Park, K. Influence of OATP1B1 genotype on the pharmacokinetics of rosuvastatin in Koreans. Clin. Pharmacol. Ther. 2008, 83, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, L.A.; Doney, A.S.F.; Tavendale, R.; Lang, C.C.; Pearson, E.R.; Colhoun, H.M.; McCarthy, M.I.; Hattersley, A.T.; Morris, A.D.; Palmer, C.N.A. Common nonsynonymous substitutions in SLCO1B1 predispose to statin intolerance in routinely treated individuals with type 2 diabetes: A go-DARTS study. Clin. Pharmacol. Ther. 2011, 89, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Nieminen, T.; Kahonen, M.; Viiri, L.E.; Gronroos, P.; Lehtimaki, T. Pharmacogenetics of apolipoprotein E gene during lipid-lowering therapy: Lipid levels and prevention of coronary heart disease. Pharmacogenomics 2008, 9, 1475–1486. [Google Scholar] [CrossRef] [PubMed]
- Postmus, I.; Verschuren, J.J.W.; de Craen, A.J.M.; Slagboom, P.E.; Westendorp, R.G.J.; Jukema, J.W.; Trompet, S. Pharmacogenetics of statins: Achievements, whole-genome analyses and future perspectives. Pharmacogenomics 2012, 13, 831–840. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.F.; Hyde, C.L.; Wood, L.S.; Paciga, S.A.; Hinds, D.A.; Cox, D.R.; Hovingh, G.K.; Kastelein, J.J.P. Comprehensive whole-genome and candidate gene analysis for response to statin therapy in the treating to new targets (TNT) cohort. Circ. Cardiovasc. Genet. 2009, 2, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.; di Angelantonio, E.; Sarwar, N.; Erqou, S.; Saleheen, D.; Dullaart, R.P.F.; Keavney, B.; Ye, Z.; Danesh, J. Association of cholesteryl ester transfer protein genotypes with CETP mass and activity, lipid levels, and coronary risk. JAMA 2008, 299, 2777–2788. [Google Scholar] [CrossRef] [PubMed]
- Regieli, J.J.; Jukema, J.W.; Grobbee, D.E.; Kastelein, J.J.P.; Kuivenhoven, J.A.; Zwinderman, A.H.; van der Graaf, Y.; Bots, M.L.; Doevendans, P.A. CETP genotype predicts increased mortality in statin-treated men with proven cardiovascular disease: An adverse pharmacogenetic interaction. Eur. Heart J. 2008, 29, 2792–2799. [Google Scholar] [CrossRef] [PubMed]
- Dupont, C.; Armant, D.R.; Brenner, C.A. Epigenetics: Definition, mechanisms and clinical perspective. Semin. Reprod. Med. 2009, 27, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Schiano, C.; Vietri, M.T.; Grimaldi, V.; Picascia, A.; Pascale, M.R.D.; Napoli, C. Epigenetic-related therapeutic challenges in cardiovascular disease. Trends Pharmacol. Sci. 2015, 36, 226–235. [Google Scholar] [CrossRef] [PubMed]
