Endocannabinoids as Guardians of Metastasis

{kind=link}
Abstract
:1. Introduction
2. Rise of 2-AG in the Tumor Environment and in Plasma
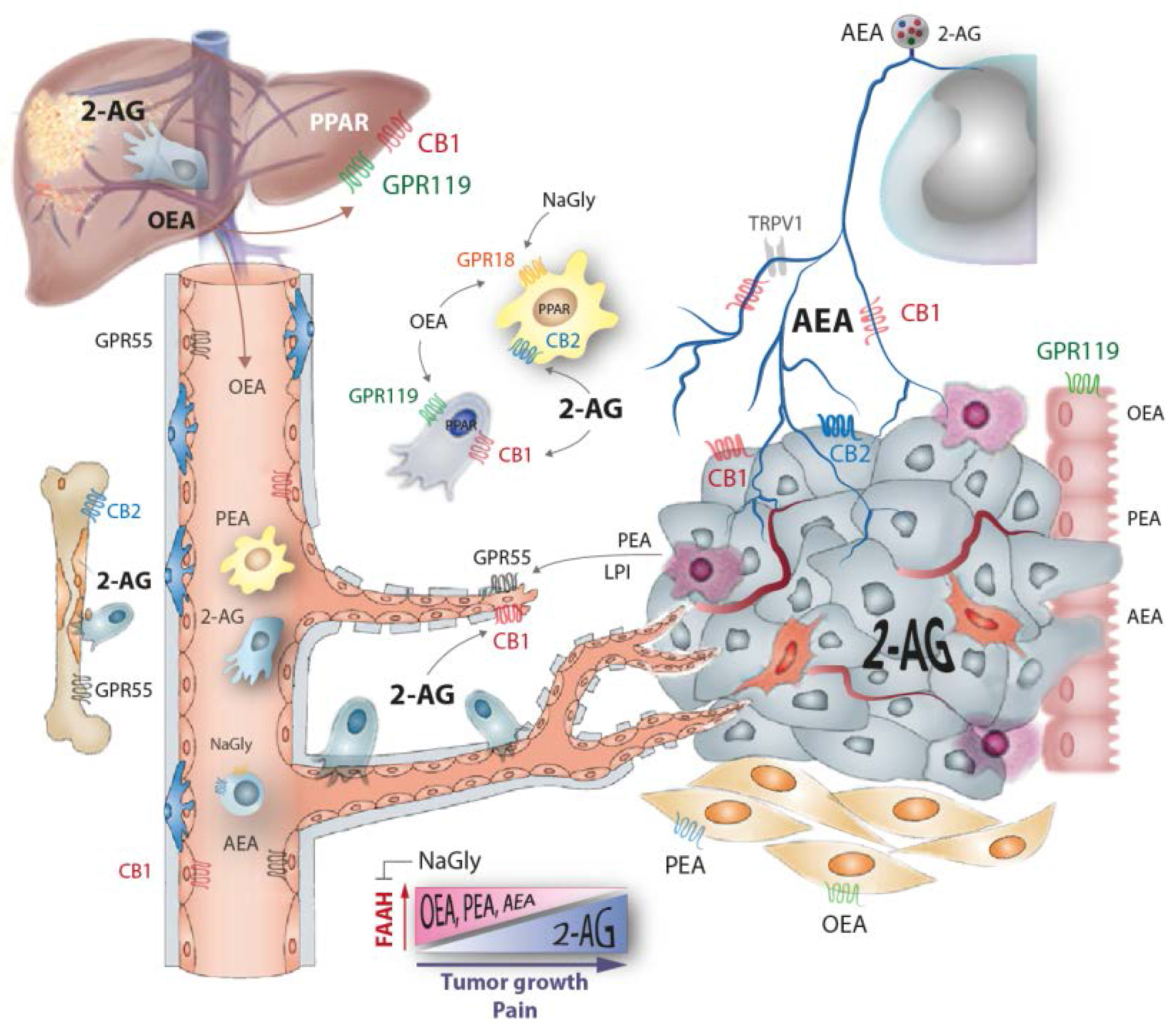
3. Loss of Ethanolamide Endocannabinoids in Tumor Environment and Plasma
4. Cannabinoid Receptors of Tumor Cells
5. Oleoylethanolamide
6. Exogenous Cannabinoids and Therapeutic Implications
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Galve-Roperh, I.; Sanchez, C.; Cortes, M.L.; del Pulgar, T.G.; Izquierdo, M.; Guzman, M. Anti-tumoral action of cannabinoids: Involvement of sustained ceramide accumulation and extracellular signal-regulated kinase activation. Nat. Med. 2000, 6, 313–319. [Google Scholar] [PubMed]
- Di Marzo, V.; Melck, D.; de Petrocellis, L.; Bisogno, T. Cannabimimetic fatty acid derivatives in cancer and inflammation. Prostaglandins Lipid Mediat. 2000, 61, 43–61. [Google Scholar] [CrossRef]
- Sharkey, K.A.; Darmani, N.A.; Parker, L.A. Regulation of nausea and vomiting by cannabinoids and the endocannabinoid system. Eur. J. Pharmacol. 2014, 722, 134–146. [Google Scholar] [CrossRef] [PubMed]
- Khasabova, I.A.; Khasabov, S.G.; Harding-Rose, C.; Coicou, L.G.; Seybold, B.A.; Lindberg, A.E.; Steevens, C.D.; Simone, D.A.; Seybold, V.S. A decrease in anandamide signaling contributes to the maintenance of cutaneous mechanical hyperalgesia in a model of bone cancer pain. J. Neurosci. 2008, 28, 11141–11152. [Google Scholar] [CrossRef] [PubMed]
- Brown, I.; Cascio, M.G.; Rotondo, D.; Pertwee, R.G.; Heys, S.D.; Wahle, K.W. Cannabinoids and omega-3/6 endocannabinoids as cell death and anticancer modulators. Prog. Lipid Res. 2013, 52, 80–109. [Google Scholar] [CrossRef] [PubMed]
- Sailler, S.; Schmitz, K.; Jaeger, E.; Ferreiros, N.; Wicker, S.; Zschiebsch, K.; Pickert, G.; Geisslinger, G.; Walter, C.; Tegeder, I.; et al. Regulation of circulating endocannabinoids associated with cancer and metastases in mice and humans. Oncoscience 2014, 1, 272–282. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, N.; Pacher, P.; Tegeder, I.; Amaya, F.; Constantin, C.E.; Brenner, G.J.; Rubino, T.; Michalski, C.W.; Marsicano, G.; Monory, K.; et al. Cannabinoids mediate analgesia largely via peripheral type 1 cannabinoid receptors in nociceptors. Nat. Neurosci. 2007, 10, 870–879. [Google Scholar] [CrossRef] [PubMed]
- Centonze, D.; Battistini, L.; Maccarrone, M. The endocannabinoid system in peripheral lymphocytes as a mirror of neuroinflammatory diseases. Curr. Pharm. Des. 2008, 14, 2370–2342. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Ungvari, Z. Pleiotropic effects of the CB2 cannabinoid receptor activation on human monocyte migration: Implications for atherosclerosis and inflammatory diseases. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H1133–H1134. [Google Scholar] [CrossRef] [PubMed]
