
Mechanical and IL-1β Responsive miR-365 Contributes to Osteoarthritis Development by Targeting Histone Deacetylase 4


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
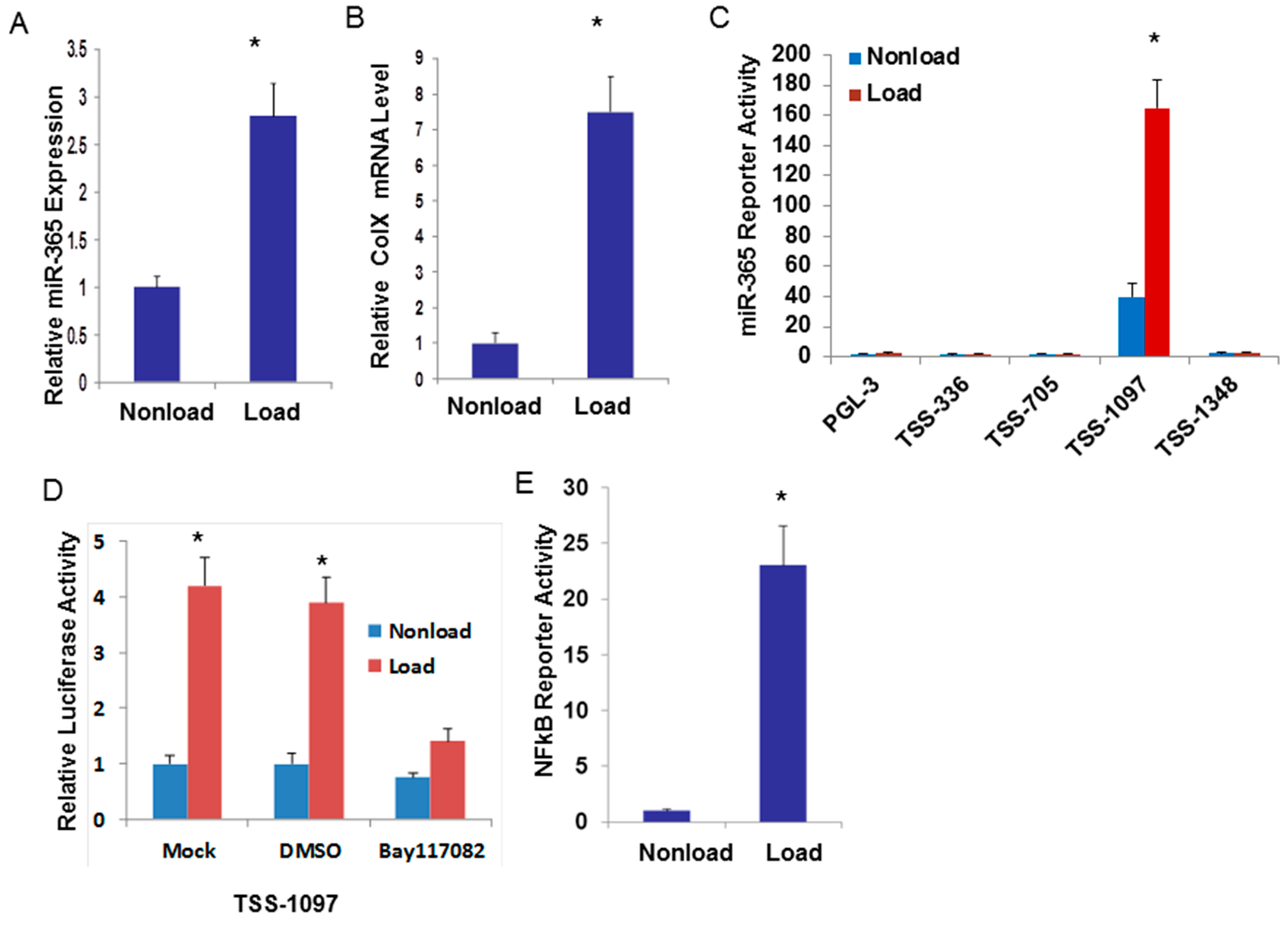
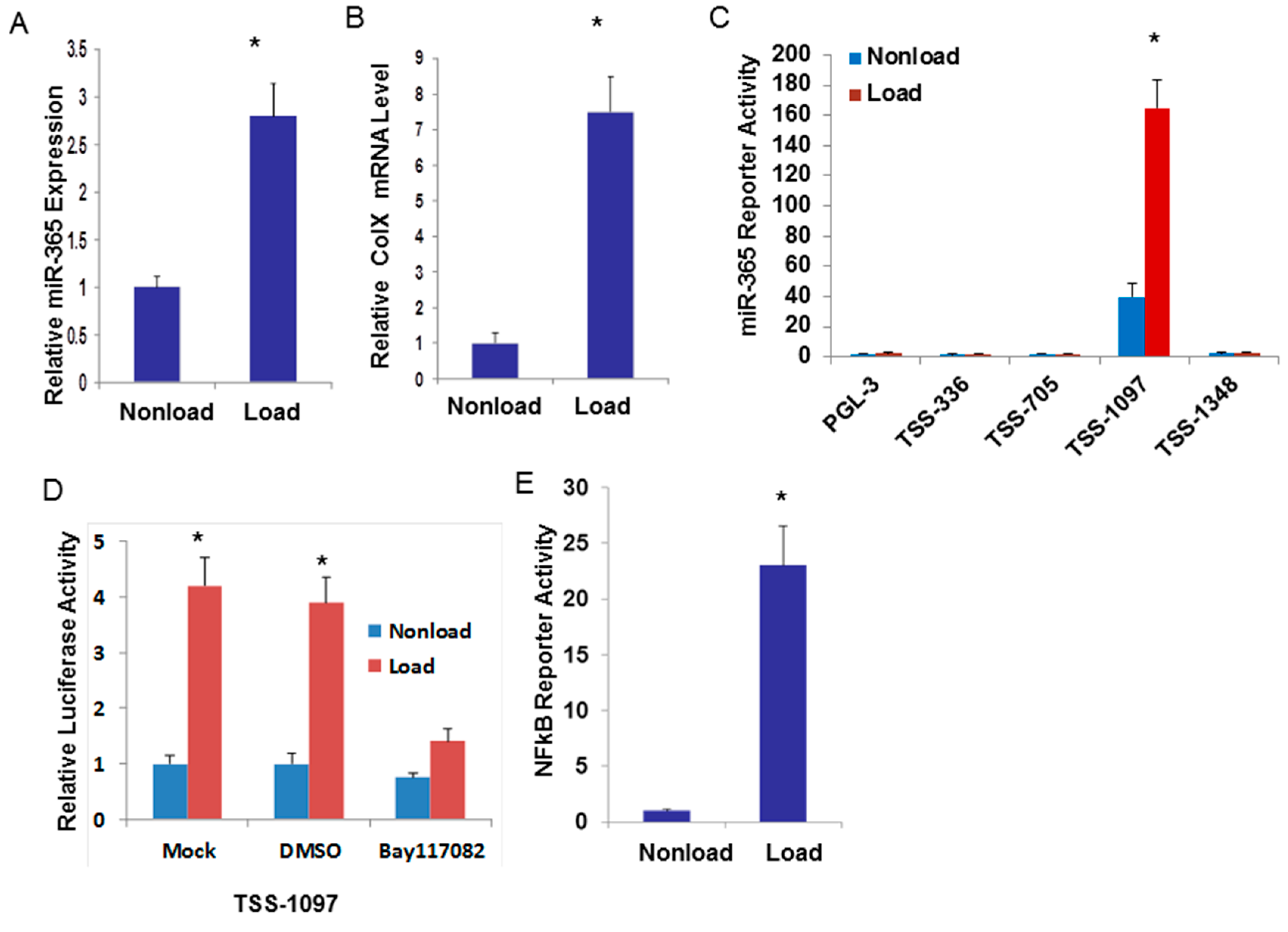
2.1. Cyclic Loading Transcriptionally Up-Regulates miR-365 through NF-кB Signaling
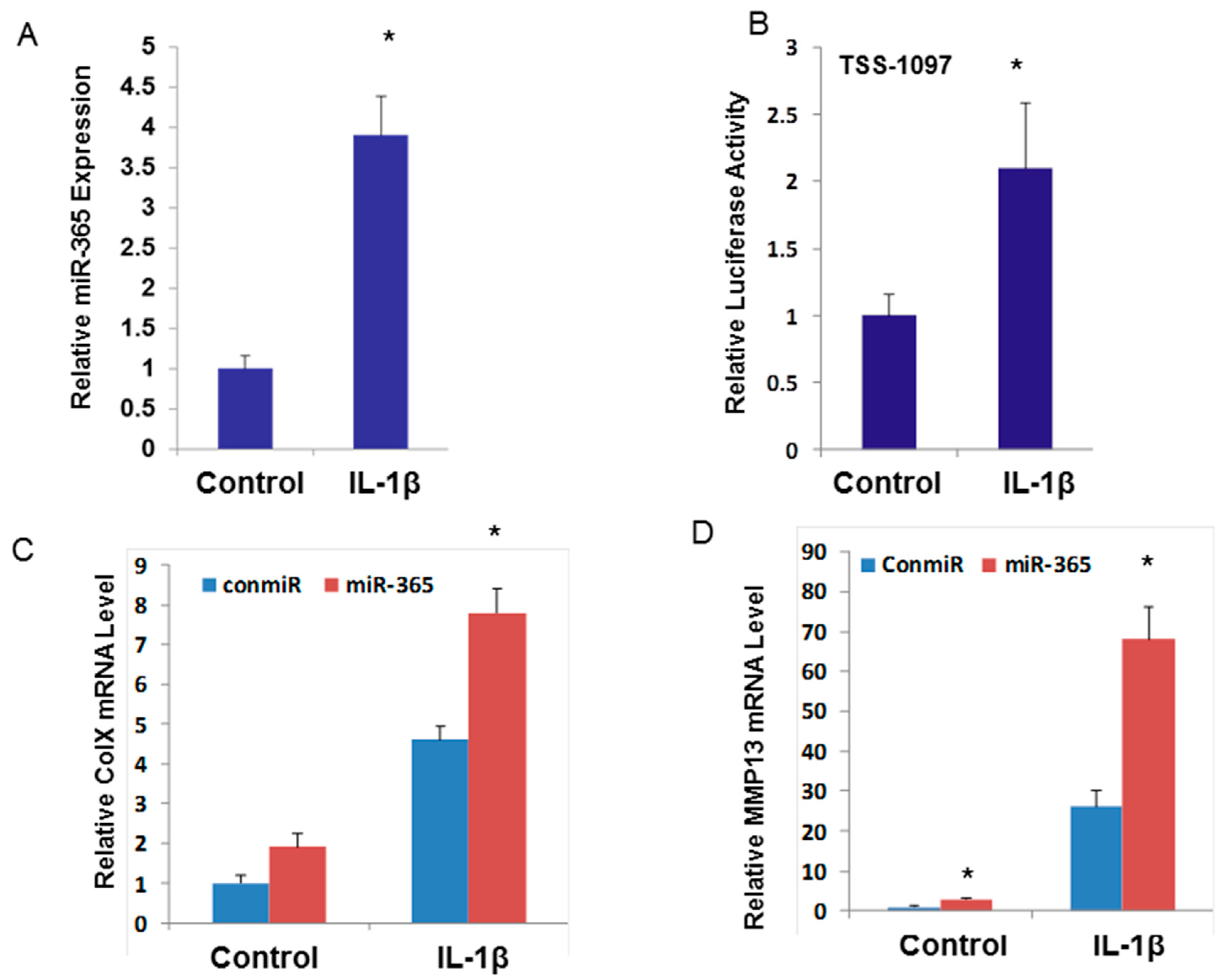
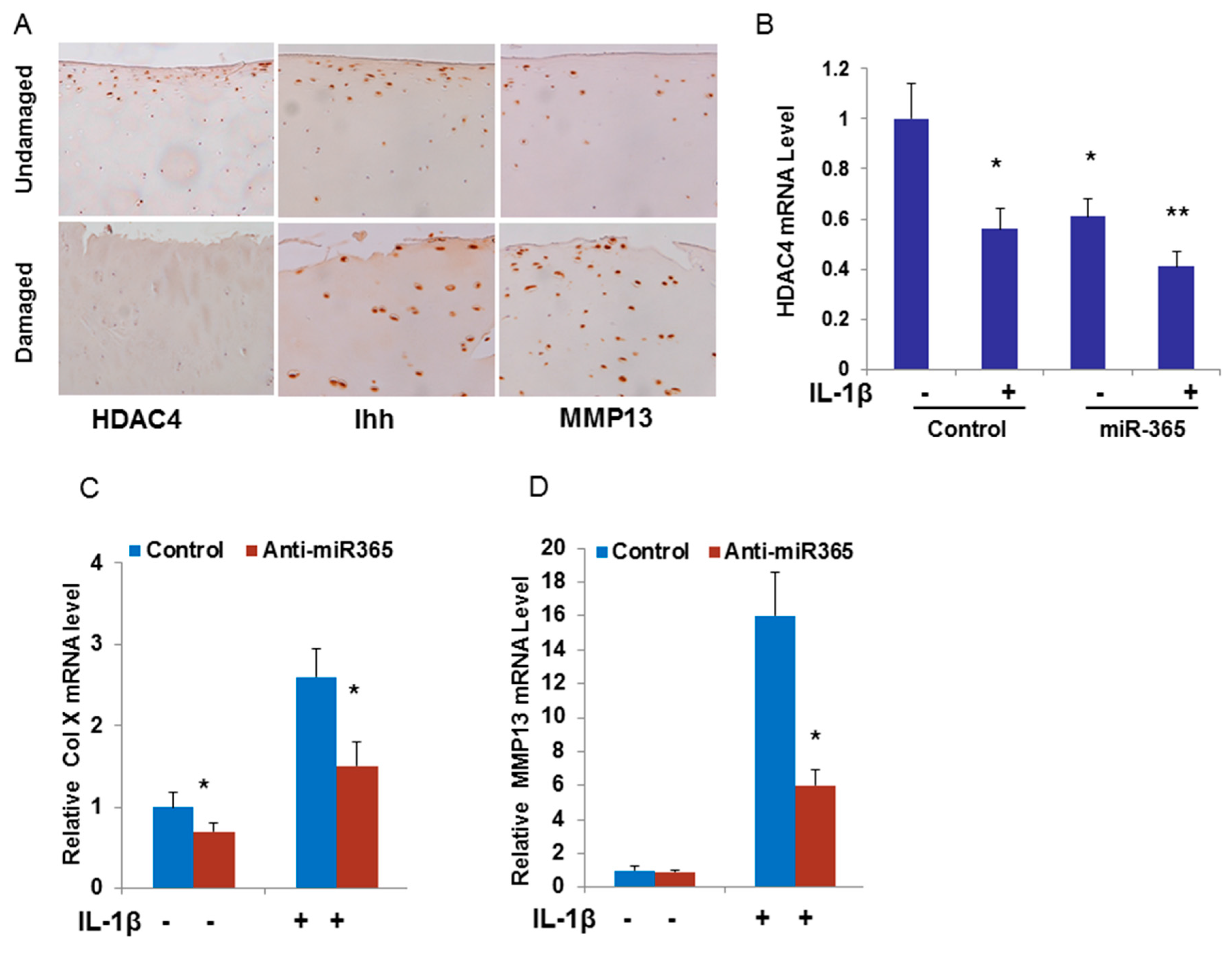
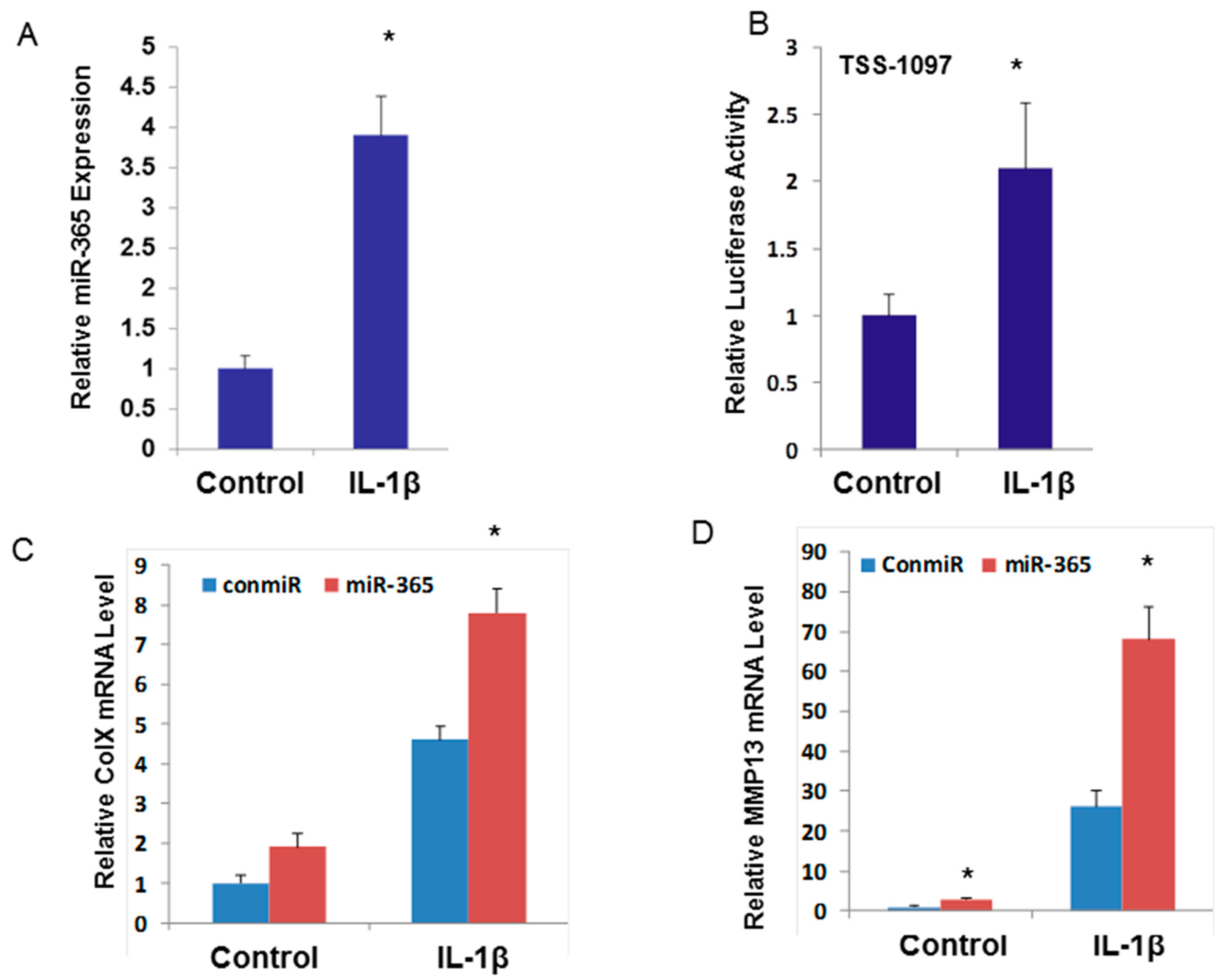
2.2. miR-365 is Up-Regulated by Pro-inflammatory Cytokine IL-1β and is Essential for Cartilage Degeneration
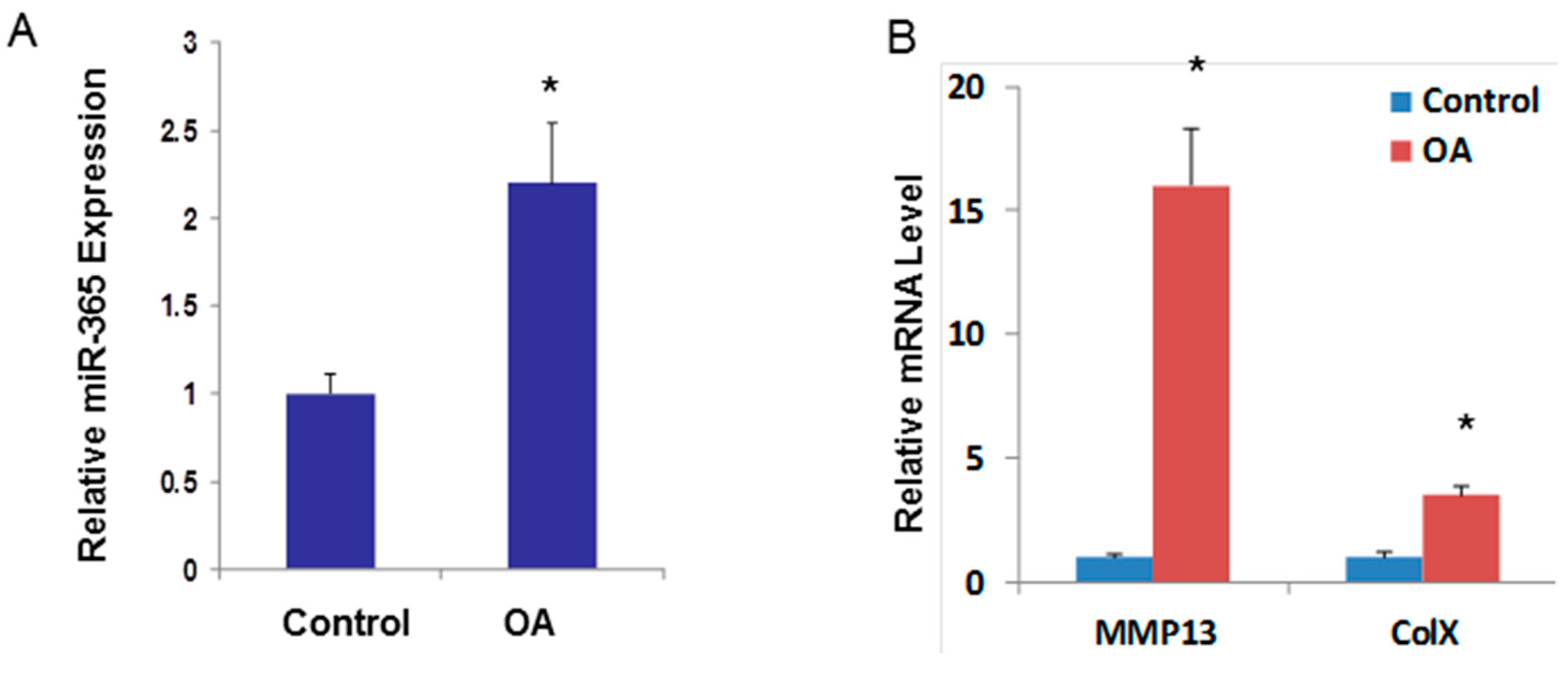
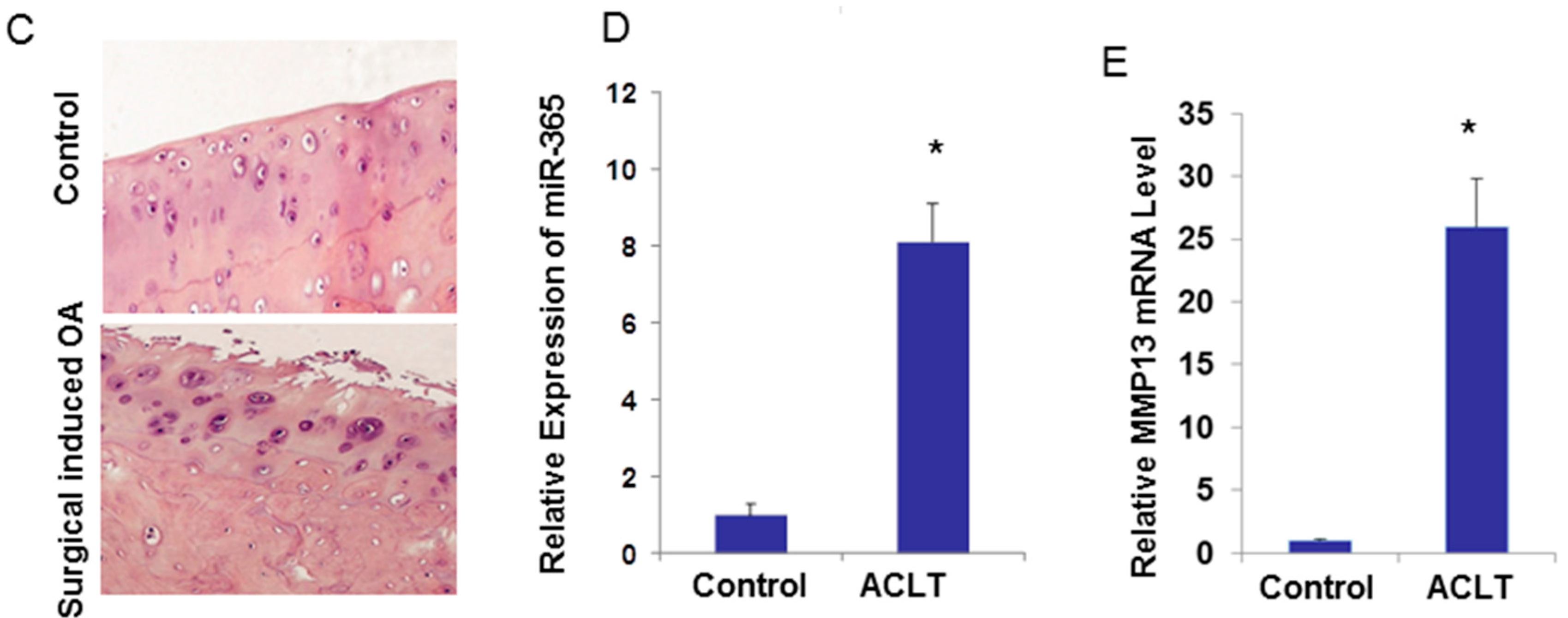
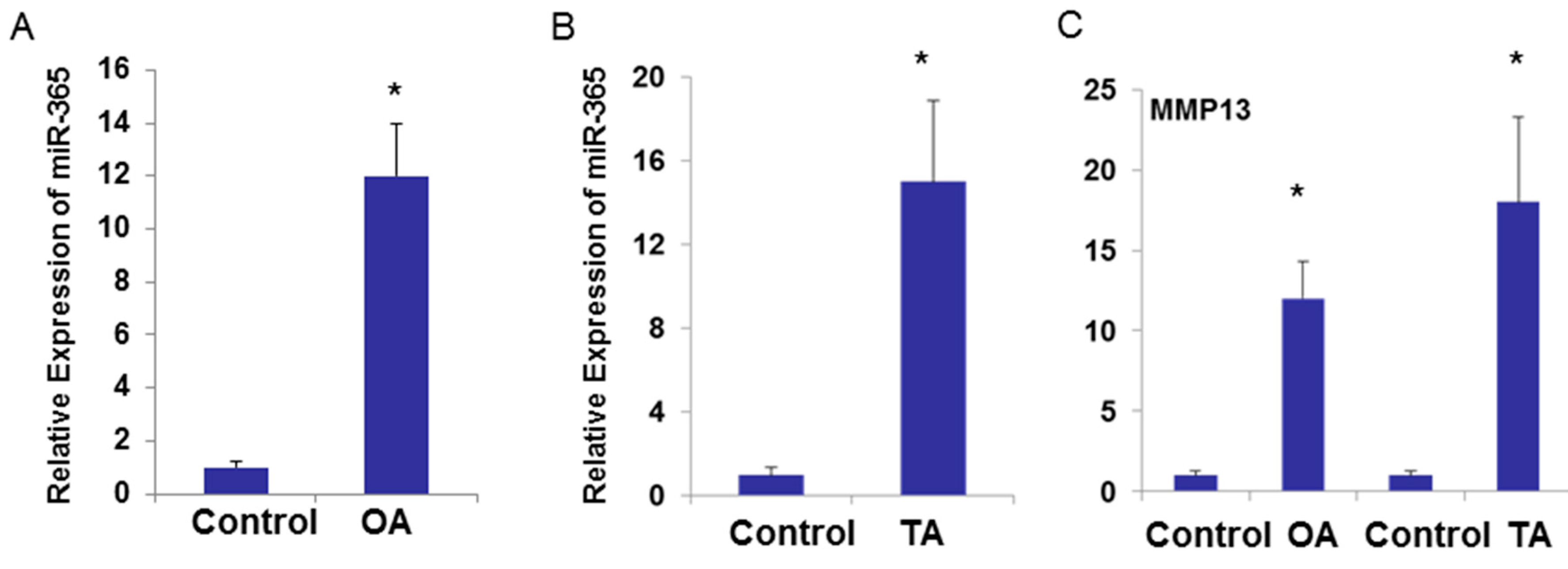
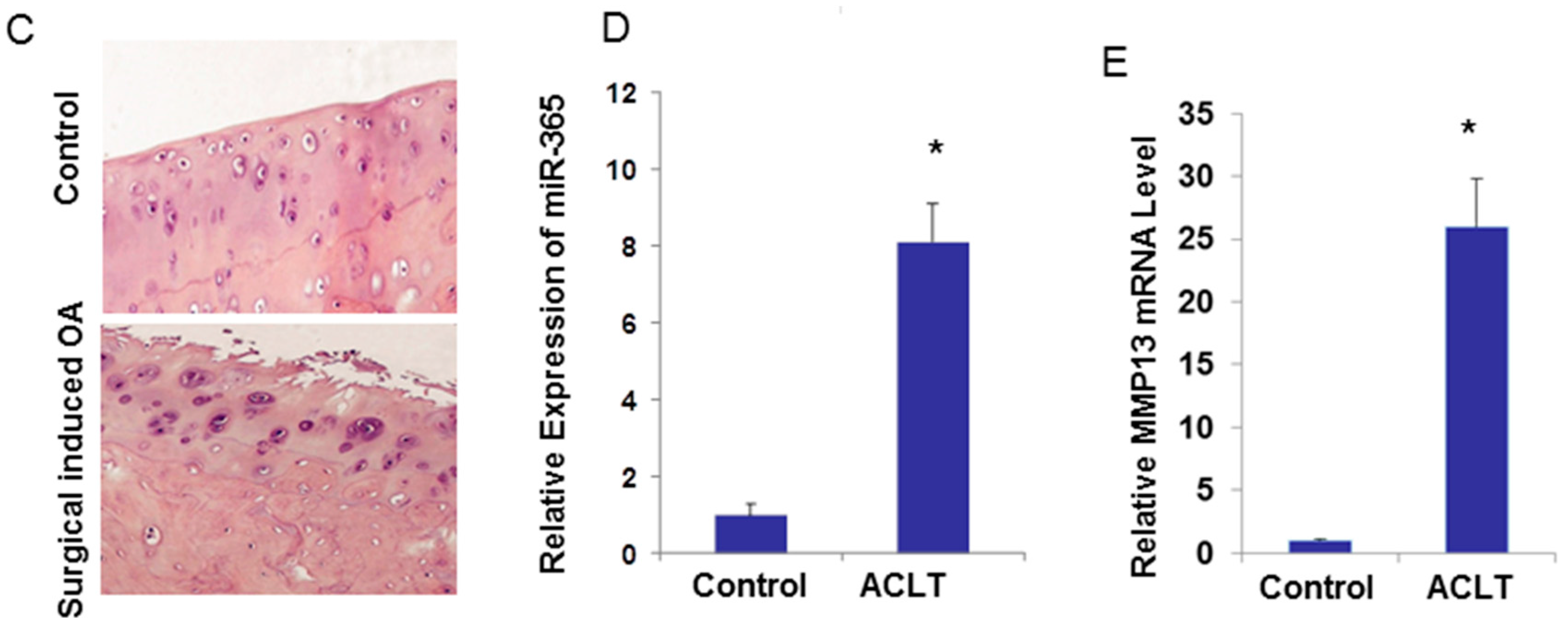
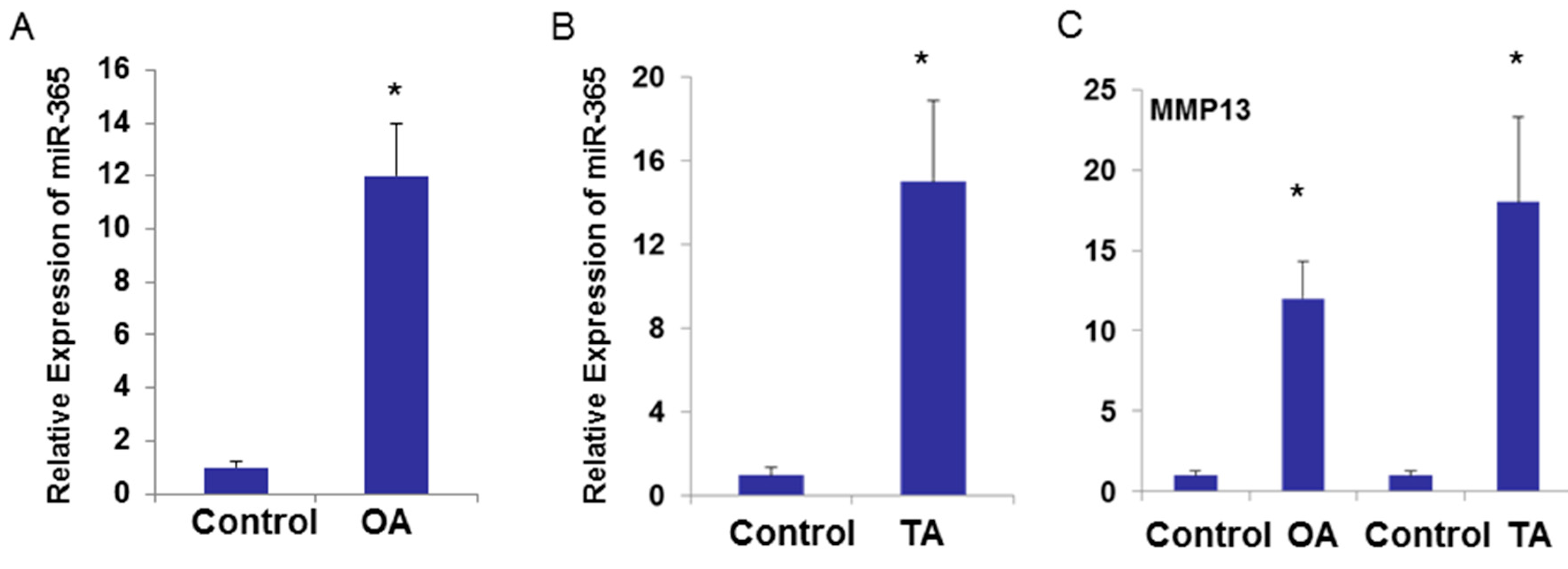
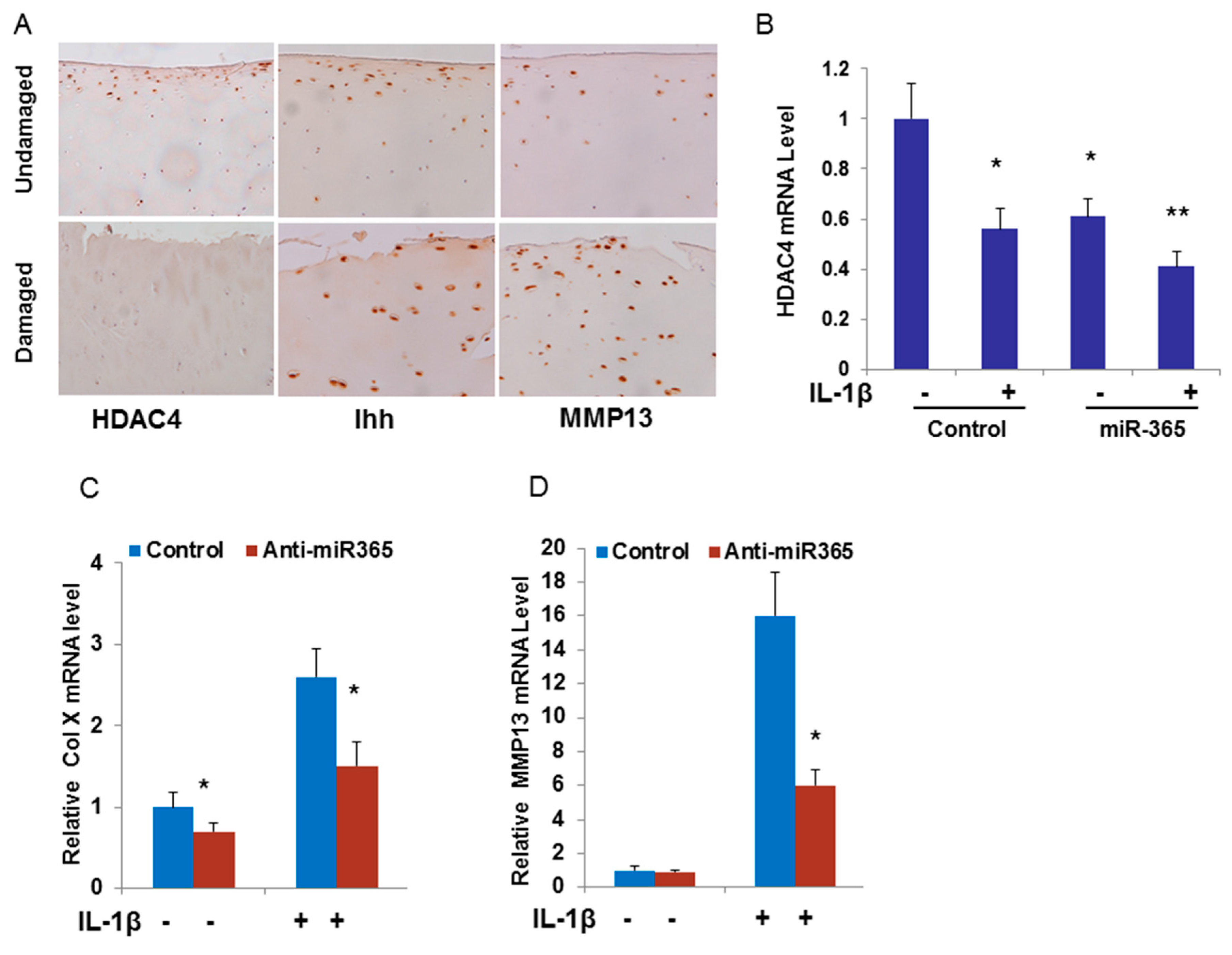
2.3. miR-365 Expression Is Increased in Osteoarthritis (OA) Cartilage
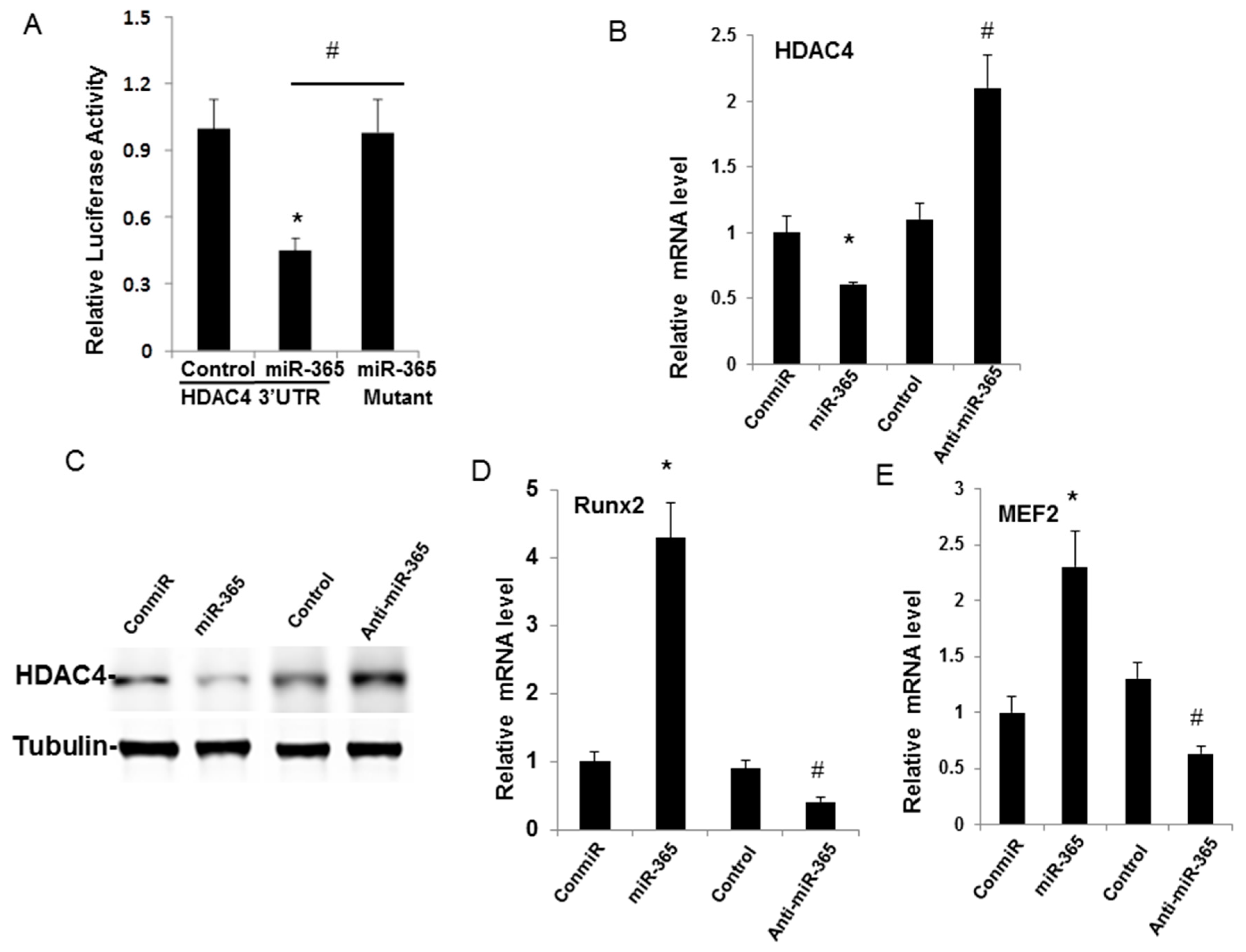
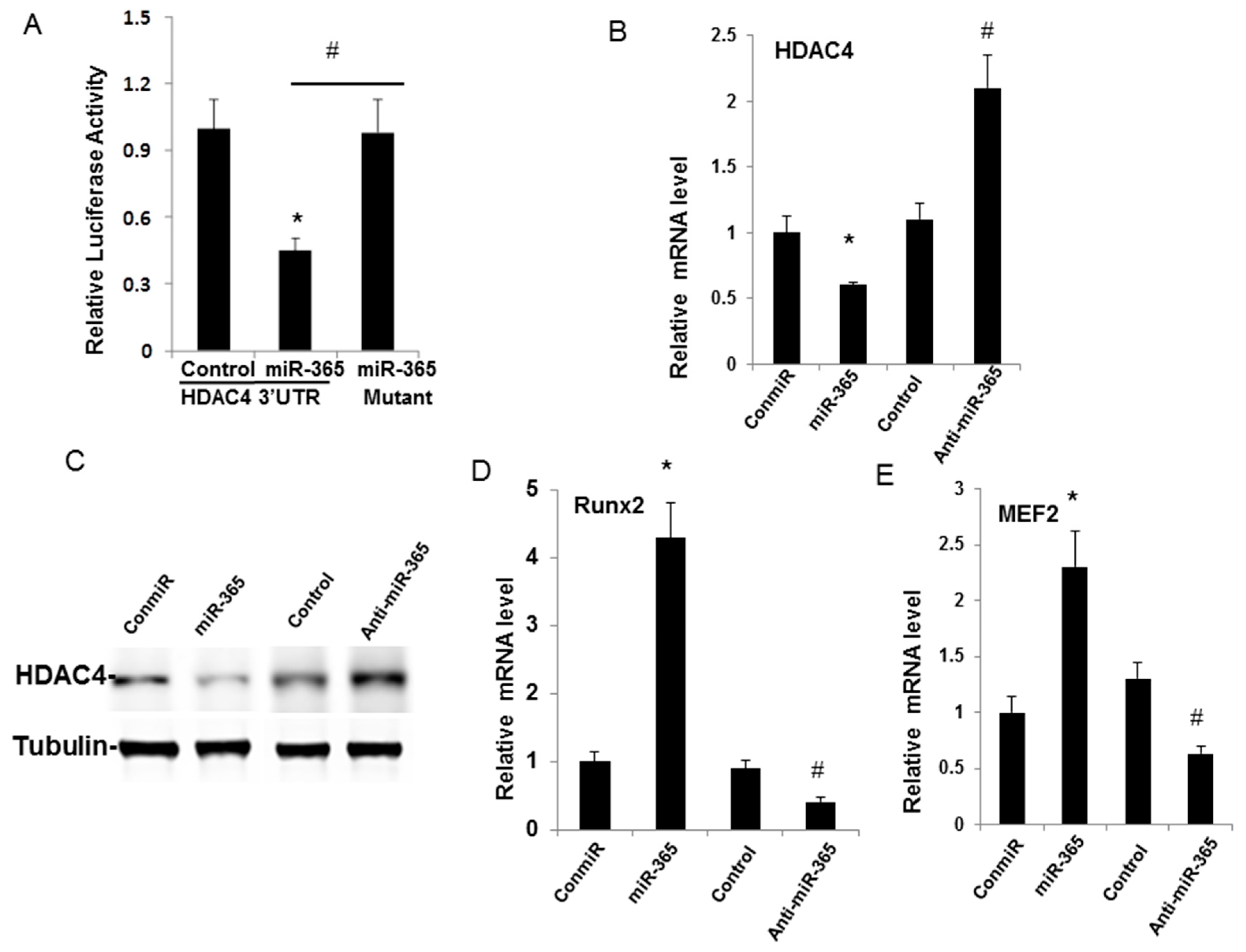
2.4. miR-365 Directly Targets HDAC4 Leading to Down-Regulation of HDAC4 Expression
2.5. Inhibition of miR-365 or Overexpression of HDAC4 Impairs IL-1β Induction of MMP13 mRNA
3. Discussion
4. Materials and Methods
4.1. Primary OA and Traumatic OA Specimen Selection
4.2. Primary Chondrocyte Culture, Transfection, IL-1β Treatment and 3D Cyclic Loading
4.3. Luciferase Assays
4.4. Quantification of mRNA and miRNA
4.5. Surgically Induced OA Model by Anterior Cruciate Ligament Transection (ACLT) in Rats
4.6. Histology and Immunohistochemistry
