
Ruthenium Complexes Induce HepG2 Human Hepatocellular Carcinoma Cell Apoptosis and Inhibit Cell Migration and Invasion through Regulation of the Nrf2 Pathway

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
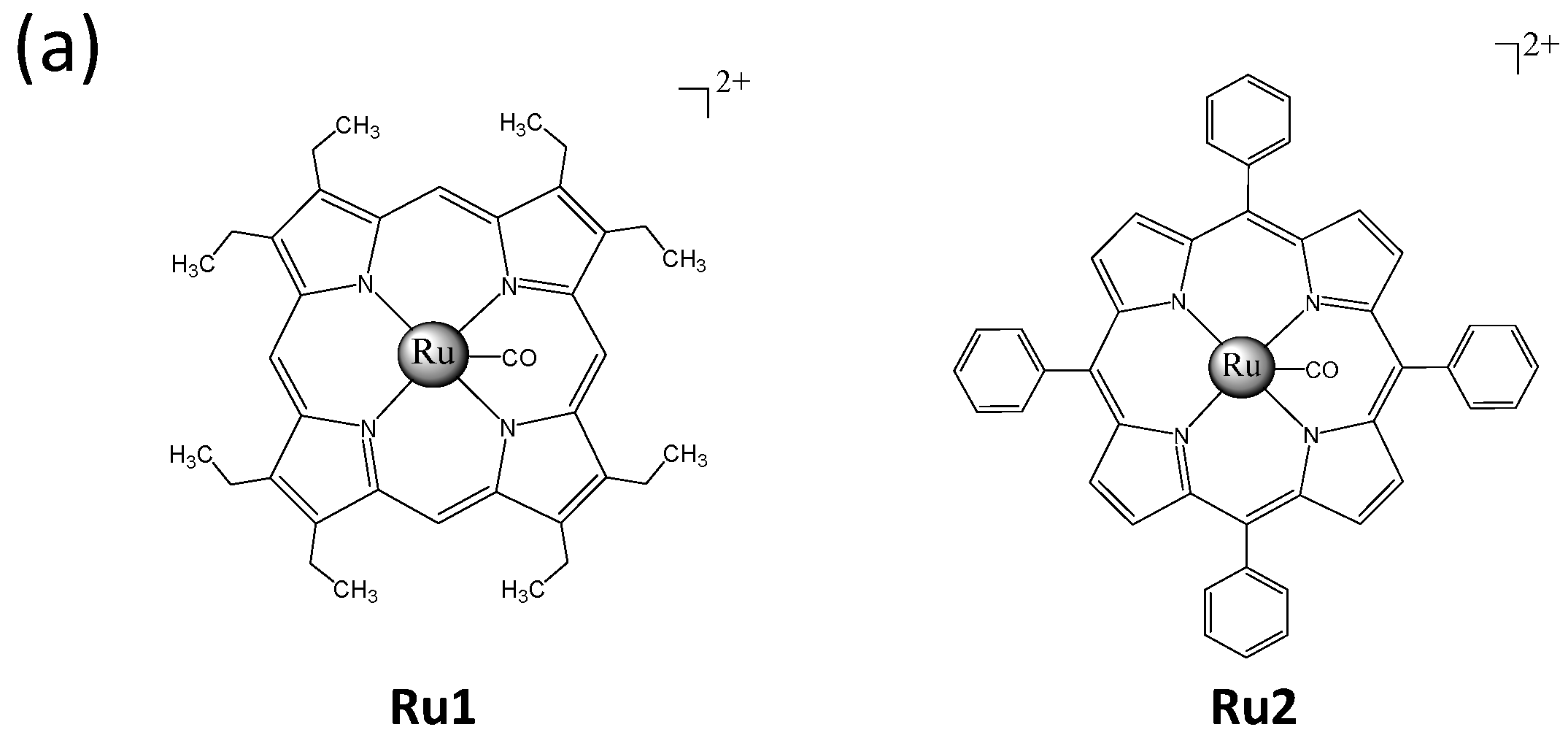
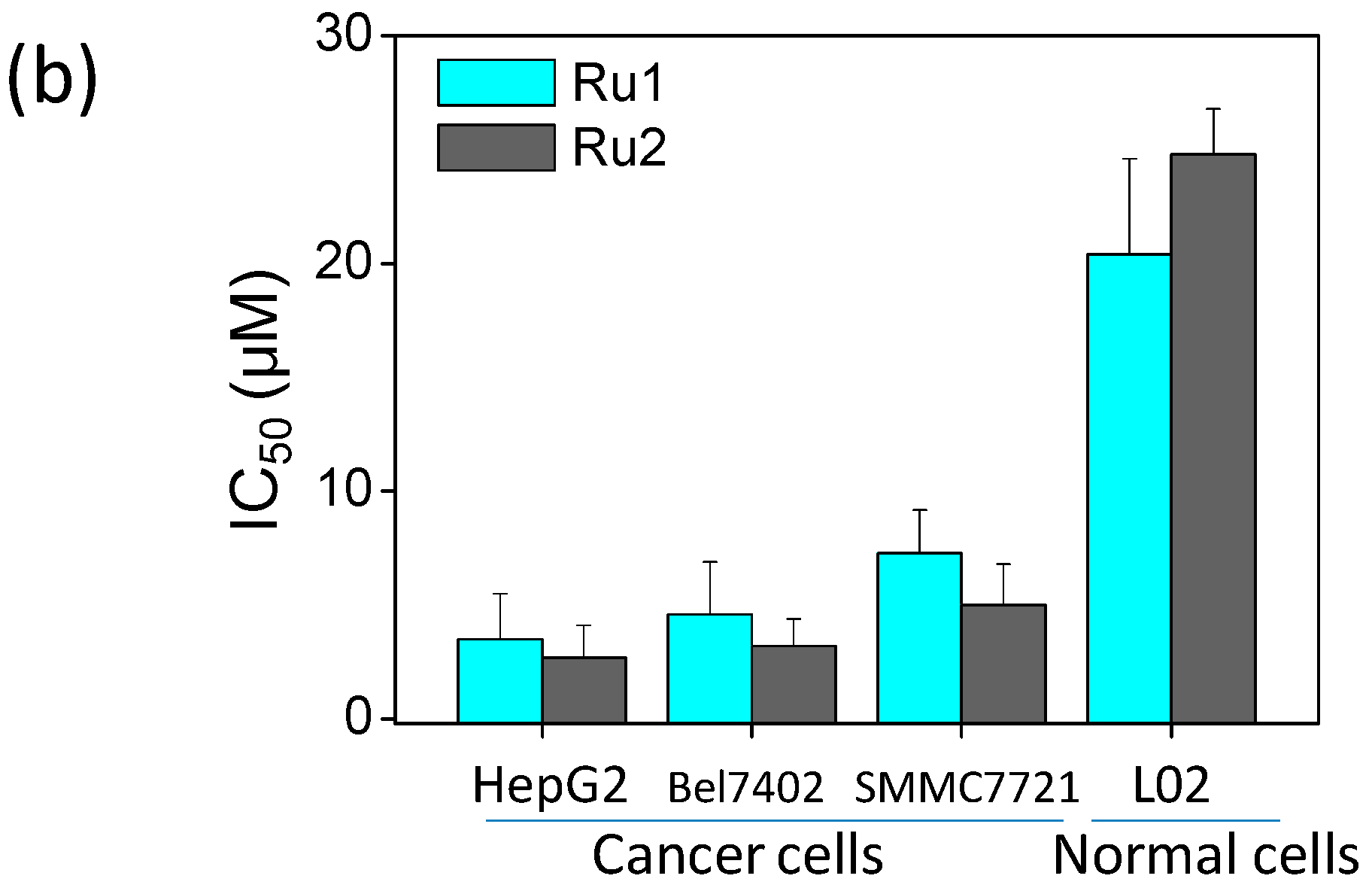
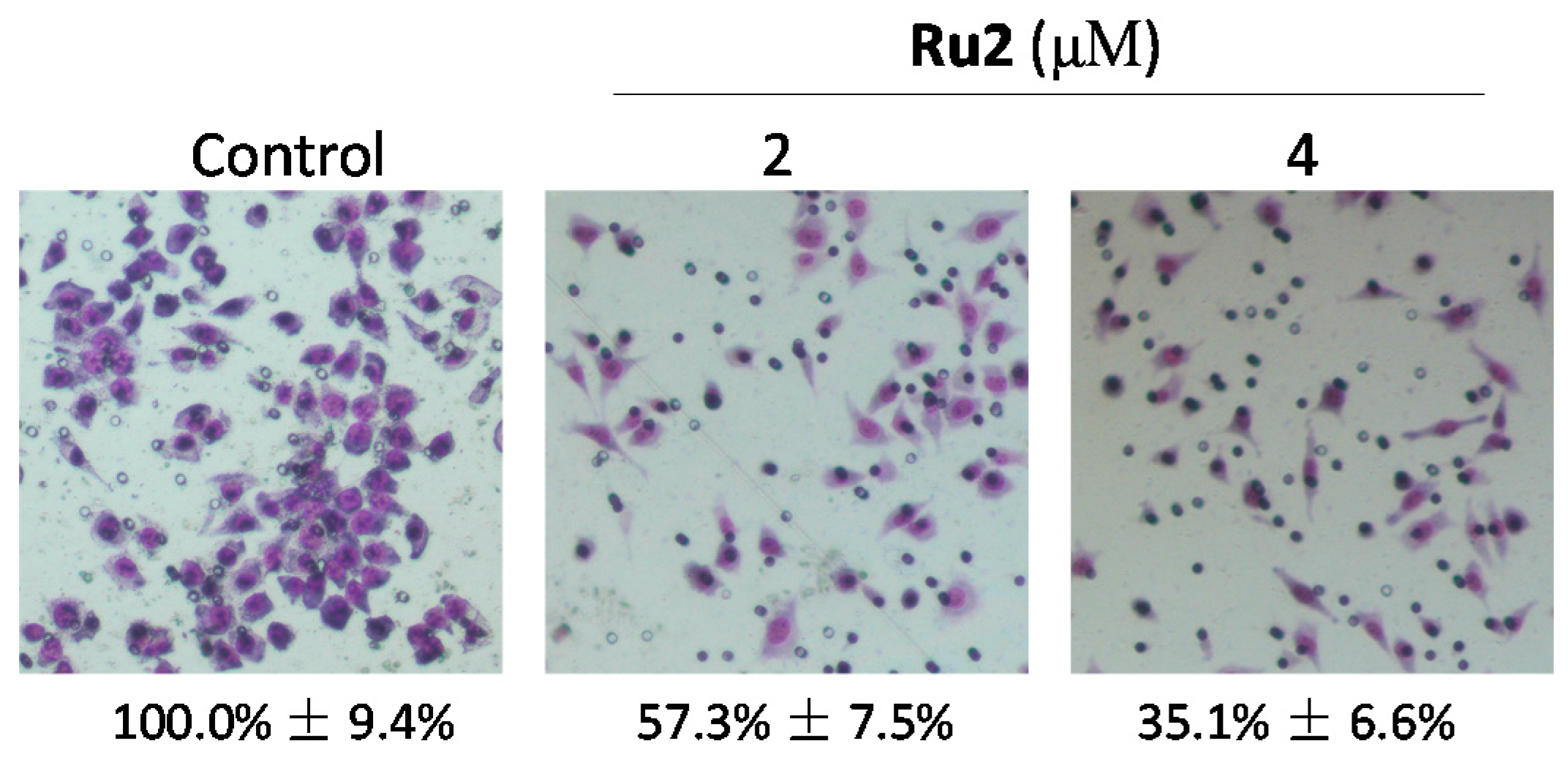
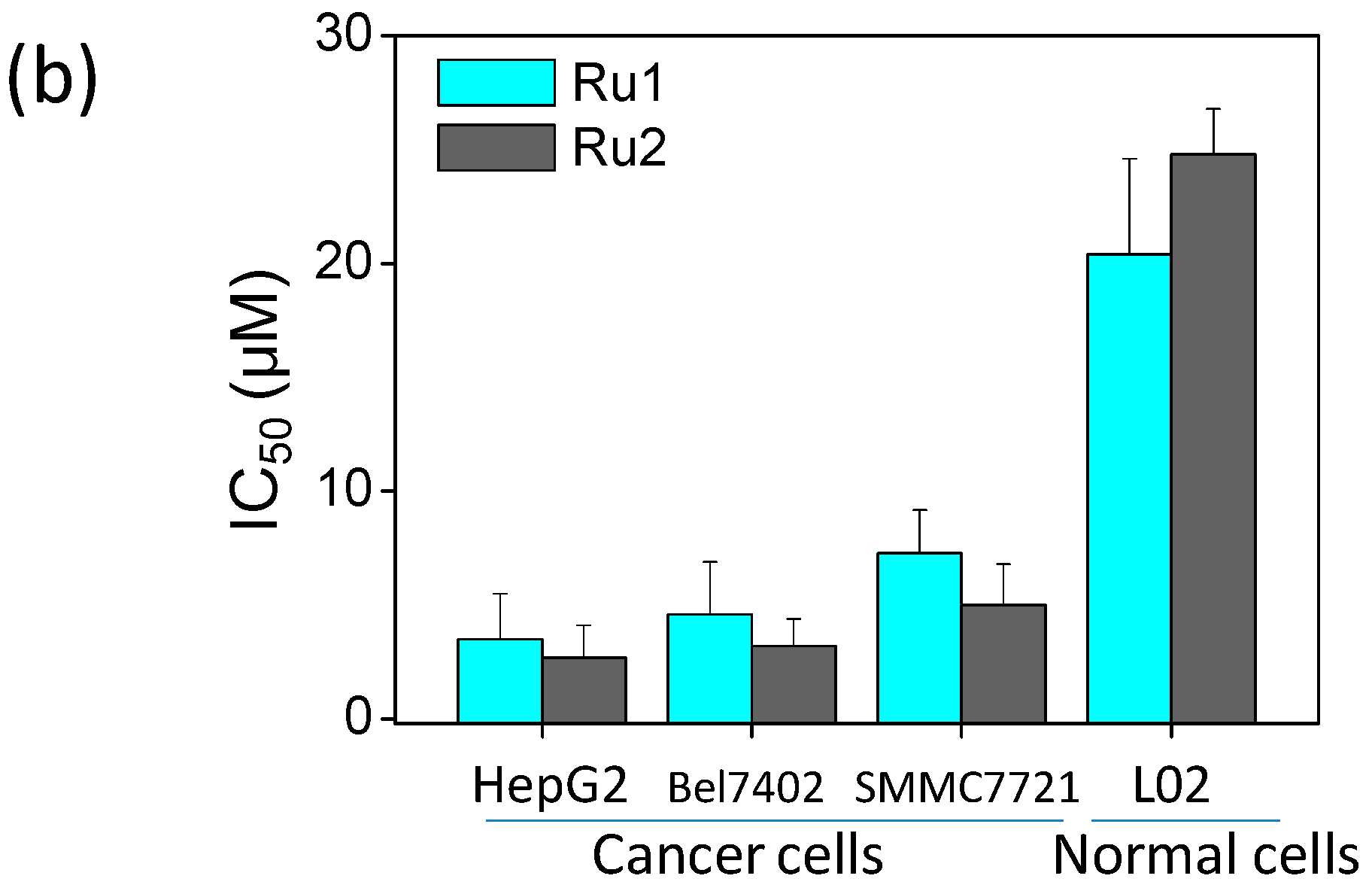
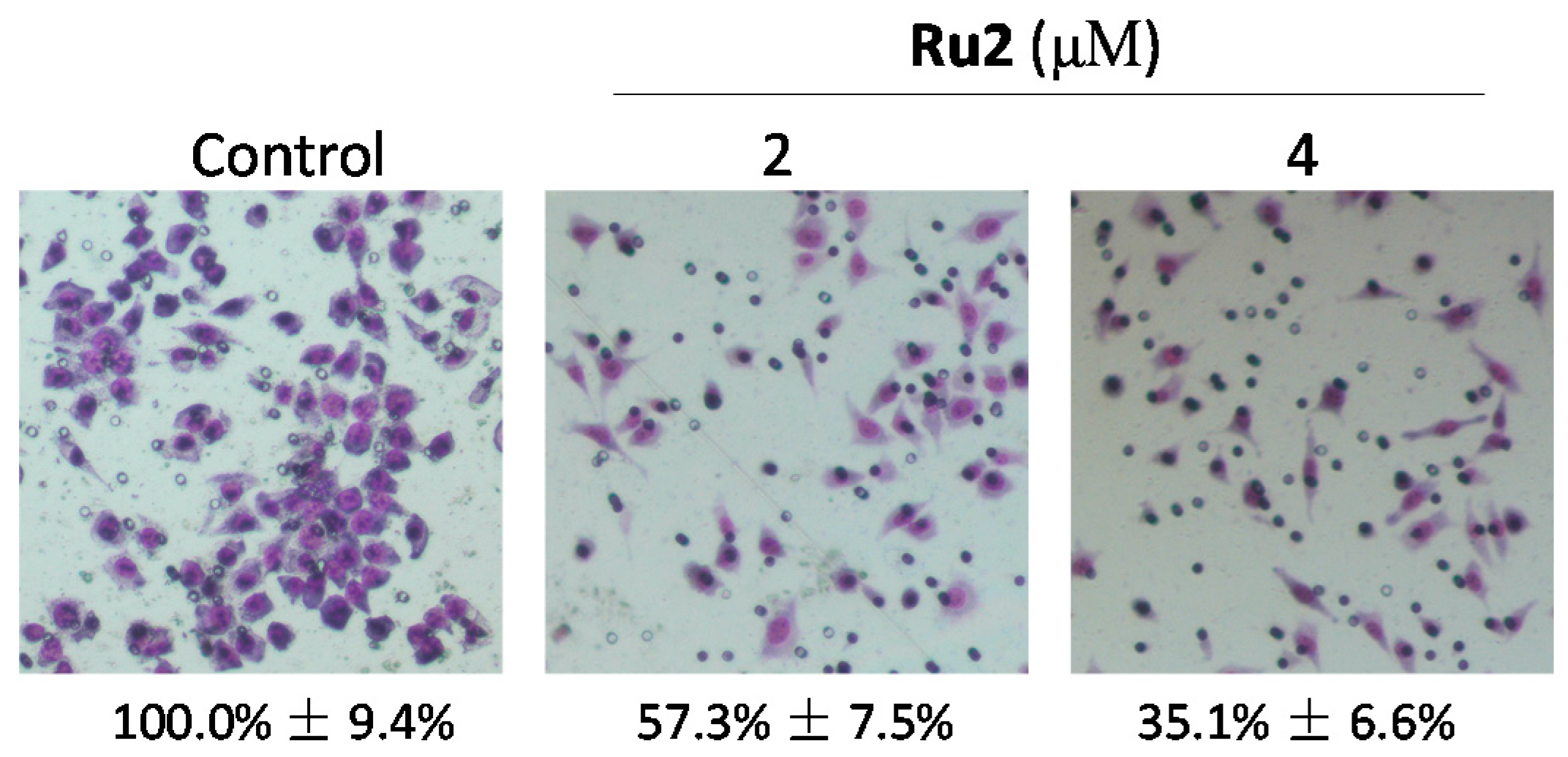
2.1. Ru Complexes Inhibit HCC Cell Growth, Migration, and Invasion
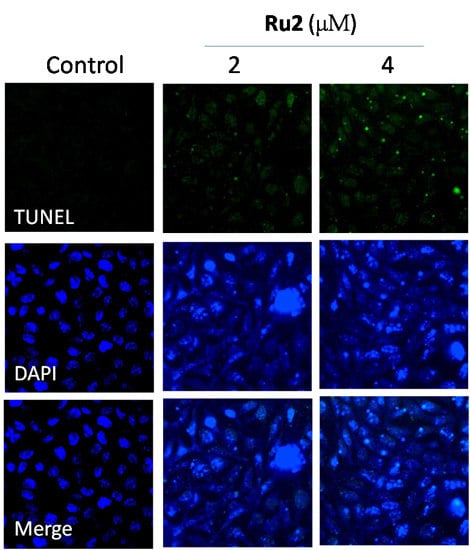
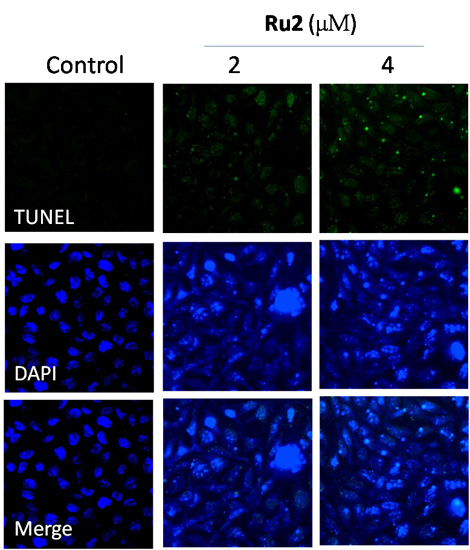
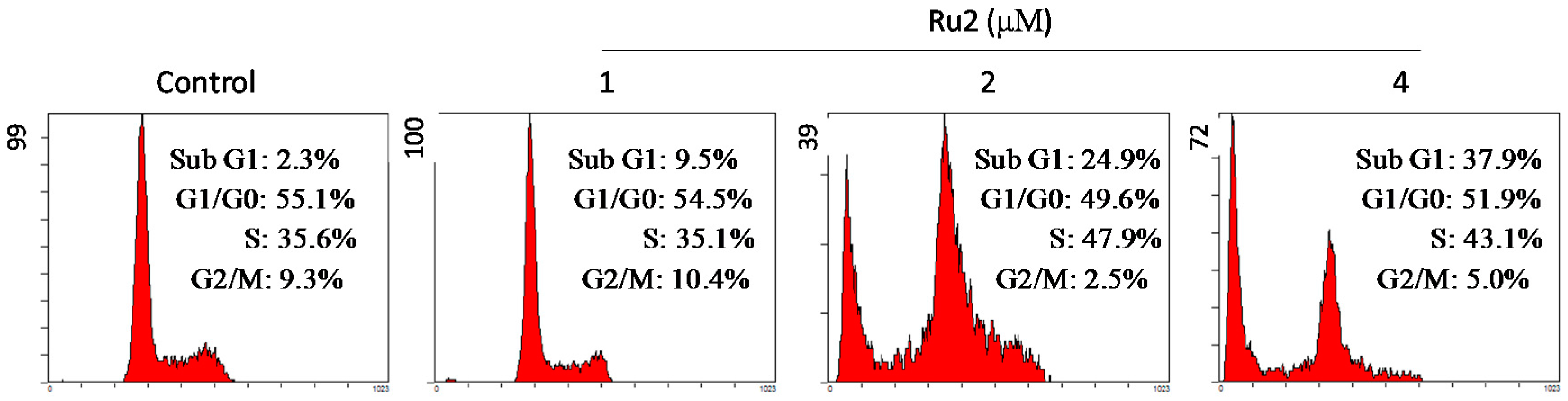
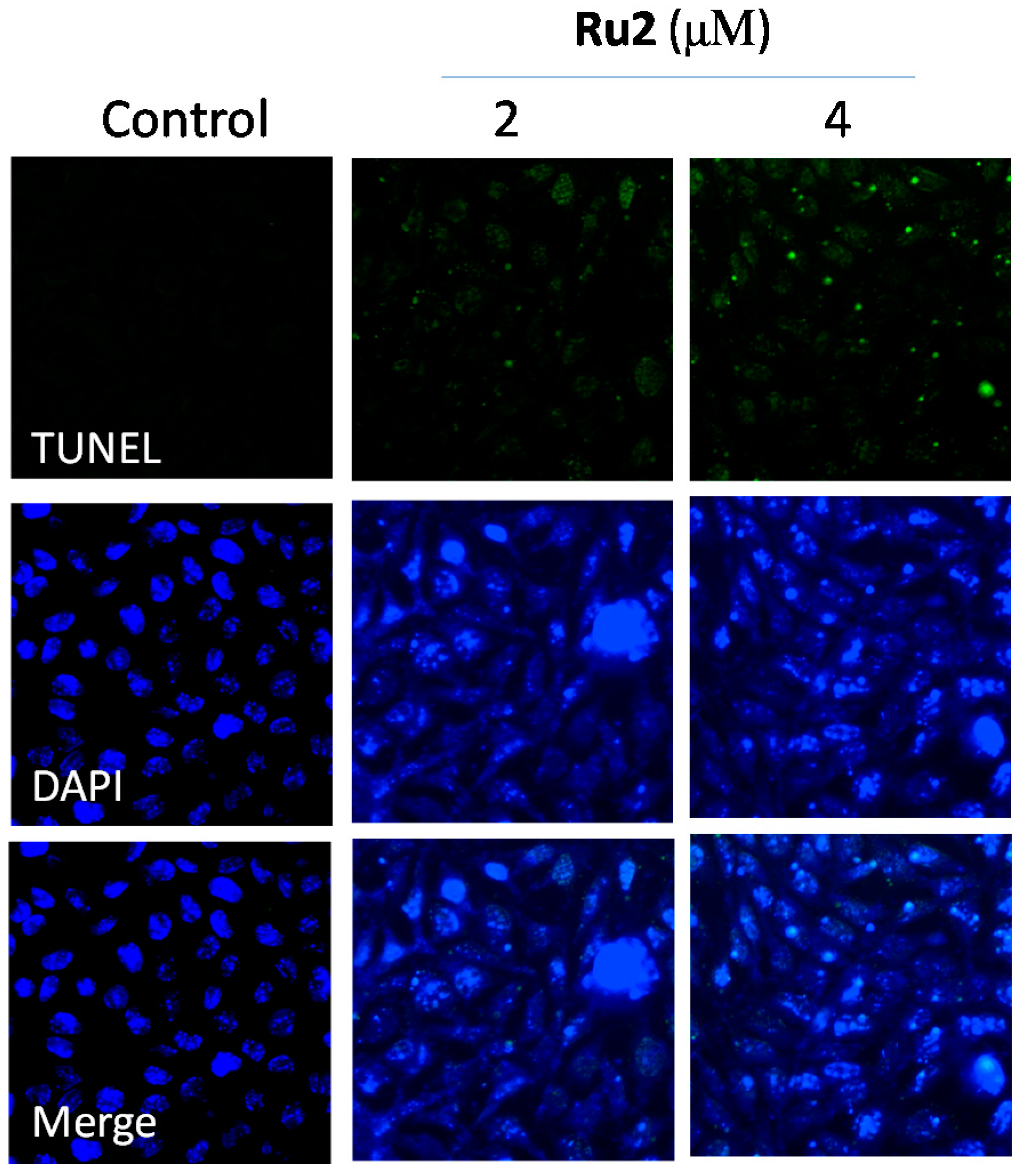
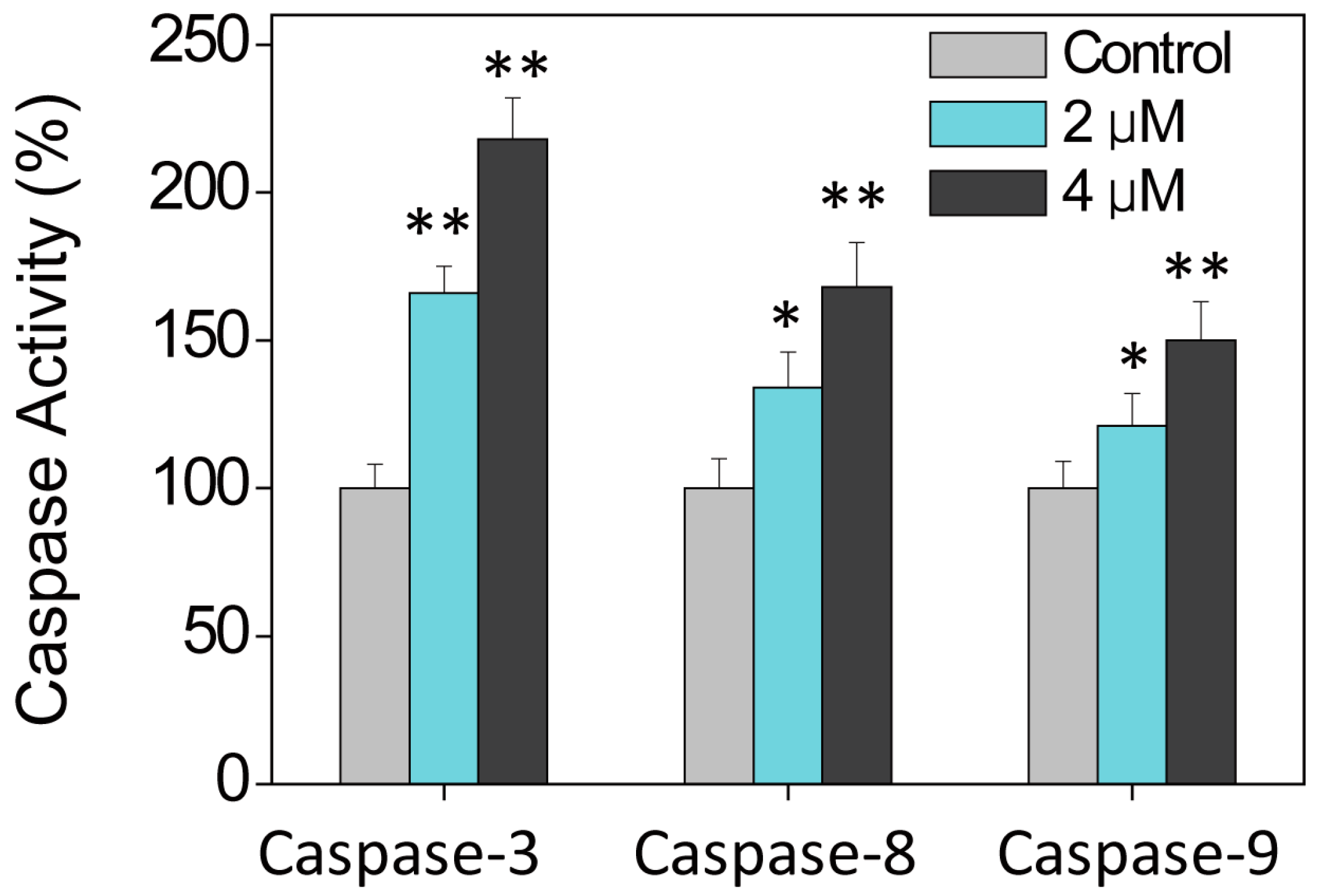
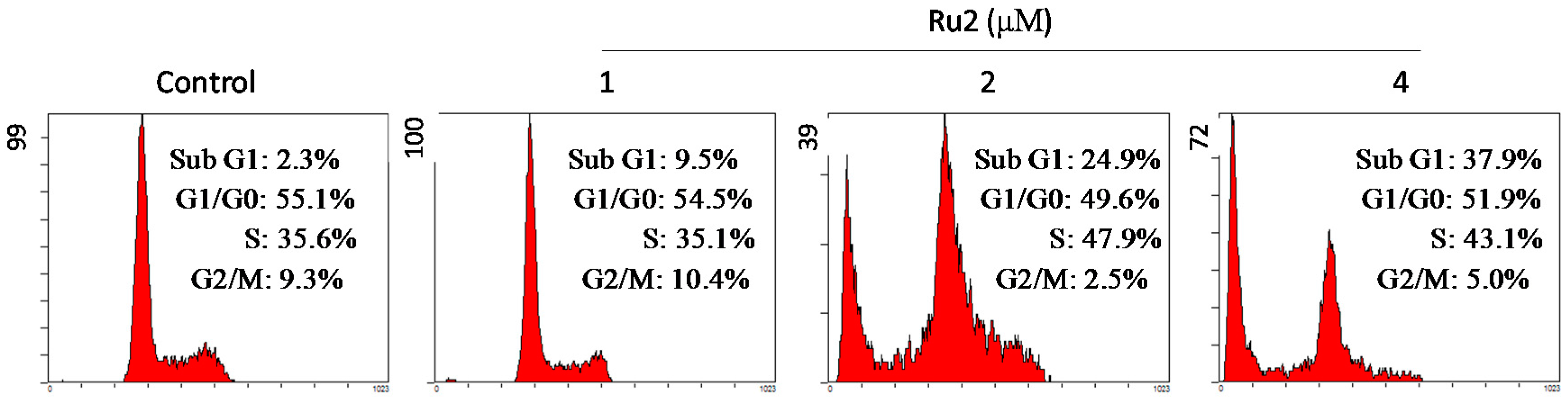
2.2. Cell Apoptosis Activation by Ru Complexes
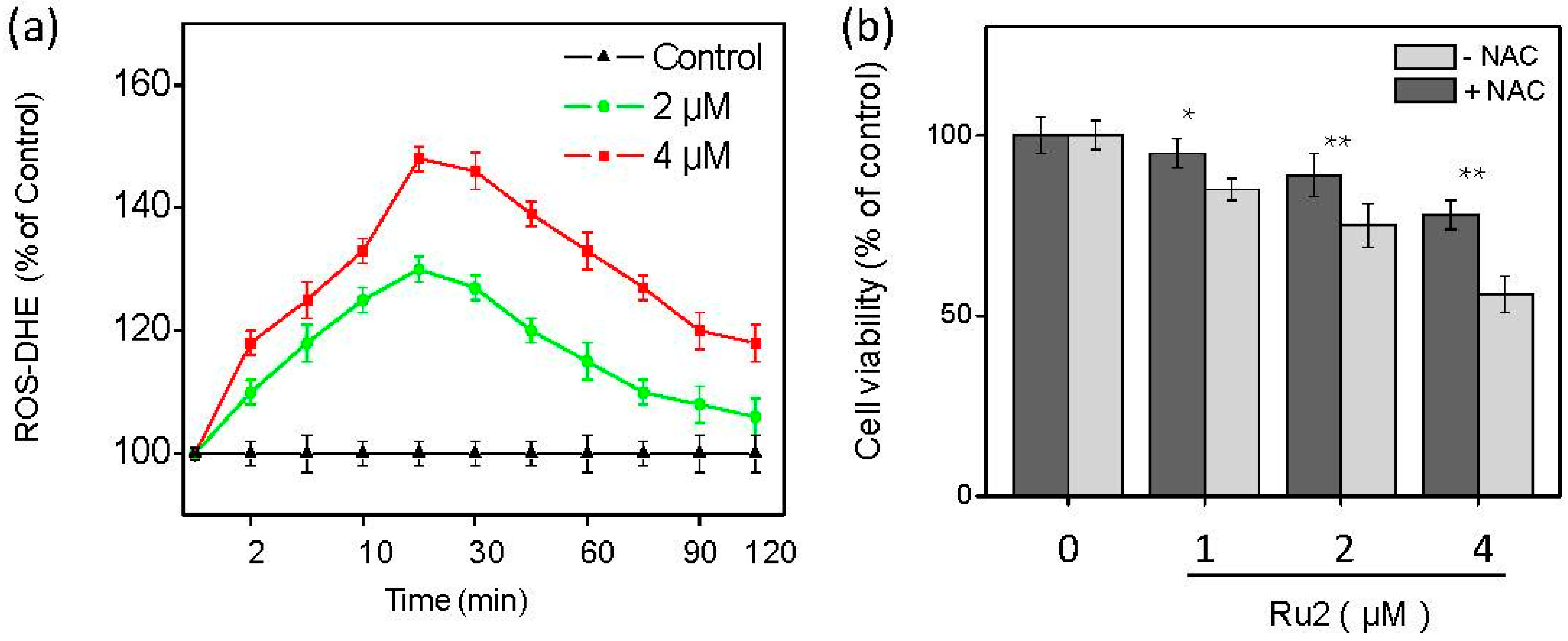
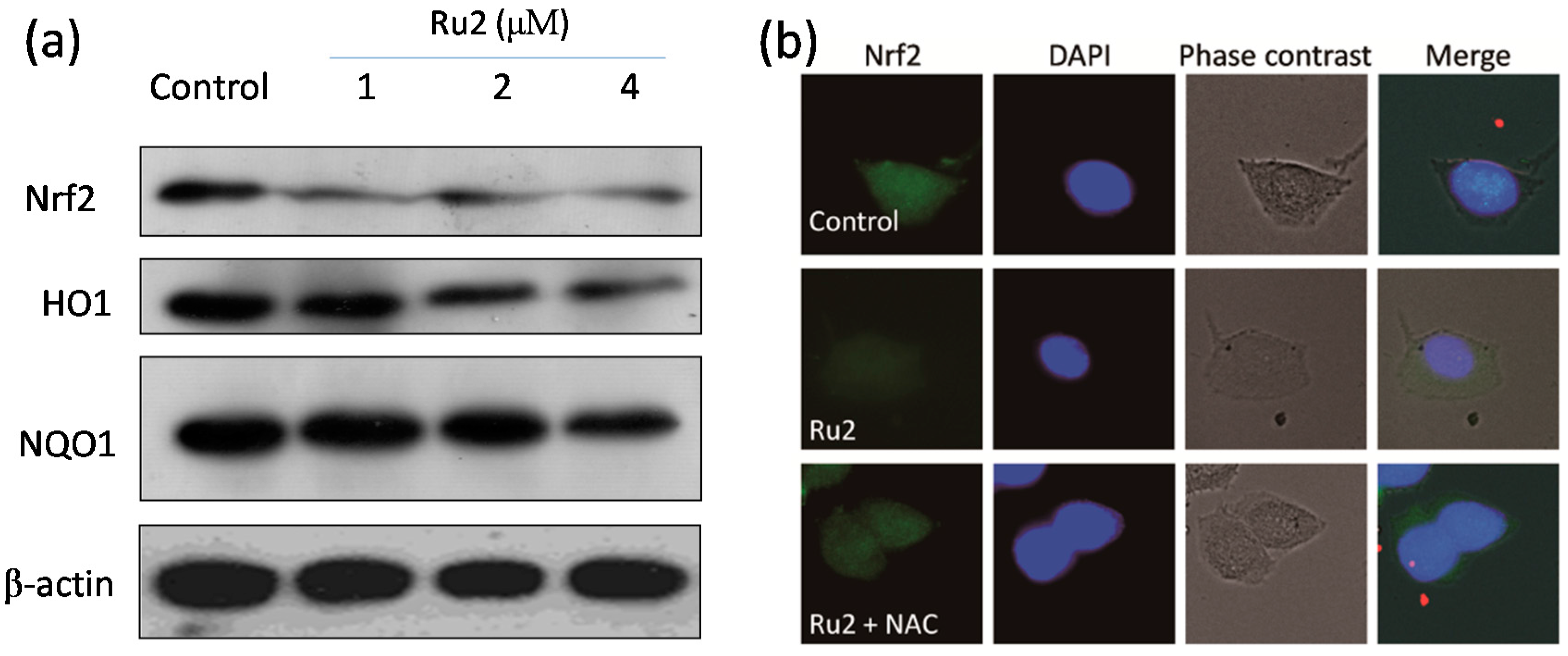
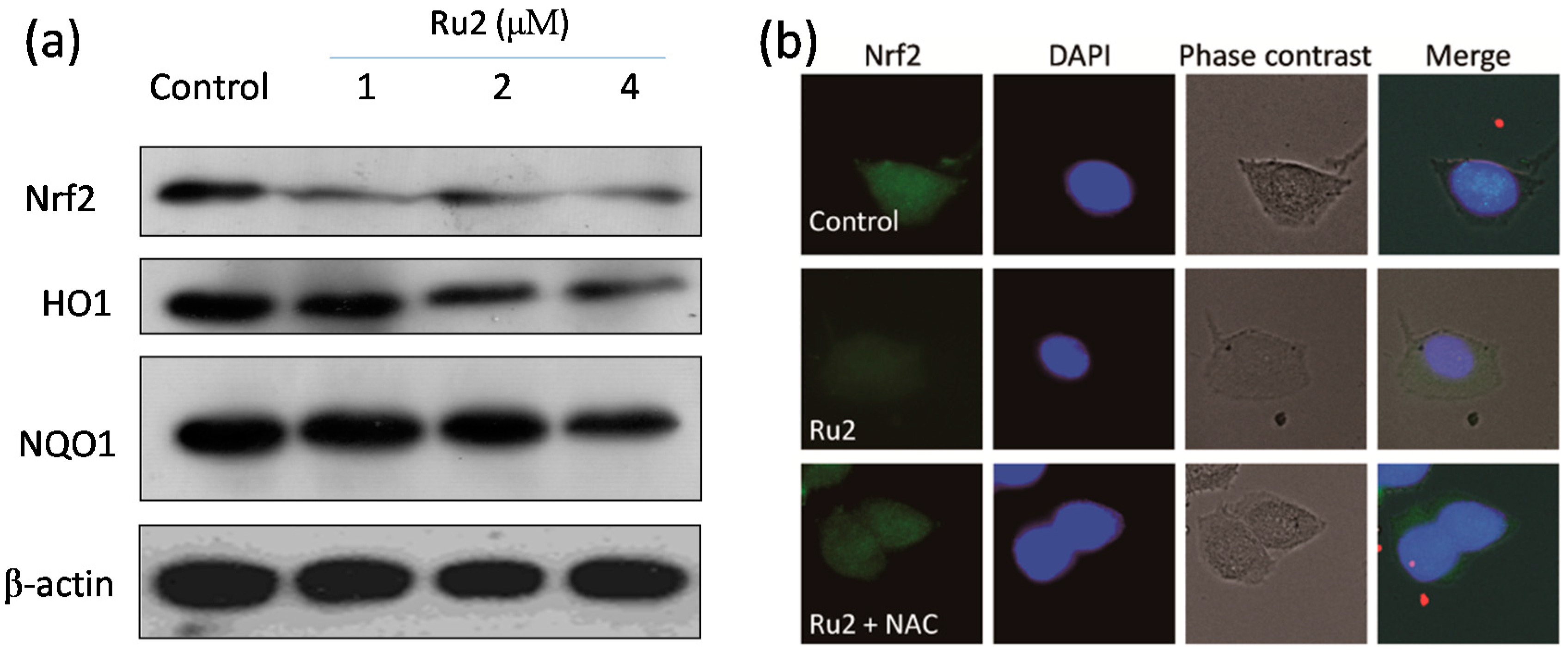
2.3. Ru Complexes Induce Reactive Oxygen Species (ROS) Overproduction via Nrf2 Pathway Regulation
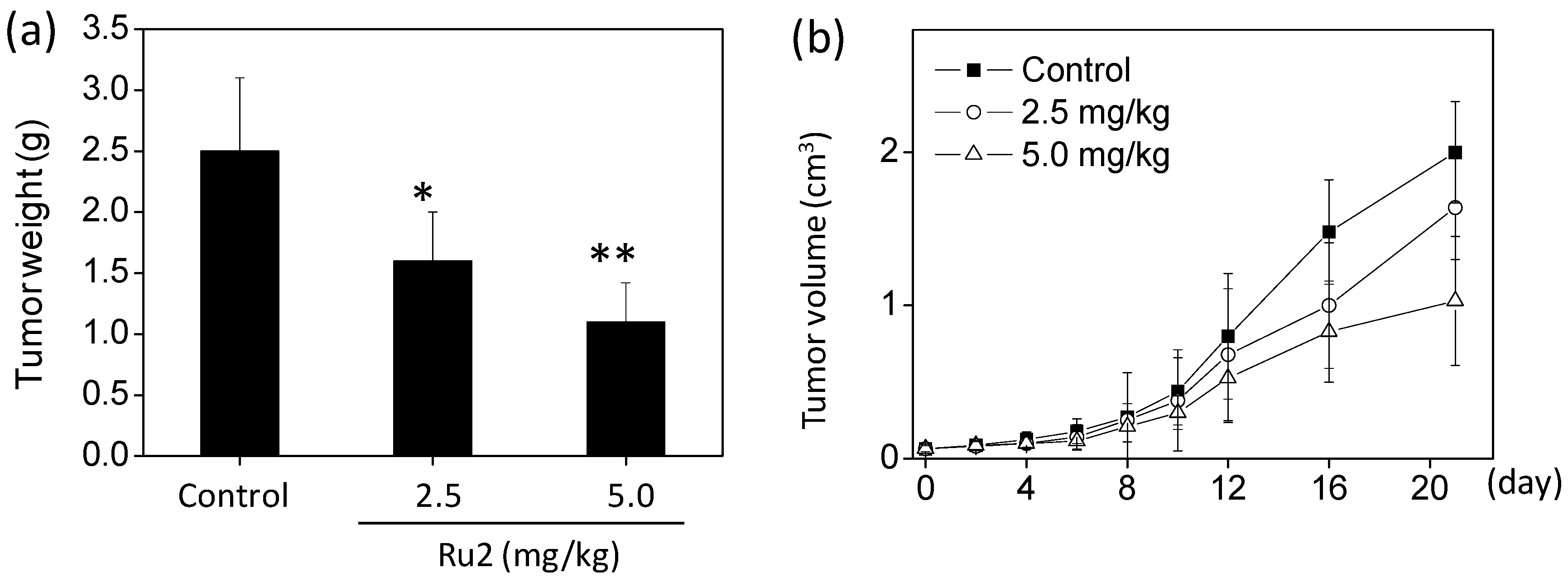
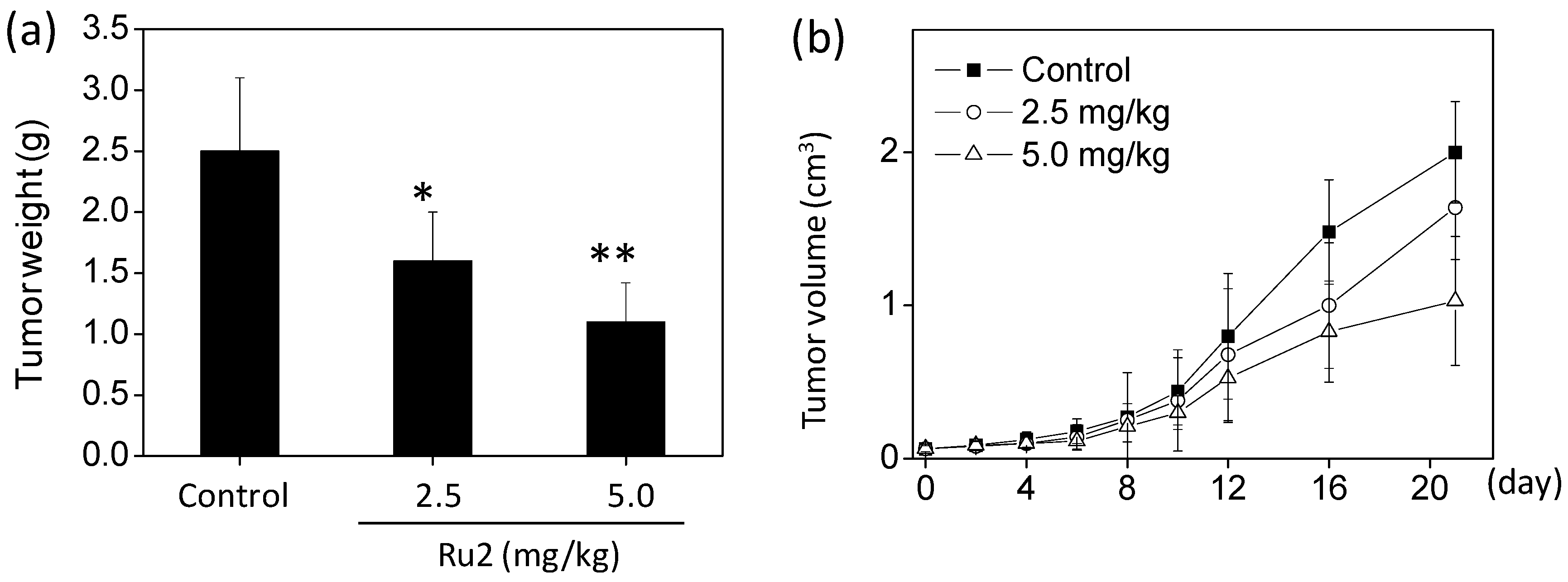
2.4. In Vivo Anticancer Activities of Ru2
3. Materials and Methods
3.1. Reagents, Cell Lines, and Cell Cultures
3.2. Cell Viability Assessment via MTT Assay
3.3. Cell Migration and Invasion
3.4. Flow Cytometric Analysis
3.5. Apoptotic DNA Fragmentation Assessment via TUNEL Staining Assays
3.6. Caspase Activation by Ru Complexes
3.7. Western Blotting
3.8. ROS Generation Assessment