- Tomar, S.; E, E.Z.; Nagarkatti, M.; Nagarkatti, P.S. Protective role of cannabinoid receptor 2 activation in galactosamine/lipopolysaccharide-induced acute liver failure through regulation of macrophage polarization and microRNAs. J. Pharmacol. Exp. Ther. 2015, 353, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Ley, S.; Weigert, A.; Heriche, J.K.; Mille-Baker, B.; Janssen, R.A.; Brune, B. RNAi screen in apoptotic cancer cell-stimulated human macrophages reveals co-regulation of IL-6/IL-10 expression. Immunobiology 2013, 218, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Ryberg, E.; Larsson, N.; Sjogren, S.; Hjorth, S.; Hermansson, N.O.; Leonova, J.; Elebring, T.; Nilsson, K.; Drmota, T.; Greasley, P.J. The orphan receptor GPR55 is a novel cannabinoid receptor. Br. J. Pharmacol. 2007, 152, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- Henstridge, C.M.; Balenga, N.A.; Ford, L.A.; Ross, R.A.; Waldhoer, M.; Irving, A.J. The GPR55 ligand l-alpha-lysophosphatidylinositol promotes RhoA-dependent Ca2+ signaling and NFAT activation. FASEB J. 2009, 23, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Ford, L.A.; Roelofs, A.J.; Anavi-Goffer, S.; Mowat, L.; Simpson, D.G.; Irving, A.J.; Rogers, M.J.; Rajnicek, A.M.; Ross, R.A. A role for L-alpha-lysophosphatidylinositol and GPR55 in the modulation of migration, orientation and polarization of human breast cancer cells. Br. J. Pharmacol. 2010, 160, 762–771. [Google Scholar] [CrossRef] [PubMed]
- Ross, R.A. The enigmatic pharmacology of GPR55. Trends Pharmacol. Sci. 2009, 30, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Oka, S.; Nakajima, K.; Yamashita, A.; Kishimoto, S.; Sugiura, T. Identification of GPR55 as a lysophosphatidylinositol receptor. Biochem. Biophys. Res. Commun. 2007, 362, 928–934. [Google Scholar] [CrossRef] [PubMed]
- Boucharaba, A.; Serre, C.M.; Gres, S.; Saulnier-Blache, J.S.; Bordet, J.C.; Guglielmi, J.; Clezardin, P.; Peyruchaud, O. Platelet-derived lysophosphatidic acid supports the progression of osteolytic bone metastases in breast cancer. J. Clin. Investig. 2004, 114, 1714–1725. [Google Scholar] [CrossRef] [PubMed]
- Fourcade, O.; Simon, M.F.; Viode, C.; Rugani, N.; Leballe, F.; Ragab, A.; Fournie, B.; Sarda, L.; Chap, H. Secretory phospholipase A2 generates the novel lipid mediator lysophosphatidic acid in membrane microvesicles shed from activated cells. Cell 1995, 80, 919–927. [Google Scholar] [CrossRef]
- Peyruchaud, O. Novel implications for lysophospholipids, lysophosphatidic acid and sphingosine 1-phosphate, as drug targets in cancer. Anti-Cancer Agents Med. Chem. 2009, 9, 381–391. [Google Scholar] [CrossRef]
- Leblanc, R.; Lee, S.C.; David, M.; Bordet, J.C.; Norman, D.D.; Patil, R.; Miller, D.; Sahay, D.; Ribeiro, J.; Clezardin, P.; et al. Interaction of platelet-derived autotaxin with tumor integrin alphaVbeta3 controls metastasis of breast cancer cells to bone. Blood 2014, 124, 3141–3150. [Google Scholar] [CrossRef] [PubMed]
- Fulkerson, Z.; Wu, T.; Sunkara, M.; Kooi, C.V.; Morris, A.J.; Smyth, S.S. Binding of autotaxin to integrins localizes lysophosphatidic acid production to platelets and mammalian cells. J. Biol. Chem. 2011, 286, 34654–34663. [Google Scholar] [CrossRef] [PubMed]
