
Challenging Density Functional Theory Calculations with Hemes and Porphyrins



Abstract
:1. Introduction
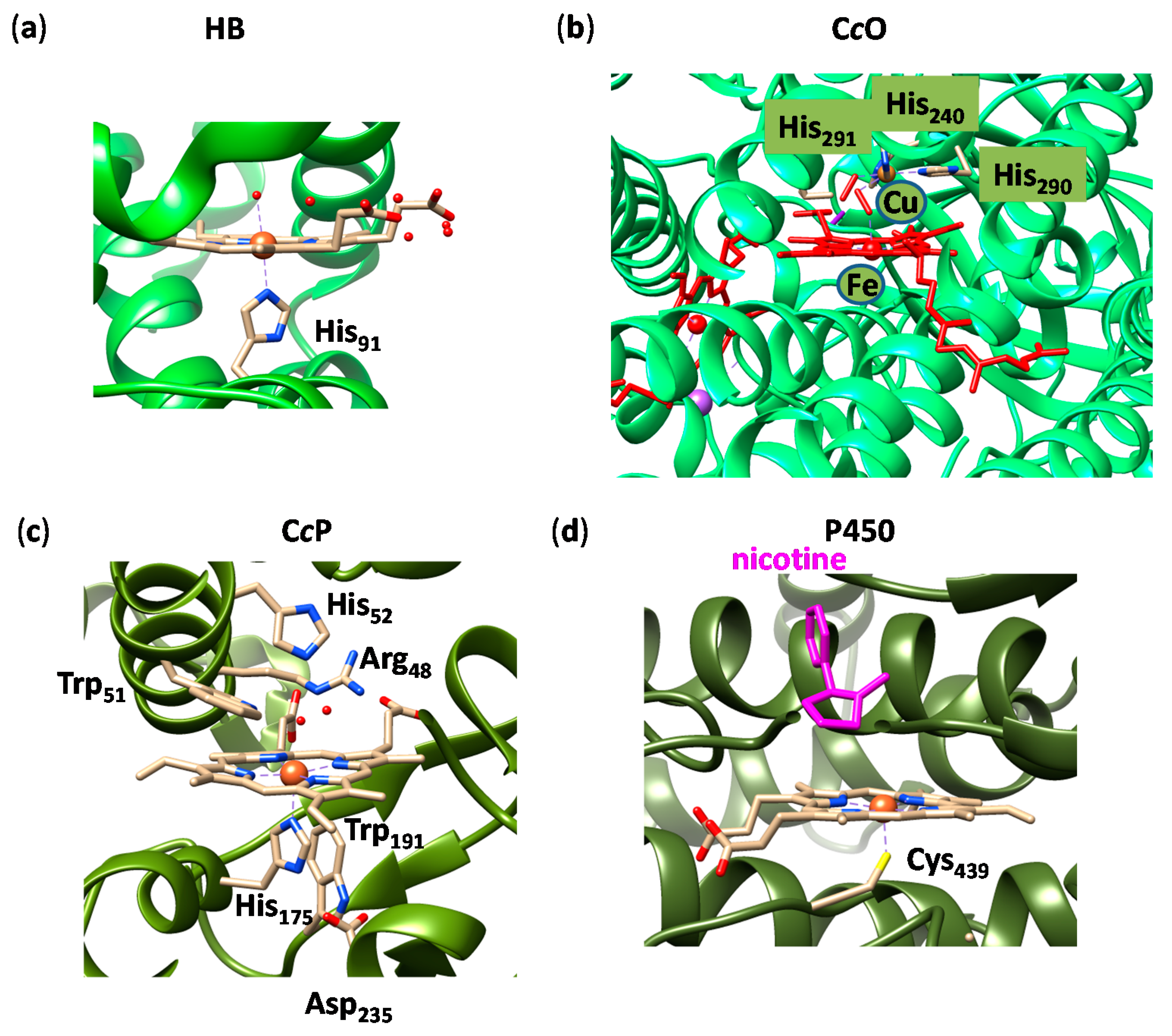
2. Results
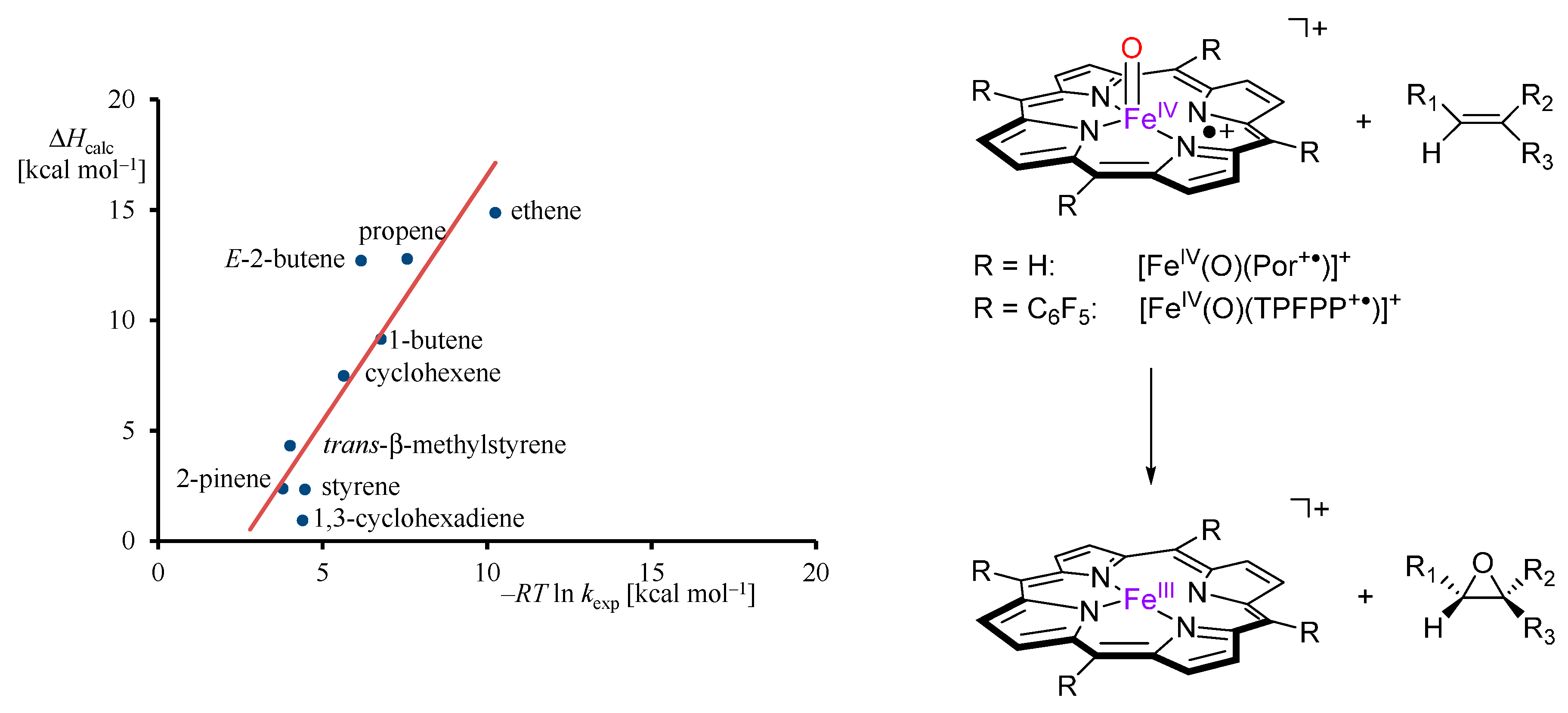
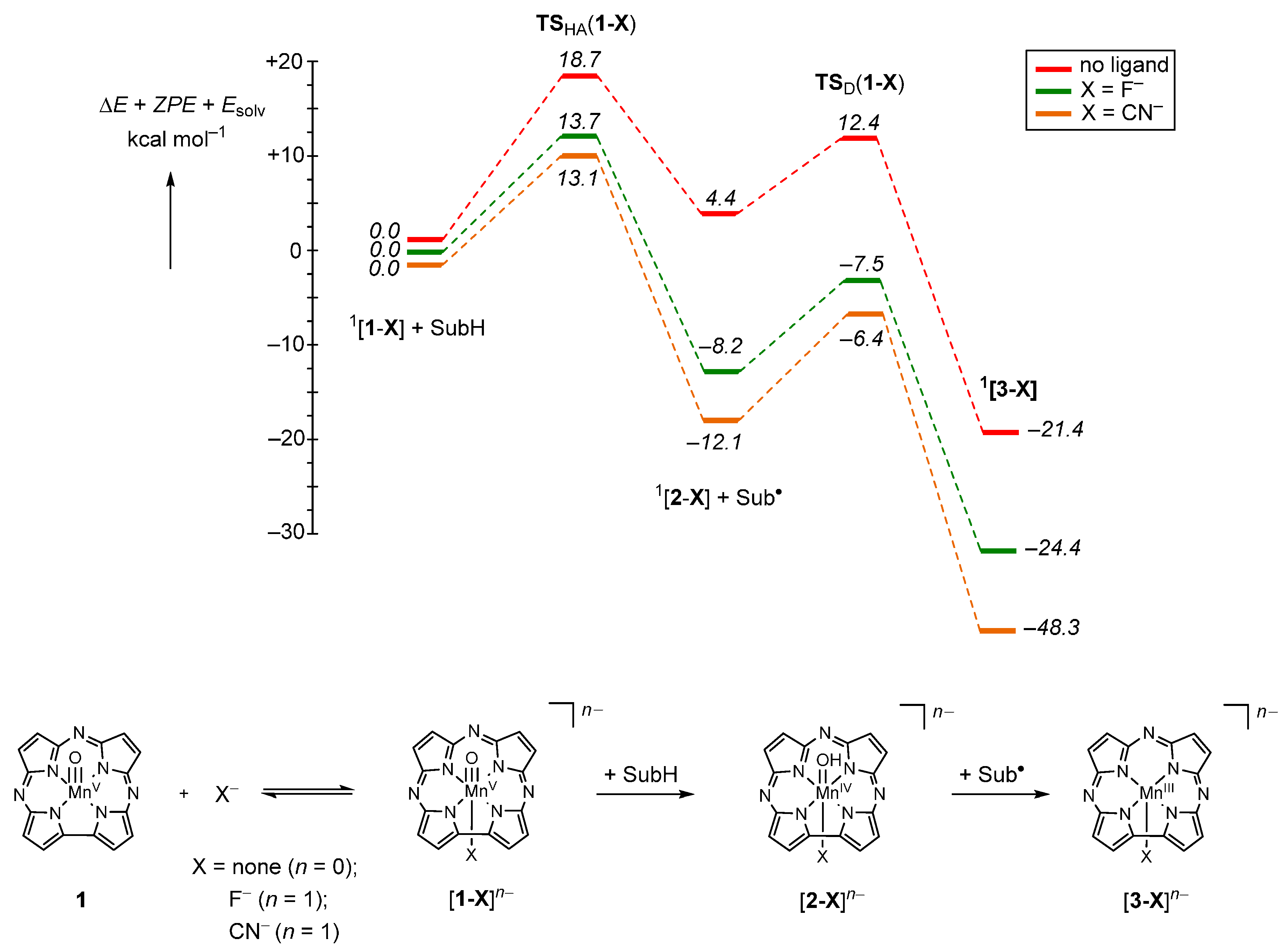
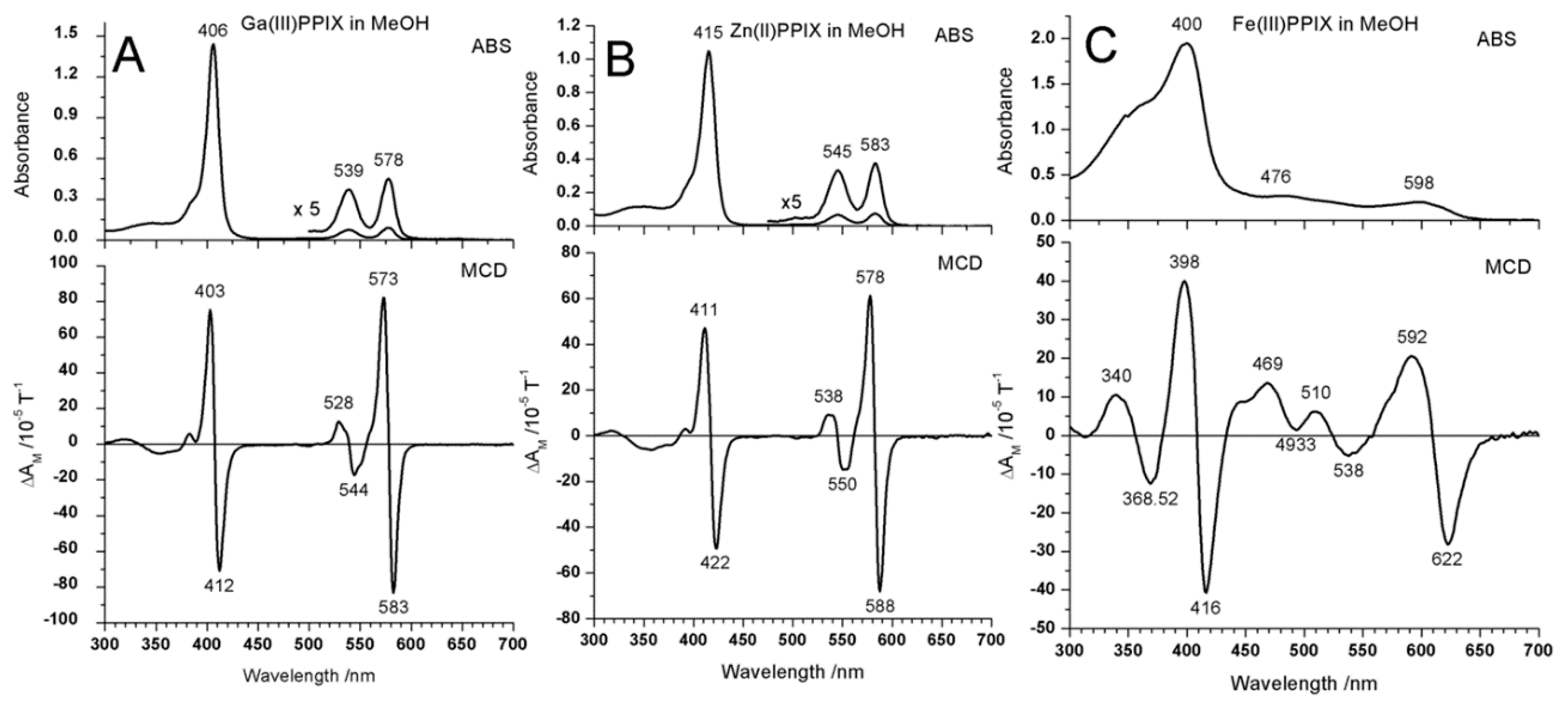
