
Fragment Screening of Human Aquaporin 1


Abstract
:
1. Introduction
2. Results
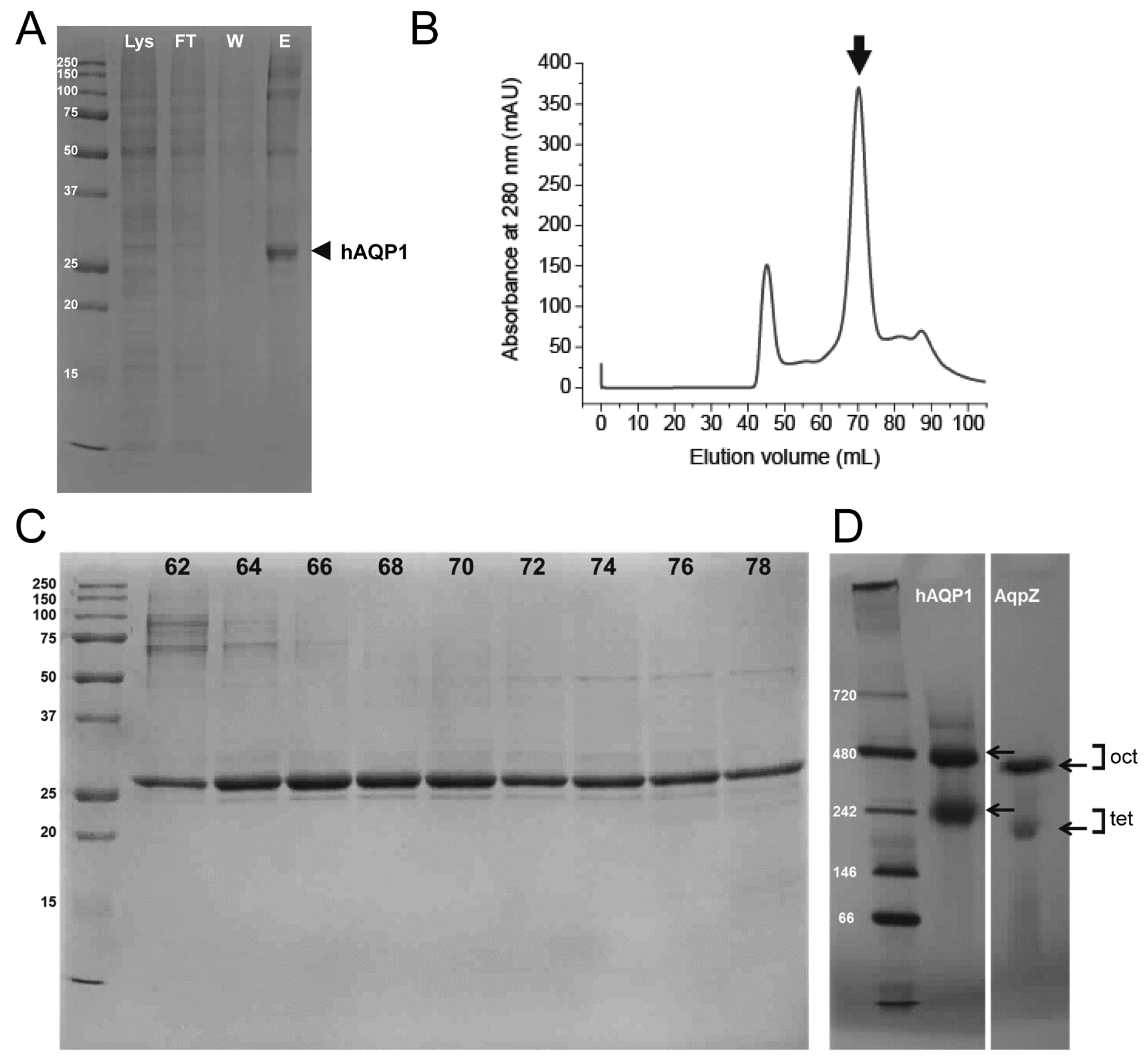
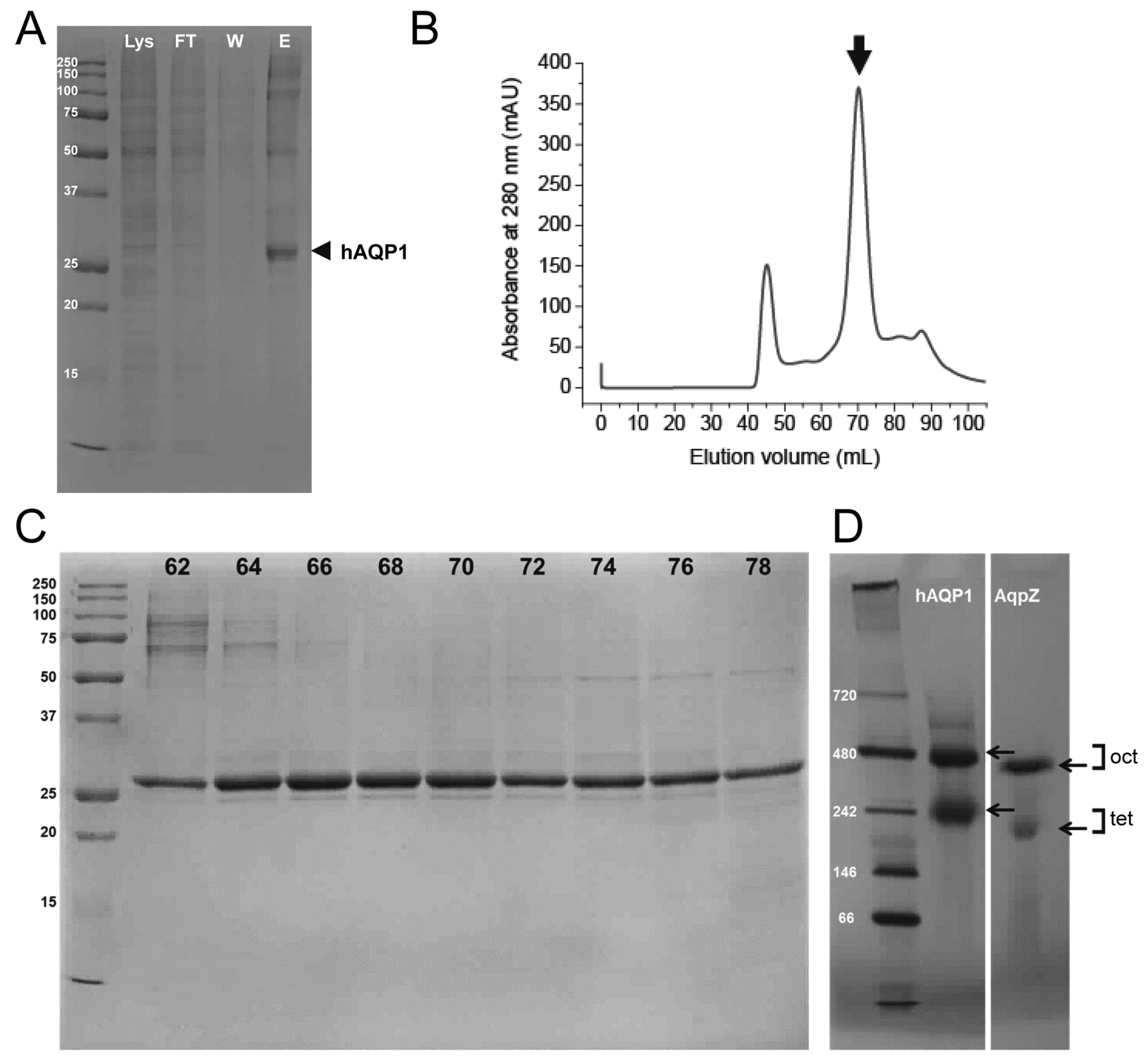
2.1. Expression and Purification of Human Aquaporin 1 (hAQP1) from Insect Cells
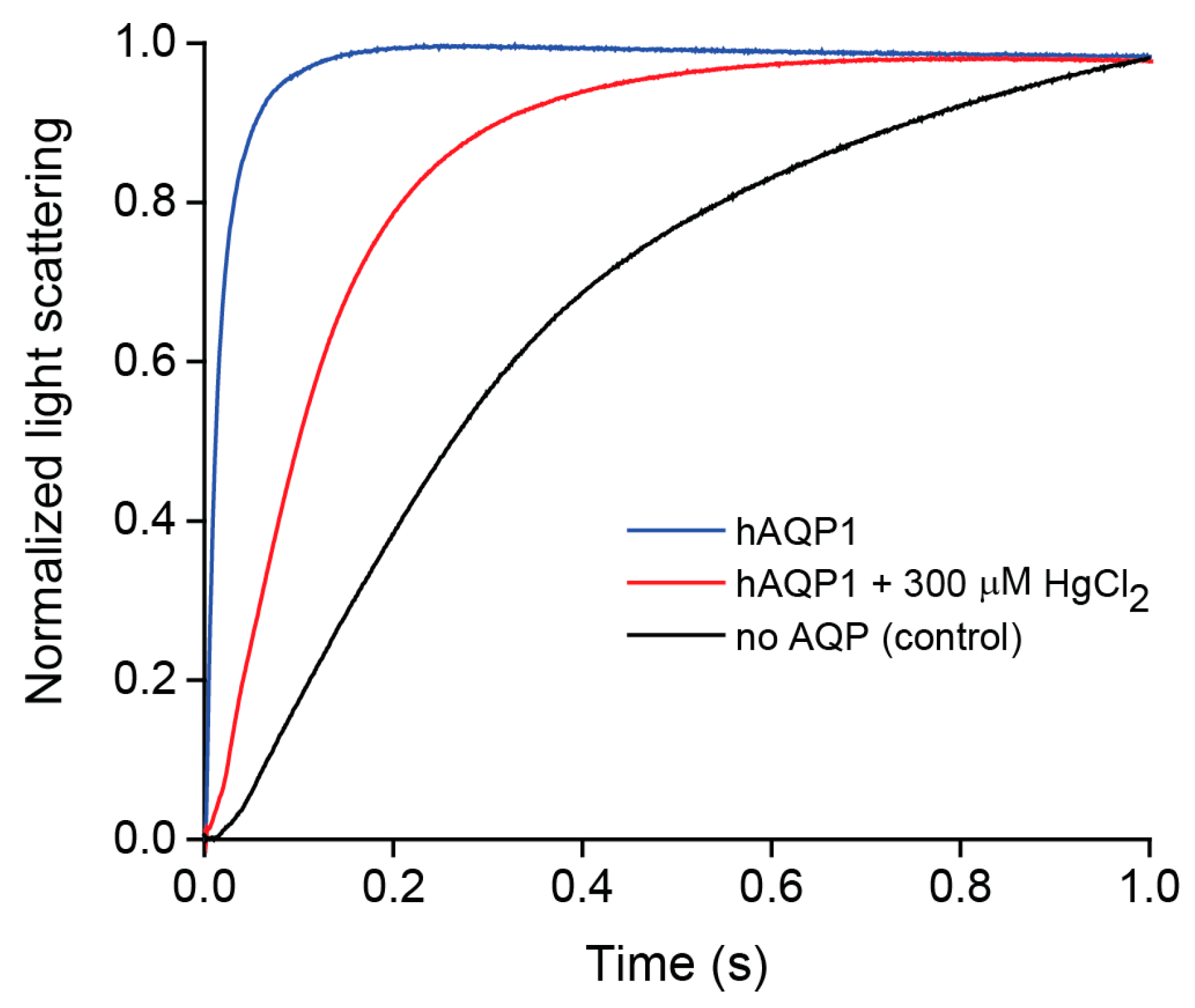
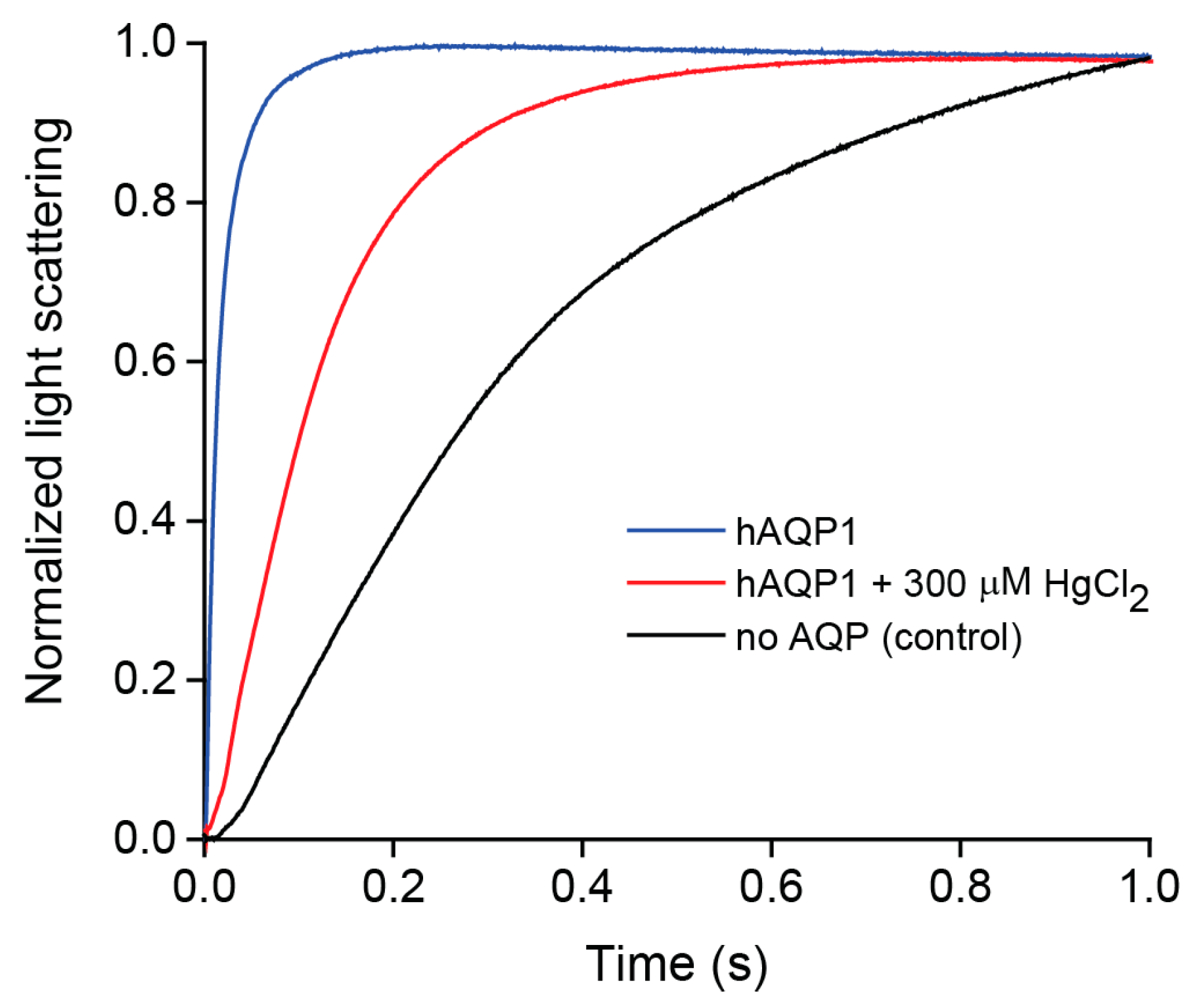
2.2. Water Permeability of Purified hAQP1
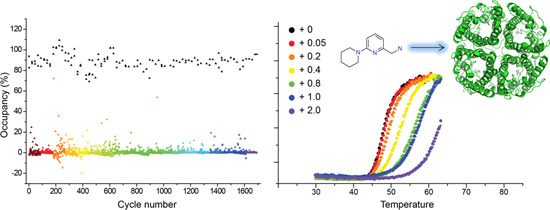
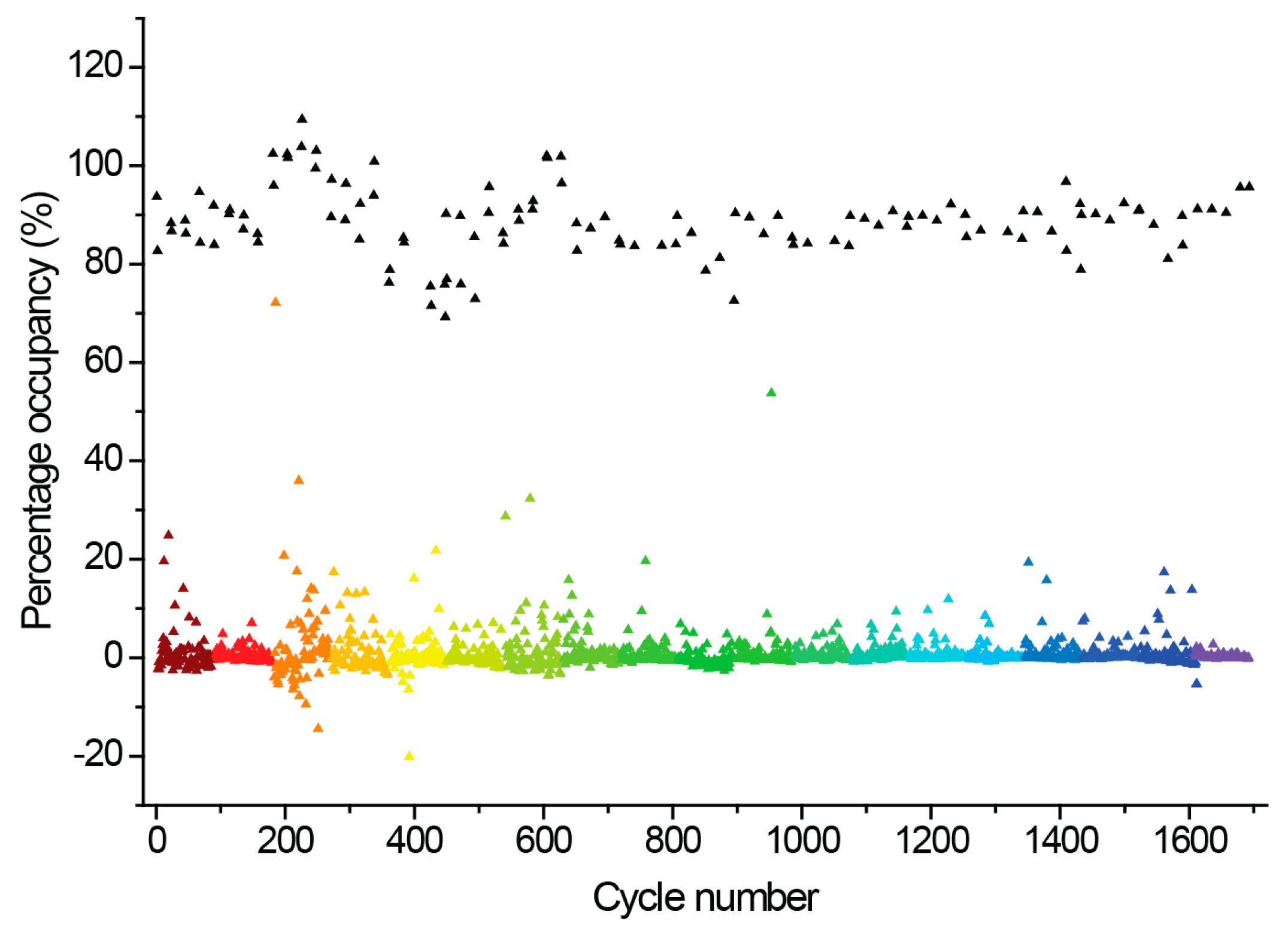
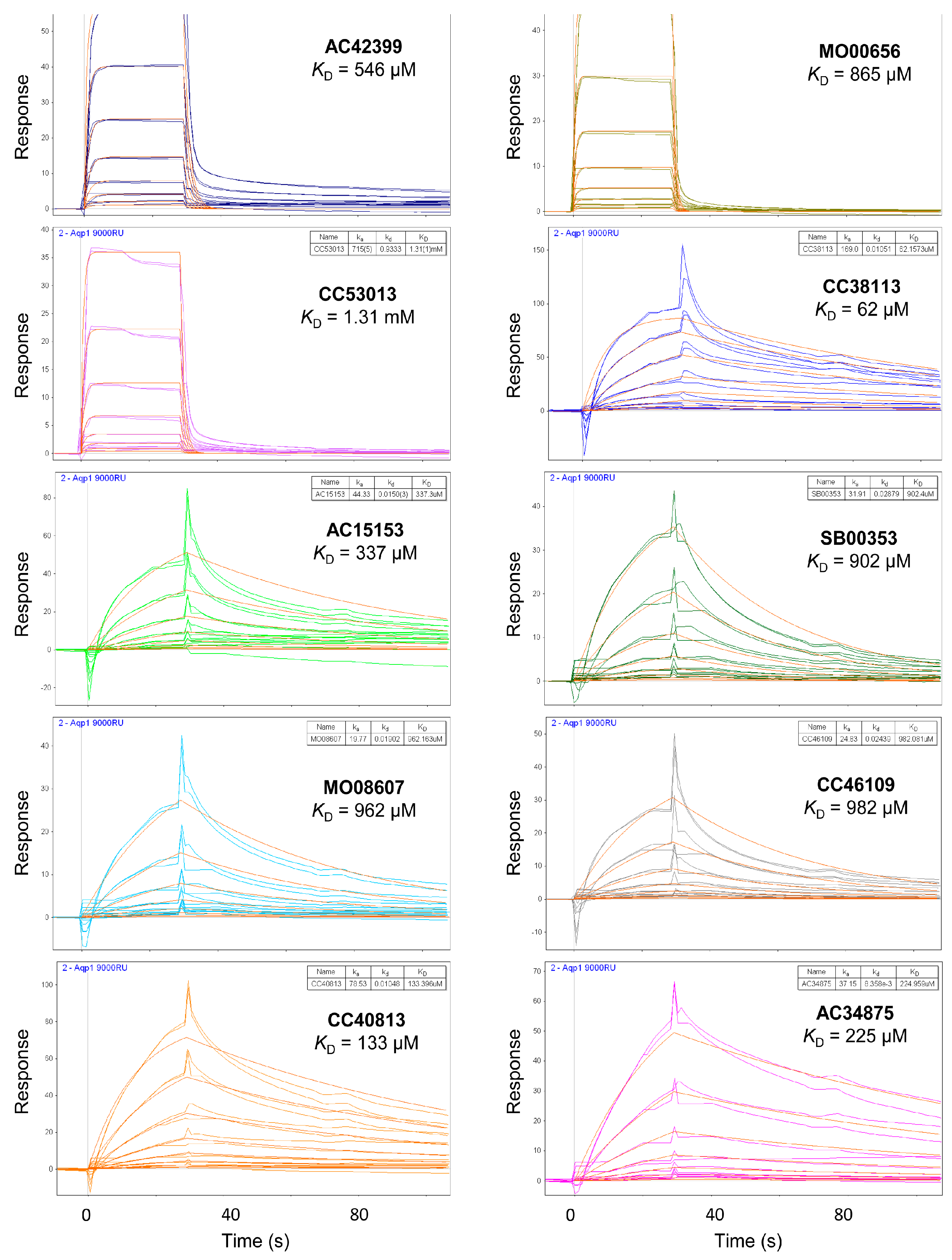
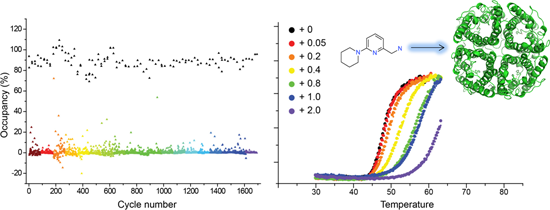
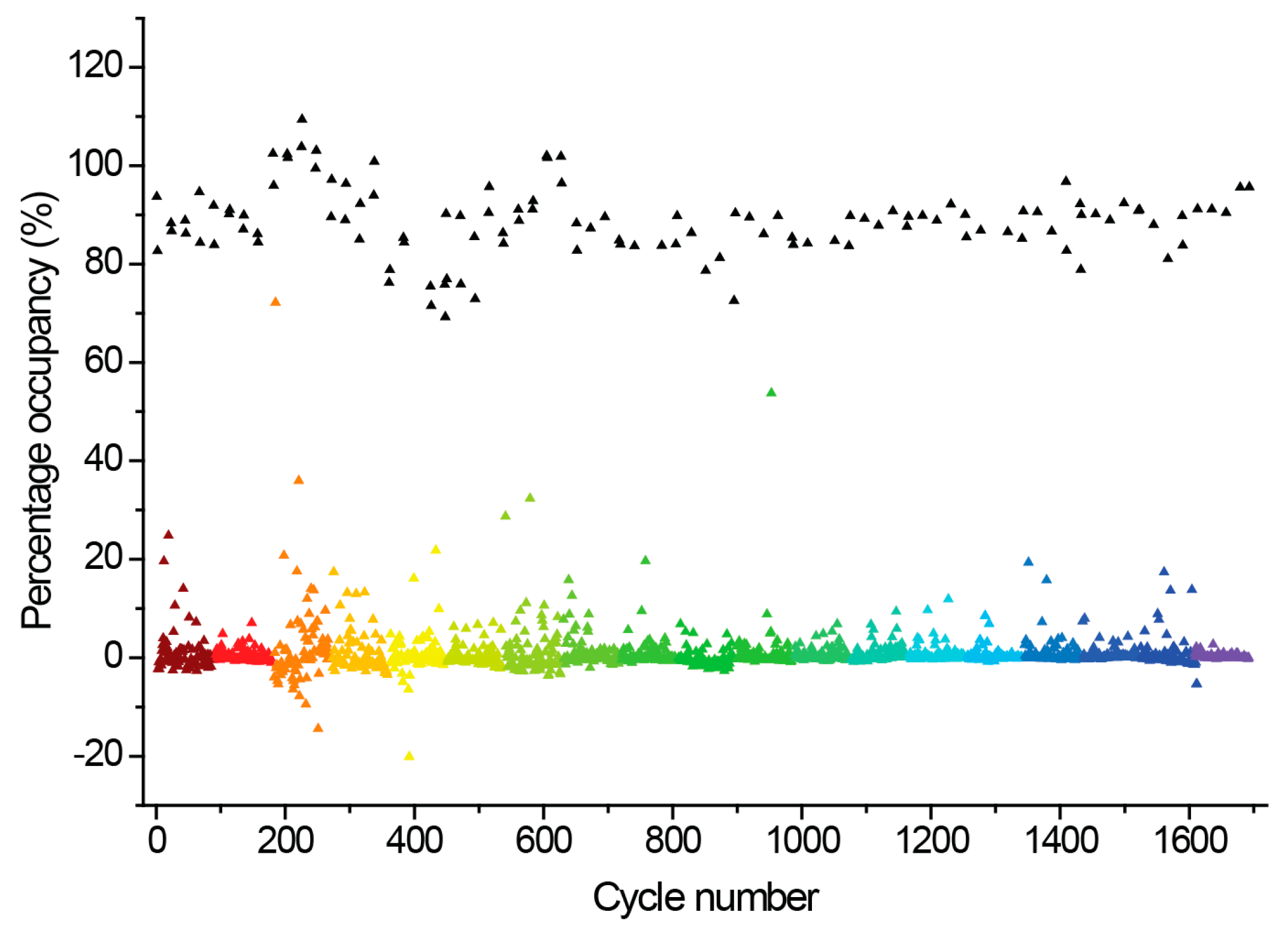
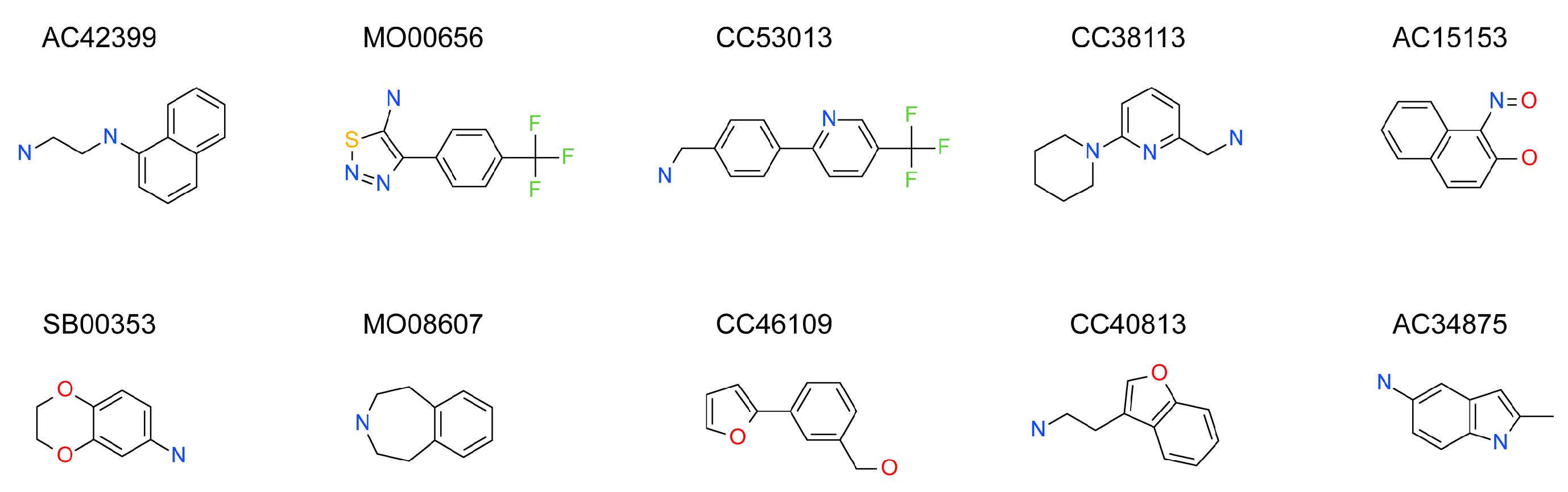
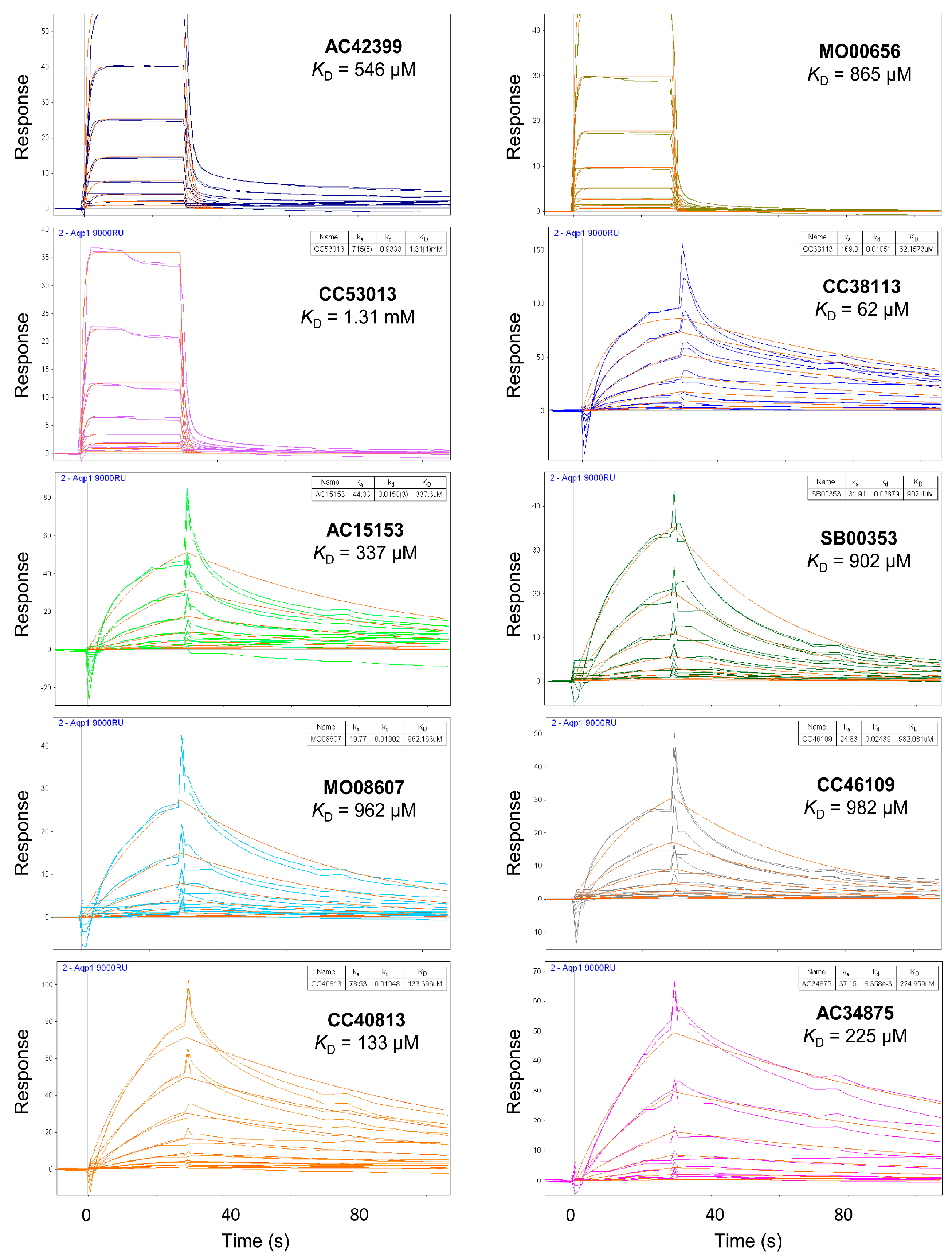
2.3. Hit Binding Measured by Surface Plasmon Resonance (SPR)
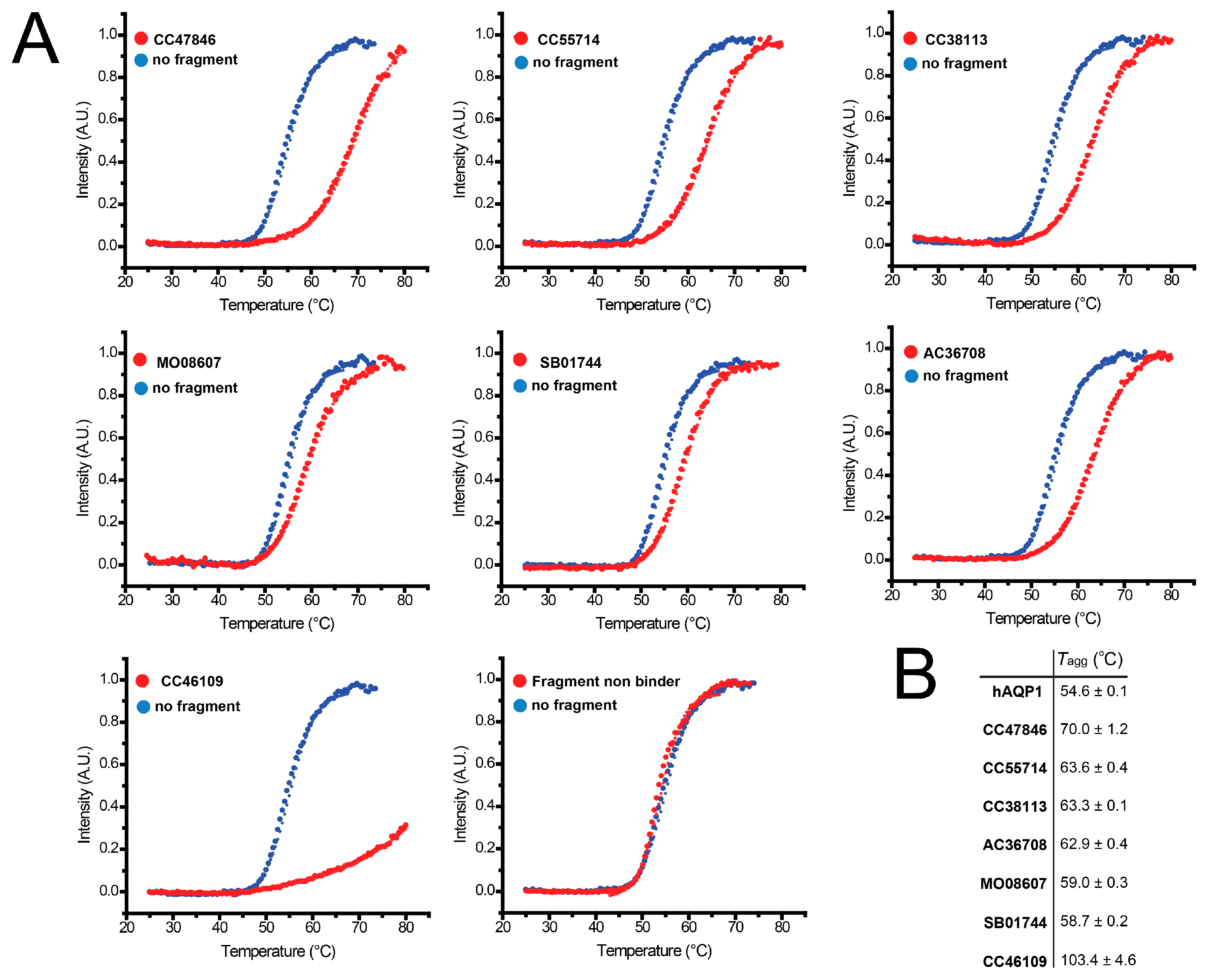
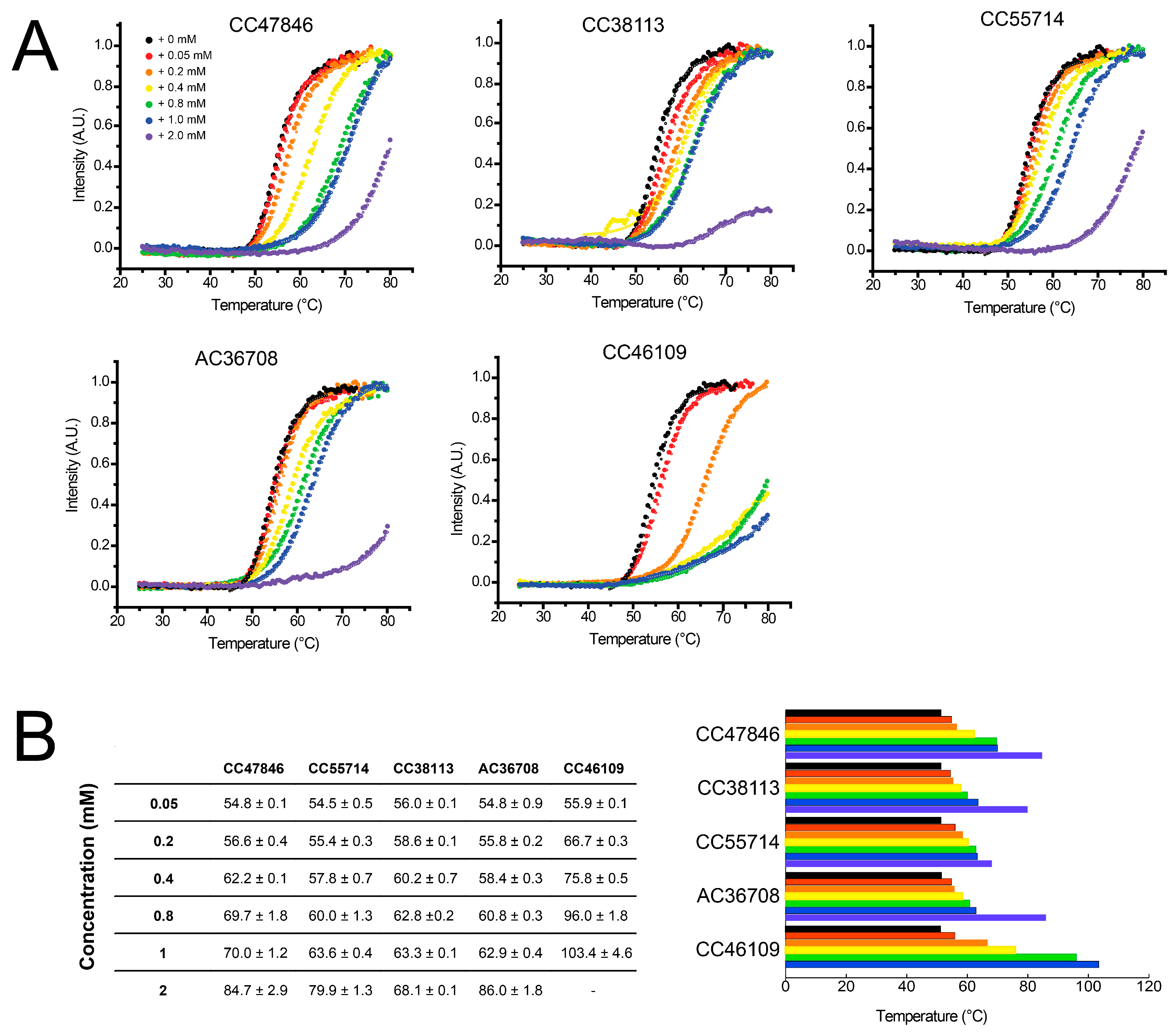
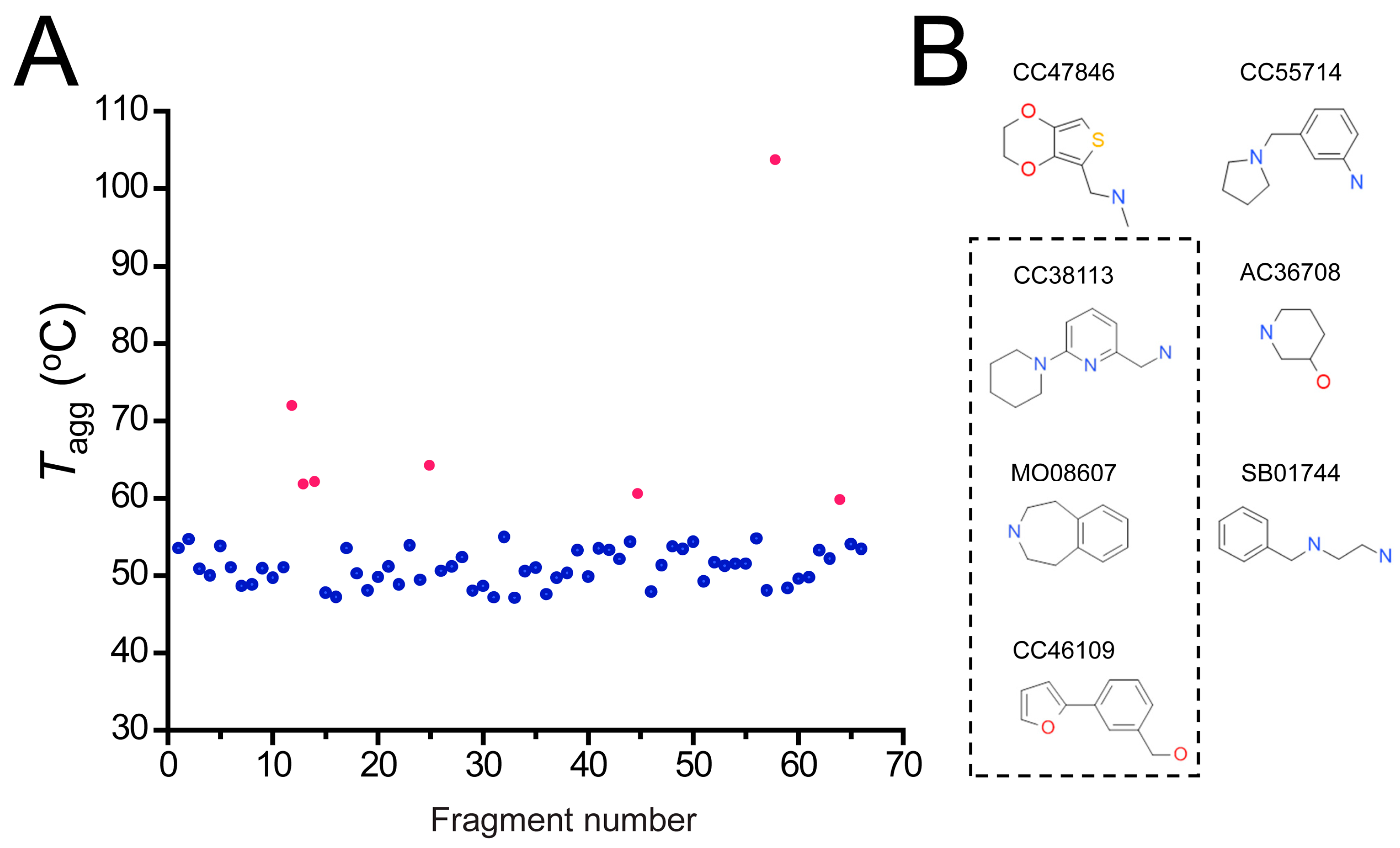
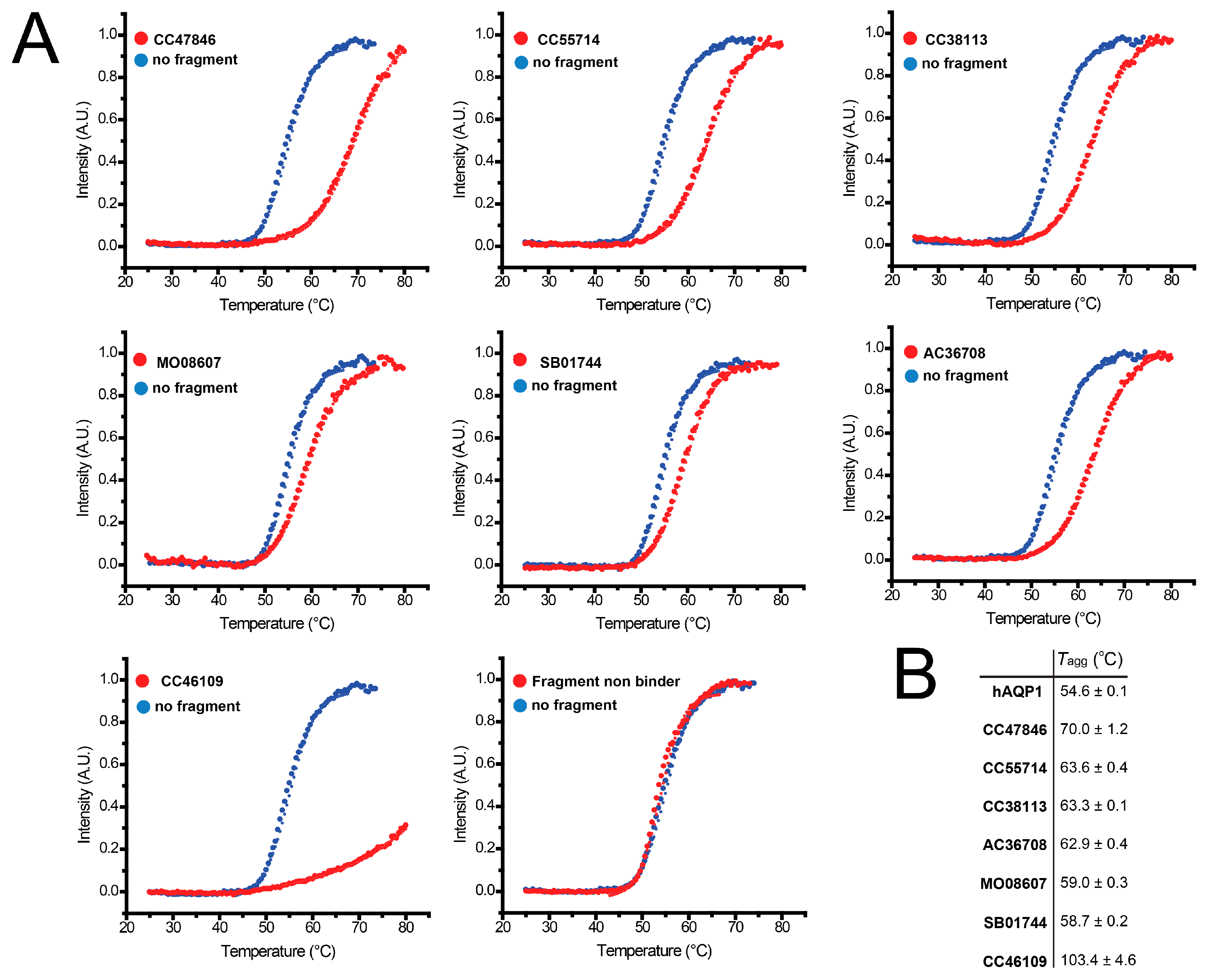
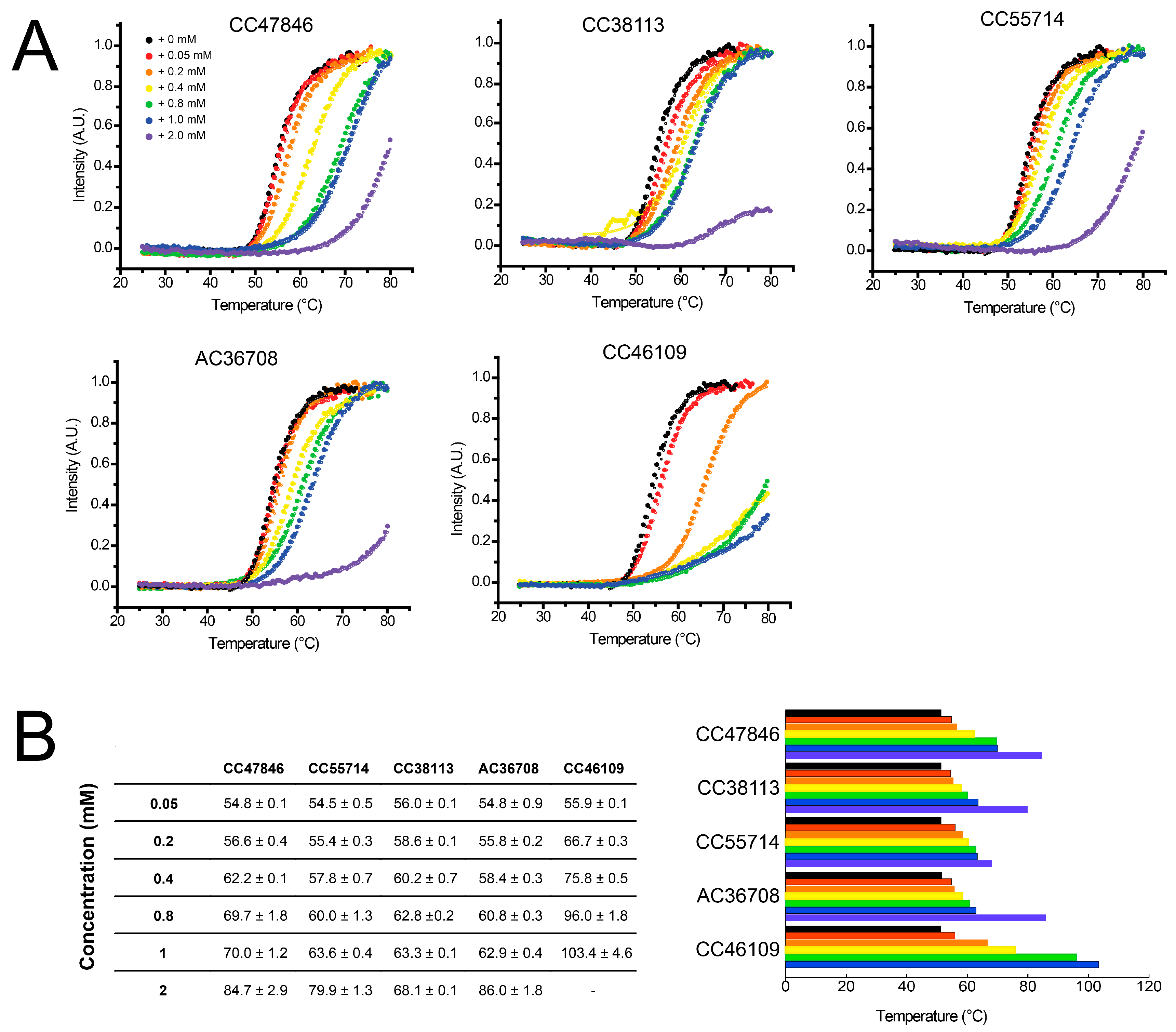
2.4. Hit Confirmation Using Thermal Aggregation Shift Assay
3. Discussion
4. Materials and Methods
4.1. Purification of hAQP1 from Insect Cells
4.2. Gel Electrophoresis
4.3. Reconstitution of Purified Aquaporin into Liposomes.
4.4. Stopped-Flow Water Permeability Assay
4.5. Light Scattering Data Analysis
4.6. Surface Plasmon Resonance (SPR)-Protein Immobilization
4.7. Fragment Screening Using SPR
4.8. Data Processing and Curve Fitting
4.9. Thermal Aggregation Shift Assay
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AQP | Aquaporin |
| DSLS | Differential Static Light Scattering |
| FBDD | Fragment-based Drug Discovery |
| HTS | High-throughput Screening |
| SPR | Surface Plasmon Resonance |
| TM | Transmembrane |
References
- Benga, G.; Popescu, O.; Borza, V.; Pop, V.I.; Muresan, A.; Mocsy, I.; Brain, A.; Wrigglesworth, J.M. Water permeability in human erythrocytes: Identification of membrane proteins involved in water transport. Eur. J. Cell Biol. 1986, 41, 252–262. [Google Scholar] [PubMed]
- Denker, B.M.; Smith, B.L.; Kuhajda, F.P.; Agre, P. Identification, purification, and partial characterization of a novel MR 28,000 integral membrane protein from erythrocytes and renal tubules. J. Biol. Chem. 1988, 263, 15634–15642. [Google Scholar] [PubMed]
- Preston, G.M.; Carroll, T.P.; Guggino, W.B.; Agre, P. Appearance of water channels in Xenopus oocytes expressing red cell CHIP28 protein. Science 1992, 256, 385–387. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, K.; Hara, S.; Kondo, S. Aquaporin water channels in mammals. Clin. Exp. Nephrol. 2009, 13, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Yasui, M.; Hazama, A.; Kwon, T.H.; Nielsen, S.; Guggino, W.B.; Agre, P. Rapid gating and anion permeability of an intracellular aquaporin. Nature 1999, 402, 184–187. [Google Scholar] [PubMed]
- Yool, A.J. Functional domains of aquaporin-1: Keys to physiology, and targets for drug discovery. Curr. Pharm. Des. 2007, 13, 3212–3221. [Google Scholar] [CrossRef] [PubMed]
- Saparov, S.M.; Liu, K.; Agre, P.; Pohl, P. Fast and selective ammonia transport by aquaporin-8. J. Biol. Chem. 2007, 282, 5296–5301. [Google Scholar] [CrossRef] [PubMed]
- Holm, L.M.; Jahn, T.P.; Moller, A.L.; Schjoerring, J.K.; Ferri, D.; Klaerke, D.A.; Zeuthen, T. NH3 and NH4+ permeability in aquaporin-expressing Xenopus oocytes. Pflugers Arch. 2005, 450, 415–428. [Google Scholar] [CrossRef] [PubMed]