4.7. Western Blot Analysis
4.8. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Buckwalter, J.A.; Anderson, D.D.; Brown, T.D.; Tochigi, Y.; Martin, J.A. The roles of mechanical stresses in the pathogenesis of osteoarthritis: Implications for treatment of joint injuries. Cartilage 2013, 4, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Goldring, M.B.; Goldring, S.R. Osteoarthritis. J. Cell. Physiol. 2007, 213, 626–634. [Google Scholar] [CrossRef] [PubMed]
- Bian, Q.; Wang, Y.J.; Liu, S.F.; Li, Y.P. Osteoarthritis: Genetic factors, animal models, mechanisms, and therapies. Front. Biosci. 2012, 4, 74–100. [Google Scholar] [CrossRef]
- Loeser, R.F.; Goldring, S.R.; Scanzello, C.R.; Goldring, M.B. Osteoarthritis: A disease of the joint as an organ. Arthritis Rheum. 2012, 64, 1697–1707. [Google Scholar] [CrossRef] [PubMed]
- Aigner, T.; Fundel, K.; Saas, J.; Gebhard, P.M.; Haag, J.; Weiss, T.; Zien, A.; Obermayr, F.; Zimmer, R.; Bartnik, E. Large-scale gene expression profiling reveals major pathogenetic pathways of cartilage degeneration in osteoarthritis. Arthritis Rheum. 2006, 54, 3533–3544. [Google Scholar] [CrossRef] [PubMed]
- Miyaki, S.; Asahara, H. Macro view of microRNA function in osteoarthritis. Nat. Rev. Rheumatol. 2012, 8, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Goldring, M.B.; Marcu, K.B. Epigenomic and microRNA-mediated regulation in cartilage development, homeostasis, and osteoarthritis. Trends Mol. Med. 2012, 18, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Lagos-Quintana, M.; Rauhut, R.; Lendeckel, W.; Tuschl, T. Identification of novel genes coding for small expressed RNAs. Science 2001, 294, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Lu, J.; Cobb, B.S.; Rodda, S.J.; McMahon, A.P.; Schipani, E.; Merkenschlager, M.; Kronenberg, H.M. Dicer-dependent pathways regulate chondrocyte proliferation and differentiation. Proc. Natl. Acad. Sci. USA 2008, 105, 1949–1954. [Google Scholar] [CrossRef] [PubMed]
- Harfe, B.D.; McManus, M.T.; Mansfield, J.H.; Hornstein, E.; Tabin, C.J. The RNaseIII enzyme Dicer is required for morphogenesis but not patterning of the vertebrate limb. Proc. Natl. Acad. Sci. USA 2005, 102, 10898–10903. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Papaioannou, G.; Mirzamohammadi, F.; Kozhemyakina, E.; Zhang, M.; Blelloch, R.; Chong, M.W. Early postnatal ablation of the microRNA-processing enzyme, Drosha, causes chondrocyte death and impairs the structural integrity of the articular cartilage. Osteoarthr. Cartil. 2015, 23, 1214–1220. [Google Scholar] [CrossRef] [PubMed]
- Abouheif, M.M.; Nakasa, T.; Shibuya, H.; Niimoto, T.; Kongcharoensombat, W.; Ochi, M. Silencing microRNA-34a inhibits chondrocyte apoptosis in a rat osteoarthritis model in vitro. Rheumatology 2010, 49, 2054–2060. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, N.; Rasheed, Z.; Ramamurthy, S.; Anbazhagan, A.N.; Voss, F.R.; Haqqi, T.M. MicroRNA-27b regulates the expression of matrix metalloproteinase 13 in human osteoarthritis chondrocytes. Arthritis Rheum. 2010, 62, 1361–1371. [Google Scholar] [CrossRef] [PubMed]
