
Linking Pesticide Exposure with Pediatric Leukemia: Potential Underlying Mechanisms

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Evidence Linking Pesticide Exposure with Pediatric Leukemia
2.1. Epidemiological Studies Supporting the Association
2.2. In Vitro Studies
3. Gene-Environment Interactions
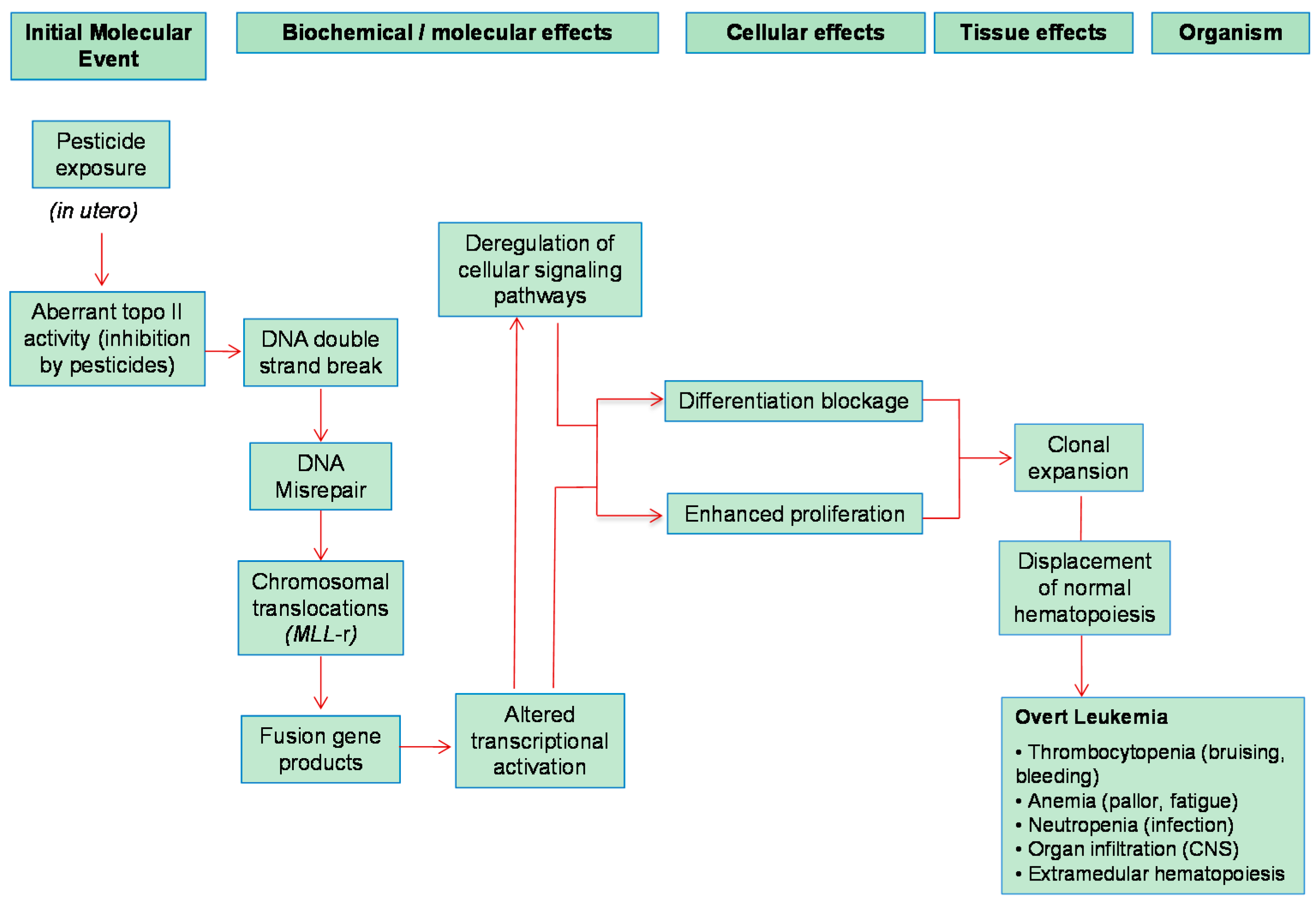
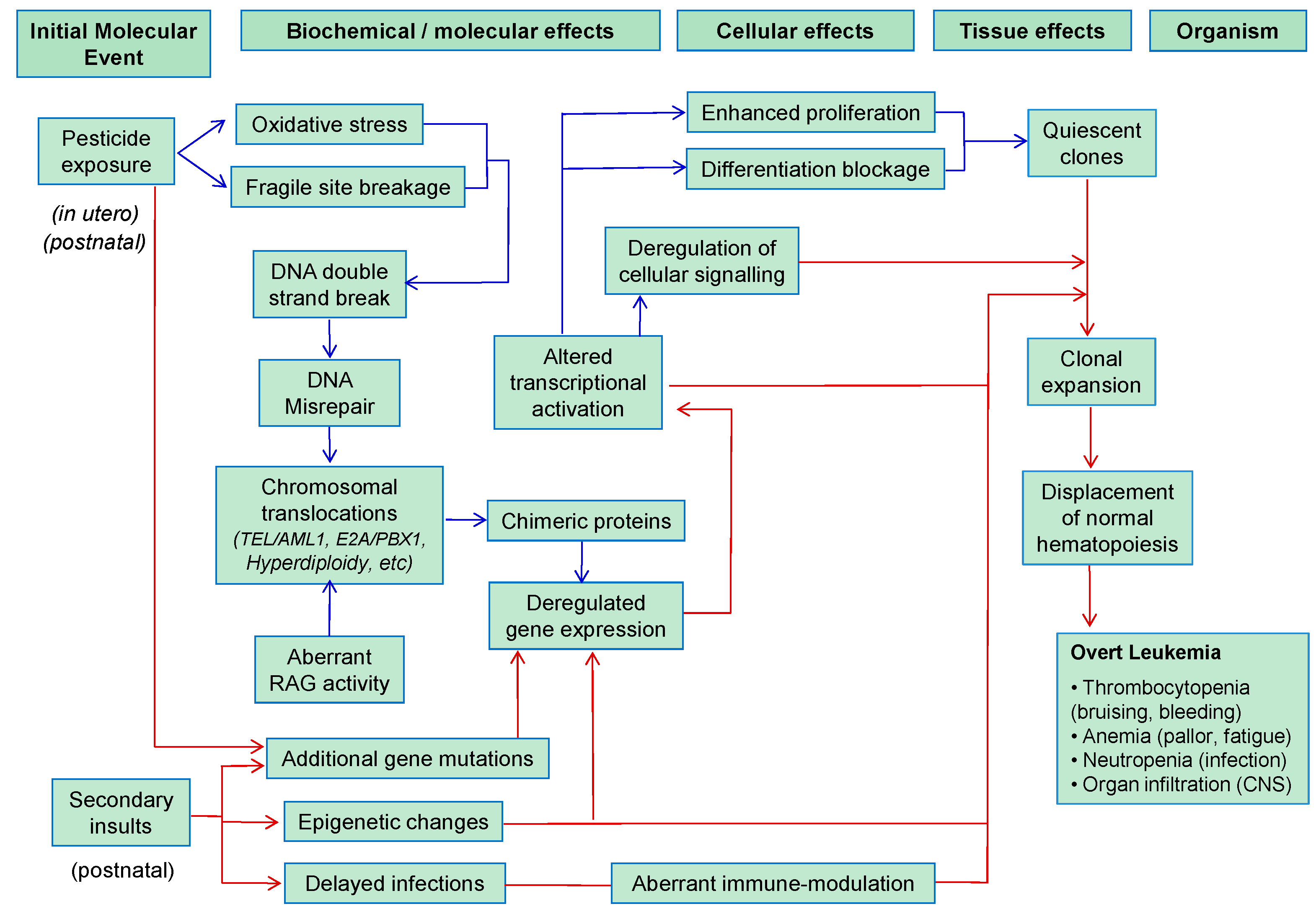
4. Early Molecular Events Involved in Pesticide-Associated Pediatric Leukemogenesis
4.1. DNA Double-Strand Breaks (DSBs)
4.2. Chromosomal Translocations
5. Pathobiology of Pediatric Leukemias
5.1. Infant Leukemia
5.2. Childhood Leukemia
6. Role of Acetylcholinesterase in Leukemogenesis
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hunger, S.P.; Mullighan, C.G. Acute lymphoblastic leukemia in children. N. Engl. J. Med. 2015, 373, 1541–1552. [Google Scholar] [PubMed]
- World Health Organization. Incidence of Childhood Leukemia, an ENHIS Fact Sheet, 2009. Available online: http://www.euro.who.int/__data/assets/pdf_file/0005/97016/4.1.-Incidence-of-childhood-leukaemia-EDITED_layouted.pdf?ua=1 (accessed on 10 February 2016).
- Wiemels, J. Perspectives on the causes of childhood leukemia. Chem. Biol. Interact. 2012, 196, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Pandita, A.; Harish, R.; Digra, S.K.; Raina, A.; Sharma, A.A.; Koul, A. Molecular cytogenetics in childhood acute lymphoblastic leukemia: A hospital-based observational study. Clin. Med. Insights Oncol. 2015, 9, 39–42. [Google Scholar] [CrossRef] [PubMed]