- King, L.S.; Kozono, D.; Agre, P. From structure to disease: The evolving tale of aquaporin biology. Nat. Rev. Mol. Cell Biol. 2004, 5, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Wysocki, R.; Chery, C.C.; Wawrzycka, D.; Van Hulle, M.; Cornelis, R.; Thevelein, J.M.; Tamas, M.J. The glycerol channel Fps1p mediates the uptake of arsenite and antimonite in Saccharomyces cerevisiae. Mol. Microbiol. 2001, 40, 1391–1401. [Google Scholar] [CrossRef] [PubMed]
- Sanders, O.I.; Rensing, C.; Kuroda, M.; Mitra, B.; Rosen, B.P. Antimonite is accumulated by the glycerol facilitator GlpF in Escherichia coli. J. Bacteriol. 1997, 179, 3365–3367. [Google Scholar] [PubMed]
- Cooper, G.J.; Boron, W.F. Effect of PCMBS on CO2 permeability of Xenopus oocytes expressing aquaporin 1 or its C189S mutant. Am. J. Physiol. Cell Physiol. 1998, 275, C1481–1486. [Google Scholar]
- Herrera, M.; Hong, N.J.; Garvin, J.L. Aquaporin-1 transports NO across cell membranes. Hypertension 2006, 48, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.I.; Loktyushkin, A.V.; Gus’kova, R.A.; Vasil’ev, N.S.; Fedorov, G.E.; Rubin, A.B. Oxygen channels of erythrocyte membrane. Doklady 2007, 414, 137–140. [Google Scholar] [CrossRef] [PubMed]
- Gonen, T.; Sliz, P.; Kistler, J.; Cheng, Y.; Walz, T. Aquaporin-0 membrane junctions reveal the structure of a closed water pore. Nature 2004, 429, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Sui, H.; Han, B.-G.; Lee, J.K.; Walian, P.; Jap, B.K. Structural basis of water-specific transport through the AQP1 water channel. Nature 2001, 414, 872–878. [Google Scholar] [CrossRef] [PubMed]
- Hiroaki, Y.; Tani, K.; Kamegawa, A.; Gyobu, N.; Nishikawa, K.; Suzuki, H.; Walz, T.; Sasaki, S.; Mitsuoka, K.; Kimura, K.; et al. Implications of the aquaporin-4 structure on array formation and cell adhesion. J. Mol. Biol. 2006, 355, 628–639. [Google Scholar] [CrossRef] [PubMed]
- Horsefield, R.; Norden, K.; Fellert, M.; Backmark, A.; Tornroth-Horsefield, S.; Terwisscha Van Scheltinga, A.C.; Kvassman, J.; Kjellbom, P.; Johanson, U.; Neutze, R. High-resolution X-ray structure of human aquaporin 5. Proc. Natl. Acad. Sci. USA 2008, 105, 13327–13332. [Google Scholar] [CrossRef] [PubMed]
- Tajkhorshid, E.; Nollert, P.; Jensen, M.Ã.; Miercke, L.J.W.; O’Connell, J.; Stroud, R.M.; Schulten, K. Control of the selectivity of the aquaporin water channel family by global orientational tuning. Science 2002, 296, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Kozono, D.; Remis, J.; Kitagawa, Y.; Agre, P.; Stroud, R.M. Structural basis for conductance by the archaeal aquaporin AqpM at 1.68 Å. Proc. Natl. Acad. Sci. USA 2005, 102, 18932–18937. [Google Scholar] [CrossRef] [PubMed]
- Savage, D.F.; Stroud, R.M. Structural Basis of Aquaporin Inhibition by Mercury. J. Mol. Biol. 2007, 368, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.B.; Skach, W.R.; Verkman, A.S. Functional independence of monomeric CHIP28 water channels revealed by expression of wild-type mutant heterodimers. J. Biol. Chem. 1994, 269, 10417–10422. [Google Scholar] [PubMed]
- Verkman, A.S. More than just water channels: Unexpected cellular roles of aquaporins. J. Cell Sci. 2005, 118, 3225–3232. [Google Scholar] [CrossRef] [PubMed]
- Manley, G.T.; Binder, D.K.; Papadopoulos, M.C.; Verkman, A.S. New insights into water transport and edema in the central nervous system from phenotype analysis of aquaporin-4 null mice. Neuroscience 2004, 129, 983–991. [Google Scholar] [CrossRef] [PubMed]