3.9. Immunofluorescence Analysis of Protein Expression in Cells
3.10. Tumor Xenograft in Nude Mice
3.11. Statistical Analysis
4. Conclusions
Author Contributions
Conflicts of Interest
References
- Rosenberg, B.; van Camp, L.; Krigas, T. Inhibition of cell division in Escherichia coli by electrolysis products from a platinum electrode. Nature 1965, 205, 698–699. [Google Scholar] [CrossRef] [PubMed]
- Kelland, L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Markman, M. Toxicities of the platinum antineoplastic agents. Expert Opin. Drug Saf. 2003, 2, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, X.; Guo, Z. Functionalization of platinum complexes for biomedical applications. Acc. Chem. Res. 2015, 48, 2622–2631. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Wong, Y.S.; Zheng, W.J.; Liu, J. Caspase- and p53-dependent apoptosis in breast carcinoma cells induced by a synthetic selenadiazole derivative. Chem. Biol. Interact. 2009, 180, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Luo, Z.; Zhao, Z.; Xie, L.; Zheng, W.; Chen, T. Cellular localization of iron(II) polypyridyl complexes determines their anticancer action mechanisms. Biomaterials 2015, 71, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Sadler, P.J. Organoiridium complexes: Anticancer agents and catalysts. Acc. Chem. Res. 2014, 47, 1174–1185. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Luo, Z.; Wu, Q.; Zheng, W.; Feng, Y.; Chen, T. Mixed-ligand ruthenium polypyridyl complexes as apoptosis inducers in cancer cells, the cellular translocation and the important role of ROS-mediated signaling. Dalton Trans. 