- Yohe, S. Molecular genetic markers in acute myeloid leukemia. J. Clin. Med. 2015, 4, 460–478. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.D.; Wagers, A.J. Dynamic niches in the origination and differentiation of haematopoietic stem cells. Nat. Rev. Mol. Cell Biol. 2011, 12, 643–655. [Google Scholar] [CrossRef] [PubMed]
- Emerenciano, M.; Koifman, S.; Pombo-de-Oliveira, M.S. Acute leukemia in early childhood. Braz. J. Med. Biol. Res. 2007, 40, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Pui, C.H.; Robison, L.L.; Look, A.T. Acute lymphoblastic leukemia. Lancet 2008, 371, 1030–1043. [Google Scholar] [CrossRef]
- Greaves, M. Infection, immune responses and the etiology of childhood leukemia. Nat. Rev. Cancer 2006, 6, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Greaves, M.; Müschen, M. Infection and the perils of B-cell activation. Cancer Discov. 2015, 5, 1244–1246. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Martín-Lorenzo, A.; Hauer, J.; Vicente-Dueñas, C.; Auer, F.; González-Herrero, I.; García-Ramírez, I.; Ginzel, S.; Thiele, R.; Constantinescu, S.N.; Bartenhagen, C.; et al. Infection exposure is a causal factor in B-cell precursor acute lymphoblastic leukemia as a result of Pax5-inherited susceptibility. Cancer Discov. 2015, 5, 1328–1343. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Chang, C.H.; Tao, L.; Lu, C. Residential exposure to pesticide during childhood and childhood cancers: A meta-analysis. Pediatrics 2015, 136, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Ntzani, E.E.; Chondrogiorgi, M.; Ntritsos, G.; Evangelou, E.; Tzoulaki, I. Literature Review on Epidemiological Studies Linking Exposure to Pesticides and Health Effects. Available online: http://www.efsa.europa.eu/sites/default/files/scientific_output/files/main_documents/497e.pdf (accessed on 25 March 2016).