- Verkman, A.S. Physiological importance of aquaporins: Lessons from knockout mice. Curr. Opin. Nephrol. Hypertens. 2000, 9, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Badaut, J.; Brunet, J.F.; Regli, L. Aquaporins in the brain: From aqueduct to “multi-duct”. Metab. Brain Dis. 2007, 22, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.; Smith, B.L.; Christensen, E.I.; Agre, P. Distribution of the aquaporin CHIP in secretory and resorptive epithelia and capillary endothelia. Proc. Natl. Acad. Sci. USA 1993, 90, 7275–7279. [Google Scholar] [CrossRef] [PubMed]
- Oshio, K.; Watanabe, H.; Song, Y.; Verkman, A.S.; Manley, G.T. Reduced cerebrospinal fluid production and intracranial pressure in mice lacking choroid plexus water channel Aquaporin-1. FASEB J. 2005, 19, 76–78. [Google Scholar] [CrossRef] [PubMed]
- Verkman, A.S. Novel roles of aquaporins revealed by phenotype analysis of knockout mice. Rev. Physiol. Biochem. Pharmacol. 2005, 155, 31–55. [Google Scholar] [PubMed]
- Nielsen, S.; Frokiaer, J.; Marples, D.; Kwon, T.H.; Agre, P.; Knepper, M.A. Aquaporins in the kidney: From molecules to medicine. Physiol. Rev. 2002, 82, 205–244. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.W.; Wang, W.; Kwon, T.H.; Knepper, M.A.; Frøkiær, J.; Nielsen, S. Increased expression of ENaC subunits and increased apical targeting of AQP2 in the kidneys of spontaneously hypertensive rats. Am. J. Physiol. Ren. Physiol. 2005, 289, F957–F968. [Google Scholar] [CrossRef] [PubMed]
- Verkman, A.S. Applications of aquaporin inhibitors. Drug News Perspect. 2001, 14, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Verkman, A.S. Role of aquaporin water channels in eye function. Exp. Eye Res. 2003, 76, 137–143. [Google Scholar] [CrossRef]
- Zhang, D.; Vetrivel, L.; Verkman, A.S. Aquaporin deletion in mice reduces intraocular pressure and aqueous fluid production. J. Gen. Physiol. 2002, 119, 561–569. [Google Scholar] [PubMed]
- Huber, V.J.; Tsujita, M.; Nakada, T. Aquaporins in drug discovery and pharmacotherapy. Mol. Asp. Med. 2012, 33, 691–703. [Google Scholar] [CrossRef] [PubMed]
- Verkman, A.S. Mammalian aquaporins: Diverse physiological roles and potential clinical significance. Expert Rev. Mol. Med. 2008, 10, e13. [Google Scholar] [CrossRef] [PubMed]
- Verkman, A.S. Aquaporins in clinical medicine. Annu. Rev. Med. 2012, 63, 303–316. [Google Scholar] [CrossRef] [PubMed]
- Castle, N.A. Aquaporins as targets for drug discovery. Drug Discov. Today 2005, 10, 485–493. [Google Scholar] [CrossRef]
- Verkman, A.S.; Anderson, M.O.; Papadopoulos, M.C. Aquaporins: Important but elusive drug targets. Nat. Rev. Drug Discov. 2014, 13, 259–277. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, M.C.; Saadoun, S.; Verkman, A.S. Aquaporins and cell migration. Pflugers Arch. 2008, 456, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Saadoun, S.; Papadopoulos, M.C.; Watanabe, H.; Yan, D.; Manley, G.T.; Verkman, A.S. Involvement of aquaporin-4 in astroglial cell migration and glial scar formation. J. Cell Sci. 2005, 118, 5691–5698. [Google Scholar] [CrossRef] [PubMed]
