
In Vitro Model for Hepatotoxicity Studies Based on Primary Human Hepatocyte Cultivation in a Perfused 3D Bioreactor System

 ,
,
Abstract
:
1. Introduction
2. Results
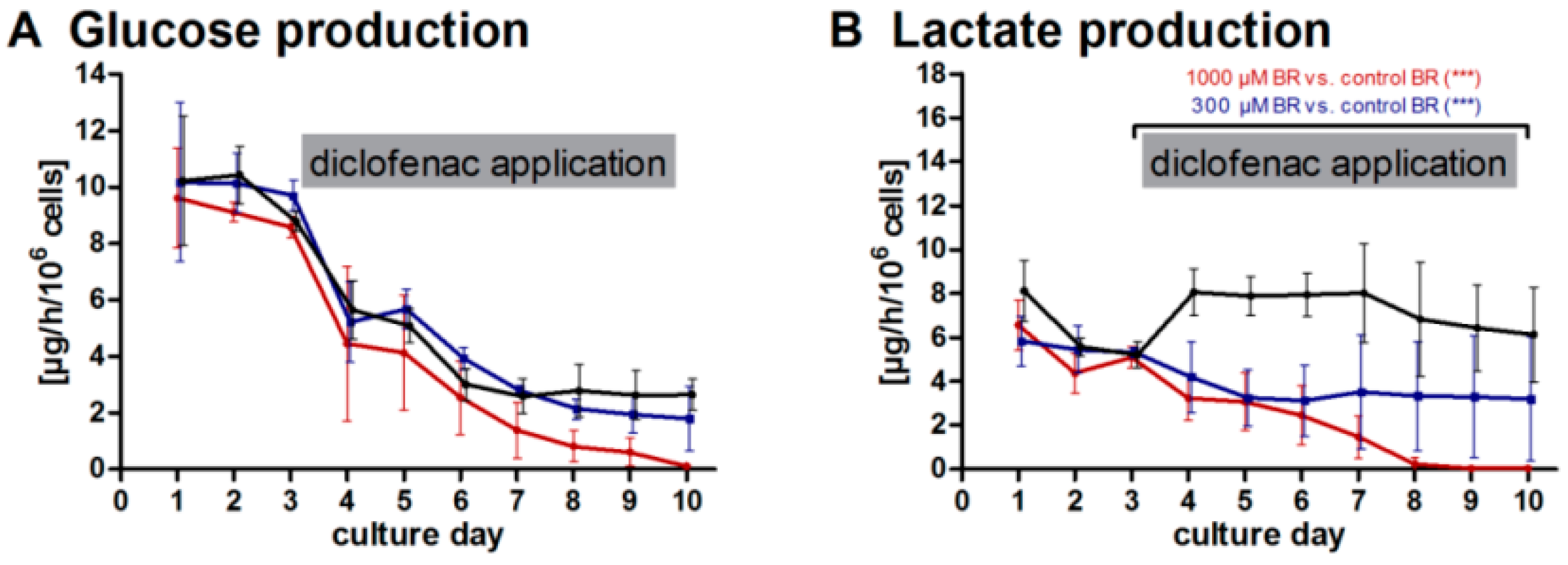
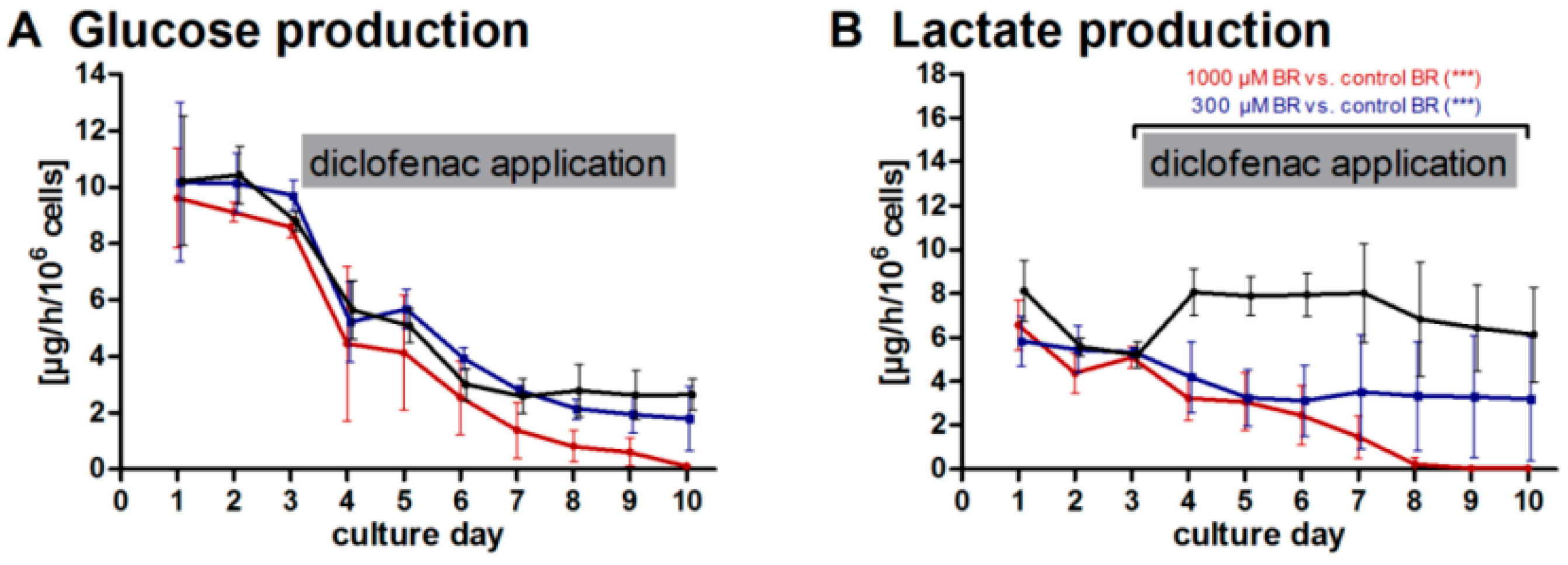
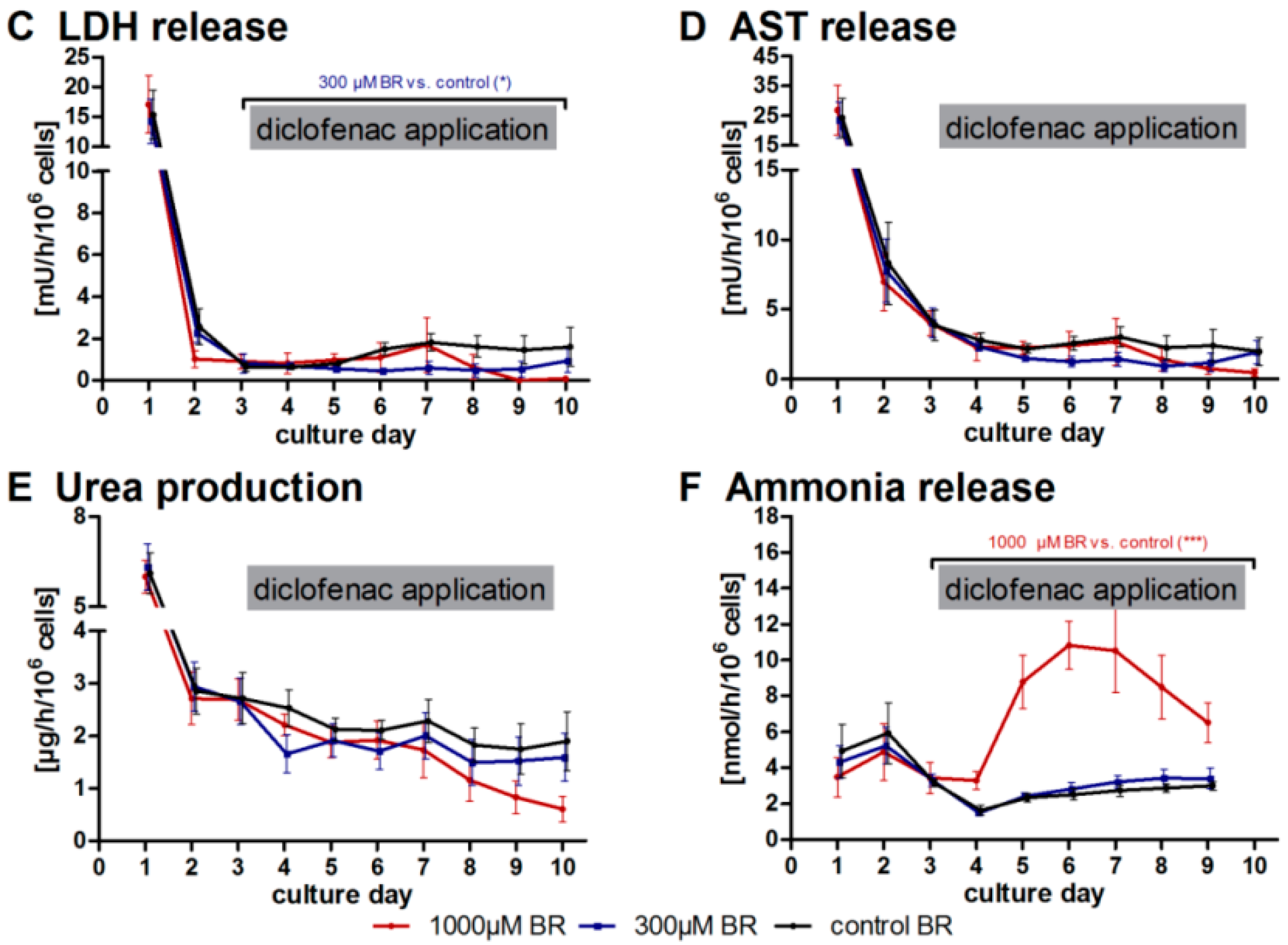
2.1. General Metabolic Activity
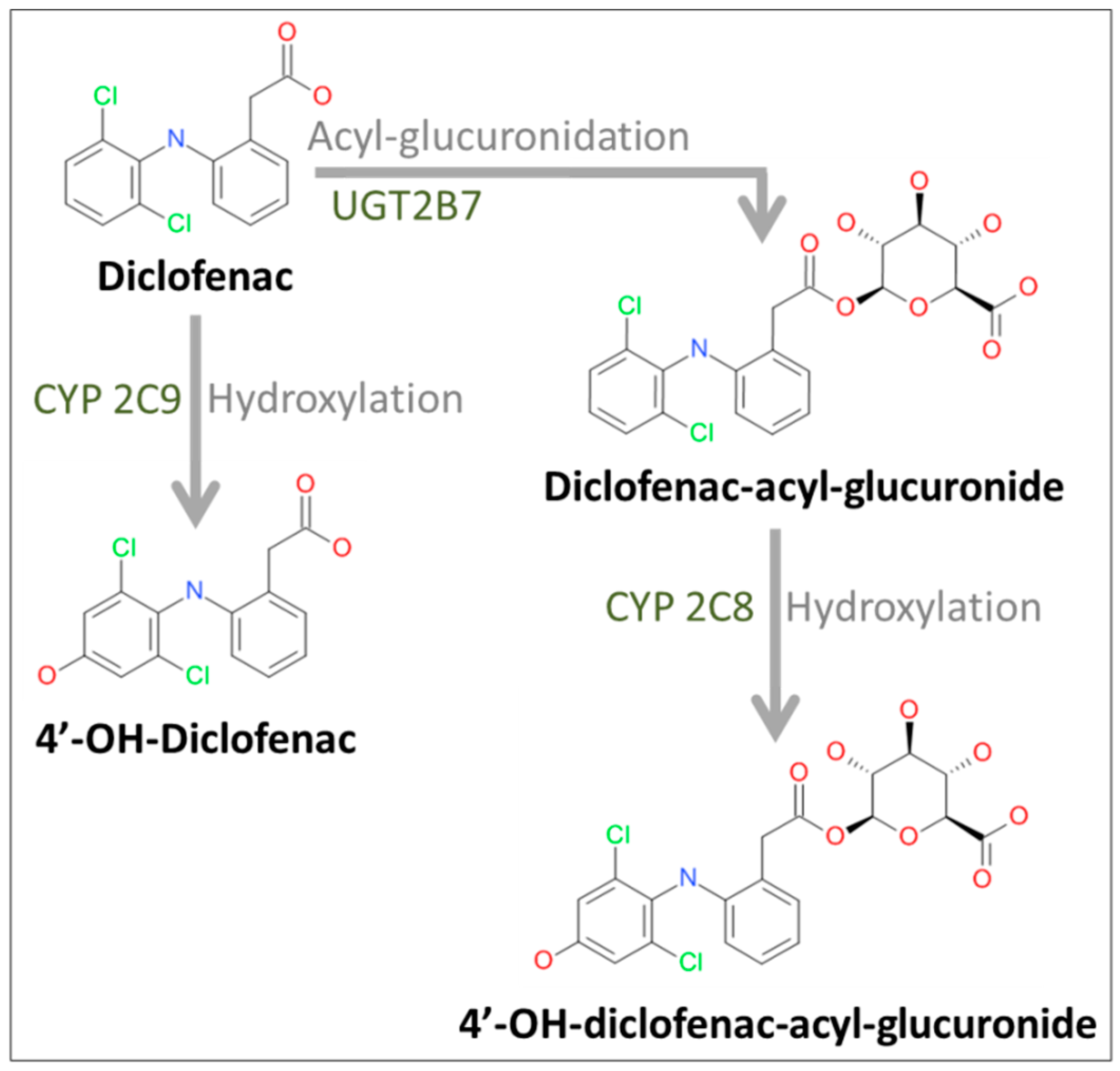
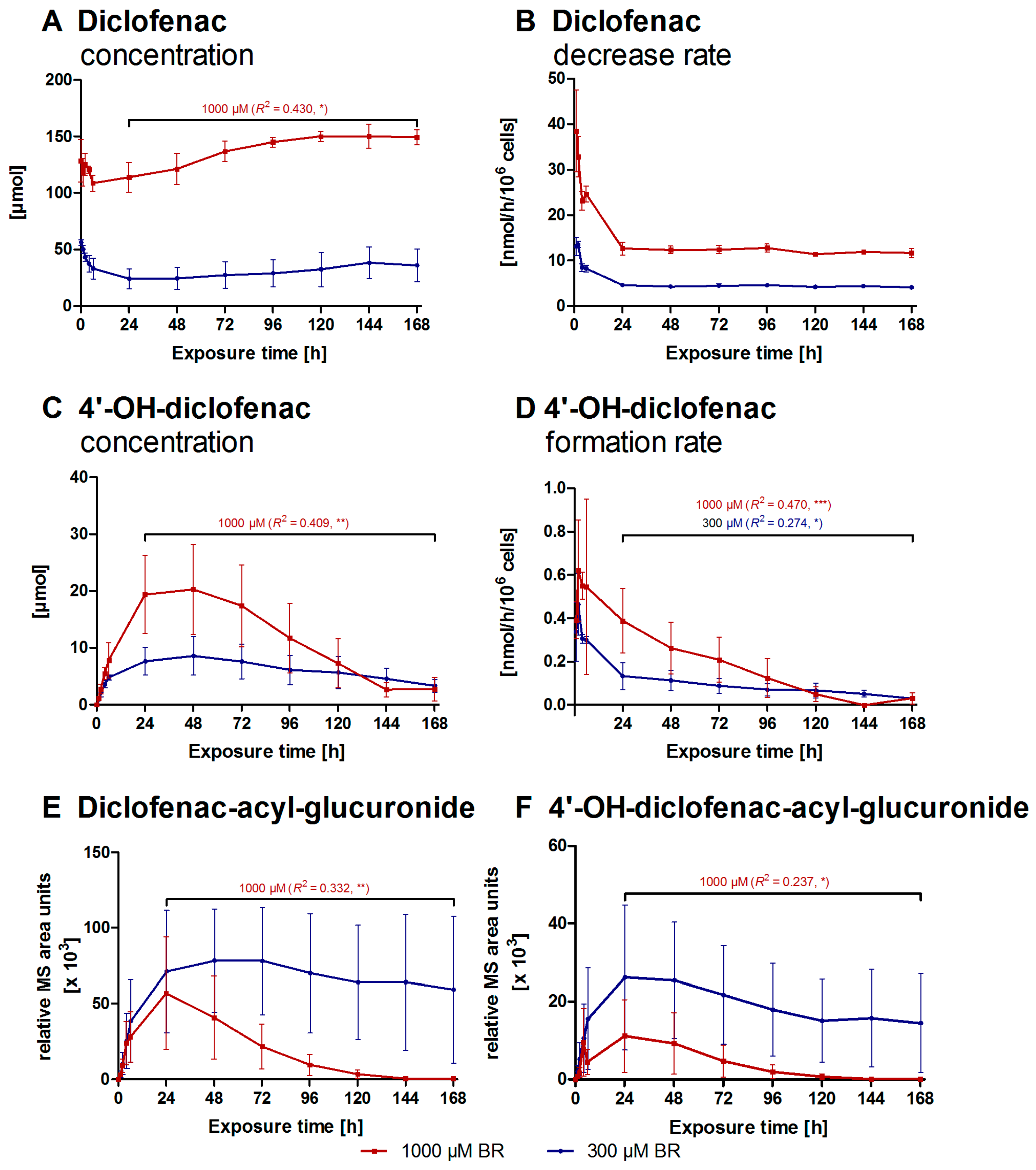
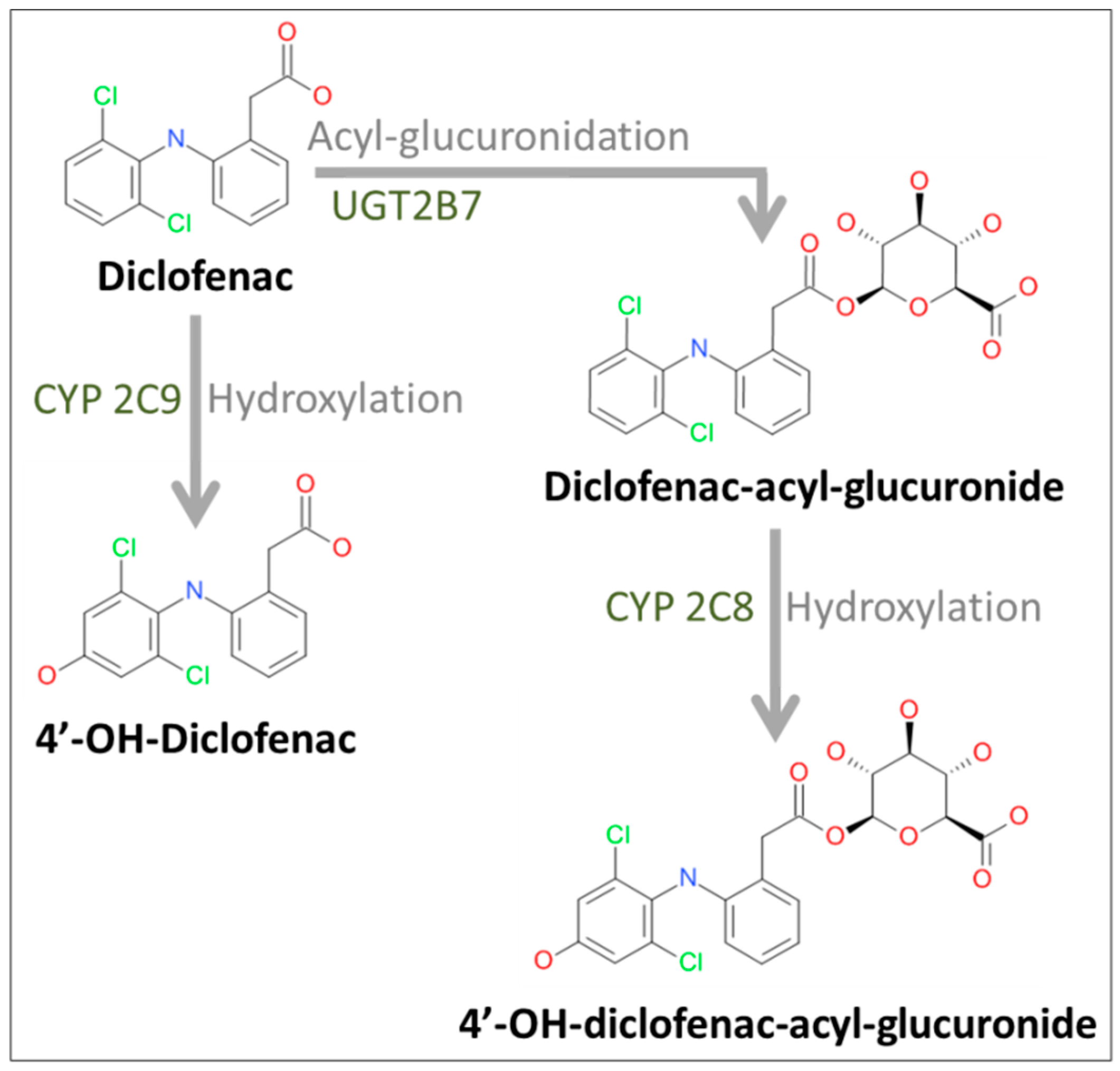
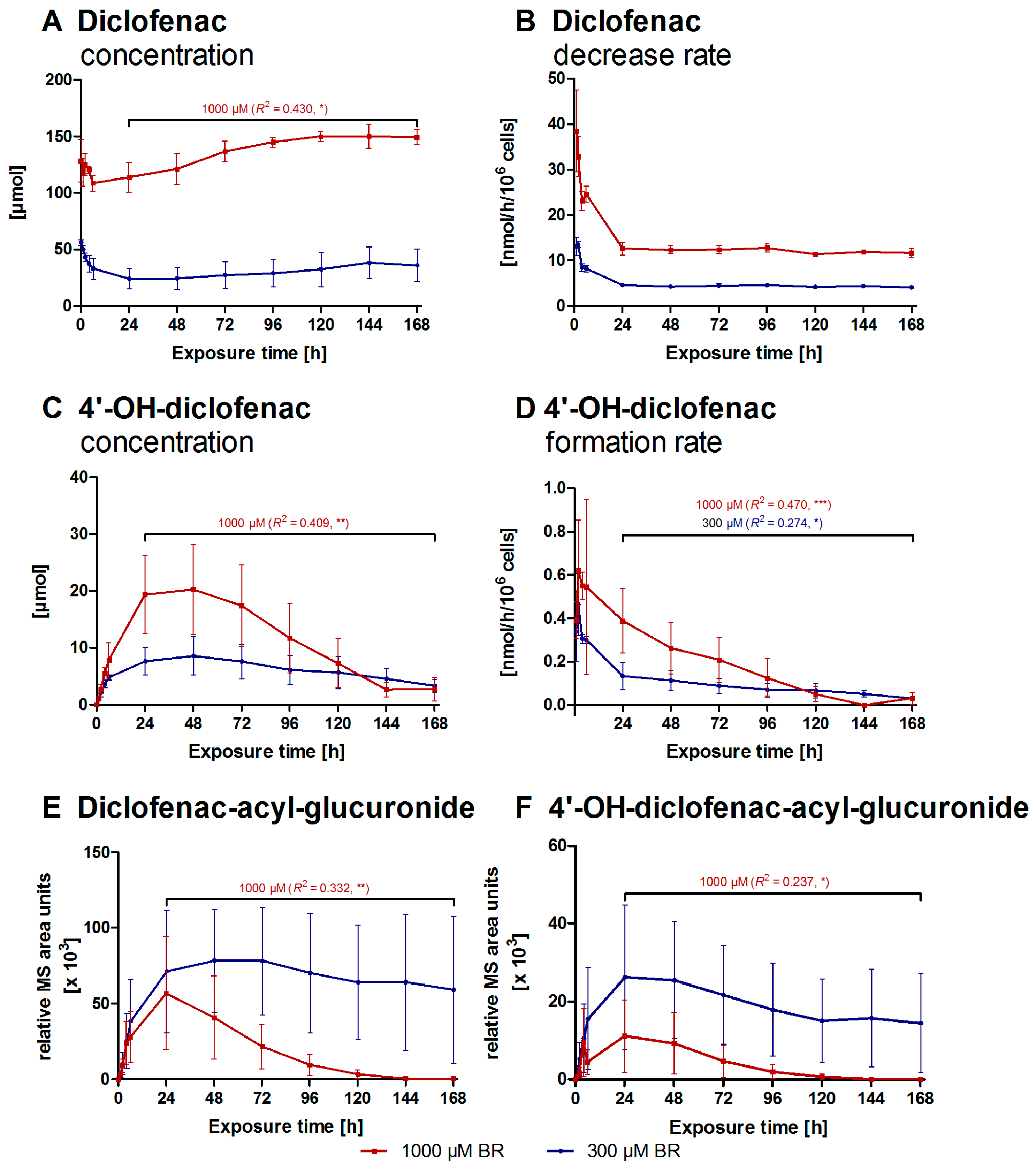
2.2. Formation of Diclofenac Metabolites
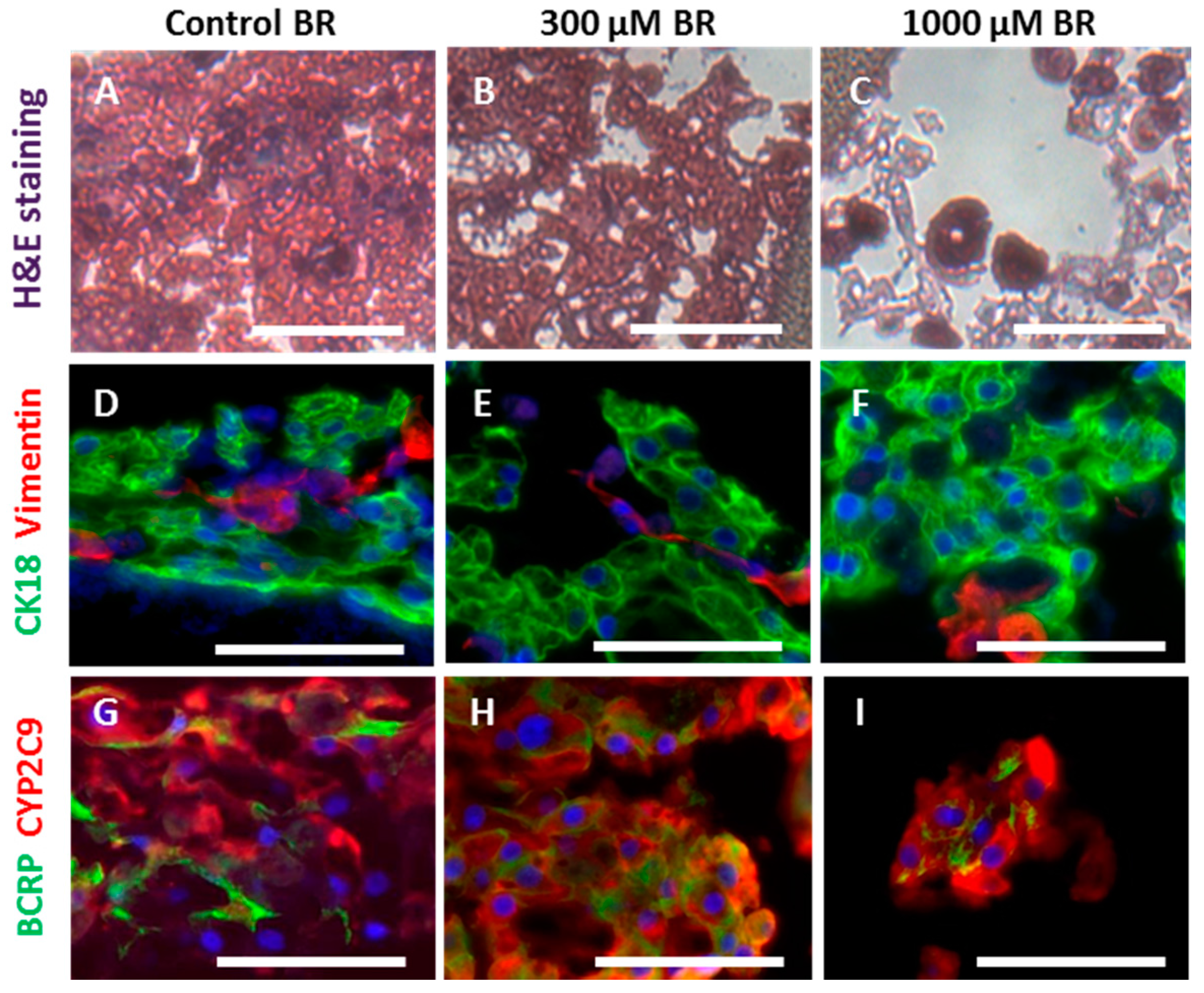
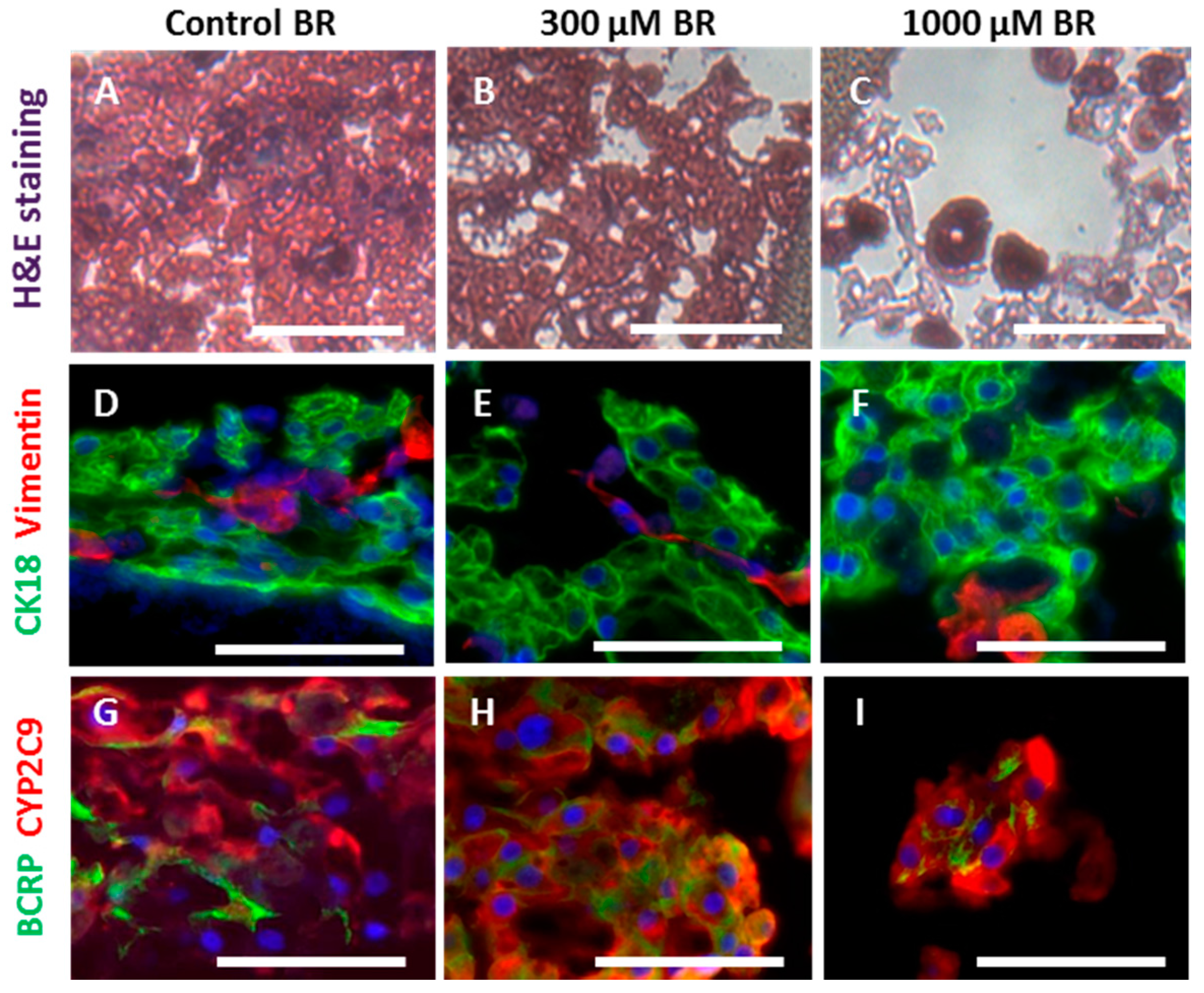
2.3. Cell Morphology and Reorganization in the Bioreactor
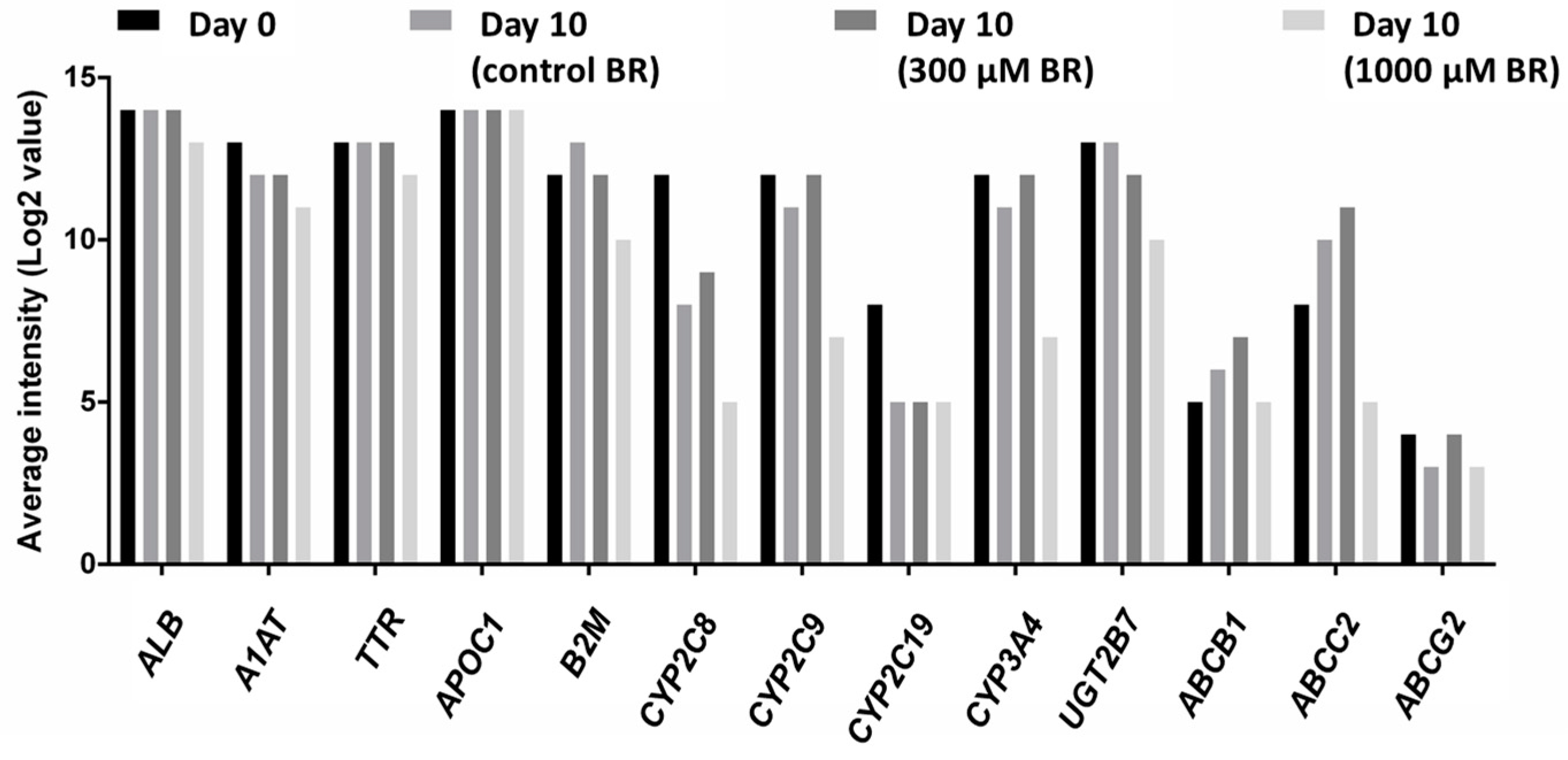
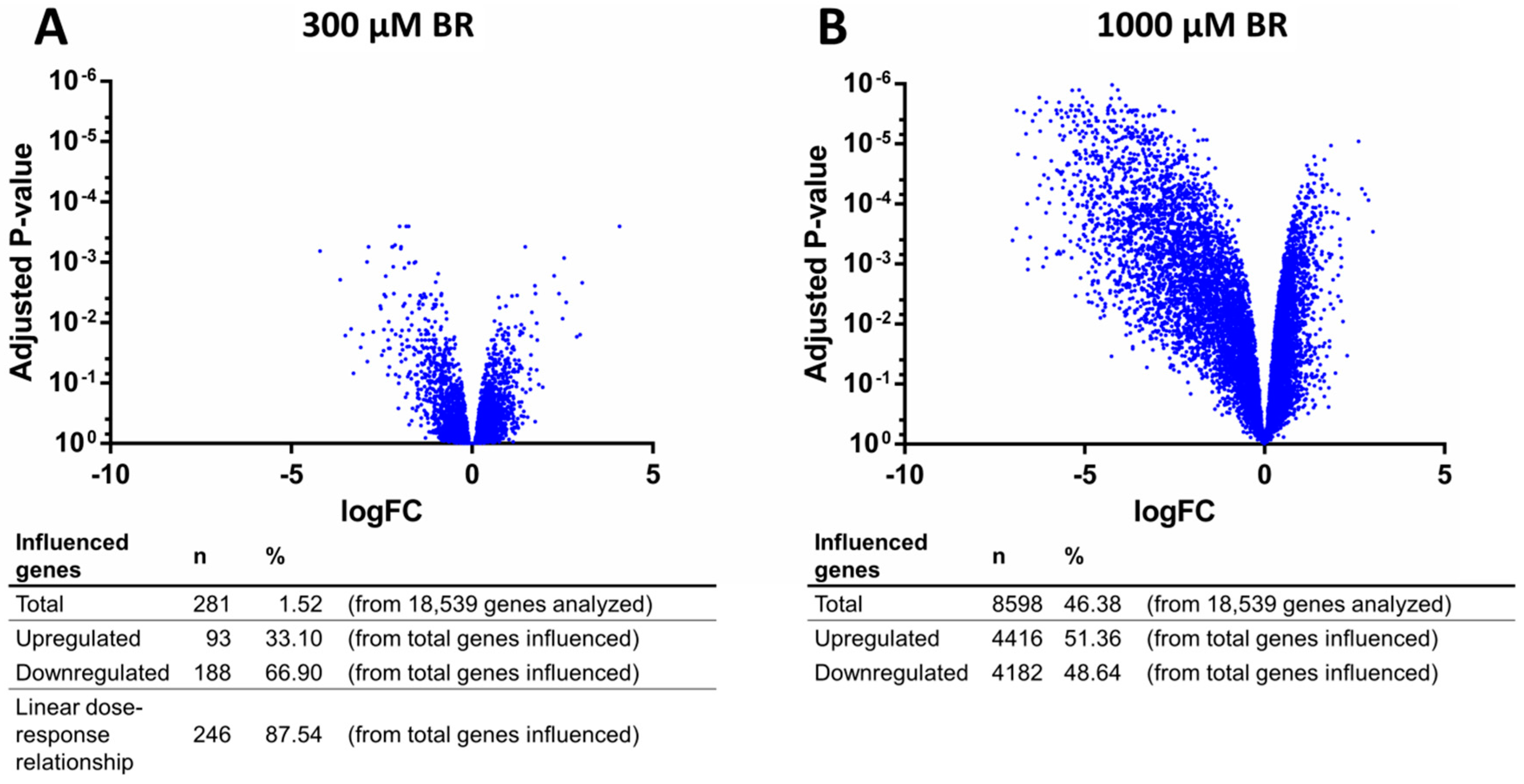
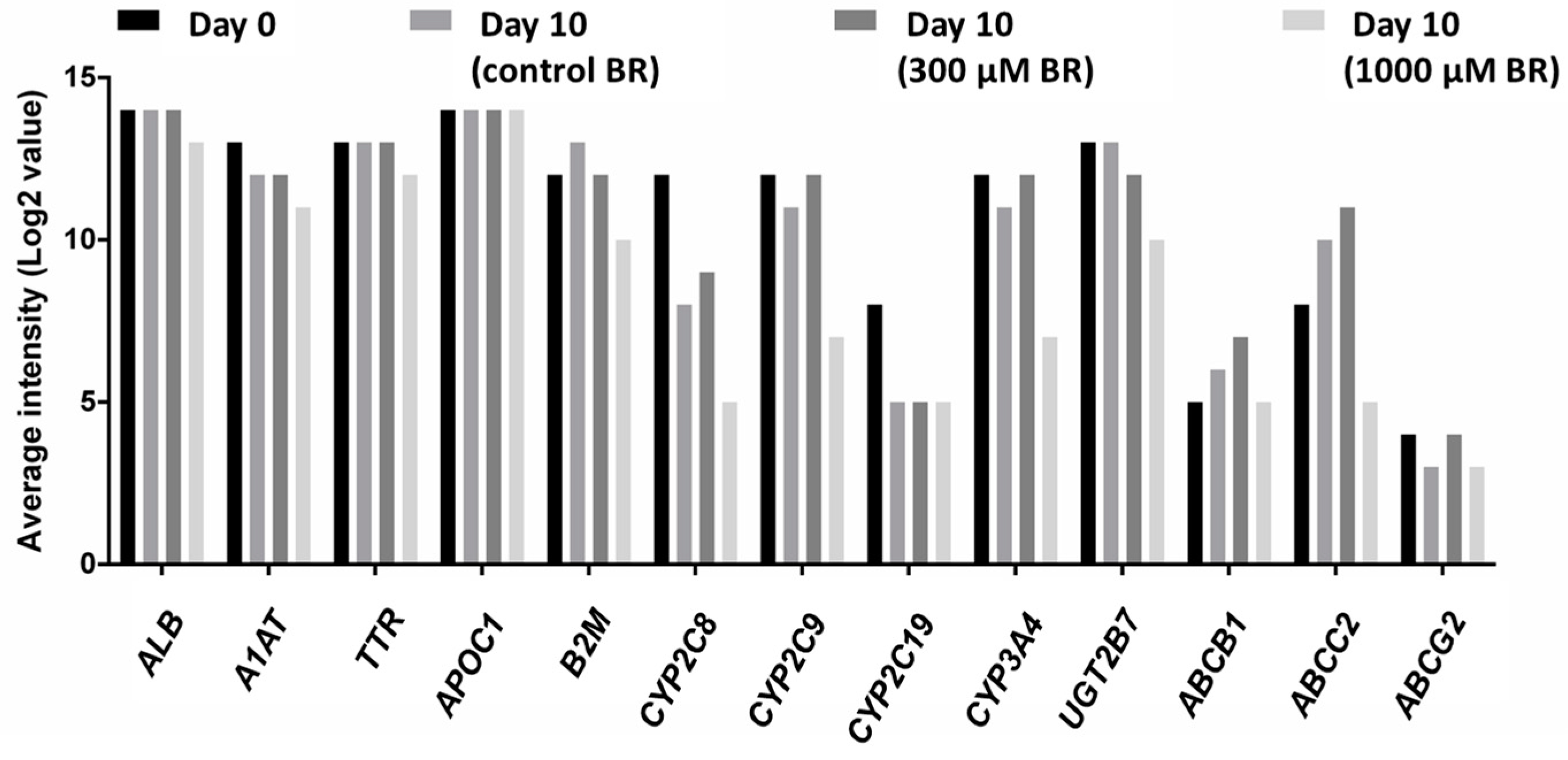
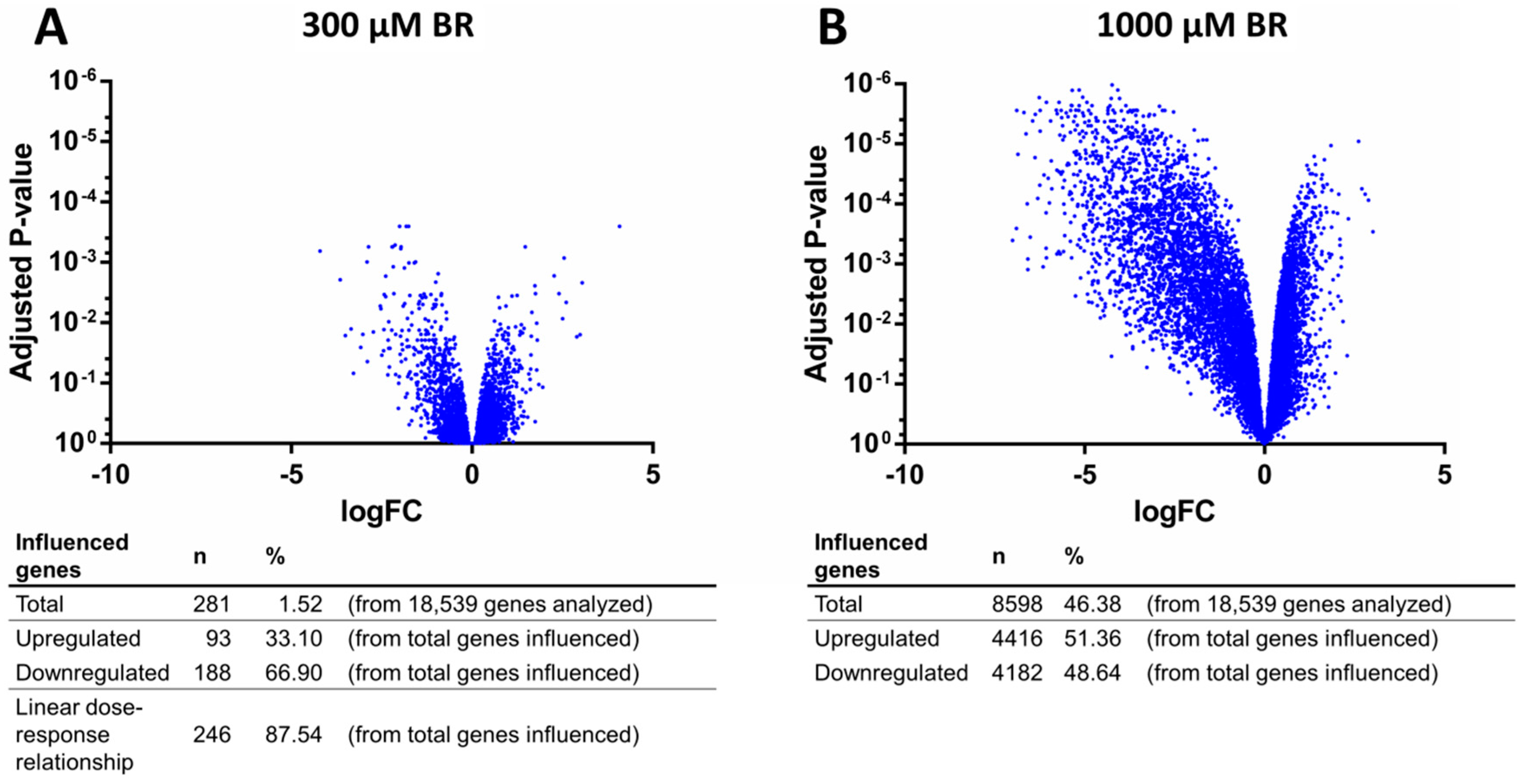
2.4. Analysis of Transcriptomic Data upon Drug Application
3. Discussion
4. Materials and Methods
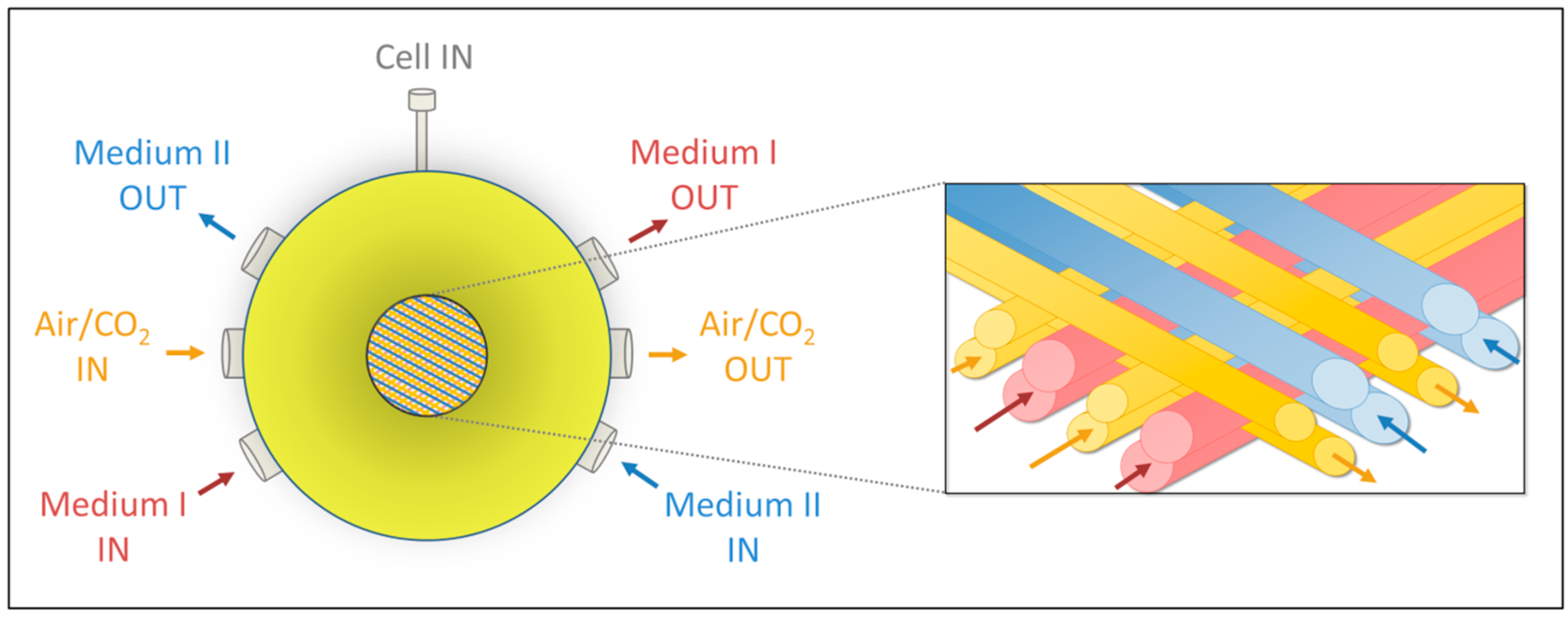
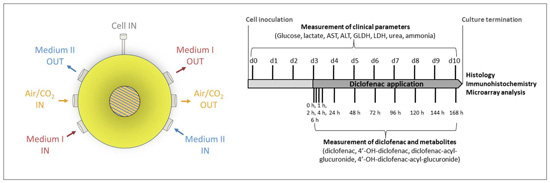
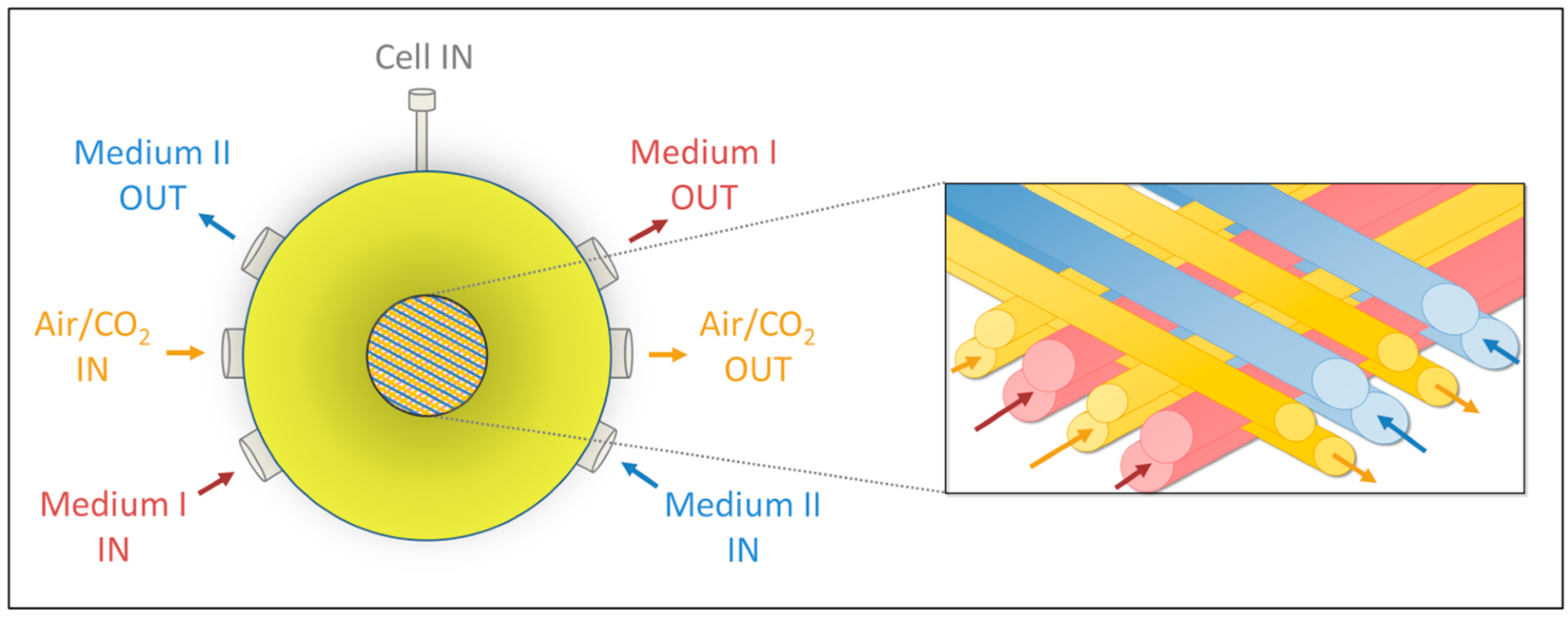
4.1. Bioreactor Technology
4.2. Isolation of Primary Human Liver Cells
4.3. 3D Culture of Primary Human Liver Cells
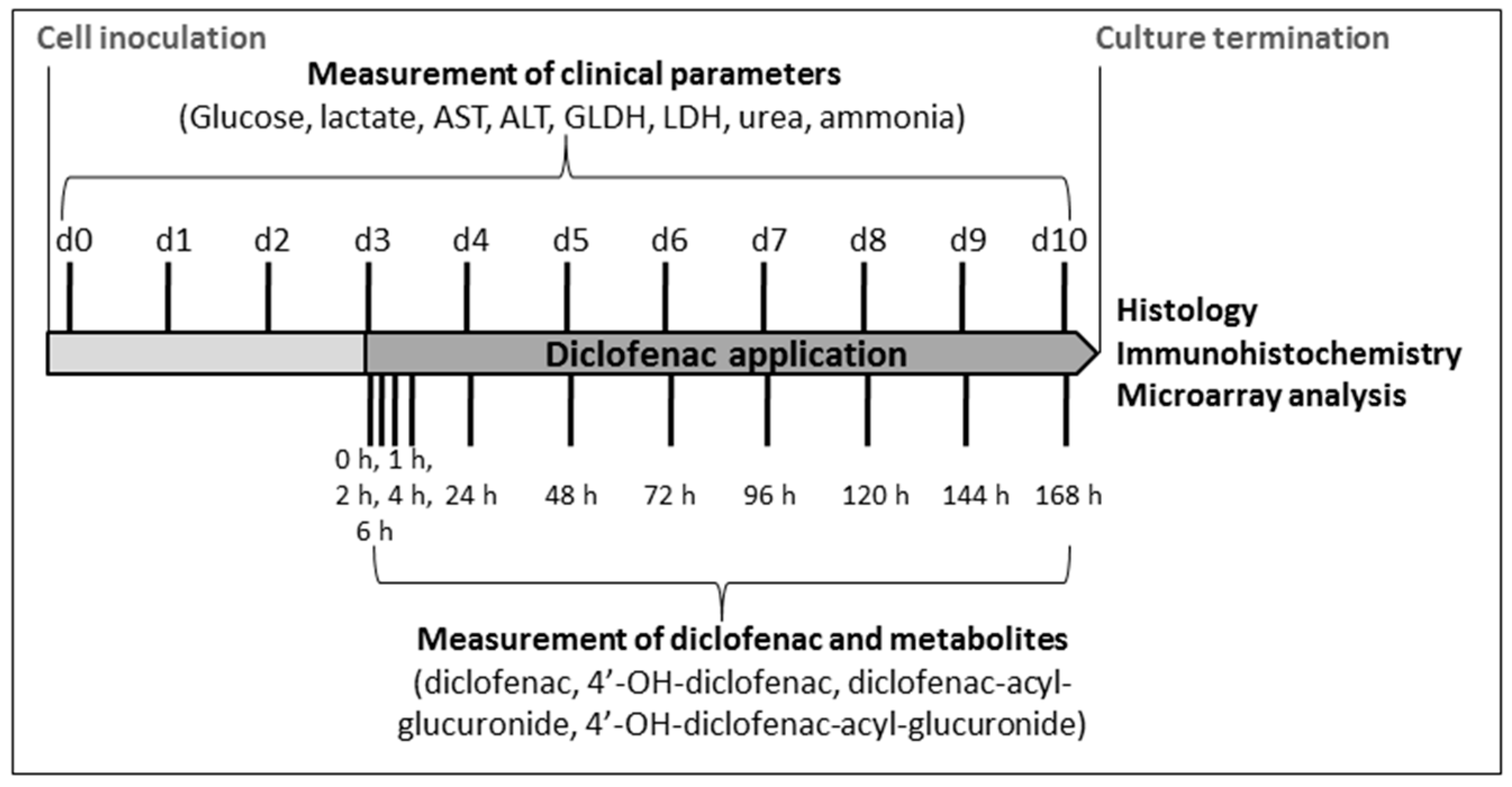
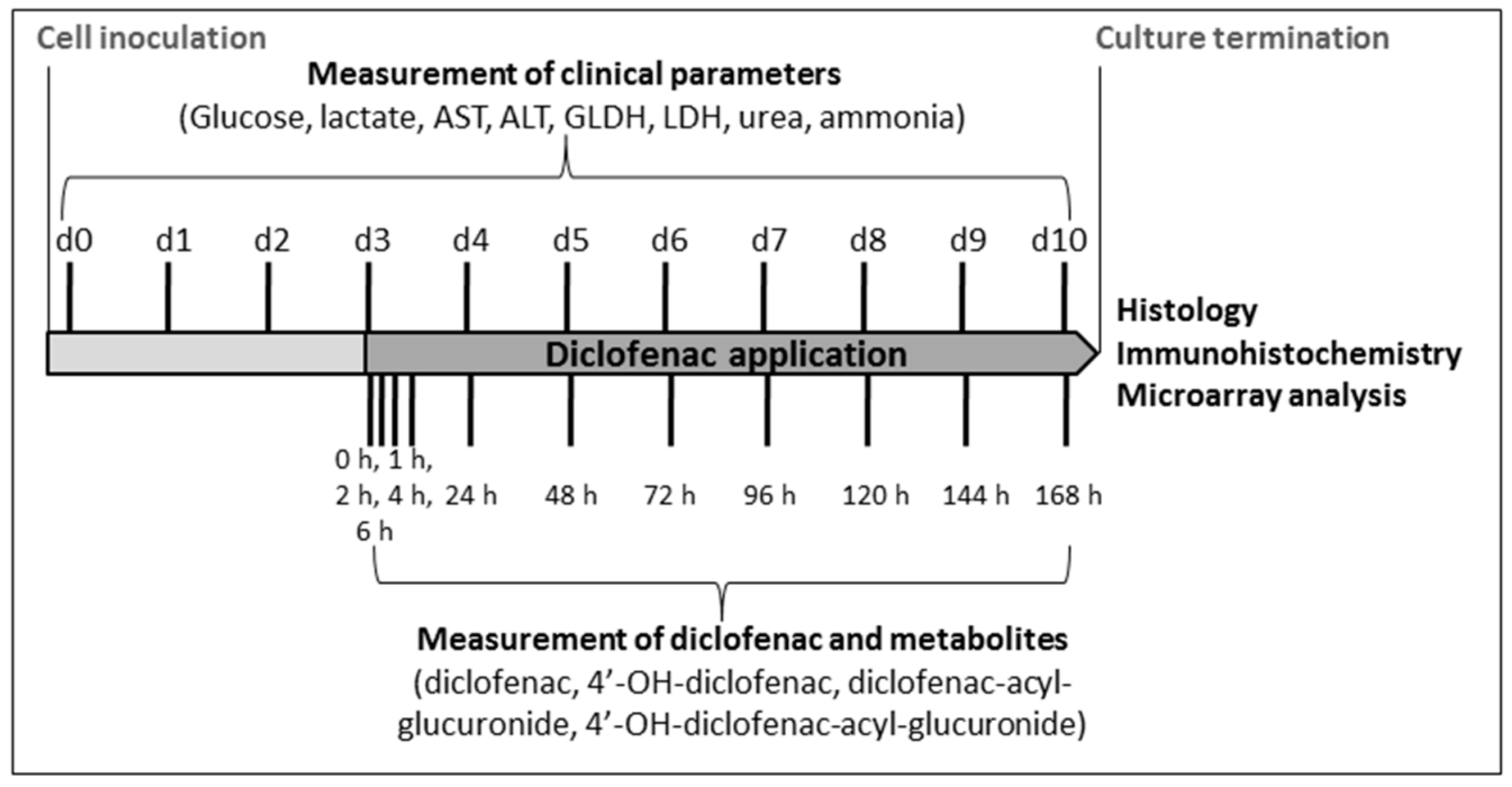
4.4. Analysis of Clinical Parameters
4.5. Analysis of Diclofenac and Its Metabolites
4.6. Histology and Immunohistochemistry
4.7. Microarray Analysis
4.8. Statistics
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| 2D | two-dimensional |
| 3D | three-dimensional |
| 300 µM BR | 300 µM bioreactors |
| 1000 µM BR | 1000 µM bioreactors |
| A1AT | α-1-antitrypsin |
| ABCB1 | ATP-binding cassette B1 |
| ABCC2 | ATP-binding cassette C2 |
| ABCG2 | ATP-binding cassette G2 |
| ALB | albumin |
| ALT | alanine transaminase |
| ARG1 | arginase 1 |
| APOC1 | apolipoprotein C-I |
| ASL | argininosuccinate lyase |
| AST | aspartate transaminase |
| ATF3 | activating transcription factor 3 |
| B2M | β -2-microglobulin |
| BCRP | breast cancer resistance protein |
| CK 18 | cytokeratin 18 |
| control BR | control bioreactors |
| CPS1 | mitochondrial carbamoyl-phosphate synthase 1 |
| CYP | cytochrome P450 |
| DILI | drug-induced liver injury |
| DMSO | dimethyl sulfoxide |
| FDR | False Discovery Rate |
| GLDH | glutamate dehydrogenase |
| GDF15 | growth differentiation factor 15 |
| LDH | lactate dehydrogenase |
| MS | mass spectrometry |
| NRF2/KEAP1 | nuclear factor (erythroid-derived 2)-like 2/kelch-like ECH-associated protein 1 |
| OTC | ornithine transcarbamoylase |
| pHH | primary human hepatocytes |
| SEM | standard errors of the mean |
| TTR | transthyretin |
| UDP | uridine diphosphate |
| UGT2B7 | UDP glucuronosyltransferase 2B7 |
References