- Lu, C.; Liu, X.; Liu, C.; Wang, J.; Li, C.; Liu, Q.; Li, Y.; Li, S.; Sun, S.; Yan, J.; et al. Chlorpyrifos induces MLL translocations through caspase 3-dependent genomic instability and topoisomerase II inhibition in human fetal liver hematopoietic stem cells. Toxicol. Sci. 2015, 147, 588–606. [Google Scholar] [CrossRef] [PubMed]
- Alexander, F.E.; Patheal, S.L.; Biondi, A.; Brandalise, S.; Cabrera, M.E.; Chan, L.C.; Chen, Z.; Cimino, G.; Cordoba, J.C.; Gu, L.J.; et al. Transplacental chemical exposure and risk of infant leukemia with MLL gene fusion. Cancer Res. 2001, 61, 2542–2546. [Google Scholar] [PubMed]
- Pombo-de-Oliveira, M.S.; Koifman, S.; Brazilian Collaborative Study Group of Infant Acute Leukemia. Infant acute leukemia and maternal exposures during pregnancy. Cancer Epidemiol. Biomark. Prev. 2006, 15, 2336–2341. [Google Scholar] [CrossRef] [PubMed]
- Cavalieri, E.L.; Stack, D.E.; Devanesan, P.D.; Todorovic, R.; Dwivedy, I.; Higginbotham, S.; Johansson, S.L.; Patil, K.D.; Gross, M.L.; Gooden, J.K.; et al. Molecular origin of cancer: Catechol estrogen-3,4-quinones as endogenous tumor initiators. Proc. Natl. Acad. Sci. USA 1997, 94, 10937–10942. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, J.D.; Couto, A.C.; Pombo-de-Oliveira, M.S.; Koifman, S.; Brazilian Collaborative Study Group of Infant Acute Leukemia. In utero pesticide exposure and leukemia in Brazilian children <2 years of age. Environ. Health Perspect. 2013, 121, 269–275. [Google Scholar] [PubMed]
- Ding, G.; Shi, R.; Gao, Y.; Zhang, Y.; Kamijima, M.; Sakai, K.; Wang, G.; Feng, C.; Tian, Y. Pyrethroid pesticide exposure and risk of childhood acute lymphocytic leukemia in Shanghai. Environ. Sci. Technol. 2012, 46, 13480–13487. [Google Scholar] [CrossRef] [PubMed]
- Metayer, C.; Colt, J.S.; Buffler, P.A.; Reed, H.D.; Selvin, S.; Crouse, V.; Ward, M.H. Exposure to herbicides in house dust and risk of childhood acute lymphoblastic leukemia. J. Expo. Sci. Environ. Epidemiol. 2013, 23, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.M.; Buffler, P.A.; Gunier, R.B.; Dahl, G.; Smith, M.T.; Reinier, K.; Reynolds, P. Critical windows of exposure to household pesticides and risk of childhood leukemia. Environ. Health Perspect. 2002, 110, 955–960. [Google Scholar] [CrossRef] [PubMed]
- Slater, M.E.; Linabery, A.M.; Spector, L.G.; Johnson, K.J.; Hilden, J.M.; Heerema, N.A.; Robison, L.L.; Ross, J.A. Maternal exposure to household chemicals and risk of infant leukemia: A report from the Children’s Oncology Group. Cancer Causes Control 2011, 22, 1197–1204. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Turner, M.C.; Wigle, D.T.; Krewski, D. Residential pesticides and childhood leukemia: A systematic review and meta-analysis. Environ. Health Perspect. 2010, 118, 33–41. [Google Scholar] [PubMed]
- Bailey, H.D.; Fritschi, L.; Infante-Rivard, C.; Glass, D.C.; Miligi, L.; Dockerty, J.D.; Lightfoot, T.; Clavel, J.; Roman, E.; Spector, L.G.; et al. Parental occupational pesticide exposure and the risk of childhood leukemia in the offspring: Findings from the childhood leukemia international consortium. Int. J. Cancer 2014, 135, 2157–2172. [Google Scholar] [CrossRef] [PubMed]
- Bailey, H.D.; Infante-Rivard, C.; Metayer, C.; Clavel, J.; Lightfoot, T.; Kaatsch, P.; Roman, E.; Magnani, C.; Spector, L.G.; Th Petridou, E.; et al. Home pesticide exposures and risk of childhood leukemia: Findings from the childhood leukemia international consortium. Int. J. Cancer 2015, 137, 2644–2663. [Google Scholar] [CrossRef] [PubMed]
- Rahden-Staron, I. The inhibitory effect of the fungicides captan and captafol on eukaryotic topoisomerases in vitro and lack of recombinagenic activity in the wing spot test of Drosophila melanogaster. Mutat. Res. 2002, 518, 205–213. [Google Scholar] [CrossRef]
- Rahden-Staroń, I.; Czeczot, H.; Kowalska-Loth, B. The ability of thiram to inhibit eukaryotic topoisomerase II and to damage DNA. Acta Biochim. Pol. 1993, 40, 51–53. [Google Scholar] [PubMed]
- Boros, L.G.; Williams, R.D. Isofenphos induced metabolic changes in K562 myeloid blast cells. Leuk. Res. 2001, 25, 883–890. [Google Scholar] [CrossRef]