- Hara-Chikuma, M.; Verkman, A.S. Aquaporin-1 facilitates epithelial cell migration in kidney proximal tubule. J. Am. Soc. Nephrol. 2006, 17, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Hara-Chikuma, M.; Verkman, A.S. Aquaporin-3 facilitates epidermal cell migration and proliferation during wound healing. J. Mol. Med. 2008, 86, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Sun, Y.; Healey, S.; Bi, Z.G.; Hu, G.; Wan, S.; Kouttab, N.; Chu, W.M.; Wan, Y.S. EGFR-mediated expression of aquaporin-3 is involved in human skin fibroblast migration. Biochem. J. 2006, 400, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Loitto, V.M.; Huang, C.; Sigal, Y.J.; Jacobson, K. Filopodia are induced by aquaporin-9 expression. Exp. Cell Res. 2007, 313, 1295–1306. [Google Scholar] [CrossRef] [PubMed]
- Loitto, V.M.; Karlsson, T.; Magnusson, K.E. Water flux in cell motility: Expanding the mechanisms of membrane protrusion. Cell Motil. Cytoskelet. 2009, 66, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, T.; Lagerholm, B.C.; Vikstrom, E.; Loitto, V.M.; Magnusson, K.E. Water fluxes through aquaporin-9 prime epithelial cells for rapid wound healing. Biochem. Biophys. Res. Commun. 2013, 430, 993–998. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, T.; Bolshakova, A.; Magalhaes, M.A.; Loitto, V.M.; Magnusson, K.E. Fluxes of water through aquaporin 9 weaken membrane-cytoskeleton anchorage and promote formation of membrane protrusions. PLoS ONE 2013, 8, e59901. [Google Scholar] [CrossRef] [PubMed]
- Vandergoot, F.G.; Ausio, J.; Wong, K.R.; Pattus, F.; Buckley, J.T. Dimerization stabilizes the pore-forming toxin aerolysin in solution. J. Biol. Chem. 1993, 268, 18272–18279. [Google Scholar]
- Verkman, A.S.; Hara-Chikuma, M.; Papadopoulos, M.C. Aquaporins—New players in cancer biology. J. Mol. Med. 2008, 86, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Huebert, R.C.; Vasdev, M.M.; Shergill, U.; Das, A.; Huang, B.Q.; Charlton, M.R.; LaRusso, N.F.; Shah, V.H. Aquaporin-1 facilitates angiogenic invasion in the pathological neovasculature that accompanies cirrhosis. Hepatology 2010, 52, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Saadoun, S.; Papadopoulos, M.C.; Hara-Chikuma, M.; Verkman, A.S. Impairment of angiogenesis and cell migration by targeted aquaporin-1 gene disruption. Nature 2005, 434, 786–792. [Google Scholar] [CrossRef] [PubMed]
- Vacca, A.; Frigeri, A.; Ribatti, D.; Nicchia, G.P.; Nico, B.; Ria, R.; Svelto, M.; Dammacco, F. Microvessel overexpression of aquaporin 1 parallels bone marrow angiogenesis in patients with active multiple myeloma. Br. J. Haematol. 2001, 113, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Verkman, A.S. Increased migration and metastatic potential of tumor cells expressing aquaporin water channels. FASEB J. 2006, 20, 1892–1894. [Google Scholar] [CrossRef] [PubMed]
- Nicchia, G.P.; Stigliano, C.; Sparaneo, A.; Rossi, A.; Frigeri, A.; Svelto, M. Inhibition of aquaporin-1 dependent angiogenesis impairs tumour growth in a mouse model of melanoma. J. Mol. Med. 2013, 91, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Nicchia, G.P.; Srinivas, M.; Li, W.; Brosnan, C.F.; Frigeri, A.; Spray, D.C. New possible roles for aquaporin-4 in astrocytes: Cell cytoskeleton and functional relationship with connexin43. FASEB J. 2005, 19, 1674–1676. [Google Scholar] [CrossRef] [PubMed]
- La Porta, C. AQP1 is not only a water channel: It contributes to cell migration through Lin7/β-catenin. Cell Adhes. Migr. 2010, 4, 204–206. [Google Scholar] [CrossRef]