2014, 43, 17017–17028. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Yu, L.; Yang, F.; Zhao, Z.; Yu, B.; Lai, H.; Wong, K.H.; Ngai, S.M.; Zheng, W.; Chen, T. Ruthenium polypyridyl complexes as inducer of ROS-mediated apoptosis in cancer cells by targeting thioredoxin reductase. Metallomics 2014, 6, 1480–1490. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Liu, Y.; Zheng, W.-J.; Liu, J.; Wong, Y.-S. Ruthenium polypyridyl complexes that induce mitochondria-mediated apoptosis in cancer cells. Inorg. Chem. 2010, 49, 6366–6368. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Zheng, W.; Chen, T. Ruthenium polypyridyl complex inhibits growth and metastasis of breast cancer cells by suppressing FAK signaling with enhancement of TRAIL-induced apoptosis. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Marquardt, J.U.; Andersen, J.B.; Thorgeirsson, S.S. Functional and genetic deconstruction of the cellular origin in liver cancer. Nat. Rev. Cancer 2015, 15, 653–667. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.F.; Qin, Q.P.; Qin, J.L.; Zhou, J.; Li, Y.L.; Li, N.; Liu, Y.C.; Liang, H. Water-soluble ruthenium(II) complexes with chiral 4-(2,3-dihydroxypropyl)-formamide oxoaporphine (FOA): In vitro and in vivo anticancer activity by stabilization of G-quadruplex DNA, inhibition of telomerase activity, and induction of tumor cell apoptosis. J. Med. Chem. 2015, 58, 4771–4789. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Lei, Z.; Wang, X.; Zhu, F.; Chen, D. Ruthenium complex ΛWH0402 induces hepatocellular carcinoma LM6 (HCCLM6) cell death by triggering the beclin-1-dependent autophagy pathway. Metallomics 2015, 7, 896–907. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.H.; Wu, D.H.; Liu, X.; Guoyiqibayi, G.; Guo, D.D.; Lv, G.; Wang, X.M.; Yan, H.; Jiang, H.; Lu, Z.H. Ligand-based neutral ruthenium(II) arene complex: Selective anticancer action. Inorg. Chem. 2009, 48, 2352–2354. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Zhang, P.; Yu, B.; Jin, C.; Ji, L.; Chao, H. Synthesis, characterization and biological evaluation of mixed-ligand ruthenium(II) complexes for photodynamic therapy. Dalton Trans. 2015, 44, 17335–17345. [Google Scholar] [CrossRef] [PubMed]
- Maiese, K.; Chong, Z.Z.; Shang, Y.C.; Wang, S. Targeting disease through novel pathways of apoptosis and autophagy. Expert Opin. Ther. Targets 2012, 16, 1203–1214. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Wong, Y.S. Selenocystine induces apoptosis of a375 human melanoma cells by activating ROS-mediated mitochondrial pathway and p53 phosphorylation. Cell. Mol. Life Sci. 2008, 65, 2763–2775. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Wong, Y.S. Selenocystine induces caspase-independent apoptosis in MCF-7 human breast carcinoma cells with involvement of p53 phosphorylation and reactive oxygen species generation. Int. J. Biochem. Cell Biol. 2009, 41, 666–676. [Google Scholar] [CrossRef] [PubMed]