- Hay, M.; Thomas, D.W.; Craighead, J.L.; Economides, C.; Rosenthal, J. Clinical development success rates for investigational drugs. Nat. Biotechnol. 2014, 32, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Li, A.P. Accurate prediction of human drug toxicity: A major challenge in drug development. Chem. Biol. Interact. 2004, 150, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Iwahori, T.; Matsuura, T.; Maehashi, H.; Sugo, K.; Saito, M.; Hosokawa, M.; Chiba, K.; Masaki, T.; Aizaki, H.; Ohkawa, K.; et al. CYP3A4 inducible model for in vitro analysis of human drug metabolism using a bioartificial liver. Hepatology 2003, 37, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Aninat, C.; Piton, A.; Glaise, D.; Le Charpentier, T.; Langouët, S.; Morel, F.; Guguen-Guillouzo, C.; Guillouzo, A. Expression of cytochromes P450, conjugating enzymes and nuclear receptors in human hepatoma HepaRG cells. Drug Metab. Dispos. 2006, 34, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Baxter, M.; Withey, S.; Harrison, S.; Segeritz, C.P.; Zhang, F.; Atkinson-Dell, R.; Rowe, C.; Gerrard, D.T.; Sison-Young, R.; Jenkins, R.; et al. Phenotypic and functional analyses show stem cell-derived hepatocyte-like cells better mimic fetal rather than adult hepatocytes. J. Hepatol. 2015, 62, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Li, A.P. Evaluation of adverse drug properties with cryopreserved human hepatocytes and the integrated discrete multiple organ co-culture (IdMOC(TM)) system. Toxicol. Res. 2015, 31, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Lechón, M.J.; Jover, R.; Donato, T.; Ponsoda, X.; Rodriguez, C.; Stenzel, K.G.; Klocke, R.; Paul, D.; Guillén, I.; Bort, R.; et al. Long-term expression of differentiated functions in hepatocytes cultured in three-dimensional collagen matrix. J. Cell. Physiol. 1998, 177, 553–562. [Google Scholar] [CrossRef]
- Rodríguez-Antona, C.; Donato, M.T.; Boobis, A.; Edwards, R.J.; Watts, P.S.; Castell, J.V.; Gómez-Lechón, M.J. Cytochrome P450 expression in human hepatocytes and hepatoma cell lines: Molecular mechanisms that determine lower expression in cultured cells. Xenobiotica 2002, 32, 505–520. [Google Scholar] [CrossRef] [PubMed]
- Schyschka, L.; Sánchez, J.J.; Wang, Z.; Burkhardt, B.; Müller-Vieira, U.; Zeilinger, K.; Bachmann, A.; Nadalin, S.; Damm, G.; Nussler, A.K. Hepatic 3D cultures but not 2D cultures preserve specific transporter activity for acetaminophen-induced hepatotoxicity. Arch. Toxicol. 2013, 87, 1581–1593. [Google Scholar] [CrossRef] [PubMed]
- Ohkura, T.; Ohta, K.; Nagao, T.; Kusumoto, K.; Koeda, A.; Ueda, T.; Jomura, T.; Ikeya, T.; Ozeki, E.; Wada, K.; et al. Evaluation of human hepatocytes cultured by three-dimensional spheroid systems for drug metabolism. Drug Metab. Pharmacokinet. 2014, 29, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Khetani, S.R.; Bhatia, S.N. Microscale culture of human liver cells for drug development. Nat. Biotechnol. 2008, 26, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, J.C.; Encke, J.; Hole, O.; Müller, C.; Ryan, C.J.; Neuhaus, P. Bioreactor for a larger scale hepatocyte in vitro perfusion. Transplantation 1994, 58, 984–988. [Google Scholar] [CrossRef]