- Bergers, G.; Hanahan, D. Modes of resistance to anti-angiogenic therapy. Nat. Rev. Cancer 2008, 8, 592–603. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P. Angiogenesis in life, disease and medicine. Nature 2005, 438, 932–936. [Google Scholar] [CrossRef] [PubMed]
- Prager, G.W.; Unseld, M.; Zielinski, C.C. Angiogenesis in cancer: Anti-VEGF escape mechanisms. Transl. Lung Cancer Res. 2012, 1, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Casanovas, O.; Hicklin, D.J.; Bergers, G.; Hanahan, D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell 2005, 8, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Van Hoek, A.N.; Biwersi, J.; Verkman, A.S. A point mutation at cysteine 189 blocks the water permeability of rat kidney water channel CHIP28k. Biochemistry 1993, 32, 2938–2941. [Google Scholar] [CrossRef] [PubMed]
- Preston, G.M.; Jung, J.S.; Guggino, W.B.; Agre, P. The mercury-sensitive residue at cysteine 189 in the CHIP28 water channel. J. Biol. Chem. 1993, 268, 17–20. [Google Scholar] [PubMed]
- Niemietz, C.M.; Tyerman, S.D. New potent inhibitors of aquaporins: Silver and gold compounds inhibit aquaporins of plant and human origin. FEBS Lett. 2002, 531, 443–447. [Google Scholar] [CrossRef]
- Yukutake, Y.; Tsuji, S.; Hirano, Y.; Adachi, T.; Takahashi, T.; Fujihara, K.; Agre, P.; Yasui, M.; Suematsu, M. Mercury chloride decreases the water permeability of aquaporin-4-reconstituted proteoliposomes. Biol. Cell 2008, 100, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Brooks, H.L.; Regan, J.W.; Yool, A.J. Inhibition of aquaporin-1 water permeability by tetraethylammonium: Involvement of the loop E pore region. Mol. Pharmacol. 2000, 57, 1021–1026. [Google Scholar] [PubMed]
- Brooks, H.L.; Regan, J.W.; Yool, A.J. Inhibition of aquaporin-1 water permeability by TEA. FASEB J. 1999, 13, A394–A394. [Google Scholar]
- Detmers, F.J.M.; de Groot, B.L.; Müller, E.M.; Hinton, A.; Konings, I.B.M.; Sze, M.; Flitsch, S.L.; Grubmüller, H.; Deen, P.M.T. Quaternary ammonium compounds as water channel blockers: Specificity, potency, and site of action. J. Biol. Chem. 2006, 281, 14207–14214. [Google Scholar] [CrossRef] [PubMed]
- Yukutake, Y.; Hirano, Y.; Suematsu, M.; Yasui, M. Rapid and reversible inhibition of aquaporin-4 by zinc. Biochemistry 2009, 48, 12059–12061. [Google Scholar] [CrossRef] [PubMed]
- Migliati, E.; Meurice, N.; DuBois, P.; Fang, J.S.; Somasekharan, S.; Beckett, E.; Flynn, G.; Yool, A.J. Inhibition of aquaporin-1 and aquaporin-4 water permeability by a derivative of the loop diuretic bumetanide acting at an internal pore-occluding binding site. Mol. Pharmacol. 2009, 76, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Ma, B.; Li, T.; Gao, J.W.; Yu, H.M.; Li, X.J. Acetazolamide inhibits aquaporin-1 protein expression and angiogenesis. Acta Pharmacol. Sin. 2004, 25, 812–816. [Google Scholar] [PubMed]
- Ma, B.; Xiang, Y.; Mu, S.M.; Li, T.; Yu, H.M.; Li, X.J. Effects of acetazolamide and anordiol on osmotic water permeability in AQP1-cRNA injected Xenopus oocyte. Acta Pharmacol. Sin. 2004, 25, 90–97. [Google Scholar] [PubMed]
- Huber, V.J.; Tsujita, M.; Yamazaki, M.; Sakimura, K.; Nakada, T. Identification of arylsulfonamides as Aquaporin 4 inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 1270–1273. [Google Scholar] [CrossRef] [PubMed]