- Araldi, E.; Schipani, E. MicroRNA-140 and the silencing of osteoarthritis. Genes Dev. 2010, 24, 1075–1080. [Google Scholar] [CrossRef] [PubMed]
- Miyaki, S.; Nakasa, T.; Otsuki, S.; Grogan, S.P.; Higashiyama, R.; Inoue, A.; Kato, Y.; Sato, T.; Lotz, M.K.; Asahara, H. MicroRNA-140 is expressed in differentiated human articular chondrocytes and modulates interleukin-1 responses. Arthritis Rheum. 2009, 60, 2723–2730. [Google Scholar] [CrossRef] [PubMed]
- Miyaki, S.; Sato, T.; Inoue, A.; Otsuki, S.; Ito, Y.; Yokoyama, S.; Kato, Y.; Takemoto, F.; Nakasa, T.; Yamashita, S.; et al. MicroRNA-140 plays dual roles in both cartilage development and homeostasis. Genes Dev. 2010, 24, 1173–1185. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Huang, J.; Dai, L.; Yu, D.; Chen, Q.; Zhang, X.; Dai, K. miR-146a, an IL-1β responsive miRNA, induces vascular endothelial growth factor and chondrocyte apoptosis by targeting Smad4. Arthritis Res. Ther. 2012, 14. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Gibson, G.; Kim, J.S.; Kroin, J.; Xu, S.; van Wijnen, A.J.; Im, H.J. MicroRNA-146a is linked to pain-related pathophysiology of osteoarthritis. Gene 2011, 480, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Zhao, J.; Jing, W.; Yan, S.; Wang, X.; Xiao, C.; Ma, B. Role of miR-146a in human chondrocyte apoptosis in response to mechanical pressure