- Zeilinger, K.; Schreiter, T.; Darnell, M.; Söderdahl, T.; Lübberstedt, M.; Dillner, B.; Knobeloch, D.; Nüssler, A.K.; Gerlach, J.C.; Andersson, T.B. Scaling down of a clinical three-dimensional perfusion multicompartment hollow fiber liver bioreactor developed for extracorporeal liver support to an analytical scale device useful for hepatic pharmacological in vitro studies. Tissue Eng. C Methods 2011, 17, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, S.A.; Müller-Vieira, U.; Biemel, K.; Knobeloch, D.; Heydel, S.; Lübberstedt, M.; Nüssler, A.K.; Andersson, T.B.; Gerlach, J.C.; Zeilinger, K. Analysis of drug metabolism activities in a miniaturized liver cell bioreactor for use in pharmacological studies. Biotechnol. Bioeng. 2012, 109, 3172–3181. [Google Scholar] [CrossRef] [PubMed]
- Lübberstedt, M.; Müller-Vieira, U.; Biemel, K.M.; Darnell, M.; Hoffmann, S.A.; Knöspel, F.; Wönne, E.C.; Knobeloch, D.; Nüssler, A.K.; Gerlach, J.C.; et al. Serum-free culture of primary human hepatocytes in a miniaturized hollow-fibre membrane bioreactor for pharmacological in vitro studies. J. Tissue Eng. Regen. Med. 2015, 9, 1017–1026. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Samuel, K.; Subramanian, R.; Braun, M.P.; Stearns, R.A.; Chiu, S.H.; Evans, D.C.; Baillie, T.A. Extrapolation of diclofenac clearance from in vitro microsomal metabolism data: Role of acyl glucuronidation and sequential oxidative metabolism of the acyl glucuronide. J. Pharmacol. Exp. Ther. 2002, 303, 969–978. [Google Scholar] [CrossRef] [PubMed]
- Boelsterli, U.A. Diclofenac-induced liver injury: A paradigm of idiosyncratic drug toxicity. Toxicol. Appl. Pharmacol. 2003, 192, 307–322. [Google Scholar] [CrossRef]
- Chalasani, N.; Björnsson, E. Risk factors for idiosyncratic drug-induced liver injury. Gastroenterology 2010, 138, 2246–2259. [Google Scholar] [CrossRef] [PubMed]
- Banks, A.T.; Zimmerman, H.J.; Ishak, K.G.; Harter, J.G. Diclofenac-associated hepatotoxicity: Analysis of 180 cases reported to the Food and Drug Administration as adverse reactions. Hepatology 1995, 22, 820–827. [Google Scholar] [CrossRef] [PubMed]
- Aithal, G.P. Diclofenac-induced liver injury: A paradigm of idiosyncratic drug toxicity. Expert Opin. Drug Saf. 2004, 3, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Syed, M.; Skonberg, C.; Hansen, S.H. Mitochondrial toxicity of diclofenac and its metabolites via inhibition of oxidative phosphorylation (ATP synthesis) in rat liver mitochondria: Possible role in drug induced liver injury (DILI). Toxicol. Vitro 2016, 31, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Masubuchi, N.; Makino, C.; Murayama, N. Prediction of in vivo potential for metabolic activation of drugs into chemically reactive intermediate: Correlation of in vitro and in vivo generation of reactive intermediates and in vitro glutathione conjugate formation in rats and humans. Chem. Res. Toxicol. 2007, 20, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Bort, R.; Ponsoda, X.; Jover, R.; Gómez-Lechón, M.J.; Castell, J.V. Diclofenac toxicity to hepatocytes: A role for drug metabolism in cell toxicity. J. Pharmacol. Exp. Ther. 1999, 288, 65–72. [Google Scholar] [PubMed]