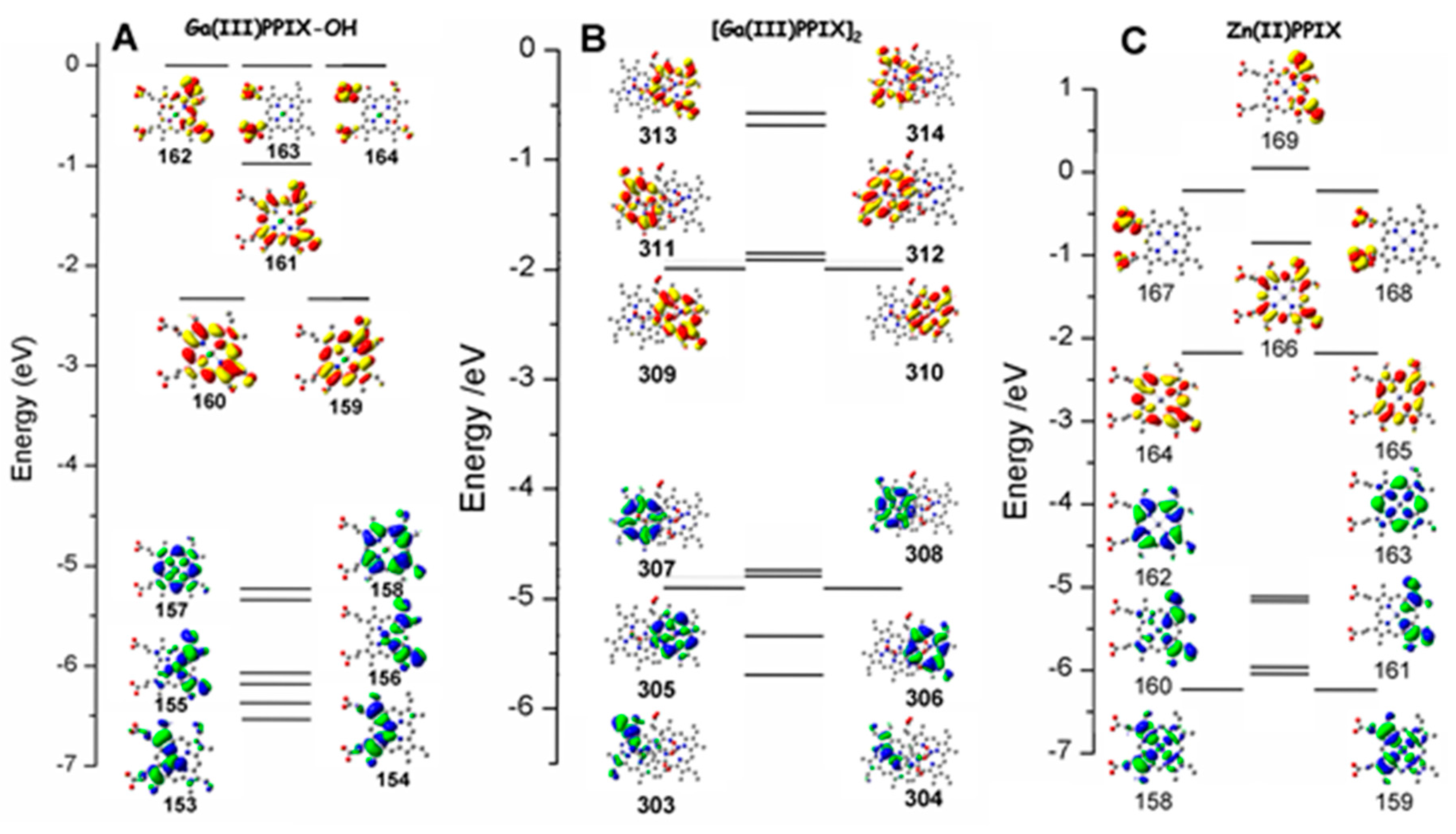
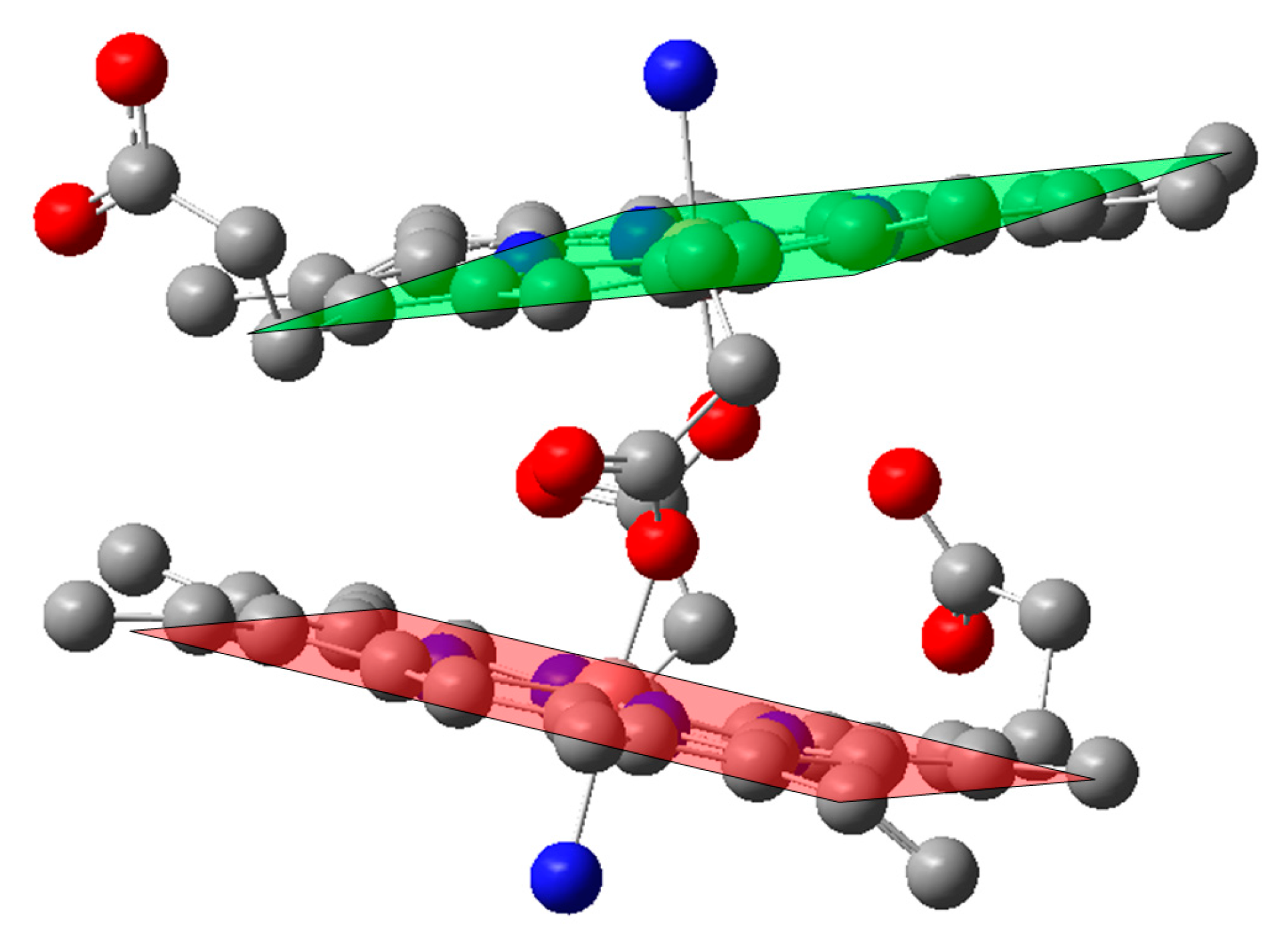
2.1. Case Study 1
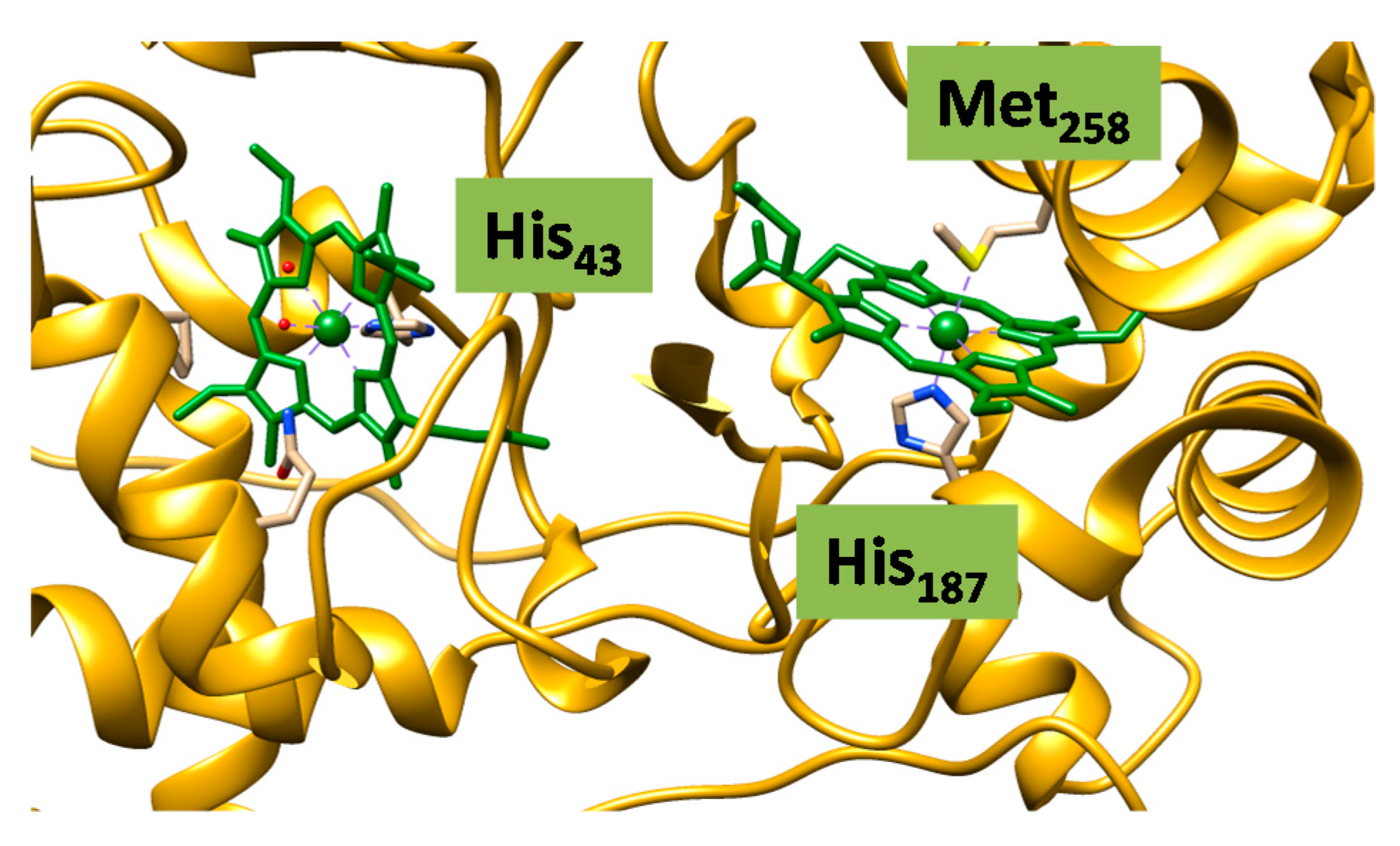
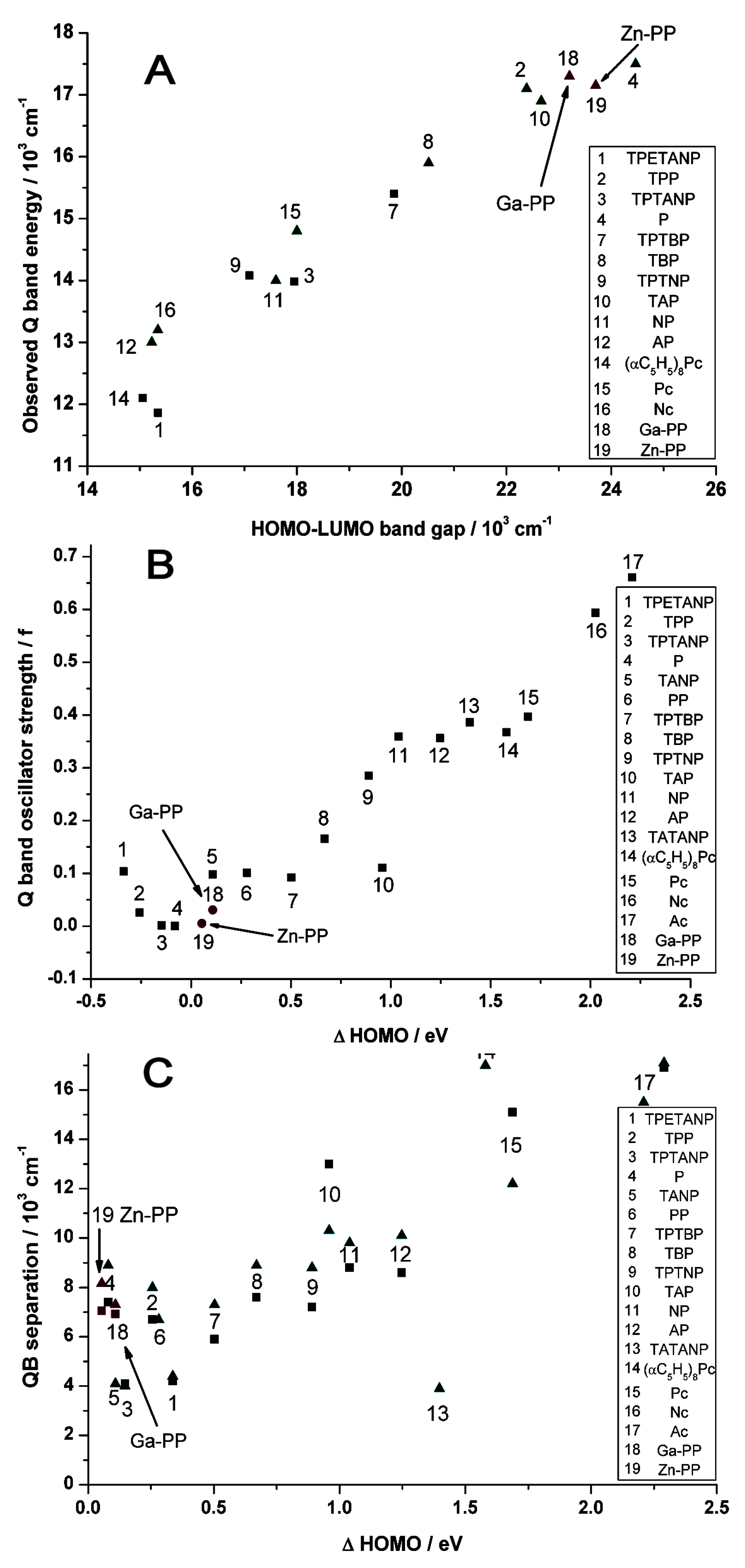
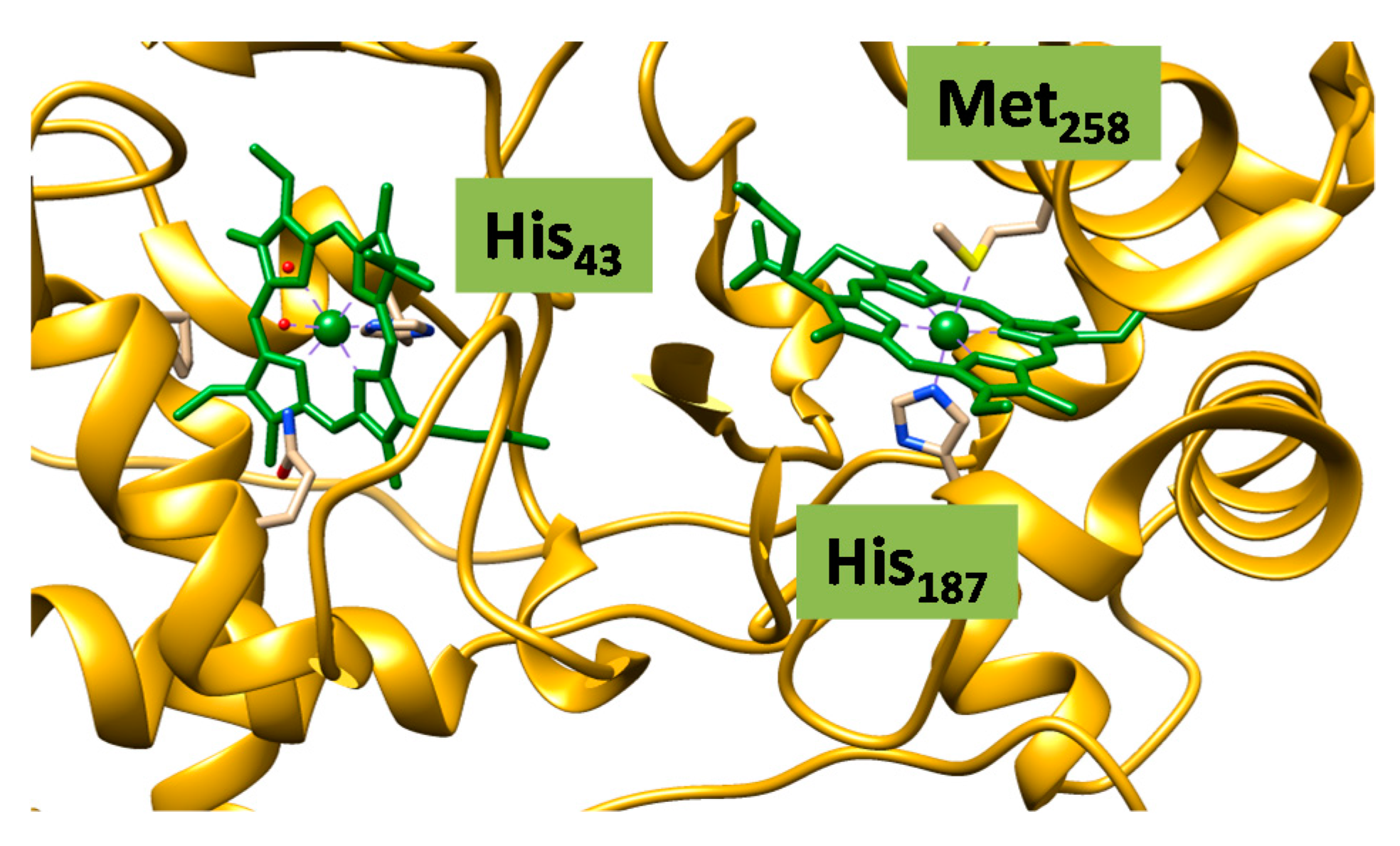
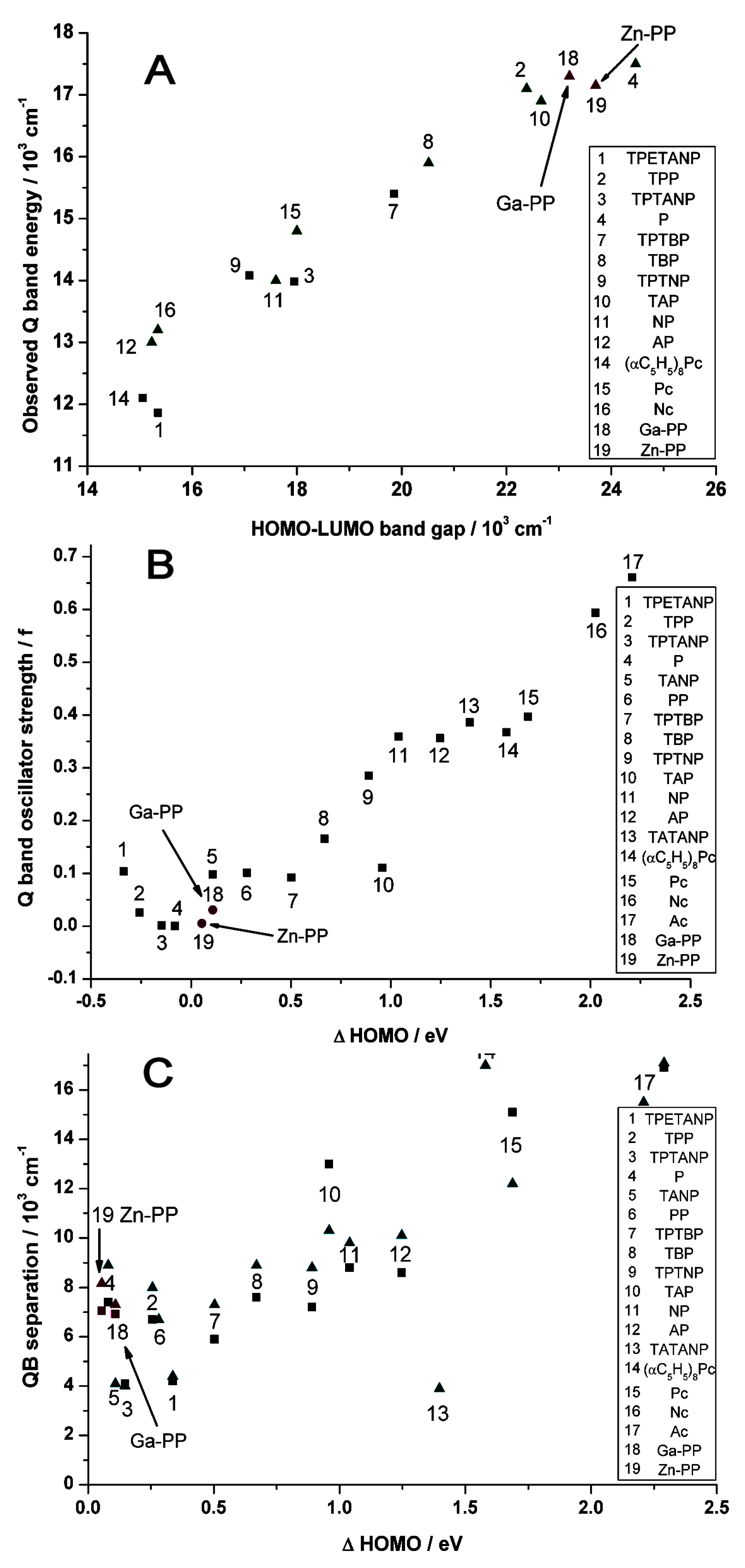
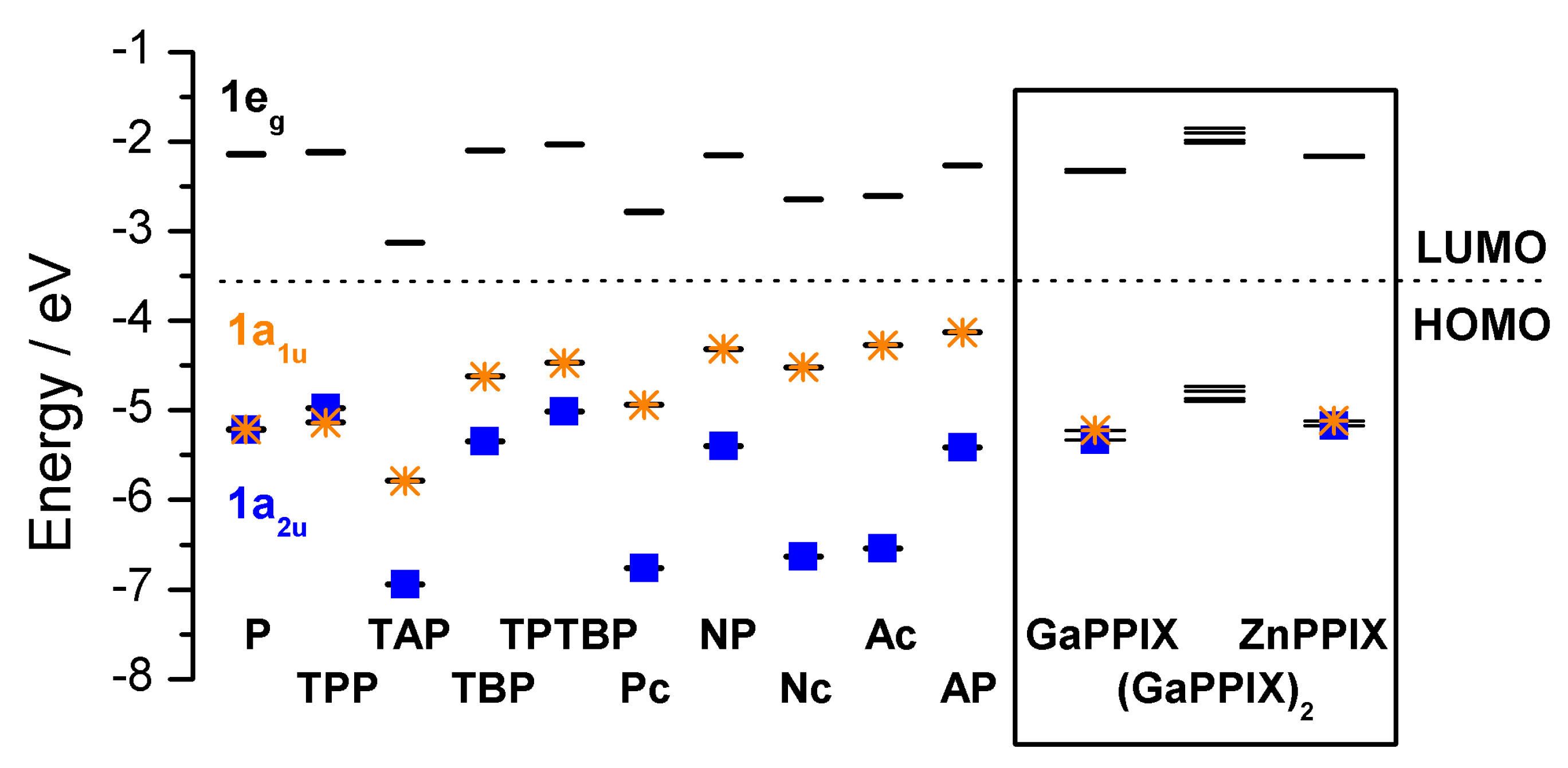