- Riedl, S.J.; Shi, Y. Molecular mechanisms of caspase regulation during apoptosis. Nat. Rev. 2004, 5, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Simon, H.-U.; Haj-Yehia, A.; Levi-Schaffer, F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis 2000, 5, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Indran, I.R.; Hande, M.P.; Pervaiz, S. Htert overexpression alleviates intracellular ROS production, improves mitochondrial function, and inhibits ROS-mediated apoptosis in cancer cells. Cancer Res. 2011, 71, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Son, Y.J.; Leong, K.W.; Yoo, H.S. Therapeutic nanorods with metallic multi-segments: Thermally inducible encapsulation of doxorubicin for anti-cancer therapy. Nano. Today 2012, 7, 76–84. [Google Scholar] [CrossRef]
- Piconi, L.; Quagliaro, L.; Assaloni, R.; Da Ros, R.; Maier, A.; Zuodar, G.; Ceriello, A. Constant and intermittent high glucose enhances endothelial cell apoptosis through mitochondrial superoxide overproduction. Diabetes Metab. Res. Rev. 2006, 22, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.H.; Iskandar, K.B.; Yadav, S.K.; Hirpara, J.L.; Loh, T.; Pervaiz, S. Simultaneous induction of non-canonical autophagy and apoptosis in cancer cells by ROS-dependent ERK and JNK activation. PLoS ONE 2010, 5, e9996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arai, M.; Shibata, Y.; Pugdee, K.; Abiko, Y.; Ogata, Y. Effects