- Mueller, D.; Müller-Vieira, U.; Biemel, K.M.; Tascher, G.; Nüssler, A.K.; Noor, F. Biotransformation of diclofenac and effects on the metabolome of primary human hepatocytes upon repeated dose exposure. Eur. J. Pharm. Sci. 2012, 45, 716–724. [Google Scholar] [CrossRef] [PubMed]
- Lauer, B.; Tuschl, G.; Kling, M.; Mueller, S.O. Species-specific toxicity of diclofenac and troglitazone in primary human and rat hepatocytes. Chem. Biol. Interact. 2009, 179, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Messner, S.; Agarkova, I.; Moritz, W.; Kelm, J.M. Multi-cell type human liver microtissues for hepatotoxicity testing. Arch. Toxicol. 2013, 87, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Lechón, M.J.; Ponsoda, X.; O’Connor, E.; Donato, T.; Castell, J.V.; Jover, R. Diclofenac induces apoptosis in hepatocytes by alteration of mitochondrial function and generation of ROS. Biochem. Pharmacol. 2003, 66, 2155–2167. [Google Scholar] [CrossRef] [PubMed]
- Prill, S.; Jaeger, M.S.; Duschl, C. Long-term microfluidic glucose and lactate monitoring in hepatic cell culture. Biomicrofluidics 2014, 8. [Google Scholar] [CrossRef] [PubMed]
- Mueller, D.; Tascher, G.; Damm, G.; Nüssler, A.K.; Heinzle, E.; Noor, F. Real-time in situ viability assessment in a 3D bioreactor with liver cells using resazurin assay. Cytotechnology 2013, 65, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Riss, T.L.; Moravec, R.A.; Niles, A.L.; Benink, H.A.; Worzella, T.J.; Minor, L. Cell viability assays; 1 May 2013 [Updated 29 June 2015]. In Assay Guidance Manual; Sittampalam, G.S., Coussens, N.P., Nelson, H., Arkin, M., Auld, D., Austin, C., Bejcek, B., Glicksman, M., Inglese, J., Iversen, P.W., et al., Eds.; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda, MD, USA, 2004. [Google Scholar]
- Pace, R.T.; Burg, K.J.L. Toxic effects of resazurin on cell cultures. Cytotechnology 2015, 67, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, W.M.; Reynolds, T.B. Serum lactic dehydrogenase in the differential diagnosis of acute hepatocellular injury. J. Clin. Gastroenterol. 1994, 19, 118–121. [Google Scholar] [CrossRef] [PubMed]
- Laine, L.; Goldkind, L.; Curtis, S.P.; Connors, L.G.; Yanqiong, Z.; Cannon, C.P. How common is diclofenac-associated liver injury? Analysis of 17,289 arthritis patients in a long-term prospective clinical trial. Am. J. Gastroenterol. 2009, 104, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.G.; Xia, T.; Yuan, J.; Yu, R.A.; Yang, K.D.; Chen, X.M.; Qu, W.; Waalkes, M.P. Effects of phenobarbital on metabolism and toxicity of diclofenac sodium in rat hepatocytes in vitro. Food Chem. Toxicol. 2004, 42, 1647–1653. [Google Scholar] [CrossRef] [PubMed]
- Borlak, J.; Chougule, A.; Singh, P.K. How useful are clinical liver function tests in in vitro human hepatotoxicity assays? Toxicol. Vitro 2014, 28, 784–795. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B.; Chevts, J.; Renner, B.; Wuttke, H.; Rau, T.; Schmidt, A.; Szelenyi, I.; Brune, K.; Werner, U. Bioavailability of diclofenac potassium at low doses. Br. J. Clin. Pharmacol 2005, 59, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Hackstein, H.; Mohl, W.; Püschel, W.; Stallmach, A.; Zeitz, M. Diclofenac-associated acute cholestatis hepatitis. Z. Gastroenterol. 1998, 36, 385–389. [Google Scholar] [PubMed]