- Huber, V.J.; Tsujita, M.; Kwee, I.L.; Nakada, T. Inhibition of aquaporin 4 by antiepileptic drugs. Biorg. Med. Chem. 2009, 17, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Kim, J.K.; Verkman, A.S. Comparative efficacy of HgCl2 with candidate aquaporin-1 inhibitors DMSO, gold, TEA+ and acetazolamide. FEBS Lett. 2006, 580, 6679–6684. [Google Scholar] [CrossRef] [PubMed]
- Seeliger, D.; Zapater, C.; Krenc, D.; Haddoub, R.; Flitsch, S.; Beitz, E.; Cerda, J.; de Groot, B.L. Discovery of novel human aquaporin-1 blockers. ACS Chem. Biol. 2013, 8, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Tornroth-Horsefield, S.; Wang, Y.; Hedfalk, K.; Johanson, U.; Karlsson, M.; Tajkhorshid, E.; Neutze, R.; Kjellbom, P. Structural mechanism of plant aquaporin gating. Nature 2006, 439, 688–694. [Google Scholar] [CrossRef] [PubMed]
- Gunnarson, E.; Zelenina, M.; Aperia, A. Regulation of brain aquaporins. Neuroscience 2004, 129, 947–955. [Google Scholar] [CrossRef] [PubMed]
- Gunnarson, E.; Zelenina, M.; Axehult, G.; Song, Y.; Bondar, A.; Krieger, P.; Brismar, H.; Zelenin, S.; Aperia, A. Identification of a molecular target for glutamate regulation of astrocyte water permeability. Glia 2008, 56, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Zelenina, M.; Zelenin, S.; Bondar, A.A.; Brismar, H.; Aperia, A. Water permeability of aquaporin-4 is decreased by protein kinase C and dopamine. Am. J. Physiol. Ren. Physiol. 2002, 283, F309–F318. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.D.; Yeh, R.; Sandstrom, A.; Chorny, I.; Harries, W.E.C.; Robbins, R.A.; Miercke, L.J.W.; Stroud, R.M. Crystal structure of human aquaporin 4 at 1.8 A and its mechanism of conductance. Proc. Natl. Acad. Sci. USA 2009, 106, 7437–7442. [Google Scholar] [CrossRef] [PubMed]
- Assentoft, M.; Kaptan, S.; Fenton, R.A.; Hua, S.Z.; de Groot, B.L.; Macaulay, N. Phosphorylation of rat aquaporin-4 at Ser111 is not required for channel gating. Glia 2013, 61, 1101–1112. [Google Scholar] [CrossRef] [PubMed]
- Sachdeva, R.; Singh, B. Phosphorylation of Ser-180 of rat aquaporin-4 shows marginal affect on regulation of water permeability: Molecular dynamics study. J. Biomol. Struct. Dyn. 2014, 32, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Reichow, S.L.; Gonen, T. Noncanonical Binding of Calmodulin to Aquaporin-0: Implications for Channel Regulation. Structure 2008, 16, 1389–1398. [Google Scholar] [CrossRef] [PubMed]
- Reichow, S.L.; Clemens, D.M.; Freites, J.A.; Németh-Cahalan, K.L.; Heyden, M.; Tobias, D.J.; Hall, J.E.; Gonen, T. Allosteric mechanism of water-channel gating by Ca2+-calmodulin. Nat. Struct. Mol. Biol. 2013, 20, 1085–1092. [Google Scholar] [CrossRef] [PubMed]
- Rees, D.C.; Congreve, M.; Murray, C.W.; Carr, R. Fragment-based lead discovery. Nat. Rev. Drug Discov. 2004, 3, 660–672. [Google Scholar] [CrossRef] [PubMed]
- Larsson, A.; Jansson, A.; Aberg, A.; Nordlund, P. Efficiency of hit generation and structural characterization in fragment-based ligand discovery. Curr. Opin. Chem. Biol. 2011, 15, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Siegal, G.; Ab, E.; Schultz, J. Integration of fragment screening and library design. Drug Discov. Today 2007, 12, 1032–1039. [Google Scholar] [CrossRef] [PubMed]
- Carr, R.A.E.; Congreve, M.; Murray, C.W.; Rees, D.C. Fragment-based lead discovery: Leads by design. Drug Discov. Today 2005, 10, 987–992. [Google Scholar] [CrossRef]