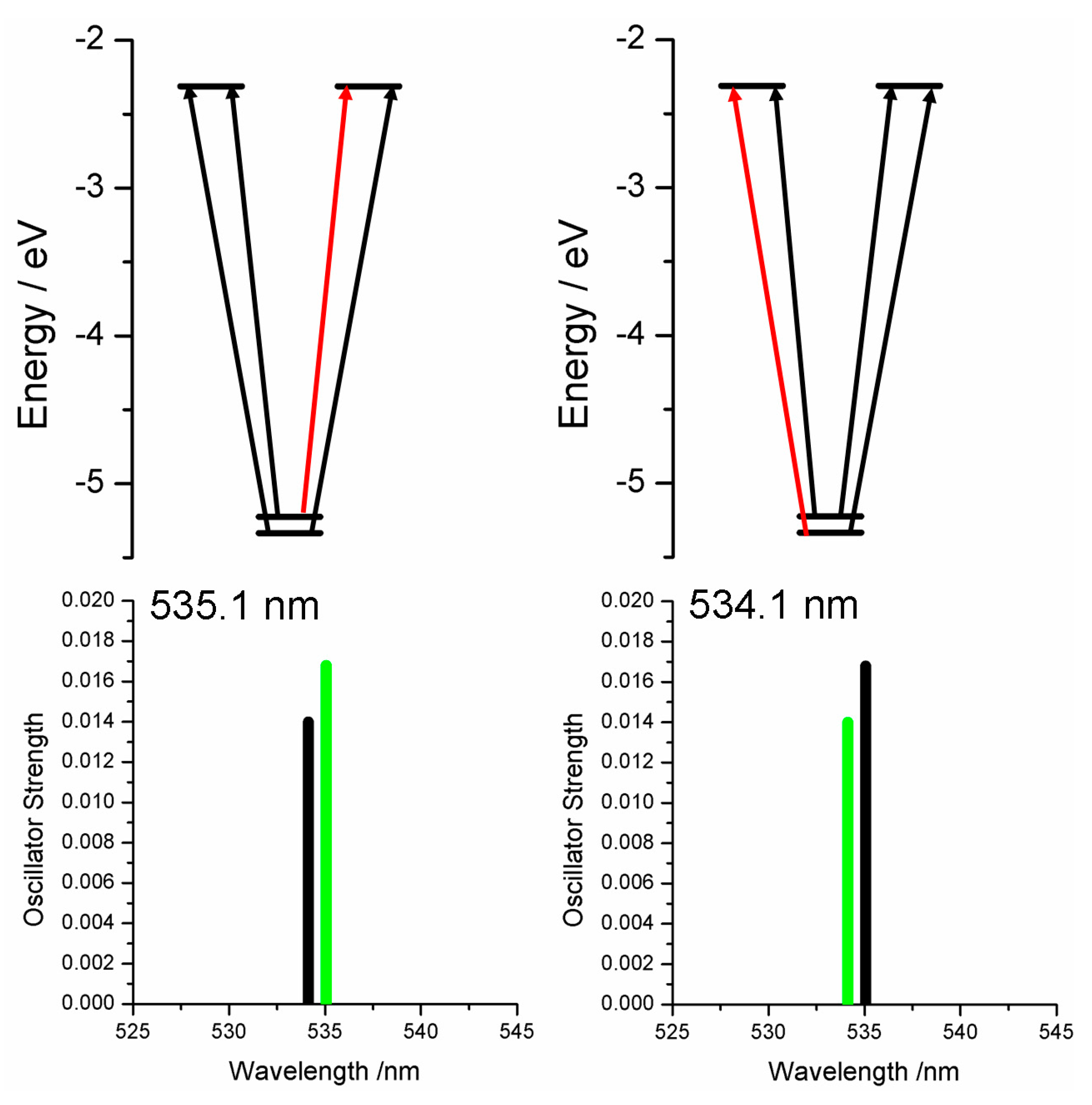
2.2. Case Study 2
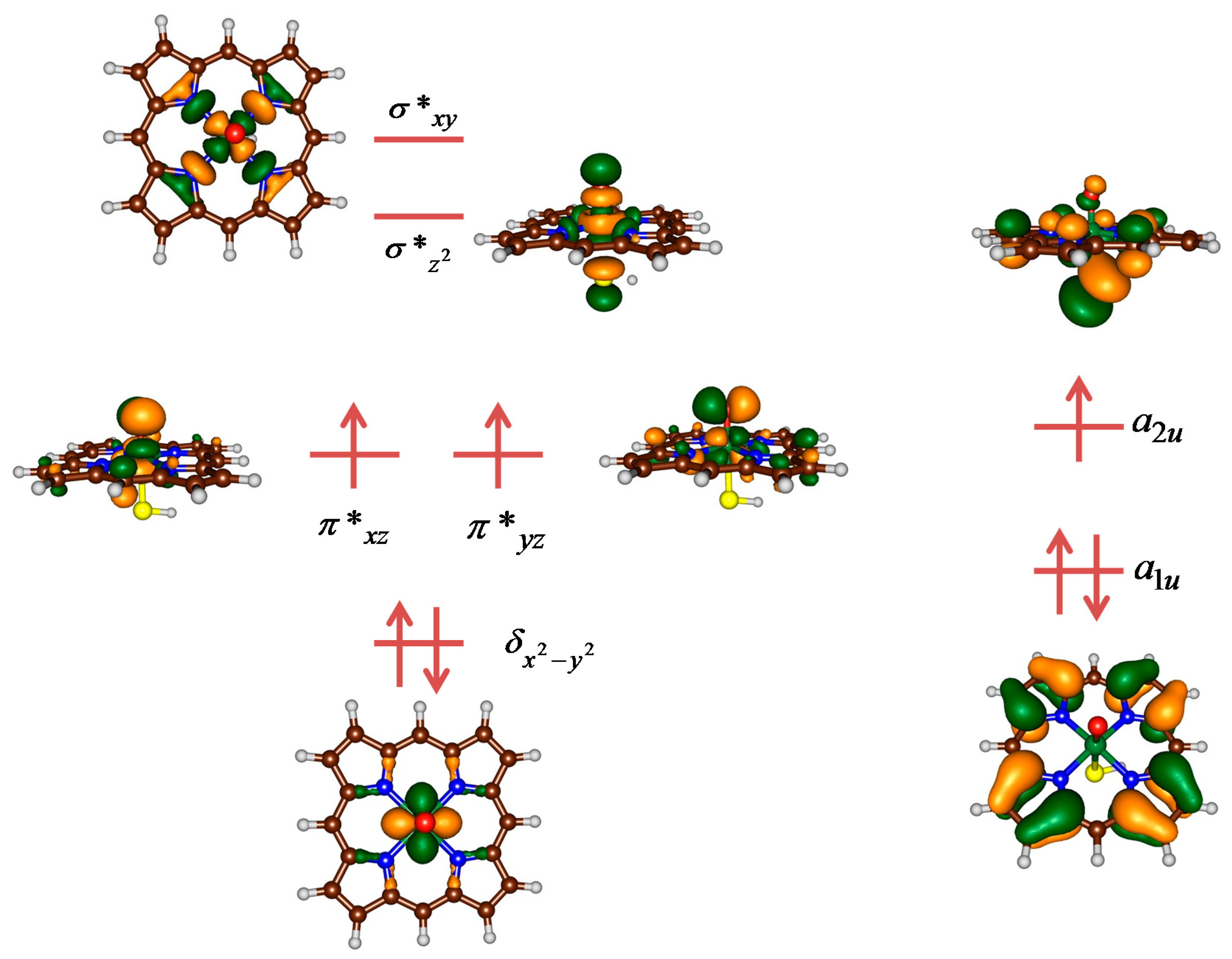
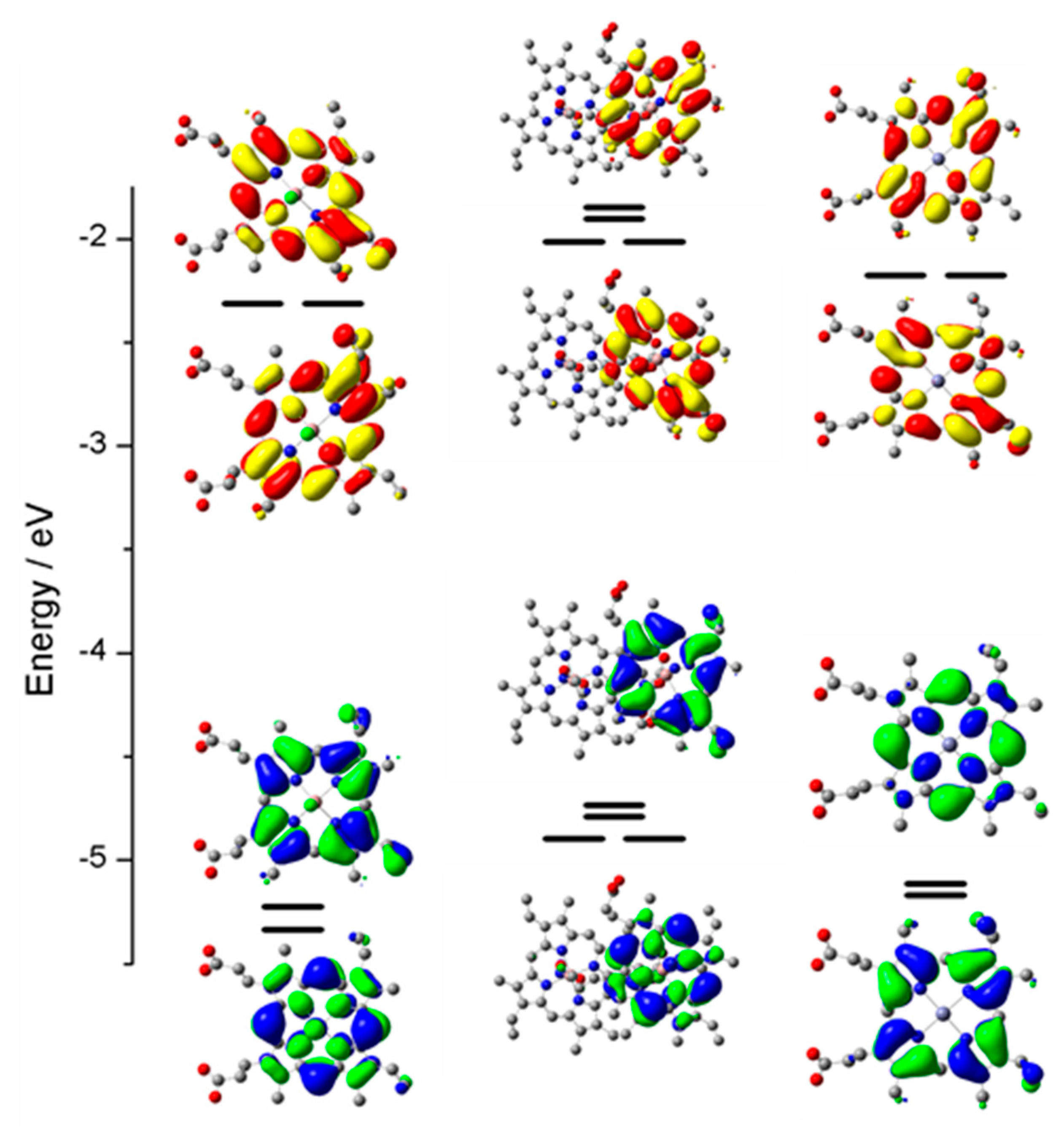
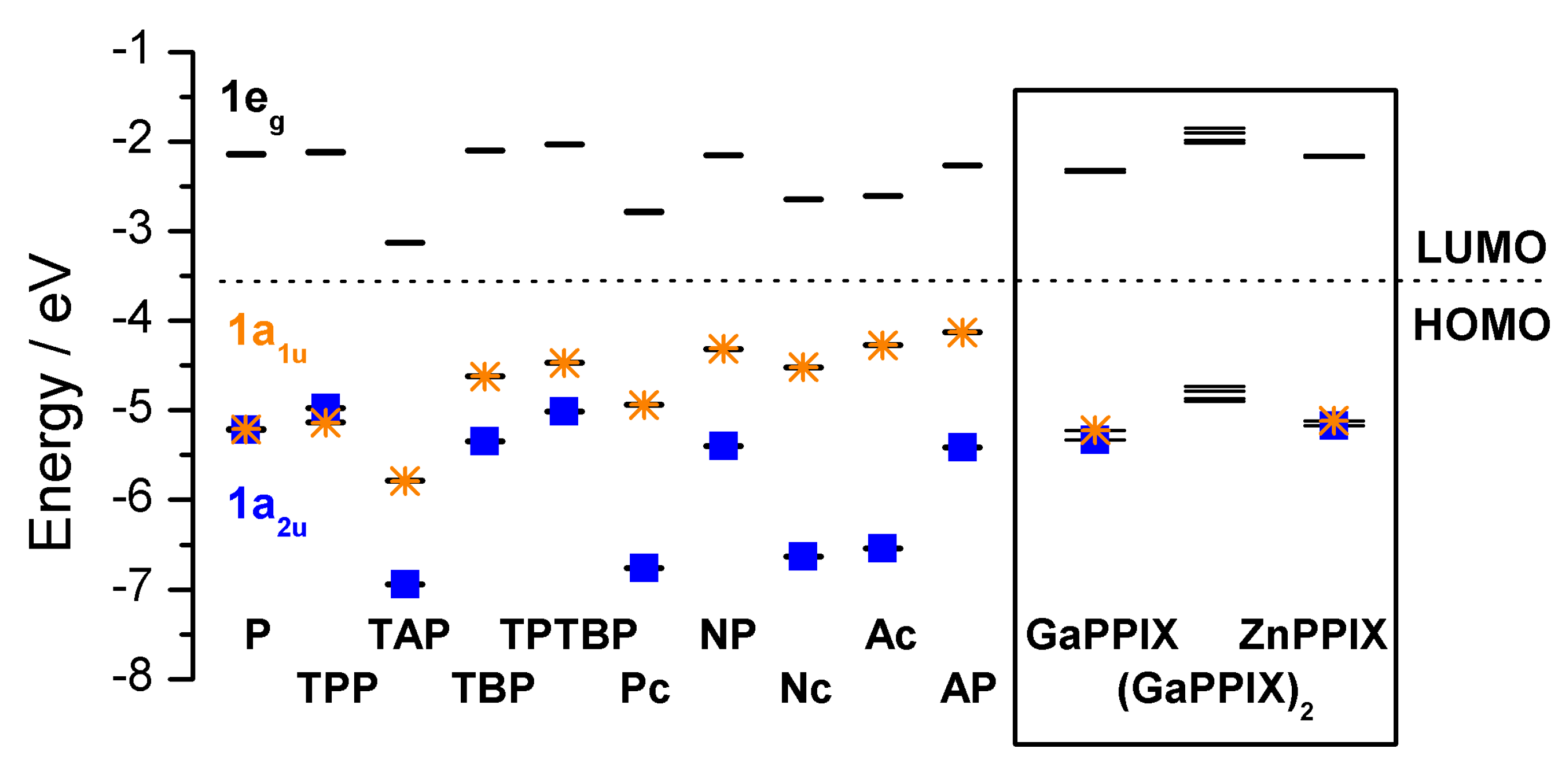
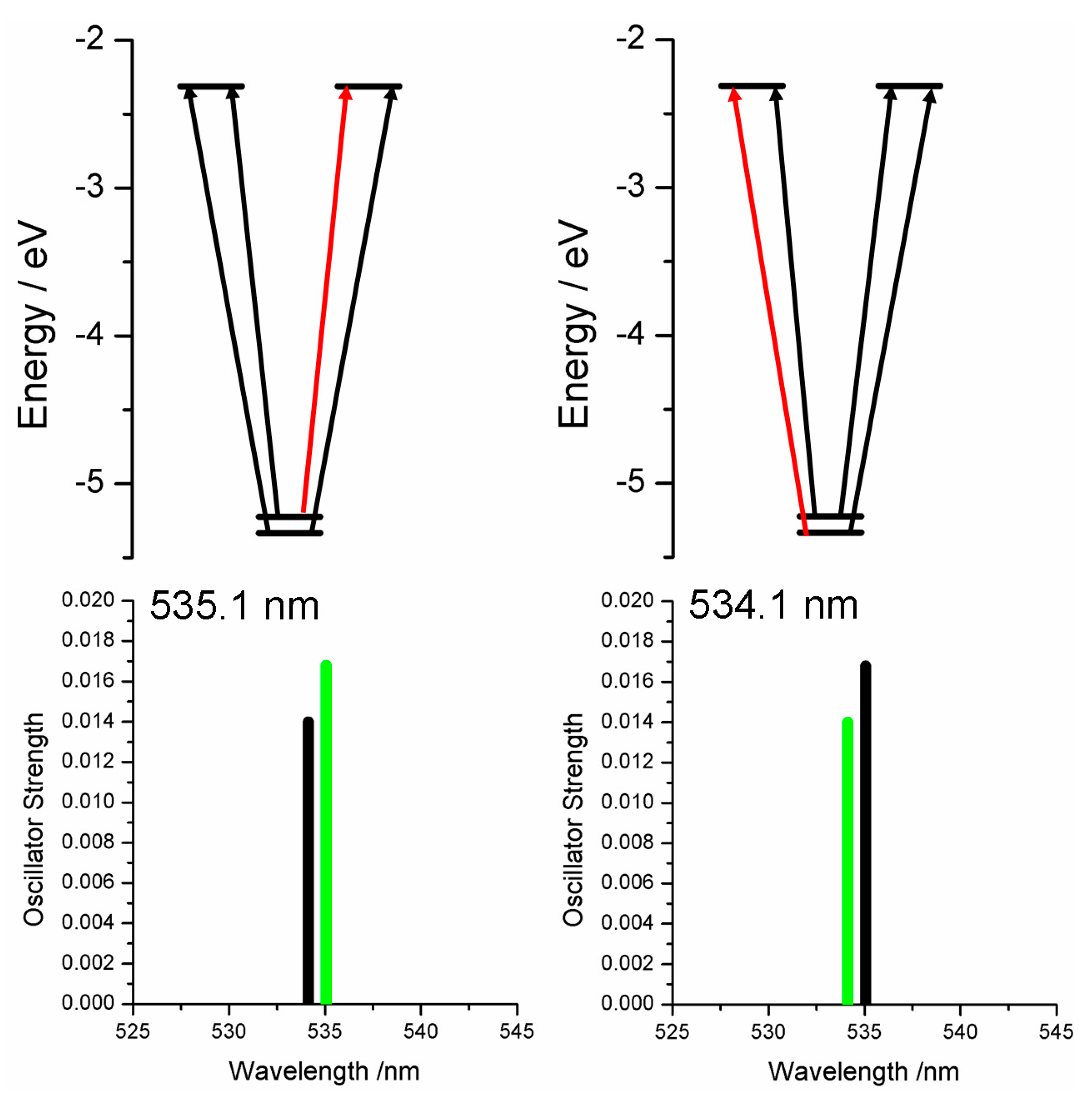
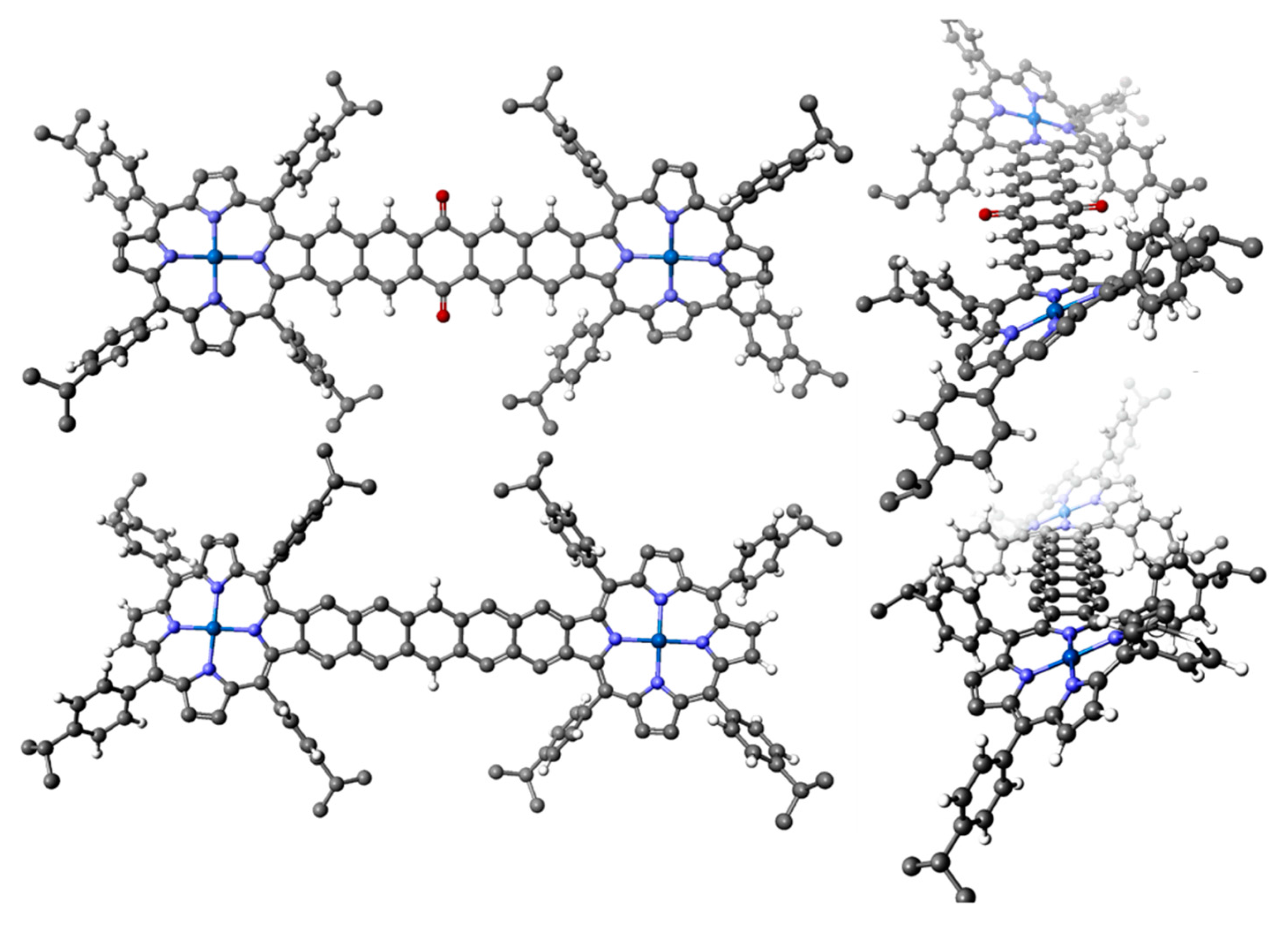