- Hama, K.; Aoki, J.; Fukaya, M.; Kishi, Y.; Sakai, T.; Suzuki, R.; Ohta, H.; Yamori, T.; Watanabe, M.; Chun, J.; et al. Lysophosphatidic acid and autotaxin stimulate cell motility of neoplastic and non-neoplastic cells through LPA1. J. Biol. Chem. 2004, 279, 17634–17639. [Google Scholar] [CrossRef] [PubMed]
- Van Meeteren, L.A.; Ruurs, P.; Stortelers, C.; Bouwman, P.; van Rooijen, M.A.; Pradere, J.P.; Pettit, T.R.; Wakelam, M.J.; Saulnier-Blache, J.S.; Mummery, C.L.; et al. Autotaxin, a secreted lysophospholipase D, is essential for blood vessel formation during development. Mol. Cell. Biol. 2006, 26, 5015–5022. [Google Scholar] [CrossRef] [PubMed]
- Hiley, C.R.; Kaup, S.S. GPR55 and the vascular receptors for cannabinoids. Br. J. Pharmacol. 2007, 152, 559–561. [Google Scholar] [CrossRef] [PubMed]
- Ligresti, A.; Bisogno, T.; Matias, I.; de Petrocellis, L.; Cascio, M.G.; Cosenza, V.; D’Argenio, G.; Scaglione, G.; Bifulco, M.; Sorrentini, I.; et al. Possible endocannabinoid control of colorectal cancer growth. Gastroenterology 2003, 125, 677–687. [Google Scholar] [CrossRef]
- Izzo, A.A.; Aviello, G.; Petrosino, S.; Orlando, P.; Marsicano, G.; Lutz, B.; Borrelli, F.; Capasso, R.; Nigam, S.; Capasso, F.; et al. Increased endocannabinoid levels reduce the development of precancerous lesions in the mouse colon. J. Mol. Med. 2008, 86, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Ramer, R.; Hinz, B. Inhibition of cancer cell invasion by cannabinoids via increased expression of tissue inhibitor of matrix metalloproteinases-1. J. Natl. Cancer Inst. 2008, 100, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Grimaldi, C.; Pisanti, S.; Laezza, C.; Malfitano, A.M.; Santoro, A.; Vitale, M.; Caruso, M.G.; Notarnicola, M.; Iacuzzo, I.; Portella, G.; et al. Anandamide inhibits adhesion and migration of breast cancer cells. Exp. Cell Res. 2006, 312, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Patsos, H.A.; Hicks, D.J.; Dobson, R.R.; Greenhough, A.; Woodman, N.; Lane, J.D.; Williams, A.C.; Paraskeva, C. The endogenous cannabinoid, anandamide, induces cell death in colorectal carcinoma cells: A possible role for cyclooxygenase 2. Gut 2005, 54, 1741–1750. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.; Niggemann, B.; Zaenker, K.S.; Entschladen, F. Anandamide is an endogenous inhibitor for the migration of tumor cells and T lymphocytes. Cancer Immunol. Immunother. 2004, 53, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Salazar, M.; Carracedo, A.; Salanueva, I.J.; Hernandez-Tiedra, S.; Lorente, M.; Egia, A.; Vazquez, P.; Blazquez, C.; Torres, S.; Garcia, S.; et al. Cannabinoid action induces autophagy-mediated cell death through stimulation of ER stress in human glioma cells. J. Clin. Investig. 2009, 119, 1359–1372. [Google Scholar] [CrossRef] [PubMed]
- Perez-Gomez, E.; Andradas, C.; Blasco-Benito, S.; Caffarel, M.M.; Garcia-Taboada, E.; Villa-Morales, M.; Moreno, E.; Hamann, S.; Martin-Villar, E.; Flores, J.M.; et al. Role of cannabinoid receptor CB2 in HER2 pro-oncogenic signaling in breast cancer. J. Natl. Cancer Inst. 2015, 107, djv077. [Google Scholar] [CrossRef] [PubMed]