- Wenlock, M.C.; Austin, R.P.; Barton, P.; Davis, A.M.; Leeson, P.D. A comparison of physiochemical property profiles of development and marketed oral drugs. J. Med. Chem. 2003, 46, 1250–1256. [Google Scholar] [CrossRef] [PubMed]
- Navratilova, I.; Hopkins, A.L. Fragment screening by surface plasmon resonance. ACS Med. Chem. Lett. 2010, 1, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Fruh, V.; Zhou, Y.; Chen, D.; Loch, C.; Ab, E.; Grinkova, Y.N.; Verheij, H.; Sligar, S.G.; Bushweller, J.H.; Siegal, G. Application of fragment-based drug discovery to membrane proteins: identification of ligands of the integral membrane enzyme DsbB. Chem. Biol. 2010, 17, 881–891. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Errey, J.C.; Heitman, L.H.; Marshall, F.H.; Ijzerman, A.P.; Siegal, G. Fragment screening of GPCRs using biophysical methods: identification of ligands of the adenosine A2A receptor with novel biological activity. ACS Chem. Biol. 2012, 7, 2064–2073. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Ranganathan, A.; AP, I.J.; Siegal, G.; Carlsson, J. Complementarity between in silico and biophysical screening approaches in fragment-based lead discovery against the A2A adenosine receptor. J. Chem. Inf. Mod. 2013, 53, 2701–2714. [Google Scholar] [CrossRef] [PubMed]
- Lambruschini, C.; Veronesi, M.; Romeo, E.; Garau, G.; Bandiera, T.; Piomelli, D.; Scarpelli, R.; Dalvit, C. Development of fragment-based n-FABS NMR screening applied to the membrane enzyme FAAH. ChemBioChem 2013, 14, 1611–1619. [Google Scholar] [CrossRef] [PubMed]
- Brandts, J.F.; Lin, L.N. Study of strong to ultratight protein interactions using differential scanning calorimetry. Biochemistry 1990, 29, 6927–6940. [Google Scholar] [CrossRef] [PubMed]
- To, J.; Yeo, C.Y.; Soon, C.H.; Torres, J. A generic high-throughput assay to detect aquaporin functional mutants: Potential application to discovery of aquaporin inhibitors. Biochim. Biophys. Acta Gen. Subj. 2015, 1850, 1869–1876. [Google Scholar] [CrossRef] [PubMed]
- Vedadi, M.; Niesen, F.H.; Allali-Hassani, A.; Fedorov, O.Y.; Finerty, P.J.; Wasney, G.A.; Yeung, R.; Arrowsmith, C.; Ball, L.J.; Berglund, H.; et al. Chemical screening methods to identify ligands that promote protein stability, protein crystallization, and structure determination. Proc. Natl. Acad. Sci. USA 2006, 103, 15835–15840. [Google Scholar] [CrossRef] [PubMed]
- Wittig, I.; Braun, H.P.; Schägger, H. Blue native PAGE. Nat. Protocols 2006, 1, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Heuberger, E.H.M.L.; Veenhoff, L.M.; Duurkens, R.H.; Friesen, R.H.E.; Poolman, B. Oligomeric state of membrane transport proteins analyzed with blue native electrophoresis and analytical ultracentrifugation. J. Mol. Biol. 2002, 317, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Gan, S.W.; Vararattanavech, A.; Nordin, N.; Eshaghi, S.; Torres, J. A cost-effective method for simultaneous homo-oligomeric size determination and monodispersity conditions for membrane proteins. Anal. Biochem. 2011, 416, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Zeidel, M.L.; Ambudkar, S.V.; Smith, B.L.; Agre, P. Reconstitution of functional water channels in liposomes containing purified red cell CHIP28 protein. Biochemistry 1992, 31, 7436–7440. [Google Scholar] [CrossRef] [PubMed]