- Breen, E.G.; McNicholl, J.; Cosgrove, E.; McCabe, J.; Stevens, F.M. Fatal hepatitis associated with diclofenac. Gut 1986, 27, 1390–1393. [Google Scholar] [CrossRef] [PubMed]
- Helfgott, S.M.; Sandberg-Cook, J.; Zakim, D.; Nestler, J. Diclofenac-associated hepatotoxicity. JAMA 1990, 264, 2660–2662. [Google Scholar] [CrossRef] [PubMed]
- Sallustio, B.C.; Holbrook, F.L. In vivo perturbation of rat hepatocyte canalicular membrane function by diclofenac. Drug Metab. Dispos. 2001, 29, 1535–1538. [Google Scholar] [PubMed]
- Kretz-Rommel, A.; Boelsterli, U.A. Cytotoxic activity of T cells and non-T cells from diclofenac-immunized mice against cultured syngeneic hepatocytes exposed to diclofenac. Hepatology 1995, 22, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Ingelman-Sundberg, M. Pharmacogenetics of cytochrome P450 and its applications in drug therapy: The past, present and future. Trends Pharmacol. Sci. 2004, 25, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Guguen-Guillouzo, C.; Guillouzo, A. General review on in vitro hepatocyte models and their applications. Methods Mol. Biol. 2010, 640, 1–40. [Google Scholar] [PubMed]
- Rodrigues, R.M.; Heymans, A.; de Boe, V.; Sachinidis, A.; Chaudhari, U.; Govaere, O.; Roskams, T.; Vanhaecke, T.; Rogiers, V.; de Kock, J. Toxicogenomics-based prediction of acetaminophen-induced liver injury using human hepatic cell systems. Toxicol. Lett. 2016, 240, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Lagas, J.S.; van der Kruijssen, C.M.; van de Wetering, K.; Beijnen, J.H.; Schinkel, A.H. Transport of diclofenac by breast cancer resistance protein (ABCG2) and stimulation of multidrug resistance protein 2 (ABCC2)-mediated drug transport by diclofenac and benzbromarone. Drug Metab. Dispos. 2009, 37, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.X.; Govindarajan, S.; Kaplowitz, N. Innate immune system plays a critical role in determining the progression and severity of acetaminophen hepatotoxicity. Gastroenterology 2004, 127, 1760–1774. [Google Scholar] [CrossRef] [PubMed]
- Campion, S.N.; Johnson, R.; Aleksunes, L.M.; Goedken, M.J.; van Rooijen, N.; Scheffer, G.L.; Cherrington, N.J.; Manautou, J.E. Hepatic Mrp4 induction following acetaminophen exposure is dependent on Kupffer cell function. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 295, G294–G304. [Google Scholar] [CrossRef] [PubMed]