- N’Guessan, P.D.; Riediger, F.; Vardarova, K.; Scharf, S.; Eitel, J.; Opitz, B.; Slevogt, H.; Weichert, W.; Hocke, A.C.; Schmeck, B.; et al. Statins control oxidized LDL-mediated histone modifications and gene expression in cultured human endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Rayner, K.J.; Suarez, Y.; Davalos, A.; Parathath, S.; Fitzgerald, M.L.; Tamehiro, N.; Fisher, E.A.; Moore, K.J.; Fernandez-Hernando, C. miR-33 contributes to the regulation of cholesterol homeostasis. Science 2010, 328, 1570–1573. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Lamon, B.D.; Moran, G.; Sun, T.; Gotto, A.M.; Hajjar, D.P. Pitavastatin differentially modulates microRNA-associated cholesterol transport proteins in macrophages. PLoS ONE 2016, 11, e0159130. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.M.; Sheu, W.H.H.; Tseng, P.C.; Lee, T.S.; Lee, W.J.; Chang, P.J.; Chiang, A.N. Modulation of microRNA expression in subjects with metabolic syndrome and decrease of cholesterol efflux from macrophages via microRNA-33-mediated attenuation of ATP-binding cassette transporter A1 expression by statins. PLoS ONE 2016, 11, e0154672. [Google Scholar] [CrossRef] [PubMed]
- Allen, R.M.; Marquart, T.J.; Albert, C.J.; Suchy, F.J.; Wang, D.Q.H.; Ananthanarayanan, M.; Ford, D.A.; Baldán, Á. miR-33 controls the expression of biliary transporters, and mediates statin- and diet-induced hepatotoxicity. EMBO Mol. Med. 2012, 4, 882–895. [Google Scholar] [CrossRef] [PubMed]
- Cerda, A.; Fajardo, C.M.; Basso, R.G.; Hirata, M.H.; Hirata, R.D.C. Role of microRNAs 221/222 on statin induced nitric oxide release in human endothelial cells. Arq. Bras. Cardiol. 2015, 104, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Minami, Y.; Satoh, M.; Maesawa, C.; Takahashi, Y.; Tabuchi, T.; Itoh, T.; Nakamura, M. Effect of atorvastatin on microRNA 221/222 expression in endothelial progenitor cells obtained from patients with coronary artery disease. Eur. J. Clin. Investig. 2009, 39, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Satoh, M.; Minami, Y.; Tabuchi, T.; Itoh, T.; Nakamura, M. Expression of miR-146a/b is associated with the Toll-like receptor 4 signal in coronary artery disease: Effect of renin-angiotensin system blockade and statins on miRNA-146a/b and Toll-like receptor 4 levels. Clin. Sci. 2010, 119, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, A.C.; Neri, E.A.; Filho, S.V.; Reboucas, N.A.; Hirata, R.D.C.; Yu, A. Atorvastatin attenuation of ABCB1 expression is mediated by microRNA miR-491–3p in Caco-2 cells. Eur. J. Pharm. Sci. 2016, 93, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Yin, Y.; Guo, T.; Sun, X.; Zhu, M.; Zhao, F.; Xu, P.; Chen, Y.; Wan, G.; Jiang, F.; et al. Inhibition of aberrant microRNA-133a expression in endothelial cells by statin prevents endothelial dysfunction by targeting GTP cyclohydrolase 1 in vivo. Circulation 2016, 134, 1752–1765. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Li, P.; Wang, Z.; Chen, J.; Lin, Z.; Liang, X.; Mo, Y. Rosuvastatin may reduce the incidence of cardiovascular events in patients with acute coronary syndromes receiving percutaneous coronary intervention by suppressing miR-155/SHIP-1 signaling pathway. Cardiovasc. Ther. 2014, 32, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Liu, H.; Li, L.; Yang, M.; Du, A. Regulation of lovastatin on a key inflammation-related microRNA in myocardial cells. Chin. Med. J. 2014, 127, 2977–2981. [Google Scholar] [PubMed]
- Miltiadous, G.; Xenophontos, S.; Bairaktari, E.; Ganotakis, M.; Cariolou, M.; Elisaf, M. Genetic and environmental factors affecting the response to statin therapy in patients with molecularly defined familial hypercholesterolaemia. Pharmacogenet. Genom. 2005, 15, 219–225. [Google Scholar] [CrossRef]
- Greenland, P.; Abrams, J.; Aurigemma, G.P.; Bond, M.G.; Clark, L.T.; Criqui, M.H.; Crouse, J.R.; Friedman, L.; Fuster, V.; Herrington, D.M.; et al. Prevention conference V: Beyond secondary prevention: Identifying the high-risk patient for primary prevention: Noninvasive tests of atherosclerotic burden: Writing group III. Circulation 2000, 101, 16–22. [Google Scholar] [CrossRef]
- Karazniewicz-Lada, M.; Glowka, A.; Mikolajewski, J.; Przyslawski, J. Genetic and non-genetic determinants of the pharmacological activity of statins. Curr. Drug Metab. 2016, in press. [Google Scholar] [CrossRef]
- Chen, Y.; Ku, H.; Zhao, L.; Wheeler, D.C.; Li, L.C.; Li, Q.; Varghese, Z.; Moorhead, J.F.; Powis, S.H.; Huang, A.; et al. Inflammatory stress induces statin resistance by disrupting 3-hydroxy-3-methylglutaryl-CoA reductase feedback regulation. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Robertson, J.; Peters, M.J.; McInnes, I.B.; Sattar, N. Changes in lipid levels with inflammation and therapy in RA: A maturing paradigm. Nat. Rev. Rheumatol. 2013, 9, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Gay, M.A.; Gonzalez-Juanatey, C. Inflammation and lipid profile in rheumatoid arthritis: Bridging an apparent paradox. Ann. Rheum. Dis. 2014, 73, 1281–1283. [Google Scholar] [CrossRef] [PubMed]
- Hirota, T.; Ieiri, I. Drug–drug interactions that interfere with statin metabolism. Exp. Opin. Drug Metab. Toxicol. 2015, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.C.; Hsieh, T.C.; Chou, C.L.; Wu, J.L.; Fang, T.C. Risks of adverse events following coprescription of statins and calcium channel blockers: A nationwide population-based study. Medicine 2016, 95, e2487. [Google Scholar] [CrossRef] [PubMed]