- Constantin, C.E.; Mair, N.; Sailer, C.A.; Andratsch, M.; Xu, Z.Z.; Blumer, M.J.; Scherbakov, N.; Davis, J.B.; Bluethmann, H.; Ji, R.R.; et al. Endogenous tumor necrosis factor alpha (TNFalpha) requires TNF receptor type 2 to generate heat hyperalgesia in a mouse cancer model. J. Neurosci. 2008, 28, 5072–5081. [Google Scholar] [CrossRef] [PubMed]
- Heyman, E.; Gamelin, F.X.; Goekint, M.; Piscitelli, F.; Roelands, B.; Leclair, E.; di Marzo, V.; Meeusen, R. Intense exercise increases circulating endocannabinoid and BDNF levels in humans-Possible implications for reward and depression. Psychoneuroendocrinology 2012, 37, 844–851. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Chu, A.; Li, W.; Wang, B.; Shelton, F.; Otero, F.; Nguyen, D.G.; Caldwell, J.S.; Chen, Y.A. Lipid G protein-coupled receptor ligand identification using beta-arrestin PathHunter assay. J. Biol. Chem. 2009, 284, 12328–12338. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, S.E. Cannabinoids go nuclear: Evidence for activation of peroxisome proliferator-activated receptors. Br. J. Pharmacol. 2007, 152, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Van der Stelt, M.; Trevisani, M.; Vellani, V.; de Petrocellis, L.; Schiano Moriello, A.; Campi, B.; McNaughton, P.; Geppetti, P.; di Marzo, V. Anandamide acts as an intracellular messenger amplifying Ca2+ influx via TRPV1 channels. Embo J. 2005, 24, 3026–3037. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, H.B.; Lee, S.H.; McHugh, D. Orphan endogenous lipids and orphan GPCRs: A good match. Prostaglandins Lipid Mediat. 2009, 89, 131–134. [Google Scholar] [CrossRef] [PubMed]
- Endsley, M.P.; Thill, R.; Choudhry, I.; Williams, C.L.; Kajdacsy-Balla, A.; Campbell, W.B.; Nithipatikom, K. Expression and function of fatty acid amide hydrolase in prostate cancer. Int. J. Cancer 2008, 123, 1318–1326. [Google Scholar] [CrossRef] [PubMed]
- Bishay, P.; Schmidt, H.; Marian, C.; Haussler, A.; Wijnvoord, N.; Ziebell, S.; Metzner, J.; Koch, M.; Myrczek, T.; Bechmann, I.; et al. R-flurbiprofen reduces neuropathic pain in rodents by restoring endogenous cannabinoids. PLoS ONE 2010, 5, e10628. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, K.; de Bruin, N.; Bishay, P.; Mannich, J.; Haussler, A.; Altmann, C.; Ferreiros, N.; Lotsch, J.; Ultsch, A.; Parnham, M.J.; et al. R-flurbiprofen attenuates experimental autoimmune encephalomyelitis in mice. EMBO Mol. Med. 2014, 6, 1398–13422. [Google Scholar] [CrossRef] [PubMed]
- Grosch, S.; Tegeder, I.; Schilling, K.; Maier, T.J.; Niederberger, E.; Geisslinger, G. Activation of c-Jun-N-terminal-kinase is crucial for the induction of a cell cycle arrest in human colon carcinoma cells caused by flurbiprofen enantiomers. FASEB J. 2003, 17, 1316–1318. [Google Scholar] [CrossRef] [PubMed]
- Wechter, W.J.; Leipold, D.D.; Murray, E.D., Jr.; Quiggle, D.; McCracken, J.D.; Barrios, R.S.; Greenberg, N.M. E-7869 (R-flurbiprofen) inhibits progression of prostate cancer in the TRAMP mouse. Cancer Res. 2000, 60, 2203–2208. [Google Scholar] [PubMed]