- Giannetti, A.M. From eperimental design to validated hits: A comprehensive walk-through of fragment lead identification using surface plasmon resonance. Methods Enzymol. 2011, 493, 169–218. [Google Scholar] [PubMed]
- Altintas, Z.; Uludag, Y.; Gurbuz, Y.; Tothill, I. Development of surface chemistry for surface plasmon resonance based sensors for the detection of proteins and DNA molecules. Anal. Chim. Acta 2012, 712, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Hann, M.M.; Leach, A.R.; Harper, G. Molecular complexity and its impact on the probability of finding leads for drug discovery. J. Chem. Inf. Comput. Sci. 2001, 41, 856–864. [Google Scholar] [CrossRef] [PubMed]
- Klein, J.; Meinecke, R.; Mayer, M.; Meyer, B. Detecting binding affinity to immobilized receptor proteins in compound libraries by HR-MAS STD NMR. J. Am. Chem. Soc. 1999, 121, 5336–5337. [Google Scholar] [CrossRef]
- Mayer, M.; Meyer, B. Characterization of ligand binding by saturation transfer difference NMR spectroscopy. Angew. Chem. Int. Ed. 1999, 38, 1784–1788. [Google Scholar] [CrossRef]
- Agre, P.; Smith, B.L.; Preston, G.M. ABH and Colton blood group antigens on aquaporin-1, the human red cell water channel protein. Transfus. Clin. Biol. 1995, 2, 303–308. [Google Scholar] [CrossRef]
- Chou, C.L.; Knepper, M.A.; Van Hoek, A.N.; Brown, D.; Yang, B.; Ma, T.; Verkman, A.S. Reduced water permeability and altered ultrastructure in thin descending limb of Henle in aquaporin-1 null mice. J. Clin. Investig. 1999, 103, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Olson, F.; Hunt, C.A.; Szoka, F.C.; Vail, W.J.; Papahadjopoulos, D. Preparation of liposomes of defined size distribution by extrusion through polycarbonate membranes. BBA Biomembr. 1979, 557, 9–23. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
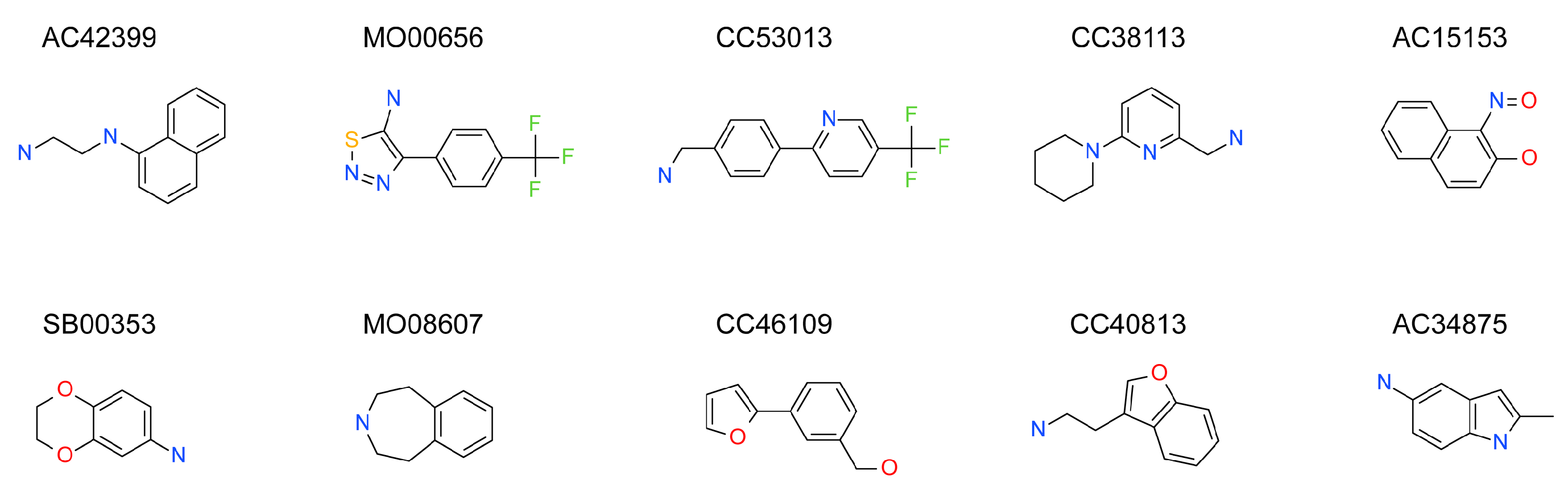
| Maybridge Code | Structure | SPR (KD) | Tagg (°C) | ΔTagg (+/−) |
|---|---|---|---|---|
| AC42399 |  | 546 µM | 48.0 | − |
| MO00656 |  | 865 µM | 51.8 | − |
| CC53013 | 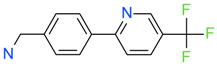 | 1.31 mM | 48.6 | − |
| CC38113 | 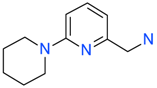 | 62 µM | 63.3 ± 0.1 | + |
| AC15153 | 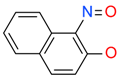 | 337 µM | n.a. | n.a. |
| SB00353 |  | 902 µM | 51.1 | − |
| MO08607 | 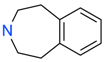 | 962 µM | 59.0 ± 0.3 | + |
| CC46109 |  | 982 µM | 103.4 ± 4.6 | + |
| CC40813 |  | 133 µM | 50.3 | − |
| AC34875 |  | 225 µM | 50.9 | − |
| CC47846 | 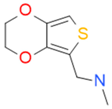 | n.a. | 70.0 ± 1.2 | + |
| CC55714 | 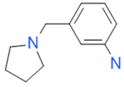 | n.a. | 63.6 ± 0.4 | + |
| AC36708 |  | n.a. | 62.9 ± 0.4 | + |
| SB01744 |  | n.a. | 58.7 ± 0.2 | + |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
To, J.; Torres, J. Fragment Screening of Human Aquaporin 1. Int. J. Mol. Sci. 2016, 17, 449. https://doi.org/10.3390/ijms17040449
To J, Torres J. Fragment Screening of Human Aquaporin 1. International Journal of Molecular Sciences. 2016; 17(4):449. https://doi.org/10.3390/ijms17040449
Chicago/Turabian StyleTo, Janet, and Jaume Torres. 2016. "Fragment Screening of Human Aquaporin 1" International Journal of Molecular Sciences 17, no. 4: 449. https://doi.org/10.3390/ijms17040449
APA StyleTo, J., & Torres, J. (2016). Fragment Screening of Human Aquaporin 1. International Journal of Molecular Sciences, 17(4), 449. https://doi.org/10.3390/ijms17040449