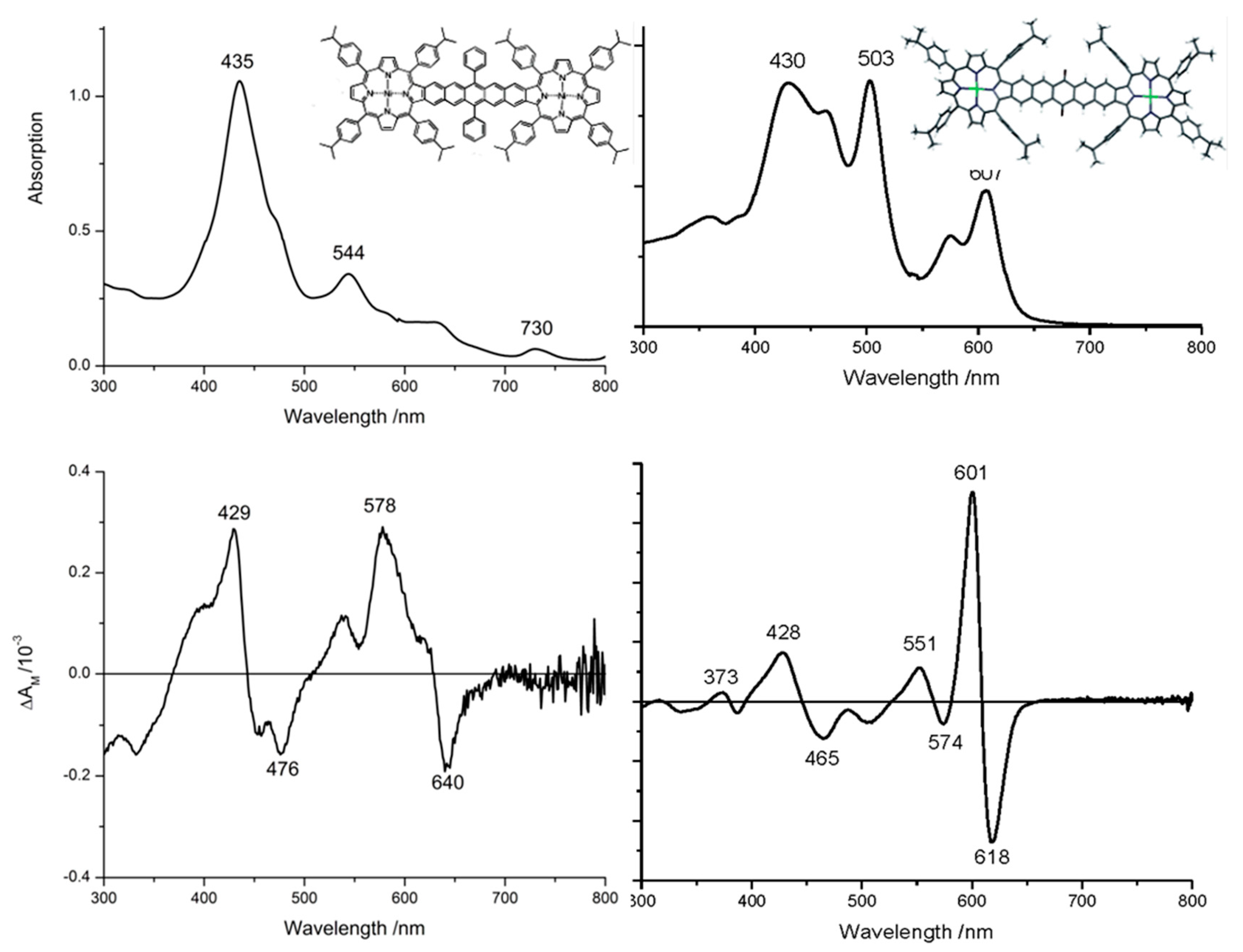
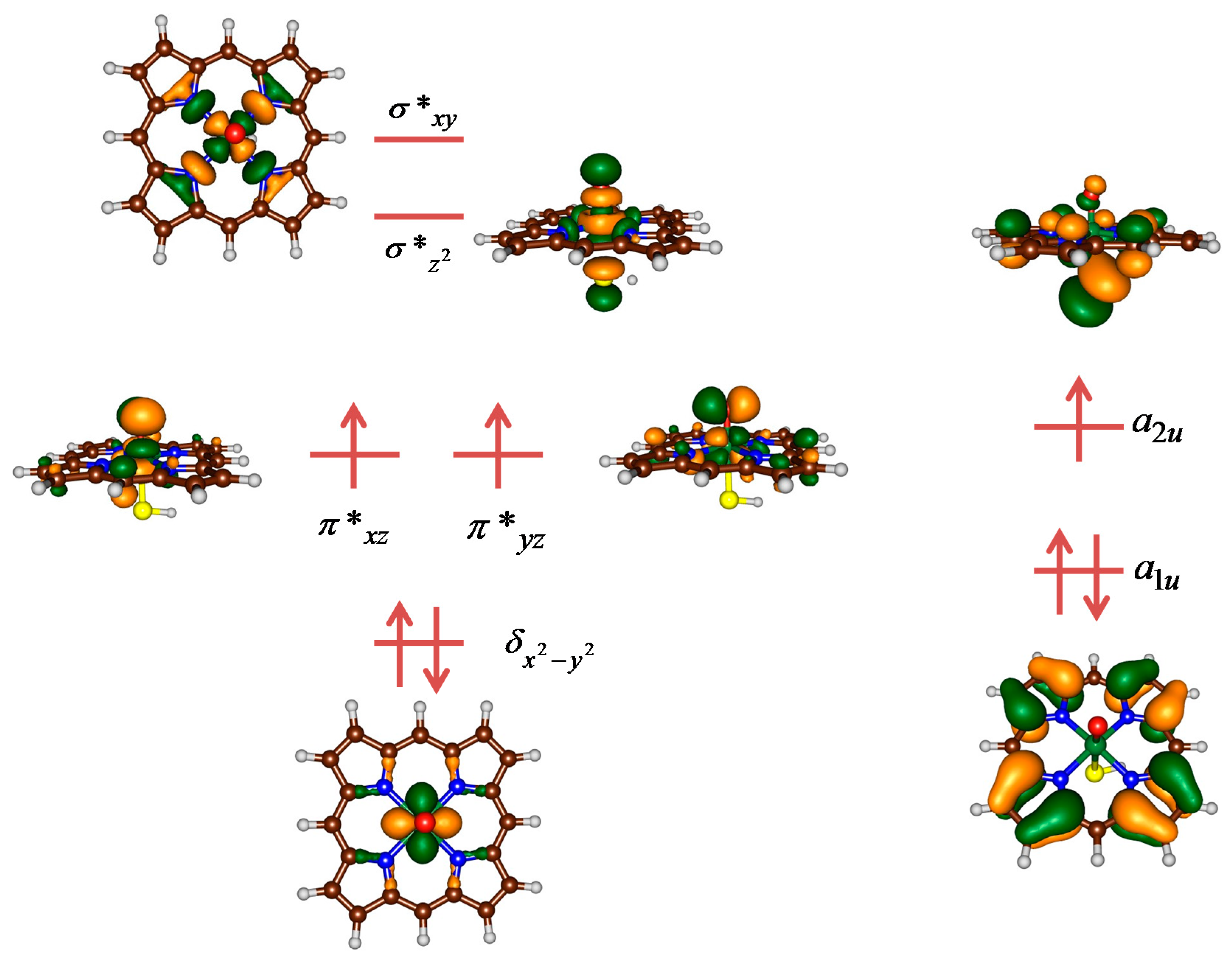
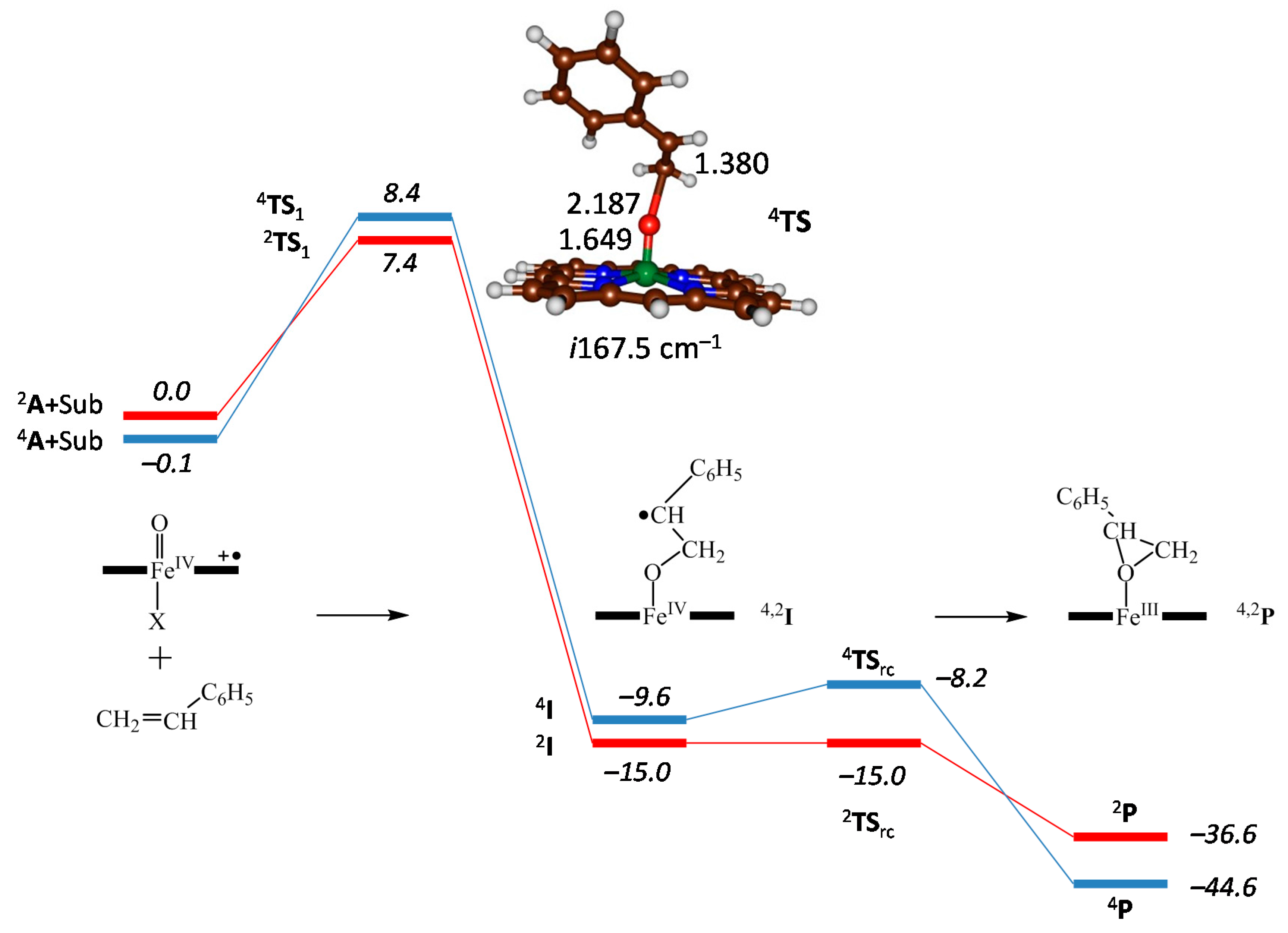
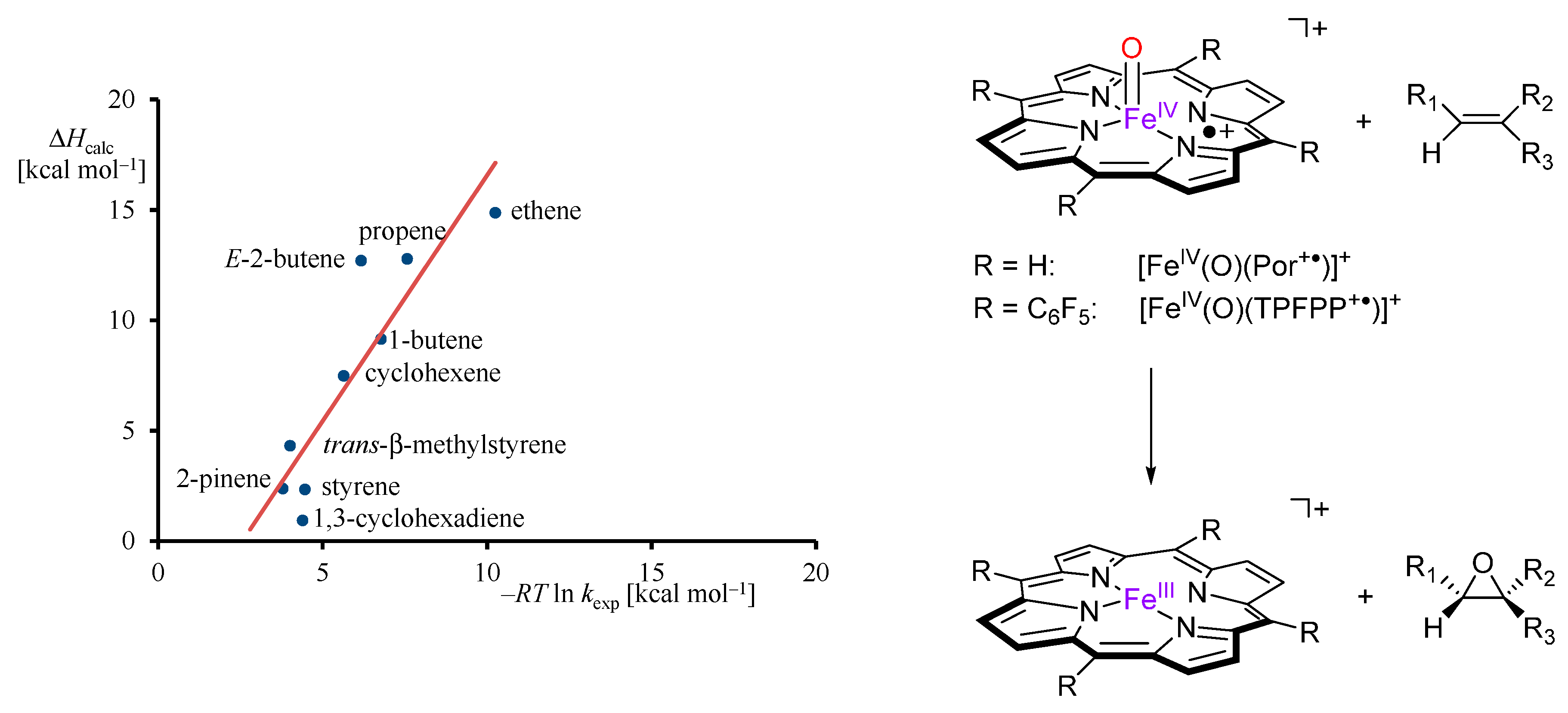
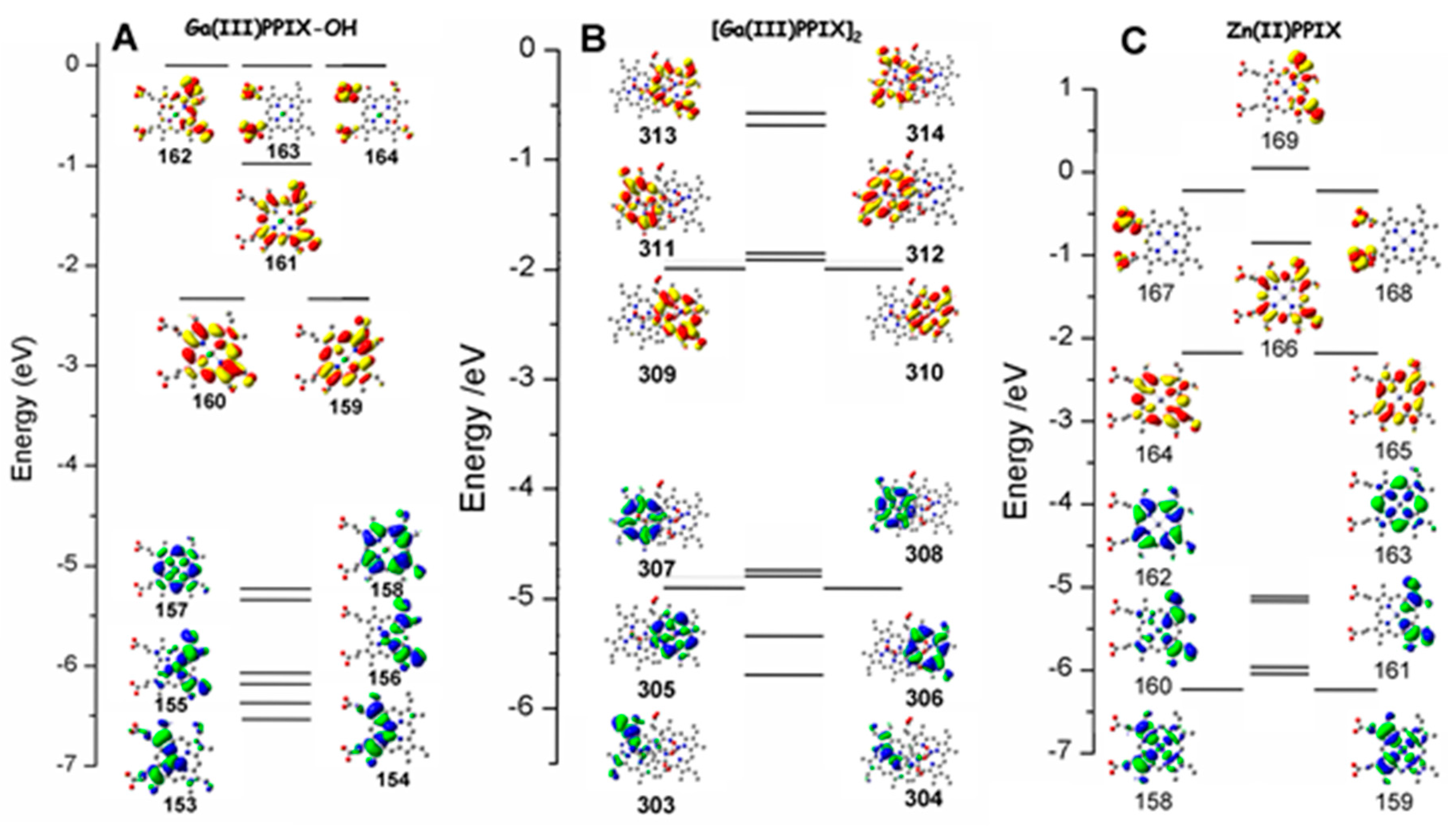
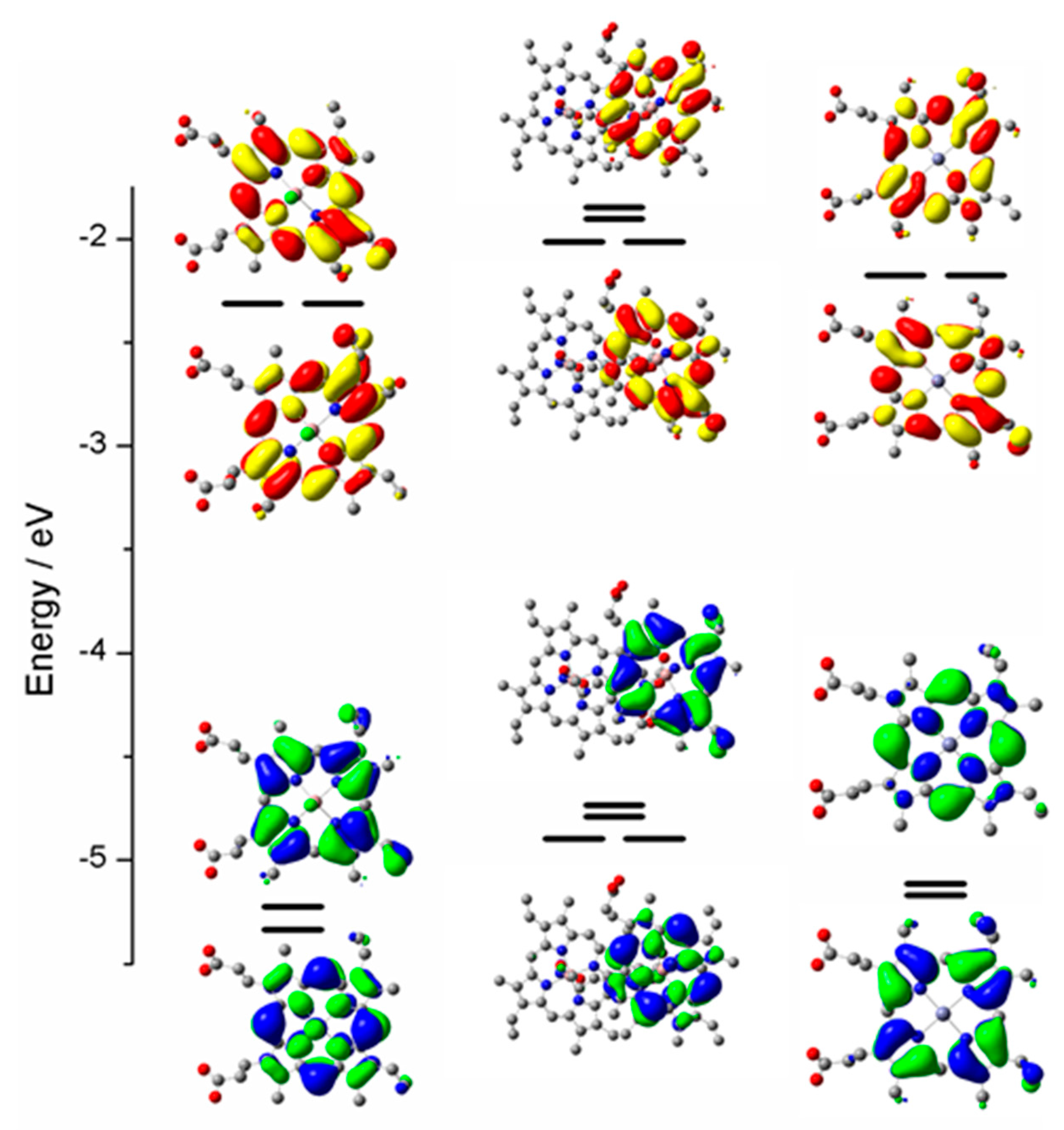
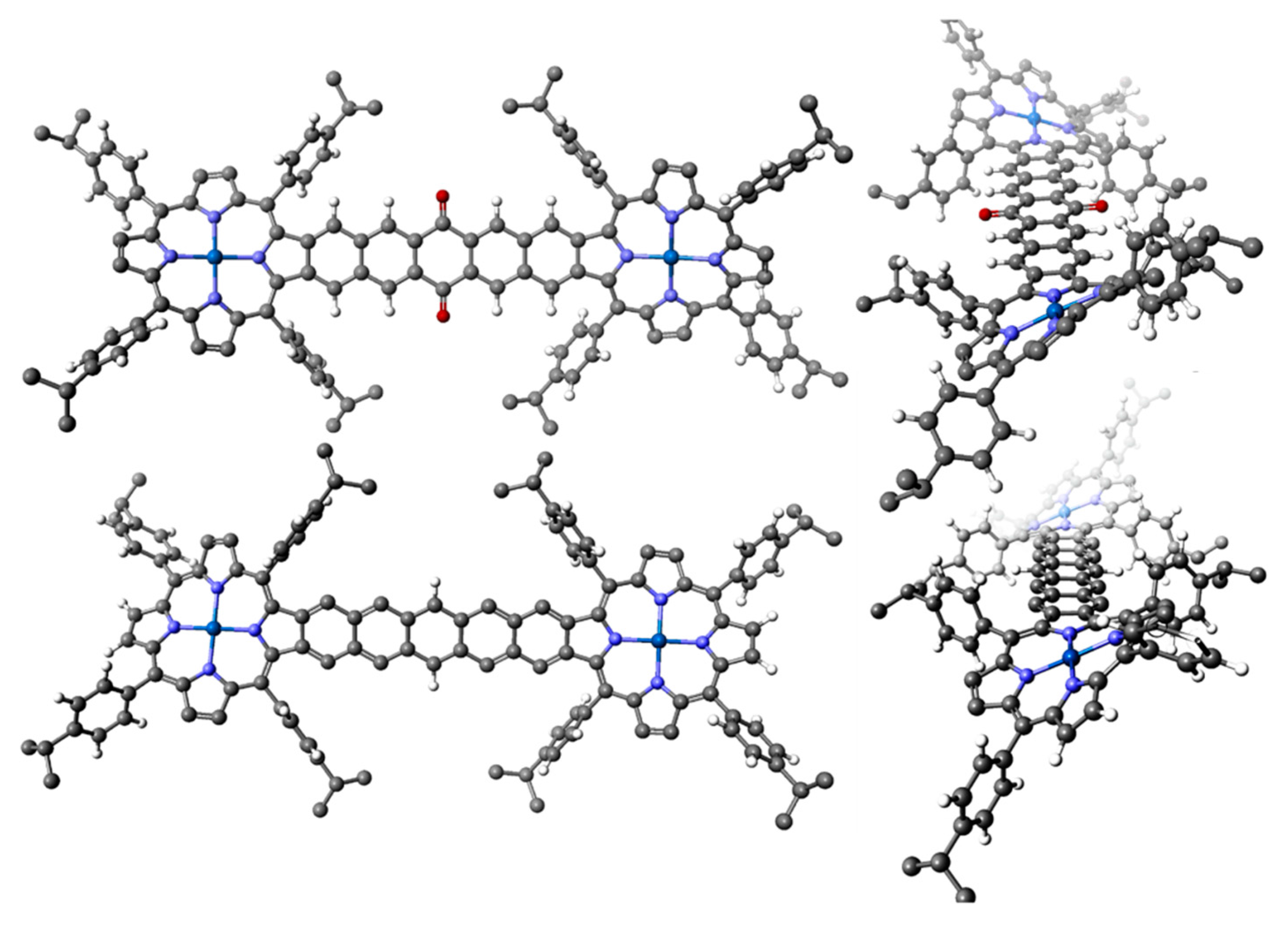