- Teng, R.; Mitchell, P.D.; Butler, K.A. Pharmacokinetic interaction studies of co-administration of ticagrelor and atorvastatin or simvastatin in healthy volunteers. Eur. J. Clin. Pharmacol. 2013, 69, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Itkonen, M.K.; Tornio, A.; Neuvonen, M.; Neuvonen, P.J.; Niemi, M.; Backman, J.T. Clopidogrel has no clinically meaningful effect on the pharmacokinetics of the organic anion transporting polypeptide 1B1 and cytochrome P450 3A4 substrate simvastatin. Drug Metab. Dispos. 2015, 43, 1655–1660. [Google Scholar] [CrossRef] [PubMed]
- Weinz, C.; Schwarz, T.; Kubitza, D.; Mueck, W.; Lang, D. Metabolism and excretion of rivaroxaban, an oral, direct factor Xa inhibitor, in rats, dogs, and humans. Drug Metab. Dispos. 2009, 37, 1056–1064. [Google Scholar] [CrossRef] [PubMed]
- Kubitza, D.; Becka, M.; Roth, A.; Mueck, W. Absence of clinically relevant interactions between rivaroxaban—An oral, direct Factor Xa inhibitor—And digoxin or atorvastatin in healthy subjects. J. Int. Med. Res. 2012, 40, 1688–1707. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.; Yoon, D.; Lim, S.G.; Hong, J.M.; Park, R.W.; Lee, J.S. Comparison of the risk of gastrointestinal bleeding among different statin exposures with concomitant administration of warfarin: Electronic health record-based retrospective cohort study. PLoS ONE 2016, 11, e0158130. [Google Scholar] [CrossRef] [PubMed]
- Staatz, C.E.; Goodman, L.K.; Tett, S.E. Effect of CYP3A and ABCB1 single nucleotide polymorphisms on the pharmacokinetics and pharmacodynamics of calcineurin inhibitors: Part I. Clin. Pharmacokinet. 2010, 49, 141–175. [Google Scholar] [CrossRef] [PubMed]
- Kalliokoski, A.; Niemi, M. Impact of OATP transporters on pharmacokinetics. Br. J. Pharmacol. 2009, 158, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Neuvonen, P.J.; Niemi, M.; Backman, J.T. Drug interactions with lipid-lowering drugs: Mechanisms and clinical relevance. Clin. Pharmacol. Ther. 2006, 80, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Herman, R.J. Drug interactions and the statins. Can. Med. Assoc. J. 1999, 161, 1281–1286. [Google Scholar]
- Wiggins, B.S.; Saseen, J.J.; Page, R.L.; Reed, B.N.; Sneed, K.; Kostis, J.B.; Lanfear, D.; Virani, S.; Morris, P.B. Recommendations for management of clinically significant drug–drug interactions with statins and select agents used in patients with cardiovascular disease: A scientific statement from the American Heart Association. Circulation 2016, 134, 468–495. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T. Physiologically based pharmacokinetic modeling of disposition and drug–drug interactions for atorvastatin and its metabolites. Eur. J. Pharm. Sci. 2015, 77, 216–229. [Google Scholar] [CrossRef] [PubMed]