- Williams, R.D.; Boros, L.G.; Kolanko, C.J.; Jackman, S.M.; Eggers, T.R. Chromosomal aberrations in human lymphocytes exposed to the anticholinesterase pesticide isofenphos with mechanisms of leukemogenesis. Leuk. Res. 2004, 28, 947–958. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wallace, A.D.; Du, P.; Lin, S.; Baccarelli, A.A.; Jiang, H.; Jafari, N.; Zheng, Y.; Xie, H.; Soares, M.B.; et al. Genome-wide study of DNA methylation alterations in response to diazinon exposure in vitro. Environ. Toxicol. Pharmacol. 2012, 34, 959–968. [Google Scholar] [CrossRef] [PubMed]
- Ukpebor, J.; Llabjani, V.; Martin, F.L.; Halsall, C.J. Sublethal genotoxicity and cell alterations by organophosphorus pesticides in MCF-7 cells: Implications for environmentally relevant concentrations. Environ. Toxicol. Chem. 2011, 30, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Sirma, S.; Agaoglu, L.; Yildiz, I.; Cayli, D.; Horgusluoglu, E.; Anak, S.; Yuksel, L.; Unuvar, A.; Celkan, T.; Apak, H.; et al. NAD(P)H:quinine oxidoreductase 1 null genotype is not associated with pediatric de novo acute leukemia. Pediatr. Blood Cancer 2004, 43, 568–570. [Google Scholar] [CrossRef] [PubMed]
- Sanjuan-Pla, A.; Bueno, C.; Prieto, C.; Acha, P.; Stam, R.W.; Marschalek, R.; Menéndez, P. Revisiting the biology of infant t(4;11)/MLL-AF4+ B-cell acute lymphoblastic leukemia. Blood 2015, 126, 2676–2685. [Google Scholar] [CrossRef] [PubMed]
- Das, P.M.; Singal, R. DNA methylation and cancer. J. Clin. Oncol. 2004, 22, 4632–4642. [Google Scholar] [CrossRef] [PubMed]
- Kamdar, K.Y.; Krull, K.R.; El-Zein, R.A.; Brouwers, P.; Potter, B.S.; Harris, L.L.; Holm, S.; Dreyer, Z.; Scaglia, F.; Etzel, C.J.; et al. Folate pathway polymorphisms predict deficits in attention and processing speed after childhood leukemia therapy. Pediatr. Blood Cancer 2011, 57, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Sharp, L.; Little, J. Polymorphisms in genes involved in folate metabolism and colorectal neoplasia: A HuGE review. Am. J. Epidemiol. 2004, 159, 423–443. [Google Scholar] [CrossRef] [PubMed]
- Duncan, T.M.; Reed, M.C.; Nijhout, H.F. The relationship between intracellular and plasma levels of folate and metabolites in the methionine cycle: A model. Mol. Nutr. Food Res. 2013, 57, 628–636. [Google Scholar] [CrossRef] [PubMed]
- Lightfoot, T.J.; Johnston, W.T.; Painter, D.; Simpson, J.; Roman, E.; Skibola, C.F.; Smith, M.T.; Allan, J.M.; Taylor, G.M.; United Kingdom Childhood Cancer Study. Genetic variation in the folate metabolic pathway and risk of childhood leukemia. Blood 2010, 115, 3923–3929. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.F.; Simões, B.P.; Tone, L.G.; Gabellini, S.M.; Zago, M.A.; Falcão, R.P. The methylenetetrahydrofolate reductase C677T gene polymorphism decreases the risk of childhood acute lymphocytic leukemia. Br. J. Haematol. 2001, 115, 616–618. [Google Scholar] [CrossRef] [PubMed]
- Garte, S.; Taioli, E.; Crosti, F.; Sainati, L.; Barisone, E.; Luciani, M.; Jankovic, M.; Biondi, A.G. Deletion of parental GST genes as a possible susceptibility factor in the etiology of infant leukemia. Leuk. Res. 2000, 24, 971–974. [Google Scholar] [CrossRef]
- Chokkalingam, A.P.; Metayer, C.; Scelo, G.A.; Chang, J.S.; Urayama, K.Y.; Aldrich, M.C.; Guha, N.; Hansen, H.M.; Dahl, G.V.; Barcellos, L.F.; et al. Variation in xenobiotic transport and metabolism genes, household chemical exposures, and risk of childhood acute lymphoblastic leukemia. Cancer Causes Control. 2012, 23, 1367–1375. [Google Scholar] [CrossRef] [PubMed]
- Buffler, P.A.; Kwan, M.L.; Reynolds, P.; Urayama, K.Y. Environmental and genetic risk factors for childhood leukemia: Appraising the evidence. Cancer Investig. 2005, 23, 60–75. [Google Scholar] [CrossRef]
- Sherborne, A.L.; Hemminki, K.; Kumar, R.; Bartram, C.R.; Stanulla, M.; Schrappe, M.; Petridou, E.; Semsei, A.F.; Szalai, C.; Sinnett, D.; et al. Rationale for an international consortium to study inherited genetic susceptibility to childhood acute lymphoblastic leukemia. Haematologica 2011, 96, 1049–1054. [Google Scholar] [CrossRef] [PubMed]