- Wechter, W.J.; Kantoci, D.; Murray, E.D., Jr.; Quiggle, D.D.; Leipold, D.D.; Gibson, K.M.; McCracken, J.D. R-flurbiprofen chemoprevention and treatment of intestinal adenomas in the APC(Min)/+ mouse model: Implications for prophylaxis and treatment of colon cancer. Cancer Res. 1997, 57, 4316–4324. [Google Scholar] [PubMed]
- Fowler, C.J. Possible involvement of the endocannabinoid system in the actions of three clinically used drugs. Trends Pharmacol. Sci. 2004, 25, 59–61. [Google Scholar] [CrossRef] [PubMed]
- Guzman, M.; Lo Verme, J.; Fu, J.; Oveisi, F.; Blazquez, C.; Piomelli, D. Oleoylethanolamide stimulates lipolysis by activating the nuclear receptor peroxisome proliferator-activated receptor alpha (PPAR-alpha). J. Biol. Chem. 2004, 279, 27849–27854. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, G.J.; Fu, J.; Astarita, G.; Li, X.; Gaetani, S.; Campolongo, P.; Cuomo, V.; Piomelli, D. The lipid messenger OEA links dietary fat intake to satiety. Cell Metab. 2008, 8, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez de Fonseca, F.; Navarro, M.; Gomez, R.; Escuredo, L.; Nava, F.; Fu, J.; Murillo-Rodriguez, E.; Giuffrida, A.; LoVerme, J.; Gaetani, S.; et al. An anorexic lipid mediator regulated by feeding. Nature 2001, 414, 209–212. [Google Scholar] [CrossRef] [PubMed]
- Soria-Gomez, E.; Guzman, K.; Pech-Rueda, O.; Montes-Rodriguez, C.J.; Cisneros, M.; Prospero-Garcia, O. Oleoylethanolamide affects food intake and sleep-waking cycle through a hypothalamic modulation. Pharmacol. Res. 2010, 61, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Parolaro, D.; Massi, P.; Rubino, T.; Monti, E. Endocannabinoids in the immune system and cancer. Prostaglandins Leukot. Essent. Fat. Acids 2002, 66, 319–332. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, H.B.; Rimmerman, N.; Hu, S.S.; Benton, V.M.; Stuart, J.M.; Masuda, K.; Cravatt, B.F.; O’Dell, D.K.; Walker, J.M. The endocannabinoid anandamide is a precursor for the signaling lipid N-arachidonyl glycine through two distinct pathways. BMC Biochem. 2009, 10, 14. [Google Scholar] [CrossRef] [PubMed]
- Burstein, S.H.; Huang, S.M.; Petros, T.J.; Rossetti, R.G.; Walker, J.M.; Zurier, R.B. Regulation of anandamide tissue levels by N-arachidonylglycine. Biochem. Pharmacol. 2002, 64, 1147–1150. [Google Scholar] [CrossRef]
- McHugh, D.; Wager-Miller, J.; Page, J.; Bradshaw, H.B. siRNA knockdown of GPR18 receptors in BV-2 microglia attenuates N-arachidonoyl glycine-induced cell migration. J. Mol. Signal. 2012, 7, 10. [Google Scholar] [CrossRef] [PubMed]
- McHugh, D.; Page, J.; Dunn, E.; Bradshaw, H.B. Delta(9)-Tetrahydrocannabinol and N-arachidonyl glycine are full agonists at GPR18 receptors and induce migration in human endometrial HEC-1B cells. Br. J. Pharmacol. 2012, 165, 2414–2424. [Google Scholar] [CrossRef] [PubMed]
- Grazia Cascio, M.; Minassi, A.; Ligresti, A.; Appendino, G.; Burstein, S.; Di Marzo, V. A structure-activity relationship study on N-arachidonoyl-amino acids as possible endogenous inhibitors of fatty acid amide hydrolase. Biochem. Biophys. Res. Commun. 2004, 314, 192–196. [Google Scholar] [CrossRef] [PubMed]