- Herpers, B.; Wink, S.; Fredriksson, L.; Di, Z.; Hendriks, G.; Vrieling, H.; de Bont, H.; van de Water, B. Activation of the Nrf2 response by intrinsic hepatotoxic drugs correlates with suppression of NF-κB activation and sensitizes toward TNFα-induced cytotoxicity. Arch. Toxicol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.D.; Berntenis, N.; Roth, A.; Ebeling, M. Data mining reveals a network of early-response genes as a consensus signature of drug-induced in vitro and in vivo toxicity. Pharmacogenom. J. 2014, 14, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Fredriksson, L.; Herpers, B.; Benedetti, G.; Matadin, Q.; Puigvert, J.C.; de Bont, H.; Dragovic, S.; Vermeulen, N.P.; Commandeur, J.N.; Danen, E.; et al. Diclofenac inhibits tumornecrosisfactor-α-induced nuclearfactor-κB activation causing synergistic hepatocyte apoptosis. Hepatology 2011, 53, 2027–2041. [Google Scholar] [CrossRef] [PubMed]
- Damm, G.; Pfeiffer, E.; Burkhardt, B.; Vermehren, J.; Nüssler, A.K.; Weiss, T.S. Human parenchymal and non-parenchymal liver cell isolation, culture and characterization. Hepatol. Int. 2013, 7, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, E.; Kegel, V.; Zeilinger, K.; Hengstler, J.G.; Nüssler, A.K.; Seehofer, D.; Damm, G. Featured Article: Isolation, characterization, and cultivation of human hepatocytes and non-parenchymal liver cells. Exp. Biol. Med. 2015, 240, 645–656. [Google Scholar] [CrossRef] [PubMed]
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef] [PubMed]
- Dai, M.; Wang, P.; Boyd, A.D.; Kostov, G.; Athey, B.; Jones, E.G.; Bunney, W.E.; Myers, R.M.; Speed, T.P.; Akil, H.; et al. Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res. 2005, 33, e175. [Google Scholar] [CrossRef] [PubMed]
- Irizarry, R.A.; Hobbs, B.; Collin, F.; Beazer-Barclay, Y.D.; Antonellis, K.J.; Scherf, U.; Speed, T.P. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 2003, 4, 249–264. [Google Scholar] [CrossRef] [PubMed]
- Smyth, G.K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004, 3, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Hochberg, Y.; Benjamini, Y. More powerful procedures for multiple significance testing. Stat. Med. 1990, 9, 811–818. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, N.; de Bondt, A.M.; Talloen, W.; Moechars, D.; Göhlmann, H.W.; Amaratunga, D. The high-level similarity of some disparate gene expression measures. Bioinformatics 2007, 23, 3032–3038. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rank | GO ID | Size | p-Value | Description | Parent |
|---|---|---|---|---|---|
| Gene Ontology Biological Processes | |||||
| 1 | GO:0072376 | 67 (90) | 6.0 × 10−33 | Protein activation cascade | |
| 2 | GO:0006956 | 47 (69) | 1.8 × 10−31 | Complement activation | 1 |
| 3 | GO:0016064 | 94 (119) | 9.8 × 10−29 | Immunoglobulin mediated immune response | |
| 4 | GO:0002455 | 43 (67) | 1.0 × 10−27 | Humoral immune response mediated by circulating immunoglobulin | 3 |
| 5 | GO:0019724 | 97 (122) | 4.9 × 10−27 | B cell mediated immunity | 3 |
| 6 | GO:0002576 | 83 (87) | 1.4 × 10−26 | Platelet degranulation | |
| 7 | GO:0002687 | 89 (90) | 3.5 × 10−26 | Positive regulation of leukocyte migration | |
| 8 | GO:0006958 | 31 (52) | 2.0 × 10−22 | Complement activation, classical pathway | 1, 2, 3, 4, 5 |
| 9 | GO:0002688 | 79 (83) | 2.7 × 10−21 | Regulation of leukocyte chemotaxis | |