- Sedelnikova, O.A.; Redon, C.E.; Dickey, J.S.; Nakamura, A.J.; Georgakilas, A.G.; Bonner, W.M. Role of oxidatively induced DNA lesions in human pathogenesis. Mutat. Res. 2010, 704, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Esperanza, M.; Cid, Á.; Herrero, C.; Rioboo, C. Acute effects of a prooxidant herbicide on the microalga Chlamydomonas reinhardtii: Screening cytotoxicity and genotoxicity endpoints. Aquat. Toxicol. 2015, 165, 210–221. [Google Scholar] [CrossRef] [PubMed]
- Guanggang, X.; Diqiu, L.; Jianzhong, Y.; Jingmin, G.; Huifeng, Z.; Mingan, S.; Liming, T. Carbamate insecticide methomyl confers cytotoxicity through DNA damage induction. Food Chem. Toxicol. 2013, 53, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Muniz, J.F.; McCauley, L.; Scherer, J.; Lasarev, M.; Koshy, M.; Kow, Y.W.; Nazar-Stewart, V.; Kisby, G.E. Biomarkers of oxidative stress and DNA damage in agricultural workers: A pilot study. Toxicol. Appl. Pharmacol. 2008, 227, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Ojha, A.; Srivastava, N. In vitro studies on organophosphate pesticides induced oxidative DNA damage in rat lymphocytes. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2014, 761, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Kryston, T.B.; Georgiev, A.B.; Pissis, P.; Georgakilas, A.G. Role of oxidative stress and DNA damage in human carcinogenesis. Mutat. Res. 2011, 711, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Hernández, A.F.; Lacasaña, M.; Gil, F.; Rodríguez-Barranco, M.; Pla, A.; López-Guarnido, O. Evaluation of pesticide-induced oxidative stress from a gene-environment interaction perspective. Toxicology 2013, 307, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Mostafalou, S.; Abdollahi, M. Pesticides and human chronic diseases: Evidences, mechanisms, and perspectives. Toxicol. Appl. Pharmacol. 2013, 268, 157–177. [Google Scholar] [CrossRef] [PubMed]
- Woodbine, L.; Brunton, H.; Goodarzi, A.A.; Shibata, A.; Jeggo, P.A. Endogenously induced DNA double strand breaks arise in heterochromatic DNA regions and require ataxia telangiectasia mutated and Artemis for their repair. Nucleic Acids Res. 2011, 39, 6986–6997. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.C.; Mishra, M.; Sharma, A.; Deepak Balaji, T.G.; Kumar, R.; Mishra, R.K.; Chowdhuri, D.K. Chlorpyrifos induces apoptosis and DNA damage in Drosophila through generation of reactive oxygen species. Ecotoxicol. Environ. Saf. 2010, 73, 1415–1423. [Google Scholar] [CrossRef] [PubMed]
- Dillon, L.W.; Burrow, A.A.; Wang, Y.H. DNA instability at chromosomal fragile sites in cancer. Curr. Genom. 2010, 11, 326–337. [Google Scholar] [CrossRef] [PubMed]
- Thys, R.G.; Lehman, C.E.; Pierce, L.C.; Wang, Y.H. Environmental and chemotherapeutic agents induce breakage at genes involved in leukemia-causing gene rearrangements in human hematopoietic stem/progenitor cells. Mutat. Res. 2015, 779, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Dillon, L.W.; Pierce, L.C.; Lehman, C.E.; Nikiforov, Y.E.; Wang, Y.H. DNA topoisomerases participate in fragility of the oncogene RET. PLoS ONE 2013, 8, e75741. [Google Scholar] [CrossRef] [PubMed]
- Deweese, J.E.; Osheroff, N. The DNA cleavage reaction of topoisomerase II: Wolf in sheep’s clothing. Nucleic Acids Res. 2009, 37, 738–748. [Google Scholar] [CrossRef] [PubMed]
- Nambiar, M.; Raghavan, S.C. How does DNA break during chromosomal translocations? Nucleic Acids Res. 2011, 39, 5813–5825. [Google Scholar] [CrossRef] [PubMed]
- Wiemels, J.L.; Greaves, M. Structure and possible mechanisms of TEL-AML1 gene fusions in childhood acute lymphoblastic leukemia. Cancer Res. 1999, 59, 4075–4082. [Google Scholar] [PubMed]
- Greaves, M.F.; Wiemels, J. Origins of chromosome translocations in childhood leukemia. Nat. Rev. Cancer 2003, 3, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Meissner, B.; Bartram, T.; Eckert, C.; Trka, J.; Panzer-Gruemayer, R.; Hermanova, I.; Ellinghaus, E.; Franke, A.; Moericke, A.; Schrauder, A.; et al. Frequent and sex-biased deletion of SLX4IP by illegitimate V(D)J-mediated recombination in childhood acute lymphoblastic leukemia. Hum. Mol. Genet. 2014, 23, 590–601. [Google Scholar] [CrossRef] [PubMed]