- Badri, P.S.; Dutta, S.; Wang, H.; Podsadecki, T.J.; Polepally, A.R.; Khatri, A.; Zha, J.; Chiu, Y.L.; Awni, W.M.; Menon, R.M. Drug interactions with the direct-acting antiviral combination of ombitasvir and paritaprevir-ritonavir. Antimicrob. Agents Chemother. 2015, 60, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Hua, W.J.; Hua, W.X.; Fang, H.J. The role of OATP1B1 and BCRP in pharmacokinetics and DDI of novel statins. Cardiovasc. Ther. 2012, 30, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Sekhar, R.V.; Balasubramanyam, A. Treatment of dyslipidemia in HIV-infected patients. Exp. Opin. Pharmacother. 2010, 11, 1845–1854. [Google Scholar] [CrossRef] [PubMed]
- Wongprikorn, A.; Sukasem, C.; Puangpetch, A.; Numthavej, P.; Thakkinstian, A.; Kiertiburanakul, S. Effects of pitavastatin on lipid profiles in HIV-infected patients with dyslipidemia and receiving atazanavir/ritonavir: A randomized, double-blind, crossover study. PLoS ONE 2016, 11, e0157531. [Google Scholar] [CrossRef] [PubMed]
- Graefe-Mody, U.; Huettner, S.; Stahle, H.; Ring, A.; Dugi, K.A. Effect of linagliptin (BI 1356) on the steady-state pharmacokinetics of simvastatin. Int. J. Clin. Pharmacol. Ther. 2010, 48, 367–374. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
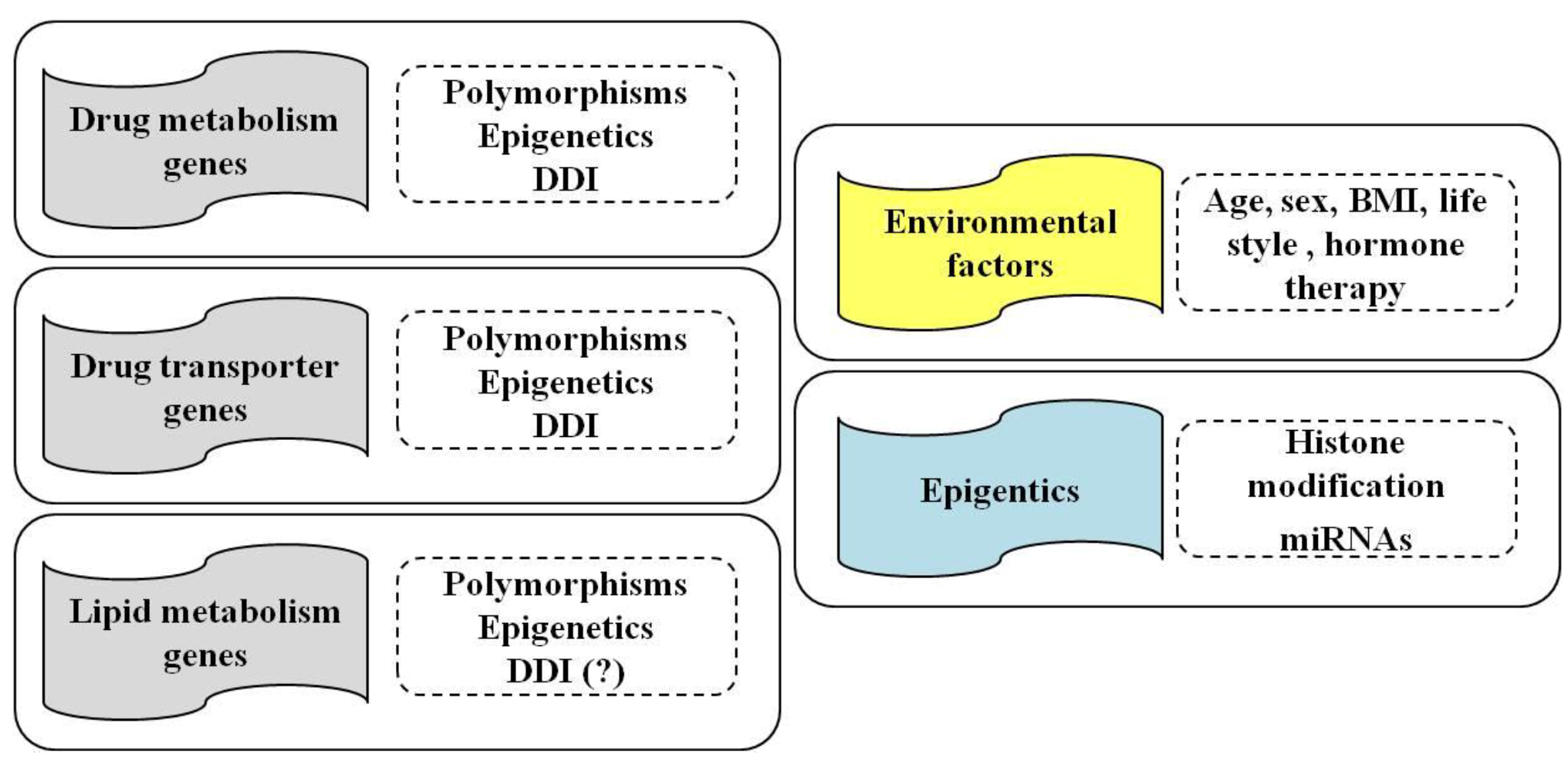{kind=link}
{kind=link}
| Gene | Location | Polymorphism | ADRs | Efficacy | References |
|---|---|---|---|---|---|
| CYP2D6 | 22q13.2 | CYP2D6*4 | √ | √ | [38,39] |
| c.1846G>A-rs3892097 | [A, S] | [A, F, P, R, S] | |||
| CYP2D6*10 | – | √ | [40] | ||
| c.188C>T-rs1065852 | [S] | ||||
| CYP2C9 | 10q23.33 | CYP2C9*2 | √ | √ | [41] |
| c.430C>T-rs1799853 | [F] | [F] | |||
| CYP2C9*3 | √ | √ | [41,42,43] | ||
| c.1075A>C-rs1057910 | [F, R] | [F, R] | |||
| CYP3A4 | 7q21.1 | CYP3A4*1B | √ | √ | [44,45] |
| c.-392A>G-rs2740574 | [A, S] | [A, S] | |||
| CYP3A4*22 | – | √ | [46,47] | ||
| c.522-191C>T-rs35599367 | [A, F, P, R, S] | ||||
| CYP3A5 | 7q21.1 | CYP3A5*3 | – | √ | [48] |
| c.6986A>G-rs776746 | [A, L, S] |
| Gene | Location | Polymorphism | ADRs | Efficacy | References |
|---|---|---|---|---|---|
| ABCB1/MDR-1 | 7q21.12 | c.1236C>T-rs1128503 | √ | √ | [49] |
| [S] | [S] | ||||
| c.2677G>T/A-rs2032582 | √ | √ | [49,50,51,52] | ||
| [S] | [A, P, S] | ||||
| c.3435C>T-rs1045642 | √ | √ | [21,45,49,53,54,55] | ||
| [A, S] | [A, S] | ||||
| Haplotype TTT | √ | √ | [49,52,56,57] | ||
| [S] | [A, R, S] | ||||
| ABCC2/MPR-2 | 10q24.2 | c.1446C>G | – | √ | [58] |
| [P] | |||||
| c.-24C>T-rs717620 | – | √ | [59,60] | ||