| 10 | GO:0002690 | 68 (69) | 2.8 × 10−18 | Positive regulation of leukocyte chemotaxis | 7, 9 |
| Gene Ontology Cellular Component | |||||
| 1 | GO:0032994 | 37 (40) | 8.7 × 10−25 | Protein-lipid complex | |
| 2 | GO:0034358 | 35 (38) | 1.8 × 10−24 | Plasma lipoprotein particle | 1 |
| 3 | GO:0042613 | 12 (16) | 4.6 × 10−24 | MHC class II protein complex | |
| 4 | GO:0042611 | 22 (27) | 1.6 × 10−20 | MHC protein complex | 3 |
| 5 | GO:0031983 | 73 (82) | 1.1 × 10−19 | Vesicle lumen | |
| 6 | GO:0060205 | 72 (81) | 2.4 × 10−19 | Cytoplasmic membrane-bounded vesicle lumen | 5 |
| 7 | GO:0030134 | 48 (52) | 6.7 × 10−17 | ER to Golgi transport vesicle | |
| 8 | GO:0012507 | 38 (42) | 1.6 × 10−15 | ER to Golgi transport vesicle membrane | 7 |
| 9 | GO:0034364 | 23 (26) | 1.0 × 10−14 | High-density lipoprotein particle | 1, 2 |
| 10 | GO:0031091 | 60 (63) | 1.3 × 10−12 | Platelet α granule | |
| Gene Ontology Molecular Functions | |||||
| 1 | GO:0042379 | 54 (58) | 2.8 × 10−9 | Chemokine receptor binding | |
| 2 | GO:0008009 | 43 (47) | 1.3 × 10−8 | Chemokine activity | 1 |
| 3 | GO:0030170 | 53 (56) | 5.3 × 10−7 | Pyridoxal phosphate binding | |
| 4 | GO:0003823 | 76 (107) | 2.0 × 10−6 | Antigen binding | |
| 5 | GO:0045236 | 17 (17) | 9.7 × 10−6 | CXCR chemokine receptor binding | 1, 2 |
| 6 | GO:0023026 | 15 (16) | 2.6 × 10−5 | MHC class II protein complex binding | 4 |
| 7 | GO:0060228 | 6 (6) | 3.1 × 10−5 | Phosphatidylcholine-sterol O-acyltransferaseactivator activity | |
| 8 | GO:0031210 | 15 (16) | 4.7 × 10−5 | Phosphatidylcholine binding | |
| 9 | GO:0019864 | 8 (12) | 1.1 × 10−4 | IgG binding | |
| 10 | GO:0023023 | 17 (19) | 1.6 × 10−4 | MHC protein complex binding | 4, 6 |
| Rank | GO ID | Size | p-Value | Description | Parent |
|---|---|---|---|---|---|
| Gene Ontology Biological Processes | |||||
| 1 | GO:0042590 | 74 (79) | 1.2 × 10−46 | Antigen processing and presentation of exogenous peptide antigen via MHC class I | |
| 2 | GO:0002479 | 70 (75) | 2.1 × 10−36 | Antigen processing and presentation of exogenous peptide antigen via MHC class I, TAP-dependent | 1 |
| 3 | GO:0006521 | 59 (62) | 1.0 × 10−31 | Regulation of cellular amino acid metabolic process | |
| 4 | GO:0033238 | 79 (82) | 2.9 × 10−26 | Regulation of cellular amine metabolic process | |
| 5 | GO:0002576 | 83 (87) | 8.9 × 10−19 | Platelet degranulation | |
| 6 | GO:0044783 | 75 (77) | 3.9 × 10−17 | G1 DNA damage check point | |
| 7 | GO:0051437 | 68 (72) | 6.7 × 10−17 | Positive regulation of ubiquitin-protein ligase activity | |
| 8 | GO:0051351 | 86 (94) | 1.4 × 10−16 | Positive regulation of ligase activity | |
| 9 | GO:0051436 | 63 (67) | 2.1 × 10−16 | Negative regulation of ubiquitin-protein ligase activity | |
| 10 | GO:0051443 | 81 (89) | 3.0 × 10−16 | Positive regulation of ubiquitin-protein transferase activity | 8 |
| Gene Ontology Cellular Component | |||||
| 1 | GO:0000502 | 63 (66) | 3.6 × 10−25 | Proteasome complex | |
| 2 | GO:1990204 | 78 (83) | 2.6 × 10−18 | Oxidoreductase complex | |