- Lieber, M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.M.; Messier, T.; Rivers, J.; O’Neill, J.P.; Walker, V.E.; Vacek, P.M.; Finette, B.A. VDJ recombinase-mediated TCR β locus gene usage and coding joint processing in peripheral T cells during perinatal and pediatric development. J. Immunol. 2012, 189, 2356–2364. [Google Scholar] [CrossRef] [PubMed]
- Pinsonneault, R.L.; Vacek, P.M.; O’Neill, J.P.; Finette, B.A. Induction of V(D)J-mediated recombination of an extrachromosomal substrate following exposure to DNA-damaging agents. Environ. Mol. Mutagen. 2007, 48, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Ernst, P.; Wang, J.; Korsmeyer, S.J. The role of MLL in hematopoiesis and leukemia. Curr. Opin. Hematol. 2002, 9, 282–287. [Google Scholar] [CrossRef] [PubMed]
- Marchesi, F.; Girardi, K.; Avvisati, G. Pathogenetic, Clinical, and Prognostic Features of Adult t(4;11)(q21;q23)/MLL-AF4 positive B-cell acute lymphoblastic leukemia. Adv. Hematol. 2011, 2011, 621627. [Google Scholar] [CrossRef] [PubMed]
- Nanya, M.; Sato, M.; Tanimoto, K.; Tozuka, M.; Mizutani, S.; Takagi, M. Dysregulation of the DNA damage response and KMT2A rearrangement in fetal liver hematopoietic cells. PLoS ONE 2015, 10, e0144540. [Google Scholar] [CrossRef] [PubMed]
- Strick, R.; Strissel, P.L.; Borgers, S.; Smith, S.L.; Rowley, J.D. Dietary bioflavonoids induce cleavage in the MLL gene and may contribute to infant leukemia. Proc. Natl. Acad. Sci. USA 2000, 97, 4790–4795. [Google Scholar] [CrossRef] [PubMed]
- Sallmyr, A.; Fan, J.; Rassool, F.V. Genomic instability in myeloid malignancies: Increased reactive oxygen species (ROS), DNA double strand breaks (DSBs) and error-prone repair. Cancer Lett. 2008, 270, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bueno, C.; Catalina, P.; Melen, G.J.; Montes, R.; Sánchez, L.; Ligero, G.; García-Pérez, J.L.; Menendez, P. Etoposide induces MLL rearrangements and other chromosomal abnormalities in human embryonic stem cells. Carcinogenesis 2009, 30, 1628–1637. [Google Scholar] [CrossRef] [PubMed]
- Bueno, C.; Montes, R.; Catalina, P.; Rodríguez, R.; Menendez, P. Insights into the cellular origin and etiology of the infant pro-B acute lymphoblastic leukemia with MLL-AF4 rearrangement. Leukemia 2011, 25, 400–410. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, M.; Eguchi-Ishimae, M.; Knight, D.; Kearney, L.; Slany, R.; Greaves, M. MLL chimeric protein activation renders cells vulnerable to chromosomal damage: An explanation for the very short latency of infant leukemia. Genes Chromosom. Cancer 2006, 45, 754–760. [Google Scholar] [CrossRef] [PubMed]
- Felix, C.A. Leukemias related to treatment with DNA topoisomerase II inhibitors. Med. Pediatr. Oncol. 2001, 36, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Menendez, P.; Catalina, P.; Rodríguez, R.; Melen, G.J.; Bueno, C.; Arriero, M.; García-Sánchez, F.; Lassaletta, A.; García-Sanz, R.; García-Castro, J. Bone marrow mesenchymal stem cells from infants with MLL-AF4+ acute leukemia harbor and express the MLL-AF4 fusion gene. J. Exp. Med. 2009, 206, 3131–3141. [Google Scholar] [CrossRef] [PubMed]
- Spector, L.G.; Xie, Y.; Robison, L.L.; Heerema, N.A.; Hilden, J.M.; Lange, B.; Felix, C.A.; Davies, S.M.; Slavin, J.; Potter, J.D.; et al. Maternal diet and infant leukemia: The DNA topoisomerase II inhibitor hypothesis: A report from the children’s oncology group. Cancer Epidemiol. Biomark. Prev. 2005, 14, 651–655. [Google Scholar] [CrossRef] [PubMed]
- Andersson, A.K.; Ma, J.; Wang, J.; Chen, X.; Gedman, A.L.; Dang, J.; Nakitandwe, J.; Holmfeldt, L.; Parker, M.; Easton, J.; et al. St. Jude Children’s Research Hospital—Washington University Pediatric Cancer Genome Project. The landscape of somatic mutations in infant MLL-rearranged acute lymphoblastic leukemias. Nat. Genet. 2015, 47, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Driessen, E.M.; van Roon, E.H.; Spijkers-Hagelstein, J.A.; Schneider, P.; de Lorenzo, P.; Valsecchi, M.G.; Pieters, R.; Stam, R.W. Frequencies and prognostic impact of RAS mutations in MLL-rearranged acute lymphoblastic leukemia in infants. Haematologica 2013, 98, 937–944. [Google Scholar] [CrossRef] [PubMed]