injury in vitro. Int. J. Mol. Med. 2014, 34, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.J.; Yang, X.; Wei, L.; Chen, Q. miR-365: A mechanosensitive microRNA stimulates chondrocyte differentiation through targeting histone deacetylase 4. FASEB J. 2011, 25, 4457–4466. [Google Scholar] [CrossRef] [PubMed]
- Cao, K.; Wei, L.; Zhang, Z.; Guo, L.; Zhang, C.; Li, Y.; Sun, C.; Sun, X.; Wang, S.; Li, P.; et al. Decreased histone deacetylase 4 is associated with human osteoarthritis cartilage degeneration by releasing histone deacetylase 4 inhibition of runt-related transcription factor-2 and increasing osteoarthritis-related genes: A novel mechanism of human osteoarthritis cartilage degeneration. Arthritis Res. Ther. 2014, 16, 1–13. [Google Scholar]
- Yang, X.; Vezeridis, P.S.; Nicholas, B.; Crisco, J.J.; Moore, D.C.; Chen, Q. Differential expression of type X collagen in a mechanically active 3-D chondrocyte culture system: A quantitative study. J. Orthop. 2006, 1. [Google Scholar] [CrossRef]
- Xu, Z.; Xiao, S.B.; Xu, P.; Xie, Q.; Cao, L.; Wang, D.; Luo, R.; Zhong, Y.; Chen, H.C.; Fang, L.R. miR-365, a novel negative regulator of interleukin-6 gene expression, is cooperatively regulated by Sp1 and NF-κB. J. Biol. Chem. 2011, 286, 21401–21412. [Google Scholar] [CrossRef] [PubMed]
- Marcu, K.B.; Otero, M.; Olivotto, E.; Borzi, R.M.; Goldring, M.B. NF-κB signaling: Multiple angles to target OA. Curr. Drug Targets 2010, 11, 599–613. [Google Scholar] [CrossRef] [PubMed]