of reactive oxygen species (ROS) on antioxidant system and osteoblastic differentiation in MC3T3-E1 cells. IUBMB Life 2007, 59, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.T.; Lee, S.H.; Li, W.; Sun, Y.N.; Yang, S.Y.; Jang, H.D.; Kim, Y.H. Evaluation of the antioxidant and anti-osteoporosis activities of chemical constituents of the fruits of prunus mume. Food Chem. 2014, 156, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.K.; Yang, L.; Meng, G.L.; Fan, J.; Chen, J.Z.; He, Q.Z.; Chen, S.; Fan, J.Z.; Luo, Z.J.; Liu, J. Protective effect of tetrahydroxystilbene glucoside against hydrogen peroxide-induced dysfunction and oxidative stress in osteoblastic MC3T3-E1 cells. Eur. J. Pharmacol. 2012, 689, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.M.; Kim, G.H.; Lee, Y.S. Protective effects of dehydrocostus lactone against hydrogen peroxide-induced dysfunction and oxidative stress in osteoblastic MC3T3-E1 cells. Toxicol. In Vitro 2009, 23, 862–867. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, W.L.; Xie, W.L.; Li, L.Z.; Sun, J.; Sun, W.J.; Gong, H.Y. Puerarin stimulates proliferation and differentiation and protects against cell death in human osteoblastic MG-63 cells via ER-dependent MEK/ERK and PI3K/Akt activation. Phytomedicine 2013, 20, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Kansanen, E.; Kuosmanen, S.M.; Leinonen, H.; Levonen, A.L. The keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol. 2013, 1, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; He, L.; Liu, W.; Fan, C.; Zheng, W.; Wong, Y.