- Dobbins, S.E.; Sherborne, A.L.; Ma, Y.P.; Bardini, M.; Biondi, A.; Cazzaniga, G.; Lloyd, A.; Chubb, D.; Greaves, M.F.; Houlston, R.S. The silent mutational landscape of infant MLL-AF4 pro-B acute lymphoblastic leukemia. Genes Chromosom.Cancer 2013, 52, 954–960. [Google Scholar] [CrossRef] [PubMed]
- Prelle, C.; Bursen, A.; Dingermann, T.; Marschalek, R. Secondary mutations in t(4;11) leukemia patients. Leukemia 2013, 27, 1425–1427. [Google Scholar] [CrossRef] [PubMed]
- Prieto, C.; Stam, R.W.; Agraz-Doblas, A.; Ballerini, P.; Camos, M.; Castaño, J.; Marschalek, R.; Bursen, A.; Varela, I.; Bueno, C.; et al. Activated KRAS cooperates with MLLAF4 to promote extramedullary engraftment and migration of cord blood CD34+ HSPC but is insufficient to initiate leukemia. Cancer Res. 2016, in press. [Google Scholar] [CrossRef] [PubMed]
- Bardini, M.; Woll, P.S.; Corral, L.; Luc, S.; Wittmann, L.; Ma, Z.; Lo Nigro, L.; Basso, G.; Biondi, A.; Cazzaniga, G.; et al. Clonal variegation and dynamic competition of leukemia-initiating cells in infant acute lymphoblastic leukemia with MLL rearrangement. Leukemia 2015, 29, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Stumpel, D.J.; Schneider, P.; van Roon, E.H.; Pieters, R.; Stam, R.W. Absence of global hypomethylation in promoter hypermethylated Mixed Lineage Leukemia-rearranged infant acute lymphoblastic leukemia. Eur. J. Cancer 2013, 49, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Stam, R.W.; Schneider, P.; Hagelstein, J.A.; van der Linden, M.H.; Stumpel, D.J.; de Menezes, R.X.; de Lorenzo, P.; Valsecchi, M.G.; Pieters, R. Gene expression profiling-based dissection of MLL translocated and MLL germline acute lymphoblastic leukemia in infants. Blood 2010, 115, 2835–2844. [Google Scholar] [CrossRef] [PubMed]
- Bhojwani, D.; Yang, J.J.; Pui, C.H. Biology of childhood acute lymphoblastic leukemia. Pediatr. Clin. N. Am. 2015, 62, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Greaves, M. In utero origins of childhood leukemia. Early Hum. Dev. 2005, 81, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Greaves, M. Childhood leukemia. BMJ 2002, 324, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanuil, E.; Rapado, I.; Li, Y.; Potter, N.E.; Wedge, D.C.; Tubio, J.; Alexandrov, L.B.; van Loo, P.; Cooke, S.L.; Marshall, J.; et al. RAG-mediated recombination is the predominant driver of oncogenic rearrangement in ETV6-RUNX1 acute lymphoblastic leukemia. Nat. Genet. 2014, 46, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Teitell, M.A.; Pandolfi, P.P. Molecular genetics of acute lymphoblastic leukemia. Annu. Rev. Pathol. 2009, 4, 175–198. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, S.; Klemm, L.; Park, E.; Papaemmanuil, E.; Ford, A.; Kweon, S.M.; Trageser, D.; Hasselfeld, B.; Henke, N.; Mooster, J.; et al. Mechanisms of clonal evolution in childhood acute lymphoblastic leukemia. Nat. Immunol. 2015, 16, 766–774. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Does, M.B.; Metayer, C.; Russo, C.; Wong, A.; Buffler, P.A. Vaccination history and risk of childhood leukemia. Int. J. Epidemiol. 2005, 34, 1100–1109. [Google Scholar] [CrossRef] [PubMed]
- Hosking, F.J.; Leslie, S.; Dilthey, A.; Moutsianas, L.; Wang, Y.; Dobbins, S.E.; Papaemmanuil, E.; Sheridan, E.; Kinsey, S.E.; Lightfoot, T.; et al. MHC variation and risk of childhood B-cell precursor acute lymphoblastic leukemia. Blood 2011, 117, 1633–1640. [Google Scholar] [CrossRef] [PubMed]
- Gilboa-Geffen, A.; Hartmann, G.; Soreq, H. Stressing hematopoiesis and immunity: An acetylcholinesterase window into nervous and immune system interactions. Front. Mol. Neurosci. 2012, 5, 30. [Google Scholar] [CrossRef] [PubMed]