- Velasco, G.; Sanchez, C.; Guzman, M. Endocannabinoids and Cancer. Handb. Exp. Pharmacol. 2015, 231, 449–472. [Google Scholar] [PubMed]
- Pisanti, S.; Picardi, P.; D’Alessandro, A.; Laezza, C.; Bifulco, M. The endocannabinoid signaling system in cancer. Trends Pharmacol. Sci. 2013, 34, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Chakravarti, B.; Ravi, J.; Ganju, R.K. Cannabinoids as therapeutic agents in cancer: Current status and future implications. Oncotarget 2014, 5, 5852–5872. [Google Scholar] [CrossRef] [PubMed]
- Bifulco, M.; Laezza, C.; Gazzerro, P.; Pentimalli, F. Endocannabinoids as emerging suppressors of angiogenesis and tumor invasion (review). Oncol. Rep. 2007, 17, 813–816. [Google Scholar] [CrossRef] [PubMed]
- Caffarel, M.M.; Andradas, C.; Mira, E.; Perez-Gomez, E.; Cerutti, C.; Moreno-Bueno, G.; Flores, J.M.; Garcia-Real, I.; Palacios, J.; Manes, S.; et al. Cannabinoids reduce ErbB2-driven breast cancer progression through Akt inhibition. Mol. Cancer 2010, 9, 196. [Google Scholar] [CrossRef] [PubMed]
- Vara, D.; Salazar, M.; Olea-Herrero, N.; Guzman, M.; Velasco, G.; Diaz-Laviada, I. Anti-tumoral action of cannabinoids on hepatocellular carcinoma: Role of AMPK-dependent activation of autophagy. Cell Death Differ. 2011, 18, 1099–1111. [Google Scholar] [CrossRef] [PubMed]
- Laezza, C.; Pisanti, S.; Crescenzi, E.; Bifulco, M. Anandamide inhibits Cdk2 and activates Chk1 leading to cell cycle arrest in human breast cancer cells. FEBS Lett. 2006, 580, 6076–6082. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, S.B.; Lindgren, T.; Jonsson, M.; Jacobsson, S.O. Cannabinoid receptor-independent cytotoxic effects of cannabinoids in human colorectal carcinoma cells: Synergism with 5-fluorouracil. Cancer Chemother. Pharmacol. 2009, 63, 691–701. [Google Scholar] [CrossRef] [PubMed]
- Theocharis, S.; Giaginis, C.; Alexandrou, P.; Rodriguez, J.; Tasoulas, J.; Danas, E.; Patsouris, E.; Klijanienko, J. Evaluation of cannabinoid CB1 and CB2 receptors expression in mobile tongue squamous cell carcinoma: Associations with clinicopathological parameters and patients’ survival. Tumour Biol. 2015, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.C.; Hammarsten, P.; Josefsson, A.; Stattin, P.; Granfors, T.; Egevad, L.; Mancini, G.; Lutz, B.; Bergh, A.; Fowler, C.J. A high cannabinoid CB(1) receptor immunoreactivity is associated with disease severity and outcome in prostate cancer. Eur. J. Cancer 2009, 45, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Orellana-Serradell, O.; Poblete, C.E.; Sanchez, C.; Castellon, E.A.; Gallegos, I.; Huidobro, C.; Llanos, M.N.; Contreras, H.R. Proapoptotic effect of endocannabinoids in prostate cancer cells. Oncol. Rep. 2015, 33, 1599–1608. [Google Scholar] [CrossRef] [PubMed]
- Preet, A.; Qamri, Z.; Nasser, M.W.; Prasad, A.; Shilo, K.; Zou, X.; Groopman, J.E.; Ganju, R.K. Cannabinoid receptors, CB1 and CB2, as novel targets for inhibition of non-small cell lung cancer growth and metastasis. Cancer Prev. Res. 2011, 4, 65–75. [Google Scholar]