| [Pi, S] | |||||
| ABCG2/BCRP | 4q22.1 | c.421C>A-rs2231142 | √ | √ | [41,52,61,62,63,64,65,66,67,68,69] |
| [A, F, R] | [A, F, R, S] | ||||
| c.34G>A-rs2231137 | – | √ | [63,66] | ||
| [R] | |||||
| SLCO1B1/OATP1B1 | 12p12.1 | SLCO1B1*1B | – | √ | [70,71,72,73] |
| c.388A>G-rs2306283 | [P] | ||||
| SLCO1B1*5 | √ | √ | [70,74,75,76,77,78,79,80,81,82,83] | ||
| c.521T>C-rs4149056 | [C, S] | [A, P, R, S] | |||
| SLCO1B1*15 | √ | √ | [75,84,85,86,87] | ||
| (c.388G-c.521C) | [L, S] | [L, P, Pi, S] | |||
| SLCO1B3/OATP1B3 | 12p12.2 | c.344T>G-rs4149117 | – | √ | [88] |
| [A, F, Pi, R] | |||||
| c.699G>A-rs7311358 | – | √ | [88] | ||
| [A, F, Pi, R] | |||||
| SLCO2B1/OATP2B1 | 11q13.4 | c.1457C>T-rs2306168 | – | √ | [81,88,89] |
| [A, R] |
| Gene | Location | Polymorphism | ADRs | Efficacy | References |
|---|---|---|---|---|---|
| APOE | 19q13.32 | ApoE ε2 | – | √ | [51,90,91,92] |
| (c.334T-c.472T) | [A, F, L, P, S] | ||||
| ApoE ε4 | √ | √ | [91,93,94,95] | ||
| (c.334C-c.472C) | [P, S] | [P, S] | |||
| HMGCoAR | 5q13.3 | SNP12 | – | √ | [96,97] |
| c.451-174A>T-rs17244841 | [P, S] | ||||
| SNP29 | √ | √ | [92,96,97,98] | ||
| c.2457 + 117T>G-rs17238540 | [P] | [A, F, P, R, S] | |||
| c.1368 + 1069G>T-rs17238484 | √ | – | [99] | ||
| CEPT | 16q13 | TaqIB | √ | √ | [1,100] |
| c.118 + 279G>A-rs708272 | [P] | [P] | |||
| LDLR | 19p13.2 | c.44857C>T-rs1433099 | √ | √ | [101,102] |
| [P] | [P] | ||||
| c.2052T>C-rs5925 | √ | √ | [103] | ||
| [P] | [P] | ||||
| KIF6 | 6p21.2 | c.2155T>C-rs20455 | √ | – | [98,104,105] |
| [P] |
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arrigoni, E.; Del Re, M.; Fidilio, L.; Fogli, S.; Danesi, R.; Di Paolo, A. Pharmacogenetic Foundations of Therapeutic Efficacy and Adverse Events of Statins. Int. J. Mol. Sci. 2017, 18, 104. https://doi.org/10.3390/ijms18010104
Arrigoni E, Del Re M, Fidilio L, Fogli S, Danesi R, Di Paolo A. Pharmacogenetic Foundations of Therapeutic Efficacy and Adverse Events of Statins. International Journal of Molecular Sciences. 2017; 18(1):104. https://doi.org/10.3390/ijms18010104
Chicago/Turabian StyleArrigoni, Elena, Marzia Del Re, Leonardo Fidilio, Stefano Fogli, Romano Danesi, and Antonello Di Paolo. 2017. "Pharmacogenetic Foundations of Therapeutic Efficacy and Adverse Events of Statins" International Journal of Molecular Sciences 18, no. 1: 104. https://doi.org/10.3390/ijms18010104
APA StyleArrigoni, E., Del Re, M., Fidilio, L., Fogli, S., Danesi, R., & Di Paolo, A. (2017). Pharmacogenetic Foundations of Therapeutic Efficacy and Adverse Events of Statins. International Journal of Molecular Sciences, 18(1), 104. https://doi.org/10.3390/ijms18010104