2.3. Case Study 3
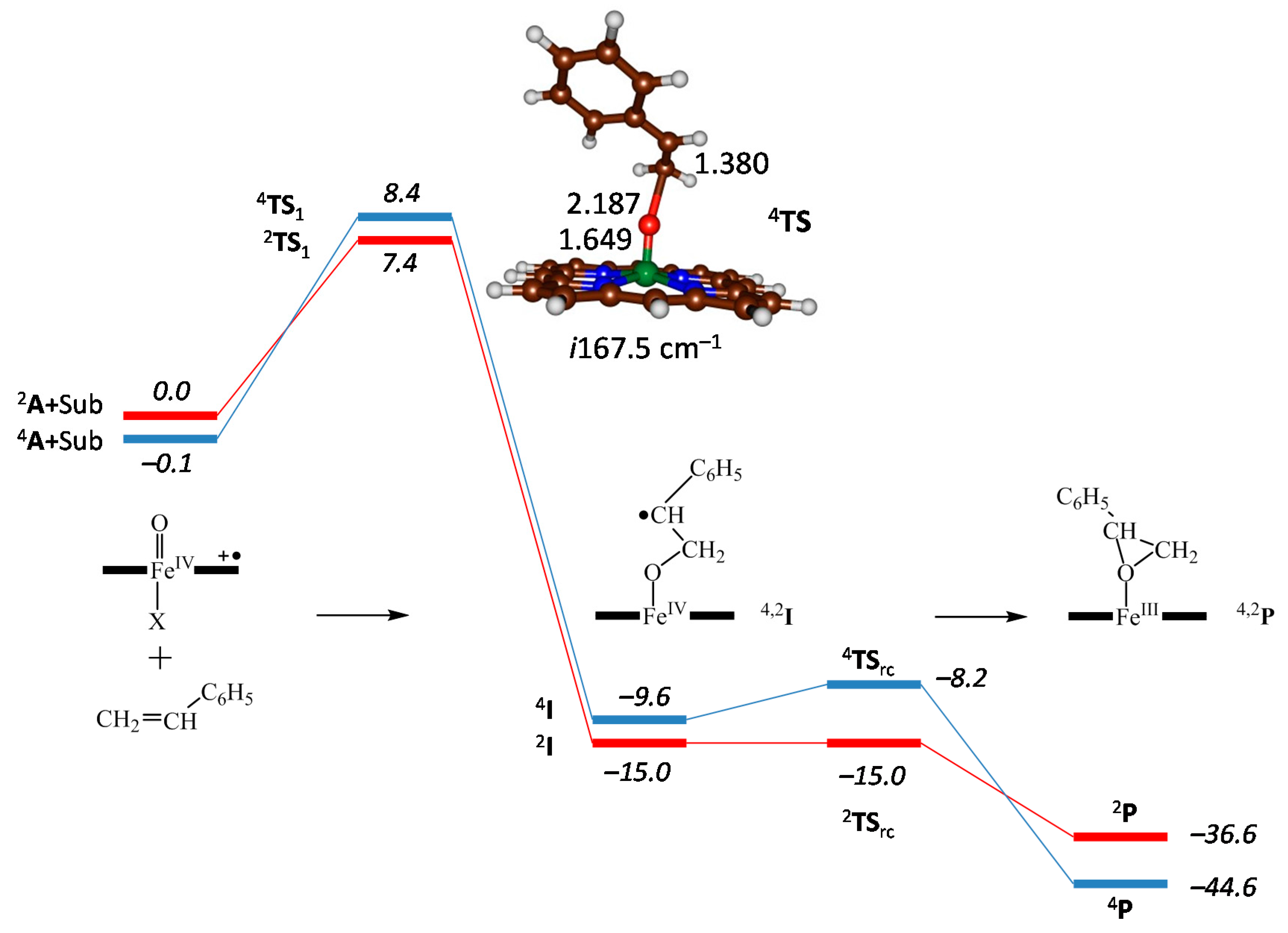
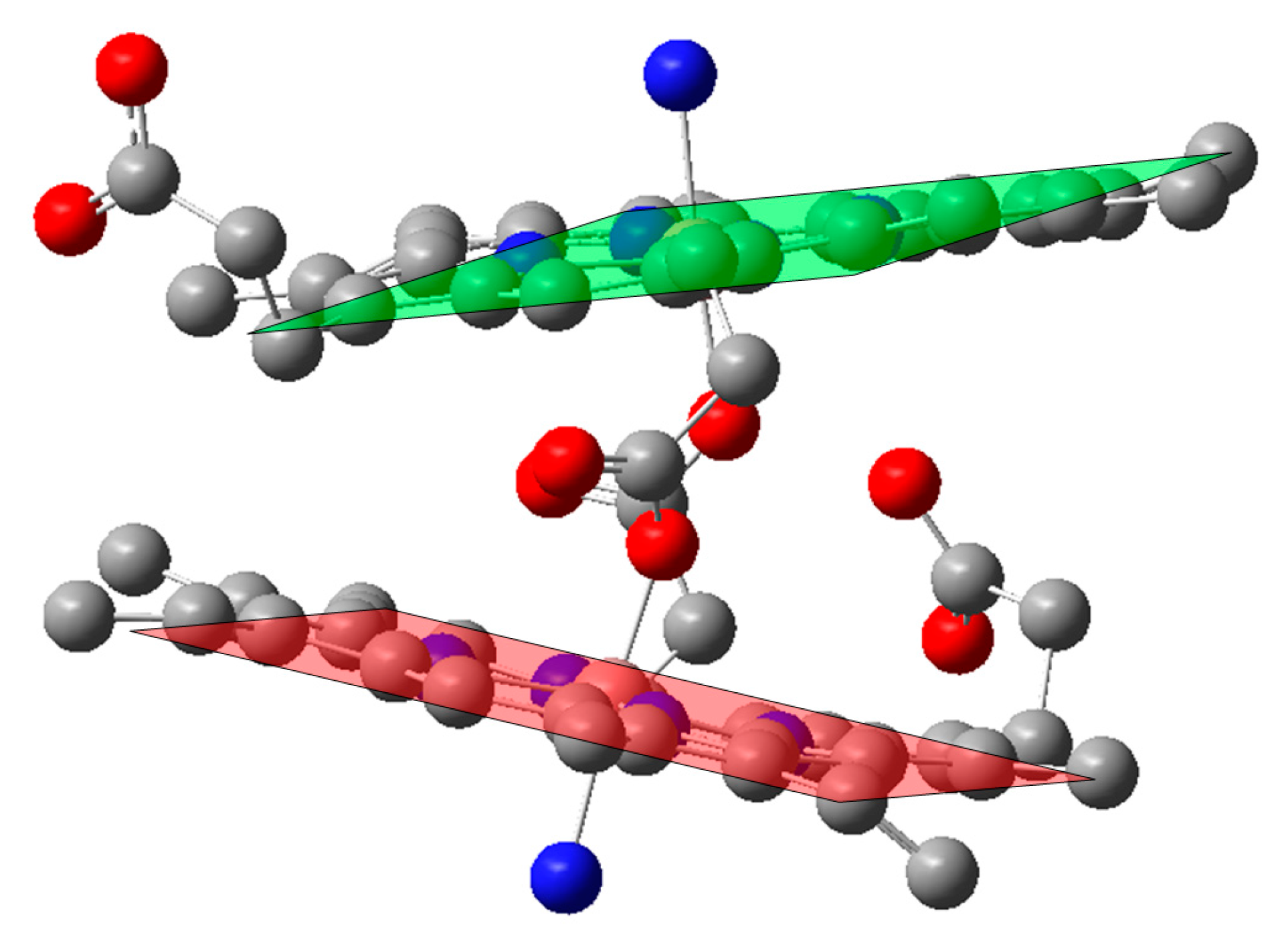
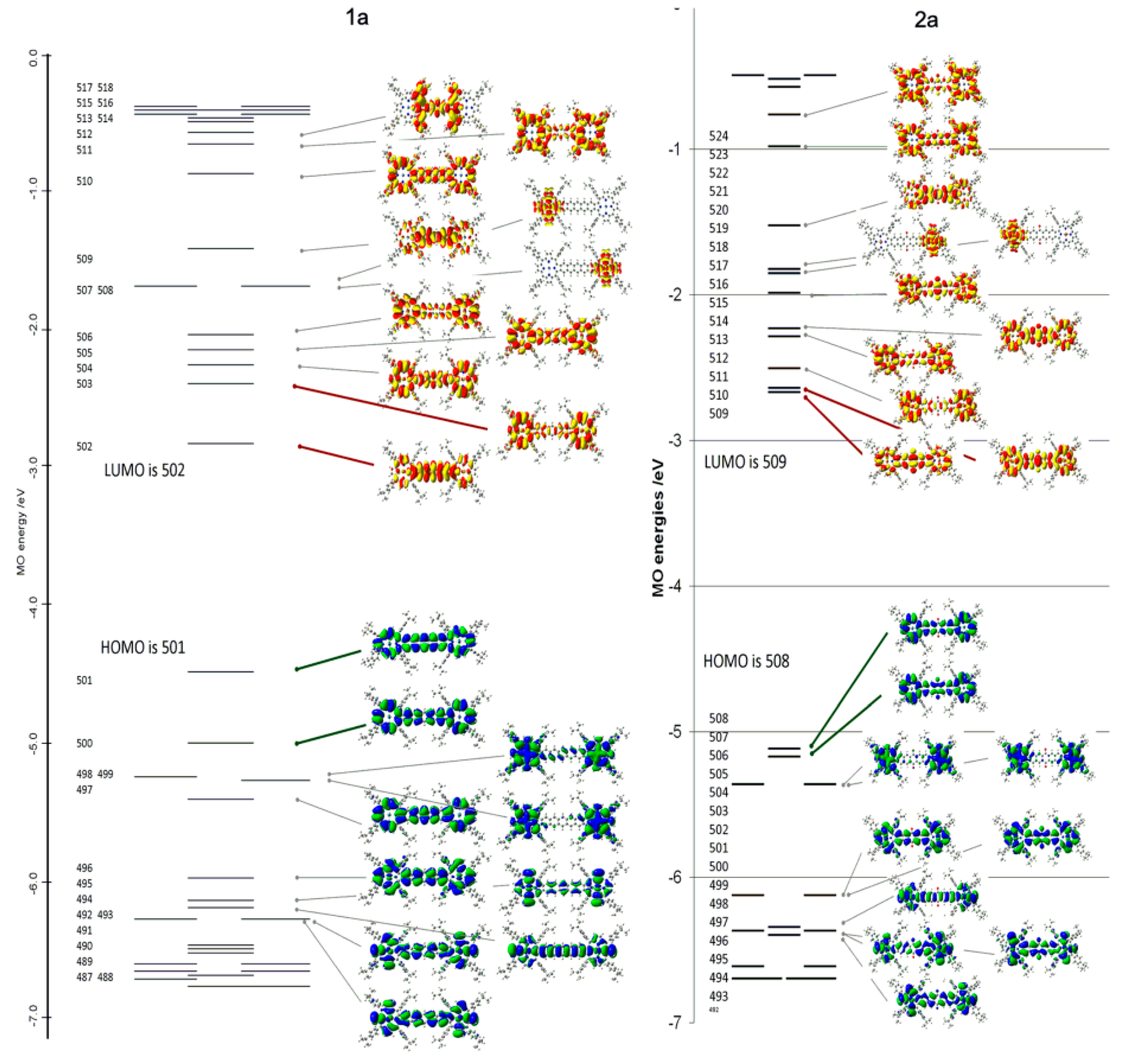
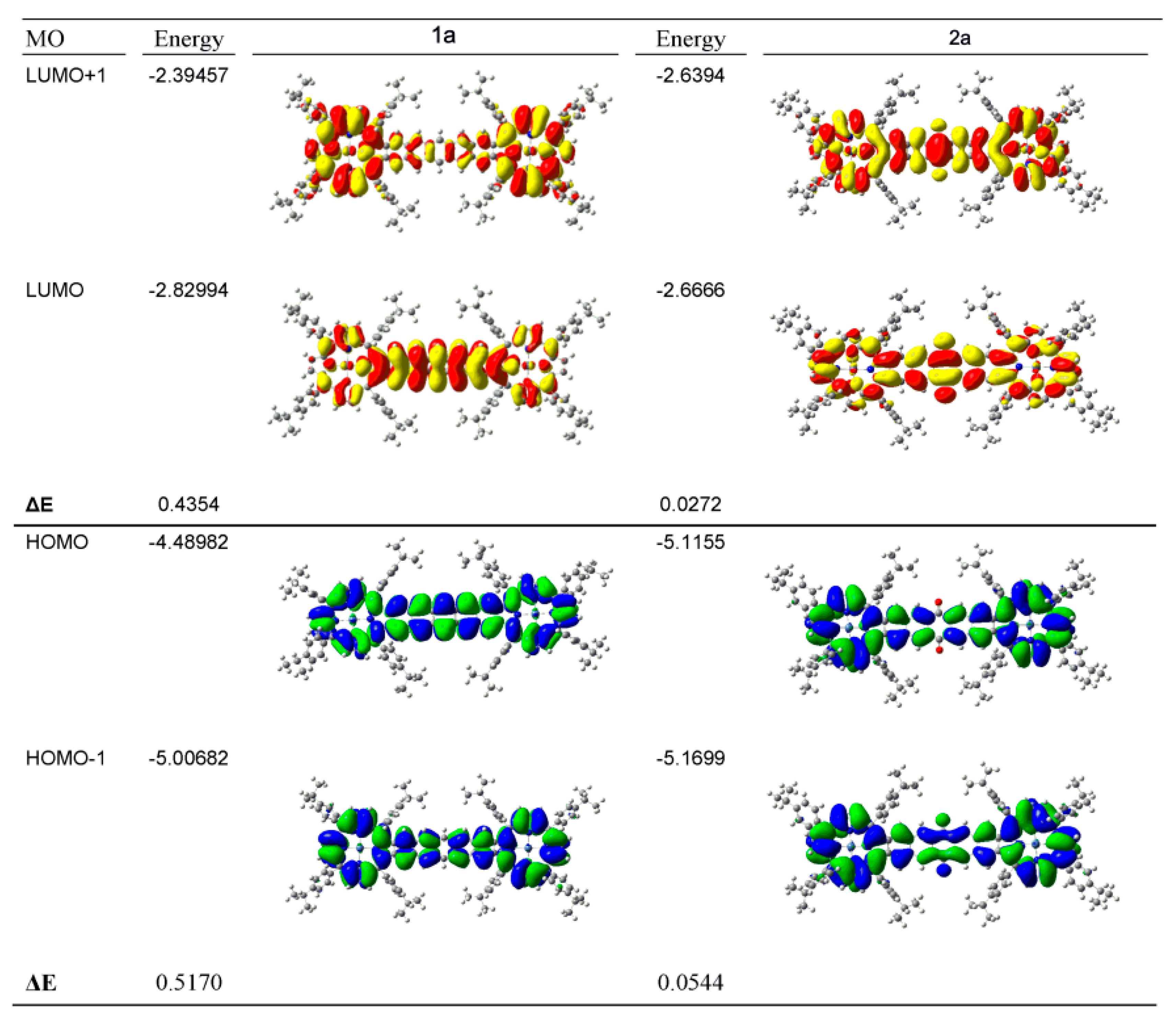
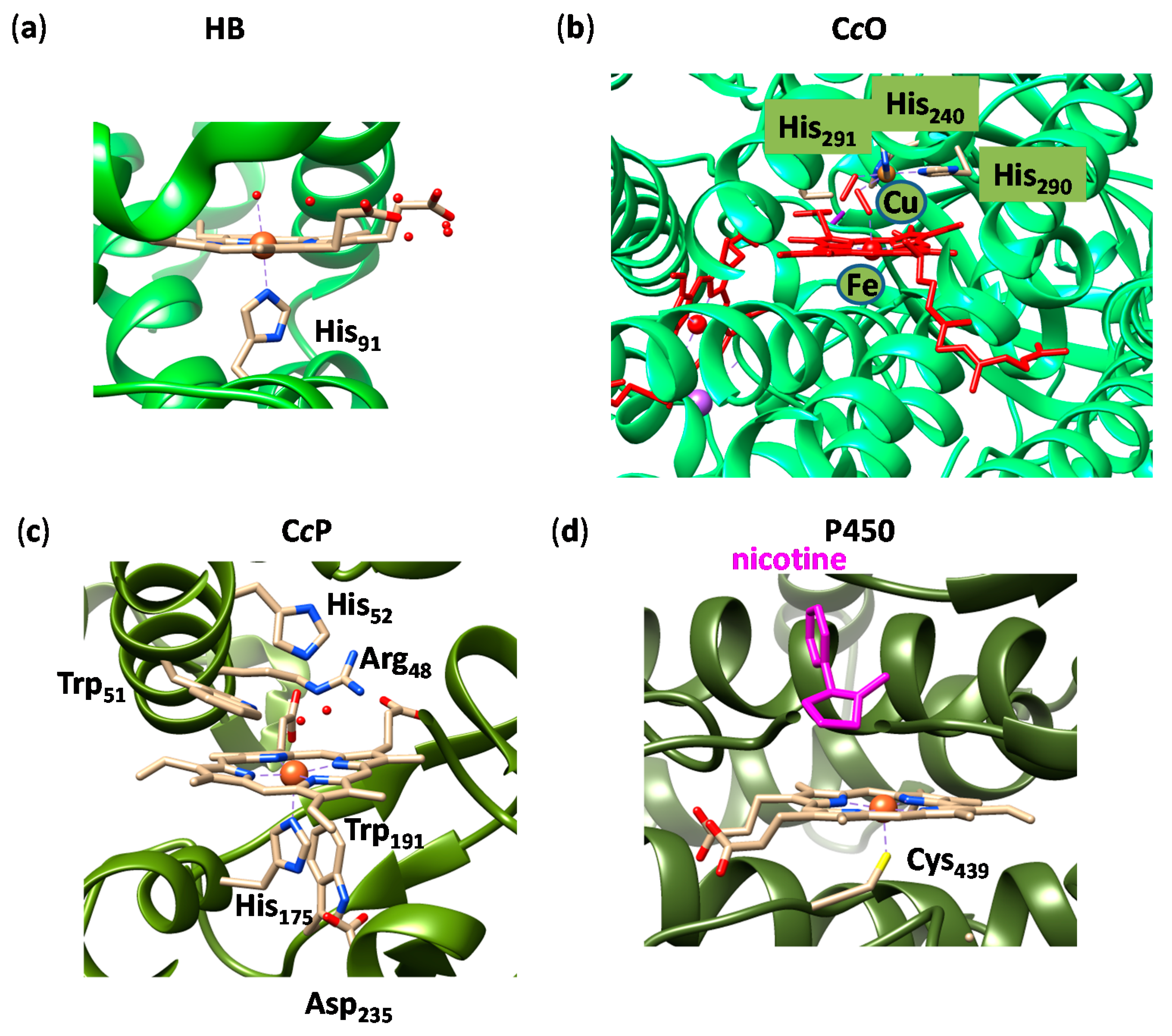
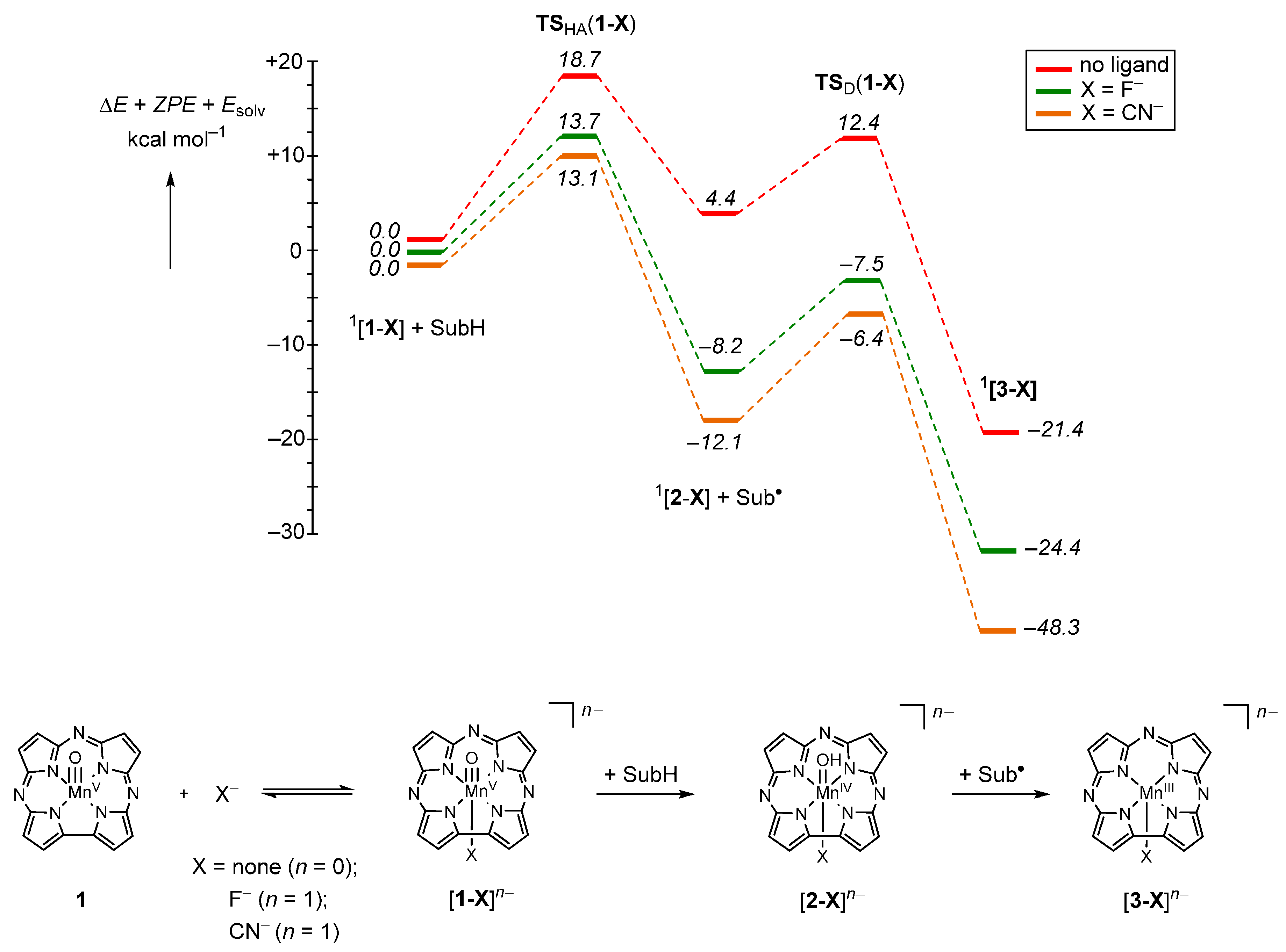