- Olivotto, E.; Otero, M.; Marcu, K.B.; Goldring, M.B. Pathophysiology of osteoarthritis: Canonical NF-κB/IKKβ-dependent and kinase-independent effects of IKKα in cartilage degradation and chondrocyte differentiation. RMD Open 2015, 1. [Google Scholar] [CrossRef] [PubMed]
- Goldring, M.B.; Otero, M.; Tsuchimochi, K.; Ijiri, K.; Li, Y. Defining the roles of inflammatory and anabolic cytokines in cartilage metabolism. Ann. Rheum. Dis. 2008, 67, iii75–iii82. [Google Scholar] [CrossRef] [PubMed]
- Rosenzweig, D.H.; Quinn, T.M.; Haglund, L. Low-frequency high-magnitude mechanical strain of articular chondrocytes activates p38 MAPK and induces phenotypic changes associated with osteoarthritis and pain. Int. J. Mol. Med. 2014, 15, 14427–14441. [Google Scholar] [CrossRef] [PubMed]
- Little, C.B.; Hunter, D.J. Post-traumatic osteoarthritis: From mouse models to clinical trials. Nat. Rev. Rheumatol. 2013, 9, 485–497. [Google Scholar] [CrossRef] [PubMed]
- Lotz, M.K.; Kraus, V.B. Posttraumatic osteoarthritis: Pathogenesis and pharmacological treatment options. Arthritis Res. Ther. 2010, 12. [Google Scholar] [CrossRef]
- Norris, K.L.; Lee, J.Y.; Yao, T.P. Acetylation goes global: The emergence of acetylation biology. Sci. Signal. 2009, 2. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef] [PubMed]
- Usui, T.; Okada, M.; Mizuno, W.; Oda, M.; Ide, N.; Morita, T.; Hara, Y.; Yamawaki, H. HDAC4 mediates development of hypertension via vascular inflammation in spontaneous hypertensive rats. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H1894–H1904. [Google Scholar] [CrossRef] [PubMed]