-S.; Chen, T. Selective cellular uptake and induction of apoptosis of cancer-targeted selenium nanoparticles. Biomaterials 2013, 34, 7106–7116. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Tang, Q.; Zhong, X.; Bai, Y.; Chen, T.; Zhang, Y.; Li, Y.; Zheng, W. Surface decoration by spirulina polysaccharide enhances the cellular uptake and anticancer efficacy of selenium nanoparticles. Int. J. Nanomed. 2012, 7, 835–844. [Google Scholar]
- Yong, K.-T.; Wang, Y.; Roy, I.; Rui, H.; Swihart, M.T.; Law, W.-C.; Kwak, S.K.; Ye, L.; Liu, J.; Mahajan, S.D. Preparation of quantum dot/drug nanoparticle formulations for traceable targeted delivery and therapy. Theranostics 2012, 2, 681–694. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhang, D.; Zhang, Q.; Chen, Y.; Zheng, D.; Hao, L.; Duan, C.; Jia, L.; Liu, G.; Liu, Y. Synergistic effect of folate-mediated targeting and verapamil-mediated P-gp inhibition with paclitaxel-polymer micelles to overcome multi-drug resistance. Biomaterials 2011, 32, 9444–9456. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, X.; Huang, Z.; Zheng, W.; Fan, C.; Chen, T. Enhancement of cell permeabilization apoptosis-inducing activity of selenium nanoparticles by atp surface decoration. Nanomedicine 2013, 9, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.; Chen, J.; Wang, Y.; Wong, Y.S.; Zhang, Y.; Zheng, W.; Cao, W.; Chen, T. Selenocystine