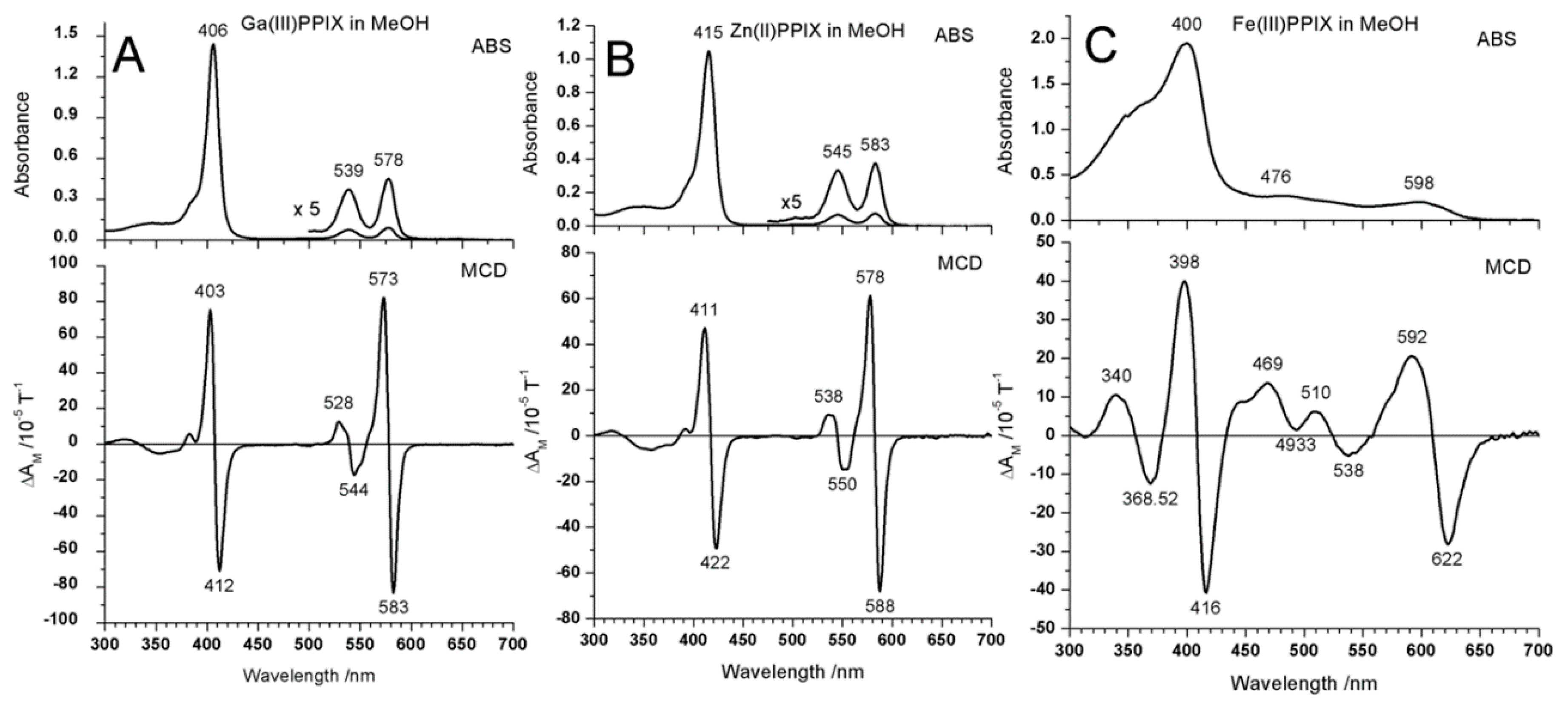
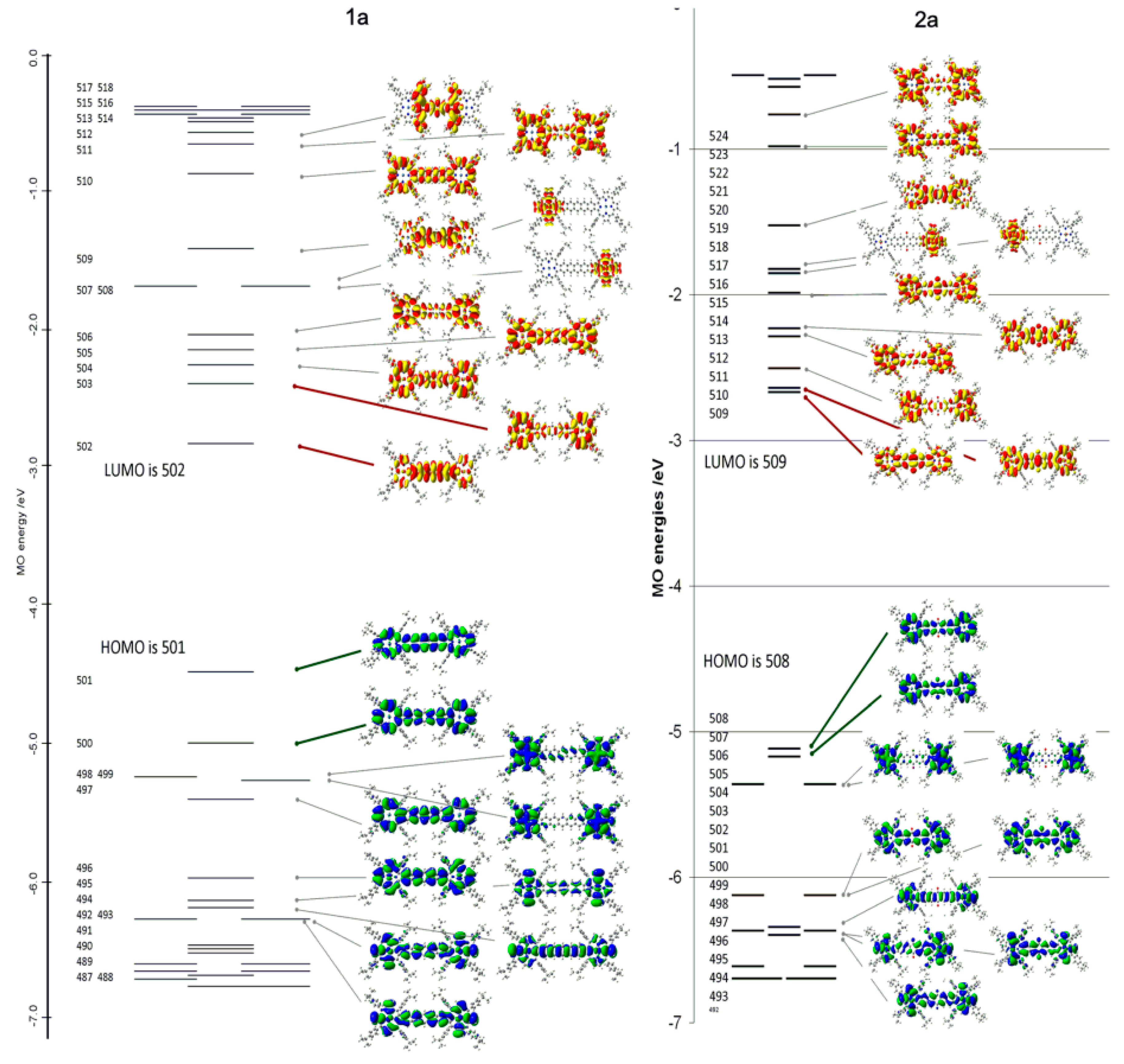
2.4. Case Study 4
3. Conclusions
Acknowledgments
Conflicts of Interest
References
- Kadish, K.M.; Smith, K.M.; Guilard, R. (Eds.) Handbook of Porphyrin Science; World Scientific Publishing Co.: Hackensack, NJ, USA, 2010.