- Pecchi, E.; Priam, S.; Gosset, M.; Pigenet, A.; Sudre, L.; Laiguillon, M.C.; Berenbaum, F.; Houard, X. Induction of nerve growth factor expression and release by mechanical and inflammatory stimuli in chondrocytes: Possible involvement in osteoarthritis pain. Arthritis Res. Ther. 2014, 16. [Google Scholar] [CrossRef] [PubMed]
- Altman, R.; Asch, E.; Bloch, D.; Bole, G.; Borenstein, D.; Brandt, K.; Christy, W.; Cooke, T.D.; Greenwald, R.; Hochberg, M.; et al. Development of criteria for the classification and reporting of osteoarthritis: Classification of osteoarthritis of the knee. Arthritis Rheum. 1986, 29, 1039–1049. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.F.; Mandel, E.M.; Thomson, J.M.; Wu, Q.; Callis, T.E.; Hammond, S.M.; Conlon, F.L.; Wang, D.Z. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat. Genet. 2006, 38, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Moore, E.E.; Bendele, A.M.; Thompson, D.L.; Littau, A.; Waggie, K.S.; Reardon, B.; Ellsworth, J.L. Fibroblast growth factor-18 stimulates chondrogenesis and cartilage repair in a rat model of injury-induced osteoarthritis. Osteoarthr. Cartil. 2005, 13, 623–631. [Google Scholar] [CrossRef] [PubMed]
- Pritzker, K.P.; Gay, S.; Jimenez, S.A.; Ostergaard, K.; Pelletier, J.P.; Revell, P.A.; Salter, D.; van den Berg, W.B. Osteoarthritis cartilage histopathology: Grading and staging. Osteoarthr. Cartil. 2006, 14, 13–29. [Google Scholar] [CrossRef] [PubMed]