potentiates cancer cell apoptosis induced by 5-fluorouracil by triggering reactive oxygen species-mediated DNA damage and inactivation of the erk pathway. Free Radic. Biol. Med. 2013, 65, 305–316. [Google Scholar] [CrossRef] [PubMed]





© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, Y.; Shen, T.; Yang, H.; Gu, W. Ruthenium Complexes Induce HepG2 Human Hepatocellular Carcinoma Cell Apoptosis and Inhibit Cell Migration and Invasion through Regulation of the Nrf2 Pathway. Int. J. Mol. Sci. 2016, 17, 775. https://doi.org/10.3390/ijms17050775
Lu Y, Shen T, Yang H, Gu W. Ruthenium Complexes Induce HepG2 Human Hepatocellular Carcinoma Cell Apoptosis and Inhibit Cell Migration and Invasion through Regulation of the Nrf2 Pathway. International Journal of Molecular Sciences. 2016; 17(5):775. https://doi.org/10.3390/ijms17050775
Chicago/Turabian StyleLu, Yiyu, Ting Shen, Hua Yang, and Weiguang Gu. 2016. "Ruthenium Complexes Induce HepG2 Human Hepatocellular Carcinoma Cell Apoptosis and Inhibit Cell Migration and Invasion through Regulation of the Nrf2 Pathway" International Journal of Molecular Sciences 17, no. 5: 775. https://doi.org/10.3390/ijms17050775
APA StyleLu, Y., Shen, T., Yang, H., & Gu, W. (2016). Ruthenium Complexes Induce HepG2 Human Hepatocellular Carcinoma Cell Apoptosis and Inhibit Cell Migration and Invasion through Regulation of the Nrf2 Pathway. International Journal of Molecular Sciences, 17(5), 775. https://doi.org/10.3390/ijms17050775