- Ortiz de Montellano, P.R. Catalytic sites of hemoprotein peroxidases. Annu. Rev. Pharmacol. Toxicol. 1992, 32, 89–107. [Google Scholar] [CrossRef] [PubMed]
- Raven, E.L. Understanding functional diversity and substrate specificity in haem peroxidases: What can we learn from ascorbate peroxidase? Nat. Prod. Rep. 2003, 20, 367–381. [Google Scholar] [CrossRef] [PubMed]
- Poulos, T.L. Thirty years of heme peroxidase structural biology. Arch. Biochem. Biophys. 2010, 500, 3–45. [Google Scholar] [CrossRef] [PubMed]
- Hollmann, F.; Arends, I.W.C.E.; Buehler, K.; Schalley, A.; Bühler, B. Enzyme-mediated oxidations for the chemist. Green Chem. 2011, 13, 226–235. [Google Scholar] [CrossRef]
- Nicholls, P.; Fita, I.; Loewen, P.C. Enzymology and structure of catalases. Adv. Inorg. Chem. 2000, 51, 51–106. [Google Scholar]
- Sono, M.; Roach, M.P.; Coulter, E.D.; Dawson, J.H. Heme-containing oxygenases. Chem. Rev. 1996, 96, 2841–2887. [Google Scholar] [CrossRef] [PubMed]
- Ortiz de Montellano, P.R. (Ed.) Cytochrome P450: Structure, Mechanism, and Biochemistry, 3rd ed.; Kluwer Academic/Plenum Publishers: New York, NY, USA, 2004.
- Meunier, B.; de Visser, S.P.; Shaik, S. Mechanism of oxidation reactions catalyzed by cytochrome P450 enzymes. Chem. Rev. 2004, 104, 3947–3980. [Google Scholar] [CrossRef] [PubMed]
- Denisov, I.G.; Makris, T.M.; Sligar, S.G.; Schlichting, I. Structure and chemistry of cytochrome P450. Chem. Rev. 2005, 105, 2253–2277. [Google Scholar] [CrossRef] [PubMed]
- Munro, A.W.; Girvan, H.M.; McLean, K.J. Variations on a (t)heme—Novel mechanisms, redox partners and catalytic functions in the cytochrome P450 superfamily. Nat. Prod. Rep. 2007, 24, 585–609. [Google Scholar] [CrossRef] [PubMed]
- Ortiz de Montellano, P.R. Hydrocarbon hydroxylation by cytochrome P450 enzymes. Chem. Rev. 2010, 110, 932–948. [Google Scholar] [CrossRef] [PubMed]
- Guengerich, F.P. Common and uncommon cytochrome P450 reactions related to metabolism and chemical toxicity. Chem. Res. Toxicol. 2001, 14, 611–650. [Google Scholar] [CrossRef] [PubMed]
- Posner, G.H.; O’Neill, P.M. Knowledge of the proposed chemical mechanism of action and cytochrome P450 metabolism of antimalarial trioxanes like artemisinin allows rational design of new antimalarial peroxides. Acc. Chem. Res. 2004, 37, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, Y.; Nakajima, M.; Yokoi, T. Cytochrome P450-mediated metabolism of estrogens and its regulation in human. Cancer Lett. 2005, 227, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Krámos, B.; Oláh, J. Enolization as an alternative proton delivery pathway in human aromatase (P450 19A1). J. Phys. Chem. B 2014, 118, 390–405. [Google Scholar] [CrossRef] [PubMed]
- Groves, J.T. The bioinorganic chemistry of iron in oxygenases and supramolecular assemblies. Proc. Natl. Acad. Sci. USA 2003, 100, 3569–3574. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Nakajima, H.; Ueno, T. Reactivities of oxo and peroxo intermediates studied by hemoprotein mutants. Acc. Chem. Res. 2007, 40, 554–562. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P.; Kumar, D. (Eds.) Iron-Containing Enzymes: Versatile Catalysts of Hydroxylation Reaction in Nature; RSC Publishing: Cambridge, UK, 2011.
- Aranda, R.; Cai, H.; Worley, C.E.; Levin, E.J.; Li, R.; Olson, J.S.; Phillips, G.N., Jr.; Richards, M.P. Structural analysis of fish versus mammalian hemoglobins: Effect of the heme pocket environment on autooxidation and hemin loss. Proteins 2009, 75, 217–230. [Google Scholar] [CrossRef] [PubMed]
- Hirata, K.; Shinzawa-Itoh, K.; Yano, N.; Takemura, S.; Kato, K.; Hatanaka, M.; Muramoto, K.; Kawahara, T.; Tsukihara, T.; Yamashita, E.; et al. Determination of damage-free crystal structure of an X-ray–sensitive protein using an XFEL. Nat. Methods 2014, 11, 734–736. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.J.; Metcalfe, C.L.; Nnamchi, C.; Moody, P.C.E.; Raven, E.L. Crystal structure of guaiacol and phenol bound to a heme peroxidase. FEBS J. 2012, 279, 1632–1639. [Google Scholar] [CrossRef] [PubMed]
- DeVore, N.M.; Scott, E.E. Nicotine and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone binding and access channel in human cytochrome P450 2A6 and 2A13 enzymes. J. Biol. Chem. 2012, 287, 26576–26585. [Google Scholar] [CrossRef] [PubMed]
- Giardina, B.; Messana, I.; Scatena, R.; Castagnola, M. The multiple functions of hemoglobin. Crit. Rev. Biochem. Mol. Biol. 1995, 30, 165–196. [Google Scholar] [CrossRef] [PubMed]
- Derat, E.; Shaik, S. Two-state reactivity, electromerism, tautomerism, and “surprise” isomers in the formation of compound II of the enzyme horseradish peroxidase from the principal species, compound I. J. Am. Chem. Soc. 2006, 128, 8185–8198. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P.; Nam, W. High-valent iron-oxo porphyrins in oxygenation reactions. In Handbook of Porphyrin Science; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; Chapter 44; World Scientific Publishing Co.: Hackensack, NJ, USA, 2010; pp. 85–140. [Google Scholar]
- Sivaraja, M.; Goodin, D.B.; Smith, M.; Hoffman, B.M. Identification by ENDOR of Trp191 as the free-radical site in cytochrome c peroxidase compound ES. Science 1989, 245, 738–740. [Google Scholar] [CrossRef] [PubMed]
- Huyett, J.E.; Doan, P.E.; Gurbiel, R.; Houseman, A.L.P.; Sivaraja, M.; Goodin, D.B.; Hoffman, B.M. Compound ES of cytochrome c peroxidase contains a Trp π-cation radical: Characterization by continuous wave and pulsed Q-band external nuclear double resonance spectroscopy. J. Am. Chem. Soc. 1995, 117, 9033–9041. [Google Scholar] [CrossRef]
- De Visser, S.P. What affects the quartet-doublet energy splitting in peroxidase enzymes? J. Phys. Chem. B 2005, 109, 11050–11057. [Google Scholar] [CrossRef] [PubMed]
- Michel, H.; Behr, J.; Harrenga, A.; Kannt, A. Cytochrome c oxidase: Structure and spectroscopy. Annu. Rev. Biophys. Biomol. Struct. 1998, 27, 329–356. [Google Scholar] [CrossRef] [PubMed]
- Capaldi, R.A. Structure and function of cytochrome c oxidase. Annu. Rev. Biochem. 1990, 59, 569–596. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.C.; Hummer, G. Cytochrome c oxidase structure and function. Biochem. Biophys. Acta 2012, 1817, 526–536. [Google Scholar] [PubMed]
- Sun, Y.; Benabbas, A.; Zeng, W.; Kleingardner, J.G.; Bren, K.L.; Champion, P.M. Investigations of heme distortion, low-frequency vibrational excitations, and electron transfer in cytochrome c. Proc. Natl. Acad. Sci. USA 2014, 111, 6570–6575. [Google Scholar] [CrossRef] [PubMed]
- Kleingardner, J.G.; Bren, K.L. Biological significance and applications of heme c proteins and peptides. Acc. Chem. Res. 2015, 48, 1845–1852. [Google Scholar] [CrossRef] [PubMed]
- Mack, J.; Stillman, M.J. Photochemical formation of the anion radical of zinc phthalocyanine and analysis of the absorption and magnetic circular dichroism spectral data. Assignment of the optical spectrum of [ZnPc(-3)]−. J. Am. Chem. Soc. 1994, 116, 1292–1304. [Google Scholar] [CrossRef]
- Poulos, T.L. Heme enzyme structure and function. Chem. Rev. 2014, 114, 3919–3962. [Google Scholar] [CrossRef] [PubMed]
- Alfonso-Prieto, M.; Biarnés, X.; Vidossich, P.; Rovira, C. The molecular mechanism of the catalase reaction. J. Am. Chem. Soc. 2009, 131, 11751–11761. [Google Scholar] [CrossRef] [PubMed]
- Dawson, J.H.; Holm, R.H.; Trudell, J.R.; Barth, G.; Linder, R.E.; Bunnenberg, E.; Djerassi, C.; Tang, S.C. Magnetic circular dichroism studies. 43. Oxidized cytochrome P-450. Magnetic circular dichroism evidence for thiolate ligation in the substrate-bound form. Implications for the catalytic mechanism. J. Am. Chem. Soc. 1976, 98, 3707–3709. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P. What external perturbations influence the electronic properties of catalase compound I? Inorg. Chem. 2006, 45, 9551–9557. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P. Trends in substrate hydroxylation reactions by heme and nonheme iron(IV)-oxo oxidants give correlations between intrinsic properties of the oxidant with barrier height. J. Am. Chem. Soc. 2010, 132, 1087–1097. [Google Scholar] [CrossRef] [PubMed]
- Gross, Z.; Nimri, S. A pronounced axial ligand effect on the reactivity of oxoiron(IV) porphyrin cation radicals. Inorg. Chem. 1994, 33, 1731–1732. [Google Scholar] [CrossRef]
- Gross, Z. The effect of axial ligands on the reactivity and stability of the oxoferryl moiety in model complexes of compound I of heme-dependent enzymes. J. Biol. Inorg. Chem. 1996, 1, 368–371. [Google Scholar] [CrossRef]
- Czarnecki, K.; Nimri, S.; Gross, Z.; Proniewicz, L.M.; Kincaid, J.R. Direct resonance Raman evidence for a trans influence on the ferryl fragment in models of compound I intermediates of heme enzymes. J. Am. Chem. Soc. 1996, 118, 2929–2935. [Google Scholar] [CrossRef]
- Green, M.T. Application of Badger’s rule to heme and non-heme iron-oxygen bonds: An examination of ferryl protonation states. J. Am. Chem. Soc. 2006, 128, 1902–1906. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.; Faponle, A.S.; Quesne, M.G.; Sainna, M.A.; Zhang, J.; Franke, A.; Kumar, D.; van Eldik, R.; Liu, W.; de Visser, S.P. Drug metabolism by cytochrome P450 enzymes: What distinguishes the pathways leading to substrate hydroxylation over desaturation? Chem. Eur. J. 2015, 21, 9083–9092. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P.; Shaik, S.; Sharma, P.K.; Kumar, D.; Thiel, W. Active species of horseradish peroxidase (HRP) and cytochrome P450: Two electronic chameleons. J. Am. Chem. Soc. 2003, 125, 15779–15788. [Google Scholar] [CrossRef] [PubMed]
- Shaik, S.; Kumar, D.; de Visser, S.P.; Altun, A.; Thiel, W. Theoretical perspective on the structure and mechanism of cytochrome P450 enzymes. Chem. Rev. 2005, 105, 2279–2328. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P.; Ogliaro, F.; Shaik, S. How does ethene inactivate cytochrome P450 en route to its epoxidation? A density functional study. Angew. Chem. Int. Ed. 2001, 40, 2871–2874. [Google Scholar] [CrossRef]
- De Visser, S.P. Substitution of hydrogen by deuterium changes the regioselectivity of ethylbenzene hydroxylation by an oxo-iron-porphyrin catalyst. Chem. Eur. J. 2006, 12, 8168–8177. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, D.; Quesne, M.G.; de Visser, S.P.; Rath, S.P. Hydrogen bonding interactions trigger a spin-flip in iron(III)-porphyrin complexes. Angew. Chem. Int. Ed. 2015, 54, 4796–4800. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Schuller, D.J.; Lanzilotta, W.N.; Sundaramoorthy, M.; Arciero, D.M.; Hooper, A.B.; Poulos, T.L. Crystal structure of Nitrosomonas europaea cytochrome c peroxidase and the structural basis for ligand switching in bacterial di-heme peroxidases. Biochemistry 2001, 40, 13483–13490. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Furuyama, T.; Tang, J.; Wu, Z.-Y.; Chen, J.-Z.; Kobayashi, N.; Zhang, J.-L. Stable iso-bacteriochlorin mimics from porpholactone: Effect of a β-oxazolone moiety on the frontier π-molecular orbitals. Inorg. Chem. Front. 2015, 2, 671–677. [Google Scholar] [CrossRef]
- Chen, M.; Schliep, M.; Willows, R.D.; Cai, Z.L.; Neilan, B.A.; Scheer, H. A red-shifted chlorophyll. Science 2010, 329, 1318–1319. [Google Scholar] [CrossRef] [PubMed]
- Linstead, R.P. Phthalocyanines Part I. A new type of synthetic colouring matters. J. Chem. Soc. 1934, 1934, 1016–1017. [Google Scholar] [CrossRef]
- Mathew, S.; Yella, A.; Gao, P.; Humphry-Baker, R.; Curchod, B.F.; Ashari-Astani, N.; Tavernelli, I.; Rothlisberger, U.; Nazeeruddin, M.K.; Grätzel, M. Dye-sensitized solar cells with 13% efficiency achieved through the molecular engineering of porphyrin sensitizers. Nat. Chem. 2014, 6, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.J.; Chen, Y.; Joshi, P.; Pera, P.; Baumann, H.; Missert, J.R.; Ohkubo, K.; Fukuzumi, S.; Nani, R.R.; Schnermann, M.J.; et al. Effect of metalation on porphyrin-based bifunctional agents in tumor imaging and photodynamic therapy. Bioconjug. Chem. 2016, 27, 667–680. [Google Scholar] [CrossRef] [PubMed]
- Zucchelli, G.; Santabarbara, S.; Jennings, R.C. The Q(y) absorption spectrum of the light-harvesting complex II as determined by structure-based analysis of chlorophyll macrocycle deformations. Biochemistry 2012, 51, 2717–2736. [Google Scholar] [CrossRef] [PubMed]