- Qamri, Z.; Preet, A.; Nasser, M.W.; Bass, C.E.; Leone, G.; Barsky, S.H.; Ganju, R.K. Synthetic cannabinoid receptor agonists inhibit tumor growth and metastasis of breast cancer. Mol. Cancer Ther. 2009, 8, 3117–3329. [Google Scholar] [PubMed]
- Morales, P.; Blasco-Benito, S.; Andradas, C.; Gomez-Canas, M.; Flores, J.M.; Goya, P.; Fernandez-Ruiz, J.; Sanchez, C.; Jagerovic, N. Selective, nontoxic CB(2) cannabinoid o-quinone with in vivo activity against triple-negative breast cancer. J. Med. Chem. 2015, 58, 2256–2264. [Google Scholar] [CrossRef] [PubMed]
- Overton, H.A.; Babbs, A.J.; Doel, S.M.; Fyfe, M.C.; Gardner, L.S.; Griffin, G.; Jackson, H.C.; Procter, M.J.; Rasamison, C.M.; Tang-Christensen, M.; et al. Deorphanization of a G protein-coupled receptor for oleoylethanolamide and its use in the discovery of small-molecule hypophagic agents. Cell Metab. 2006, 3, 167–175. [Google Scholar] [PubMed]
- Godlewski, G.; Offertaler, L.; Wagner, J.A.; Kunos, G. Receptors for acylethanolamides-GPR55 and GPR119. Prostaglandins Lipid Mediat. 2009, 89, 105–111. [Google Scholar]
- Pang, X.; Wei, Y.; Zhang, Y.; Zhang, M.; Lu, Y.; Shen, P. Peroxisome proliferator-activated receptor-gamma activation inhibits hepatocellular carcinoma cell invasion by upregulating plasminogen activator inhibitor-1. Cancer Sci. 2013, 104, 672–680. [Google Scholar] [CrossRef] [PubMed]
- Apostoli, A.J.; Skelhorne-Gross, G.E.; Rubino, R.E.; Peterson, N.T.; Di Lena, M.A.; Schneider, M.M.; Sengupta, S.K.; Nicol, C.J. Loss of PPARgamma expression in mammary secretory epithelial cells creates a pro-breast tumorigenic environment. Int. J. Cancer 2013, 134, 1055–1066. [Google Scholar] [CrossRef] [PubMed]
- Pignatelli, M.; Cortes-Canteli, M.; Lai, C.; Santos, A.; Perez-Castillo, A. The peroxisome proliferator-activated receptor gamma is an inhibitor of ErbBs activity in human breast cancer cells. J. Cell Sci. 2001, 114, 4117–4126. [Google Scholar] [PubMed]
- Nomura, D.K.; Lombardi, D.P.; Chang, J.W.; Niessen, S.; Ward, A.M.; Long, J.Z.; Hoover, H.H.; Cravatt, B.F. Monoacylglycerol lipase exerts dual control over endocannabinoid and fatty acid pathways to support prostate cancer. Chem. Biol. 2011, 18, 846–856. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.R.; Lian, Y.F.; Peng, L.X.; Lei, J.J.; Deng, C.C.; Xu, M.; Feng, Q.S.; Chen, L.Z.; Bei, J.X.; Zeng, Y.X. Monoacylglycerol lipase promotes metastases in nasopharyngeal carcinoma. Int. J. Clin. Exp. Pathol. 2014, 7, 3704–3713. [Google Scholar] [PubMed]
- Takeda, S.; Yamaori, S.; Motoya, E.; Matsunaga, T.; Kimura, T.; Yamamoto, I.; Watanabe, K. Delta(9)-Tetrahydrocannabinol enhances MCF-7 cell proliferation via cannabinoid receptor-independent signaling. Toxicology 2008, 245, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Di Marzo, V.; Melck, D.; Orlando, P.; Bisogno, T.; Zagoory, O.; Bifulco, M.; Vogel, Z.; de Petrocellis, L. Palmitoylethanolamide inhibits the expression of fatty acid amide hydrolase and enhances the anti-proliferative effect of anandamide in human breast cancer cells. Biochem. J. 2001, 358, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Thors, L.; Bergh, A.; Persson, E.; Hammarsten, P.; Stattin, P.; Egevad, L.; Granfors, T.; Fowler, C.J. Fatty acid amide hydrolase in prostate cancer: Association with disease severity and outcome, CB1 receptor expression and regulation by IL-4. PLoS ONE 2010, 5, e12275. [Google Scholar] [CrossRef] [PubMed]