- Rosas-Ballina, M.; Olofsson, P.S.; Ochani, M.; Valdés-Ferrer, S.I.; Levine, Y.A.; Reardon, C.; Tusche, M.W.; Pavlov, V.A.; Andersson, U.; Chavan, S.; et al. Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science 2011, 334, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Hoogduijn, M.J.; Cheng, A.; Genever, P.G. Functional nicotinic and muscarinic receptors on mesenchymal stem cells. Stem Cells Dev. 2009, 18, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, J.; Czepulkowski, B.; Hirst, W.; Mufti, G.J. Deletion of the acetylcholinesterase locus at 7q22 associated with myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML). Leuk. Res. 1996, 20, 235–241. [Google Scholar] [CrossRef]
- Grisaru, D.; Pick, M.; Perry, C.; Sklan, E.H.; Almog, R.; Goldberg, I.; Naparstek, E.; Lessing, J.B.; Soreq, H.; Deutsch, V. Hydrolytic and nonenzymatic functions of acetylcholinesterase comodulate hemopoietic stress responses. J. Immunol. 2006, 176, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Xi, H.J.; Wu, R.P.; Liu, J.J.; Zhang, L.J.; Li, Z.S. Role of acetylcholinesterase in lung cancer. Thorac. Cancer 2015, 6, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, V.R.; Pick, M.; Perry, C.; Grisaru, D.; Hemo, Y.; Golan-Hadari, D.; Grant, A.; Eldor, A.; Soreq, H. The stress-associated acetylcholinesterase variant AChE-R is expressed in human CD34+ hematopoietic progenitors and its C-terminal peptide ARP promotes their proliferation. Exp. Hematol. 2002, 30, 1153–1161. [Google Scholar] [CrossRef]
- Soreq, H.; Patinkin, D.; Lev-Lehman, E.; Grifman, M.; Ginzberg, D.; Eckstein, F.; Zakut, H. Antisense oligonucleotide inhibition of acetylcholinesterase gene expression induces progenitor cell expansion and suppresses hematopoietic apoptosis ex vivo. Proc. Natl. Acad. Sci. USA 1994, 91, 7907–7911. [Google Scholar] [CrossRef] [PubMed]
- Shapira, M.; Grant, A.; Korner, M.; Soreq, H. Genomic and transcriptional characterization of the human ACHE locus: Complex involvement with acquired and inherited diseases. Isr. Med. Assoc. J. 2000, 2, 470–473. [Google Scholar] [PubMed]
- Perry, C.; Sklan, E.H.; Birikh, K.; Shapira, M.; Trejo, L.; Eldor, A.; Soreq, H. Complex regulation of acetylcholinesterase gene expression in human brain tumors. Oncogene 2002, 21, 8428–8441. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.; Cotter, F.E. Monosomy 7 and 7q—Associated with myeloid malignancy. Blood Rev. 1997, 11, 46–55. [Google Scholar] [CrossRef]
- Heerema, N.A.; Nachman, J.B.; Sather, H.N.; La, M.K.; Hutchinson, R.; Lange, B.J.; Bostrom, B.; Steinherz, P.G.; Gaynon, P.S.; Uckun, F.M.; et al. Deletion of 7p or monosomy 7 in pediatric acute lymphoblastic leukemia is an adverse prognostic factor: A report from the Children’s Cancer Group. Leukemia 2004, 18, 939–947. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Jiang, H.; Wan, Y.H.; Du, A.Y.; Guo, K.J.; Liu, T.; Ye, W.Y.; Niu, X.; Wu, J.; Dong, X.Q.; et al. Induction of a 55 kDa acetylcholinesterase protein during apoptosis and its negative regulation by the Akt pathway. J. Mol. Cell. Biol. 2011, 3, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.J.; Greenberg, D.S. Acetylcholinesterase involvement in apoptosis. Front. Mol. Neurosci. 2012, 5, 40. [Google Scholar] [CrossRef] [PubMed]
- Martelli, A.M.; Evangelisti, C.; Chiarini, F.; McCubrey, J.A. The phosphatidylinositol 3-kinase/Akt/mTOR signaling network as a therapeutic target in acute myelogenous leukemia patients. Oncotarget 2010, 1, 89–103. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hernández, A.F.; Menéndez, P. Linking Pesticide Exposure with Pediatric Leukemia: Potential Underlying Mechanisms. Int. J. Mol. Sci. 2016, 17, 461. https://doi.org/10.3390/ijms17040461
Hernández AF, Menéndez P. Linking Pesticide Exposure with Pediatric Leukemia: Potential Underlying Mechanisms. International Journal of Molecular Sciences. 2016; 17(4):461. https://doi.org/10.3390/ijms17040461
Chicago/Turabian StyleHernández, Antonio F., and Pablo Menéndez. 2016. "Linking Pesticide Exposure with Pediatric Leukemia: Potential Underlying Mechanisms" International Journal of Molecular Sciences 17, no. 4: 461. https://doi.org/10.3390/ijms17040461
APA StyleHernández, A. F., & Menéndez, P. (2016). Linking Pesticide Exposure with Pediatric Leukemia: Potential Underlying Mechanisms. International Journal of Molecular Sciences, 17(4), 461. https://doi.org/10.3390/ijms17040461