- Jornet-Somoza, J.; Alberdi-Rodriguez, J.; Milne, B.F.; Andrade, X.; Marques, M.A.; Nogueira, F.; Oliveira, M.J.; Stewart, J.J.; Rubio, A. Insights into colour-tuning of chlorophyll optical response in green plants. Phys. Chem. Chem. Phys. 2015, 17, 26599–26606. [Google Scholar] [CrossRef] [PubMed]
- Mack, J.; Stillman, M.J.; Kobayashi, N. Application of MCD spectroscopy to porphyrinoids. Coord. Chem. Rev. 2007, 251, 429–453. [Google Scholar] [CrossRef]
- Sainna, M.A.; Kumar, S.; Kumar, D.; Fornarini, S.; Crestoni, M.E.; de Visser, S.P. A comprehensive test set of epoxidation rate constants by iron(IV)-oxo porphyrin complexes. Chem. Sci. 2015, 6, 1516–1529. [Google Scholar] [CrossRef]
- Kumar, D.; de Visser, S.P.; Shaik, S. Multistate reactivity in styrene epoxidation by compound I of cytochrome P450: Mechanisms of products and side products formation. Chem. Eur. J. 2005, 11, 2825–2835. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Latifi, R.; Kumar, S.; Rybak-Akimova, E.V.; Sainna, M.A.; de Visser, S.P. Rationalization of the barrier height for para-Z-styrene epoxidation by iron(IV)-oxo porphyrins with variable axial ligands. Inorg. Chem. 2013, 52, 7968–7979. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P.; Ogliaro, F.; Harris, N.; Shaik, S. Multi-state epoxidation of ethene by cytochrome P450: A quantum chemical study. J. Am. Chem. Soc. 2001, 123, 3037–3047. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P.; Ogliaro, F.; Sharma, P.K.; Shaik, S. What factors affect the regioselectivity of oxidation by cytochrome P450? A DFT study of allylic hydroxylation and double bond epoxidation in a model reaction. J. Am. Chem. Soc. 2002, 124, 11809–11826. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P.; Ogliaro, F.; Sharma, P.K.; Shaik, S. Hydrogen bonding modulates the selectivity of enzymatic oxidation by P450: A chameleon oxidant behavior of compound I. Angew. Chem. Int. Ed. 2002, 41, 1947–1951. [Google Scholar] [CrossRef]
- De Visser, S.P. The axial ligand effect of oxo-iron porphyrin catalysts. How does chloride compare to thiolate? J. Biol. Inorg. Chem. 2006, 11, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Karamzadeh, B.; Sastry, G.N.; de Visser, S.P. What factors influence the rate constant of substrate epoxidation by compound I of cytochrome P450 and analogous iron(IV)-oxo oxidants. J. Am. Chem. Soc. 2010, 132, 7656–7667. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P.; Tan, L.S. Is the bound substrate in nitric oxide synthase protonated or neutral and what is the active oxidant that performs substrate hydroxylation? J. Am. Chem. Soc. 2008, 130, 12961–12974. [Google Scholar] [CrossRef] [PubMed]
- Groves, J.T.; Avaria-Neisser, G.E.; Fish, K.M.; Imachi, M.; Kuczkowski, R.L. Hydrogen-deuterium exchange during propylene epoxidation by cytochrome P-450. J. Am. Chem. Soc. 1986, 108, 3837–3838. [Google Scholar] [CrossRef]
- Vardhaman, A.K.; Sastri, C.V.; Kumar, D.; de Visser, S.P. Nonheme ferric hydroperoxo intermediates are efficient oxidants of bromide oxidation. Chem. Commun. 2011, 47, 11044–11046. [Google Scholar] [CrossRef] [PubMed]
- Vardhaman, A.K.; Barman, P.; Kumar, S.; Sastri, C.V.; Kumar, D.; de Visser, S.P. Comparison of the reactivity of nonheme iron(IV)-oxo versus iron(IV)-imido complexes: Which is the better oxidant? Angew. Chem. Int. Ed. 2013, 52, 12288–12292. [Google Scholar] [CrossRef] [PubMed]
- Vardhaman, A.K.; Barman, P.; Kumar, S.; Sastri, C.V.; Kumar, D.; de Visser, S.P. Mechanistic insight into halide oxidation by non-heme iron complexes. Haloperoxidase versus halogenase activity. Chem. Commun. 2013, 49, 10926–10928. [Google Scholar] [CrossRef] [PubMed]
- Faponle, A.S.; Quesne, M.G.; Sastri, C.V.; Banse, F.; de Visser, S.P. Differences and comparisons of the properties and reactivities of iron(III)-hydroperoxo complexes with saturated coordination sphere. Chem. Eur. J. 2015, 21, 1221–1236. [Google Scholar] [CrossRef] [PubMed]
- Pratter, S.M.; Konstantinovics, C.; DiGiuro, C.L.M.; Leitner, E.; Kumar, D.; de Visser, S.P.; Grogan, G.; Straganz, G.D. Inversion of enantio-selectivity of a mononuclear non-heme iron(II)-dependent hydroxylase by tuning the interplay of metal-center geometry and protein structure. Angew. Chem. Int. Ed. 2013, 52, 9677–9681. [Google Scholar] [CrossRef] [PubMed]
- Gross, Z. High-valent corrole metal complexes. J. Biol. Inorg. Chem. 2001, 6, 733–738. [Google Scholar] [CrossRef] [PubMed]
- Abu-Omar, M.M.; Loaiza, A.; Hontzeas, N. Reaction mechanisms of mononuclear non-heme iron oxygenases. Chem. Rev. 2005, 105, 2227–2252. [Google Scholar] [CrossRef] [PubMed]
- Palmer, J.H. Transition Metal Corrole Coordination Chemistry. In Structure and Bonding; Springer: Berlin, Germany; Heidelberg, Germany, 2012. [Google Scholar]
- Neu, H.M.; Baglia, R.A.; Goldberg, D.P. A balancing act: Stability versus reactivity of Mn(O) complexes. Acc. Chem. Res. 2015, 48, 2754–2764. [Google Scholar] [CrossRef] [PubMed]
- İşci, Ü.; Faponle, A.S.; Afanasiev, P.; Albrieux, F.; Briois, V.; Ahsen, V.; Dumoulin, F.; Sorokin, A.B.; de Visser, S.P. Site-selective formation of an iron(IV)-oxo species at the more electron-rich iron atom of heteroleptic µ-nitrido diiron Phthalocyanines. Chem. Sci. 2015, 6, 5063–5075. [Google Scholar] [CrossRef]
- Prokop, K.A.; de Visser, S.P.; Goldberg, D.P. Unprecedented rate enhancements of hydrogen-atom transfer to a manganese(V)-oxo corrolazine complex. Angew. Chem. Int. Ed. 2010, 49, 5091–5095. [Google Scholar] [CrossRef] [PubMed]
- Prokop, K.A.; Neu, H.M.; de Visser, S.P.; Goldberg, D.P. A manganese(V)-oxo π-cation radical complex: Influence of one-electron oxidation on oxygen-atom transfer. J. Am. Chem. Soc. 2011, 133, 15874–15877. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Faponle, A.S.; Barman, P.; Vardhaman, A.K.; Sastri, C.V.; Kumar, D.; de Visser, S.P. Long-range electron transfer triggers mechanistic differences between iron(IV)-oxo and iron(IV)-imido oxidants. J. Am. Chem. Soc. 2014, 136, 17102–17115. [Google Scholar] [CrossRef] [PubMed]
- Leeladee, P.; Baglia, R.A.; Prokop, K.A.; Latifi, R.; de Visser, S.P.; Goldberg, D.P. Valence tautomerism in a high-valent manganese-oxo porphyrinoid complex induced by a Lewis acid. J. Am. Chem. Soc. 2012, 134, 10397–10400. [Google Scholar] [CrossRef] [PubMed]
- Neu, H.M.; Quesne, M.G.; Yang, T.; Prokop-Prigge, K.A.; Lancaster, K.M.; Donohoe, J.; DeBeer, S.; de Visser, S.P.; Goldberg, D.P. Dramatic influence of an anionic donor on the oxygen-atom-transfer reactivity of an Mn(V)-oxo complex. Chem. Eur. J. 2014, 20, 14584–14588. [Google Scholar] [CrossRef] [PubMed]
- Neu, H.M.; Yang, T.; Baglia, R.A.; Yosca, T.H.; Green, M.T.; Quesne, M.G.; de Visser, S.P.; Goldberg, D.P. Oxygen-atom transfer reactivity of axially ligated Mn(V)–Oxo complexes: Evidence for enhanced electrophilic and nucleophilic pathways. J. Am. Chem. Soc. 2014, 136, 13845–13852. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P.; Quesne, M.G.; Martin, B.; Comba, P.; Ryde, U. Computational modelling of oxygenation processes in enzymes and biomimetic model complexes. Chem. Commun. 2014, 50, 262–282. [Google Scholar] [CrossRef] [PubMed]
- Quesne, M.G.; Borowski, T.; de Visser, S.P. Quantum mechanics/molecular mechanics modelling of enzymatic processes: Caveats and breakthroughs. Chem. Eur. J. 2016, 22, 2562–2581. [Google Scholar] [CrossRef] [PubMed]
- Mack, J.; Stillman, M.J. Band deconvolution analysis of the absorption and magnetic circular dichroism spectral data of ZnPc(-2) recorded at cryogenic temperatures. J. Phys. Chem. 1995, 99, 7935–7945. [Google Scholar] [CrossRef]
- Coutsolelos, A.; Guilard, R.; Bayeul, D.; Lecomte, C. Gallium(III) porphyrins: Synthesis and physicochemical characteristics of halogeno gallium(III) porphyrins—X-ray crystal structure of chloro-(5,10,15,20-tetraphenylporphyrinato) gallium(III). Polyhedron 1986, 5, 1157–1164. [Google Scholar] [CrossRef]
- Arasasingham, R.D.; Balch, A.L.; Olmstead, M.M.; Philips, S.L. Paramagnetic iron(III) and diamagnetic gallium(III) porphyrin complexes with axial allyl and vinyl ligands. Inorg. Chim. Acta 1997, 263, 161–170. [Google Scholar] [CrossRef]
- Bohle, D.S.; Dodd, E.L.; Pinter, T.B.J.; Stillman, M.J. Soluble diamagnetic model for malaria pigment: Coordination chemistry of gallium(III)protoporphyrin-IX. Inorg. Chem. 2012, 51, 10747–10761. [Google Scholar] [CrossRef] [PubMed]
- Agadjanian, H.; Ma, J.; Rentsendorj, A.; Valluripalli, V.; Hwang, J.Y.; Mahammad, A.; Farkas, D.L.; Gray, H.B.; Gross, Z.; Medina-Kauwe, L.K. Tumor detection and elimination by a targeted gallium corrole. Proc. Natl. Acad. Sci. USA 2009, 106, 6105–6110. [Google Scholar] [CrossRef] [PubMed]
- Arciero, D.M.; Hooper, A.B. A di-heme cytochrome c peroxidase from Nitrosomonas europaea catalytically active in both the oxidized and half-reduced states. J. Biol. Chem. 1994, 269, 11878–11886. [Google Scholar] [PubMed]
- Harvey, P.D.; Proulx, N.; Martin, G.; Drouin, M.; Nurco, D.J.; Smith, K.M.; Bolze, F.; Gros, C.P.; Guilard, R. Preparation, characterization, and luminescence properties of gallium−metal face-to-face diporphyrins (M = H2, GaL, Ru(CO)(OH), Co). Inorg. Chem. 2001, 40, 4134–4142. [Google Scholar] [CrossRef] [PubMed]
- Dostert, C.; Guarda, G.; Romero, J.F.; Menu, P.; Gross, O.; Tardivel, A.; Suva, M.-L.; Stehle, J.-C.; Kopf, M.; Stamenkovic, I.; et al. Malarial hemozoin is a NALP3 inflammasome activating danger signal. PLoS ONE 2009, 4, e6510. [Google Scholar] [CrossRef] [PubMed]
- Coronado, L.M.; Nadovich, C.T.; Spadafora, C. Malarial hemozoin: From target to tool. Biochem. Biophys. Acta 2014, 1840, 2032–2041. [Google Scholar] [CrossRef] [PubMed]
- Boura, M.; Frita, R.; Góis, A.; Carvalho, T.; Hänscheid, T. The hemozoin conundrum: Is malaria pigment immune-activating, inhibiting, or simply a bystander? Trends Parasitol. 2013, 29, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Pinter, T.B.; Dodd, E.L.; Bohle, D.S.; Stillman, M.J. Spectroscopic and theoretical studies of Ga(III) protoporphyrin-IX and its reactions with myoglobin. Inorg. Chem. 2012, 51, 3743–3753. [Google Scholar] [CrossRef] [PubMed]
- Mack, J.; Stillman, M.J. Assignment of the optical spectra of metal phthalocyanines through spectral band deconvolution analysis and zindo calculations. Coord. Chem. Rev. 2001, 219, 993–1032. [Google Scholar] [CrossRef]
- Mack, J.; Asano, Y.; Kobayashi, N.; Stillman, M.J. Application of MCD spectroscopy and TD−DFT to a highly non-planar porphyrinoid ring system. New insights on red−shifted porphyrinoid spectral bands. J. Am. Chem. Soc. 2005, 127, 17697–17711. [Google Scholar] [CrossRef] [PubMed]
- Mack, J.; Stillman, M.J. Electronic structures of metal phthalocyanine and porphyrin complexes from analysis of the UV-visible absorption and magnetic circular dichroism spectra and molecular orbital calculations. In Handbook of Porphyrin Science; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; World Scientific Publishing Co.: Hackensack, NJ, USA, 2010. [Google Scholar]
- Jiang, L.; Engle, J.T.; Zaenglein, R.A.; Matus, A.; Ziegler, C.J.; Wang, H.; Stillman, M.J. Pentacene-fused diporphyrins. Chem. Eur. J. 2014, 20, 13865–13870. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
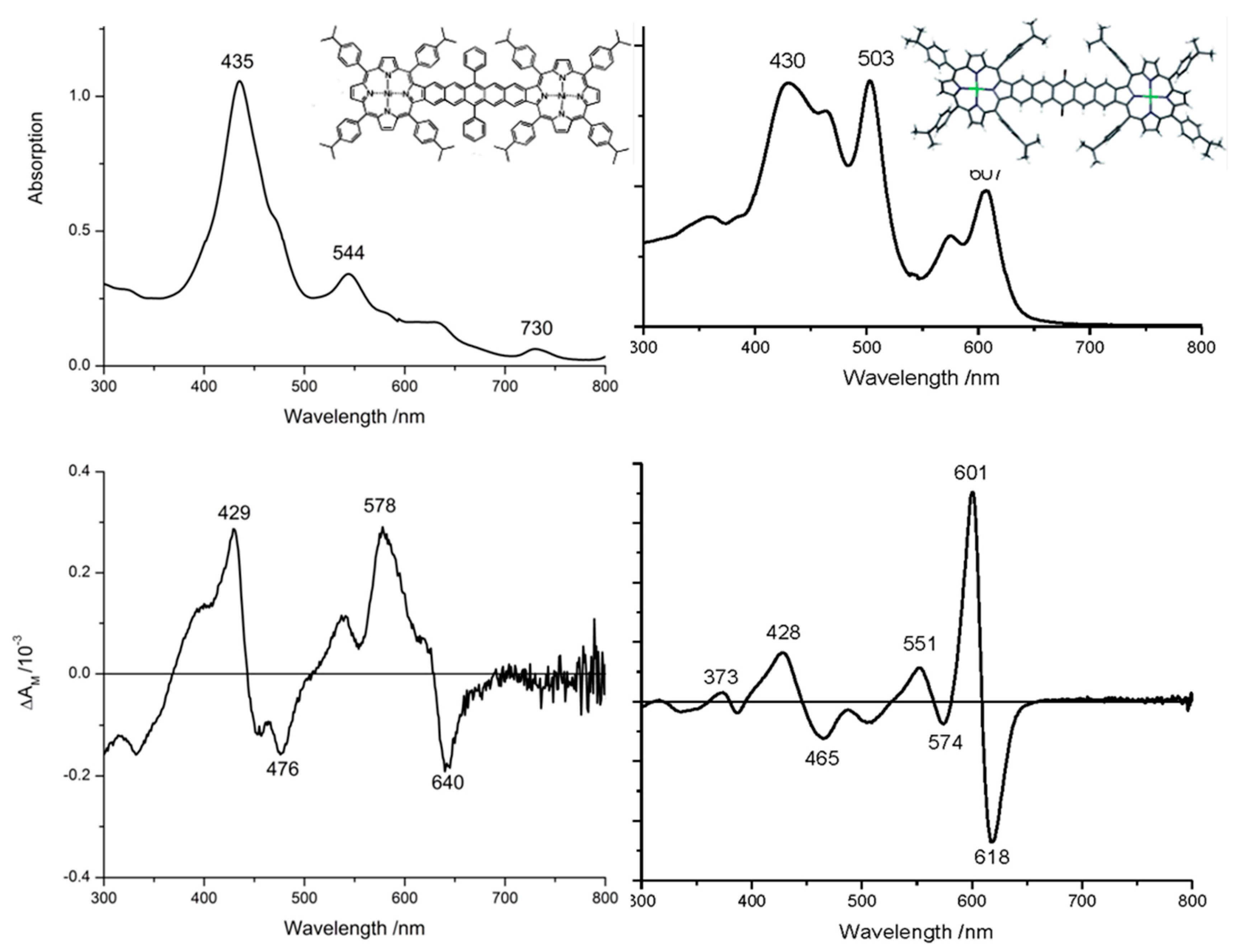{kind=link}
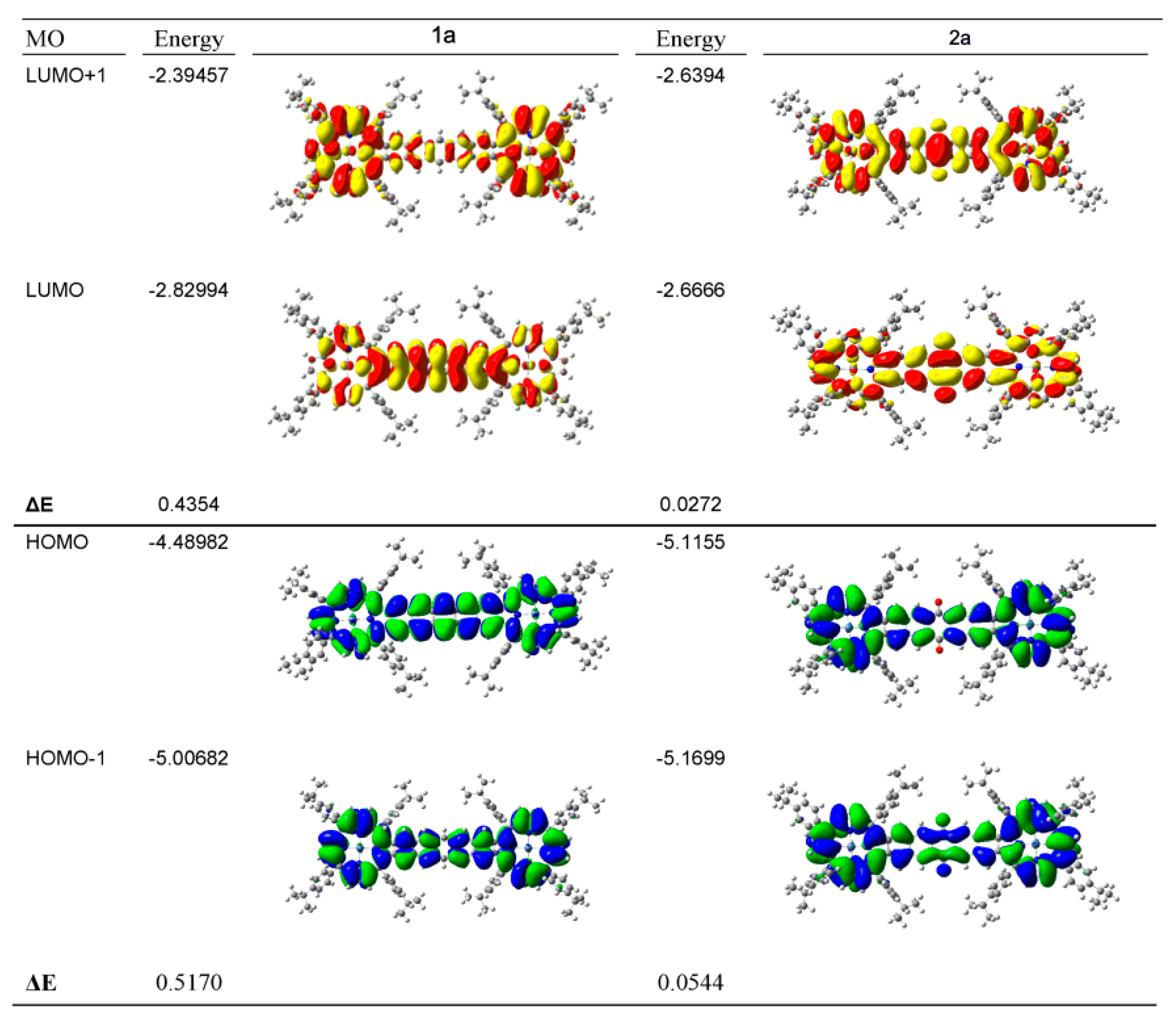
| Compound 1 | Δ{HOMO − (HOMO − 1)} | Δ{(LUMO + 1) − (LUMO)} | Δ{LUMO − HOMO} |
|---|---|---|---|
| Ga(III)-PPIX | 0.109 | ~0 | 23.20 |
| Zn(II)-PPIX | 0.054 | ~0 | 23.70 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Visser, S.P.; Stillman, M.J. Challenging Density Functional Theory Calculations with Hemes and Porphyrins. Int. J. Mol. Sci. 2016, 17, 519. https://doi.org/10.3390/ijms17040519
De Visser SP, Stillman MJ. Challenging Density Functional Theory Calculations with Hemes and Porphyrins. International Journal of Molecular Sciences. 2016; 17(4):519. https://doi.org/10.3390/ijms17040519
Chicago/Turabian StyleDe Visser, Sam P., and Martin J. Stillman. 2016. "Challenging Density Functional Theory Calculations with Hemes and Porphyrins" International Journal of Molecular Sciences 17, no. 4: 519. https://doi.org/10.3390/ijms17040519
APA StyleDe Visser, S. P., & Stillman, M. J. (2016). Challenging Density Functional Theory Calculations with Hemes and Porphyrins. International Journal of Molecular Sciences, 17(4), 519. https://doi.org/10.3390/ijms17040519