- Nomura, D.K.; Long, J.Z.; Niessen, S.; Hoover, H.S.; Ng, S.W.; Cravatt, B.F. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell 2010, 140, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Jiang, L.; Luo, X.; Jin, W.; He, Q.; An, J.; Lui, K.; Shi, J.; Rong, R.; Su, W.; et al. Potential tumor-suppressive role of monoglyceride lipase in human colorectal cancer. Oncogene 2013, 32, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Fowler, C.J. Delta(9)-tetrahydrocannabinol and cannabidiol as potential curative agents for cancer: A critical examination of the preclinical literature. Clin. Pharmacol. Ther. 2015, 97, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Torres, S.; Lorente, M.; Rodriguez-Fornes, F.; Hernandez-Tiedra, S.; Salazar, M.; Garcia-Taboada, E.; Barcia, J.; Guzman, M.; Velasco, G. A combined preclinical therapy of cannabinoids and temozolomide against glioma. Mol. Cancer Ther. 2011, 10, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Hernan Perez de la Ossa, D.; Lorente, M.; Gil-Alegre, M.E.; Torres, S.; Garcia-Taboada, E.; Aberturas Mdel, R.; Molpeceres, J.; Velasco, G.; Torres-Suarez, A.I. Local delivery of cannabinoid-loaded microparticles inhibits tumor growth in a murine xenograft model of glioblastoma multiforme. PLoS ONE 2013, 8, e54795. [Google Scholar] [CrossRef]
- Armstrong, J.L.; Hill, D.S.; McKee, C.S.; Hernandez-Tiedra, S.; Lorente, M.; Lopez-Valero, I.; Eleni Anagnostou, M.; Babatunde, F.; Corazzari, M.; Redfern, C.P.; et al. Exploiting cannabinoid-induced cytotoxic autophagy to drive melanoma cell death. J. Investig. Dermatol. 2015, 135, 1629–1637. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.R.; Lossignol, D.; Burnell-Nugent, M.; Fallon, M.T. An open-label extension study to investigate the long-term safety and tolerability of THC/CBD oromucosal spray and oromucosal THC spray in patients with terminal cancer-related pain refractory to strong opioid analgesics. J. Pain Symptom Manag. 2013, 46, 207–218. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tegeder, I. Endocannabinoids as Guardians of Metastasis. Int. J. Mol. Sci. 2016, 17, 230. https://doi.org/10.3390/ijms17020230
Tegeder I. Endocannabinoids as Guardians of Metastasis. International Journal of Molecular Sciences. 2016; 17(2):230. https://doi.org/10.3390/ijms17020230
Chicago/Turabian StyleTegeder, Irmgard. 2016. "Endocannabinoids as Guardians of Metastasis" International Journal of Molecular Sciences 17, no. 2: 230. https://doi.org/10.3390/ijms17020230
APA StyleTegeder, I. (2016). Endocannabinoids as Guardians of Metastasis. International Journal of Molecular Sciences, 17(2), 230. https://doi.org/10.3390/ijms17020230