- Kleemann, R.U.; Krocker, D.; Cedraro, A.; Tuischer, J.; Duda, G.N. Altered cartilage mechanics and histology in knee osteoarthritis: Relation to clinical assessment (ICRS Grade). Osteoarthr. Cartil. 2005, 13, 958–963. [Google Scholar] [CrossRef] [PubMed]

© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, X.; Guan, Y.; Tian, S.; Wang, Y.; Sun, K.; Chen, Q. Mechanical and IL-1β Responsive miR-365 Contributes to Osteoarthritis Development by Targeting Histone Deacetylase 4. Int. J. Mol. Sci. 2016, 17, 436. https://doi.org/10.3390/ijms17040436
Yang X, Guan Y, Tian S, Wang Y, Sun K, Chen Q. Mechanical and IL-1β Responsive miR-365 Contributes to Osteoarthritis Development by Targeting Histone Deacetylase 4. International Journal of Molecular Sciences. 2016; 17(4):436. https://doi.org/10.3390/ijms17040436
Chicago/Turabian StyleYang, Xu, Yingjie Guan, Shaoqi Tian, Yuanhe Wang, Kang Sun, and Qian Chen. 2016. "Mechanical and IL-1β Responsive miR-365 Contributes to Osteoarthritis Development by Targeting Histone Deacetylase 4" International Journal of Molecular Sciences 17, no. 4: 436. https://doi.org/10.3390/ijms17040436
APA StyleYang, X., Guan, Y., Tian, S., Wang, Y., Sun, K., & Chen, Q. (2016). Mechanical and IL-1β Responsive miR-365 Contributes to Osteoarthritis Development by Targeting Histone Deacetylase 4. International Journal of Molecular Sciences, 17(4), 436. https://doi.org/10.3390/ijms17040436