| 3 | GO:0043202 | 72 (73) | 2.5 × 10−16 | Lysosomal lumen | |
| 4 | GO:0005775 | 73 (77) | 1.4 × 10−15 | Vacuolar lumen | 3 |
| 5 | GO:0031091 | 60 (63) | 3.1 × 10−15 | Platelet α granule | |
| 6 | GO:0070469 | 65 (72) | 3.1 × 10−15 | Respiratory chain | |
| 7 | GO:0005746 | 59 (65) | 9.6 × 10−13 | Mitochondrial respiratory chain | 6 |
| 8 | GO:0022624 | 22 (24) | 3.9 × 10−12 | Proteasome accessory complex | 1 |
| 9 | GO:0031902 | 93 (99) | 3.1 × 10−11 | Late endosome membrane | |
| 10 | GO:0005839 | 20 (20) | 1.6 × 10−10 | Proteasome core complex | 1 |
| Gene Ontology Molecular Functions | |||||
| 1 | GO:0009055 | 89 (99) | 8.2 × 10−21 | Electron carrier activity | |
| 2 | GO:0003743 | 48 (52) | 1.1 × 10−14 | Translation initiation factor activity | |
| 3 | GO:0016651 | 87 (95) | 9.7 × 10−14 | Oxidoreductase activity, acting on NAD(P)H | |
| 4 | GO:0016655 | 49 (53) | 1.6 × 10−12 | Oxidoreductase activity, acting on NAD(P)H, quinon or similar compound as acceptor | 3 |
| 5 | GO:0008135 | 75 (83) | 2.9 × 10−11 | Translation factor activity, nucleic acid binding | |
| 6 | GO:0004298 | 21 (21) | 4.3 × 10−8 | Threonine-type endopeptidase activity | |
| 7 | GO:0070003 | 21 (21) | 4.3 × 10−8 | Threonine-type peptidase activity | 6 |
| 8 | GO:0016830 | 45 (47) | 1.1 × 10−6 | Carbon-carbon lyase activity | |
| 9 | GO:0003954 | 36 (39) | 2.4 × 10−6 | NADH dehydrogenase activity | 3, 4 |
| 10 | GO:0008137 | 36 (39) | 2.4 × 10−6 | NADH dehydrogenase (ubiquinone) activity | 3, 4, 9 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Knöspel, F.; Jacobs, F.; Freyer, N.; Damm, G.; De Bondt, A.; Van den Wyngaert, I.; Snoeys, J.; Monshouwer, M.; Richter, M.; Strahl, N.; et al. In Vitro Model for Hepatotoxicity Studies Based on Primary Human Hepatocyte Cultivation in a Perfused 3D Bioreactor System. Int. J. Mol. Sci. 2016, 17, 584. https://doi.org/10.3390/ijms17040584
Knöspel F, Jacobs F, Freyer N, Damm G, De Bondt A, Van den Wyngaert I, Snoeys J, Monshouwer M, Richter M, Strahl N, et al. In Vitro Model for Hepatotoxicity Studies Based on Primary Human Hepatocyte Cultivation in a Perfused 3D Bioreactor System. International Journal of Molecular Sciences. 2016; 17(4):584. https://doi.org/10.3390/ijms17040584
Chicago/Turabian StyleKnöspel, Fanny, Frank Jacobs, Nora Freyer, Georg Damm, An De Bondt, Ilse Van den Wyngaert, Jan Snoeys, Mario Monshouwer, Marco Richter, Nadja Strahl, and et al. 2016. "In Vitro Model for Hepatotoxicity Studies Based on Primary Human Hepatocyte Cultivation in a Perfused 3D Bioreactor System" International Journal of Molecular Sciences 17, no. 4: 584. https://doi.org/10.3390/ijms17040584
APA StyleKnöspel, F., Jacobs, F., Freyer, N., Damm, G., De Bondt, A., Van den Wyngaert, I., Snoeys, J., Monshouwer, M., Richter, M., Strahl, N., Seehofer, D., & Zeilinger, K. (2016). In Vitro Model for Hepatotoxicity Studies Based on Primary Human Hepatocyte Cultivation in a Perfused 3D Bioreactor System. International Journal of Molecular Sciences, 17(4), 584. https://doi.org/10.3390/ijms17040584