Identification of Novel Pathways in Plant Lectin-Induced Cancer Cell Apoptosis


Abstract

1. Introduction
2. Results and Discussion
2.1. Global Human Protein-Protein Interaction (PPI) Network Construction
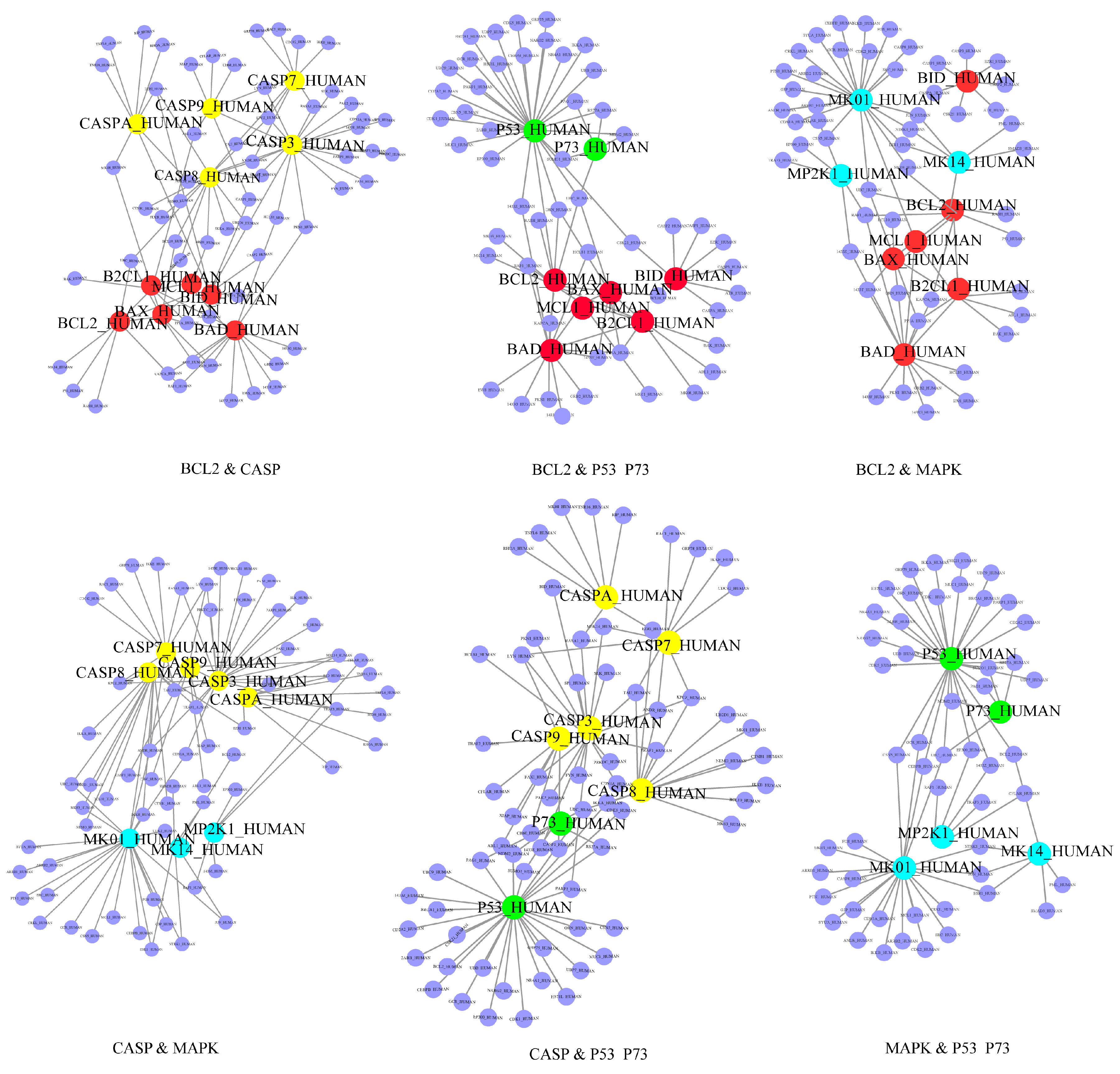
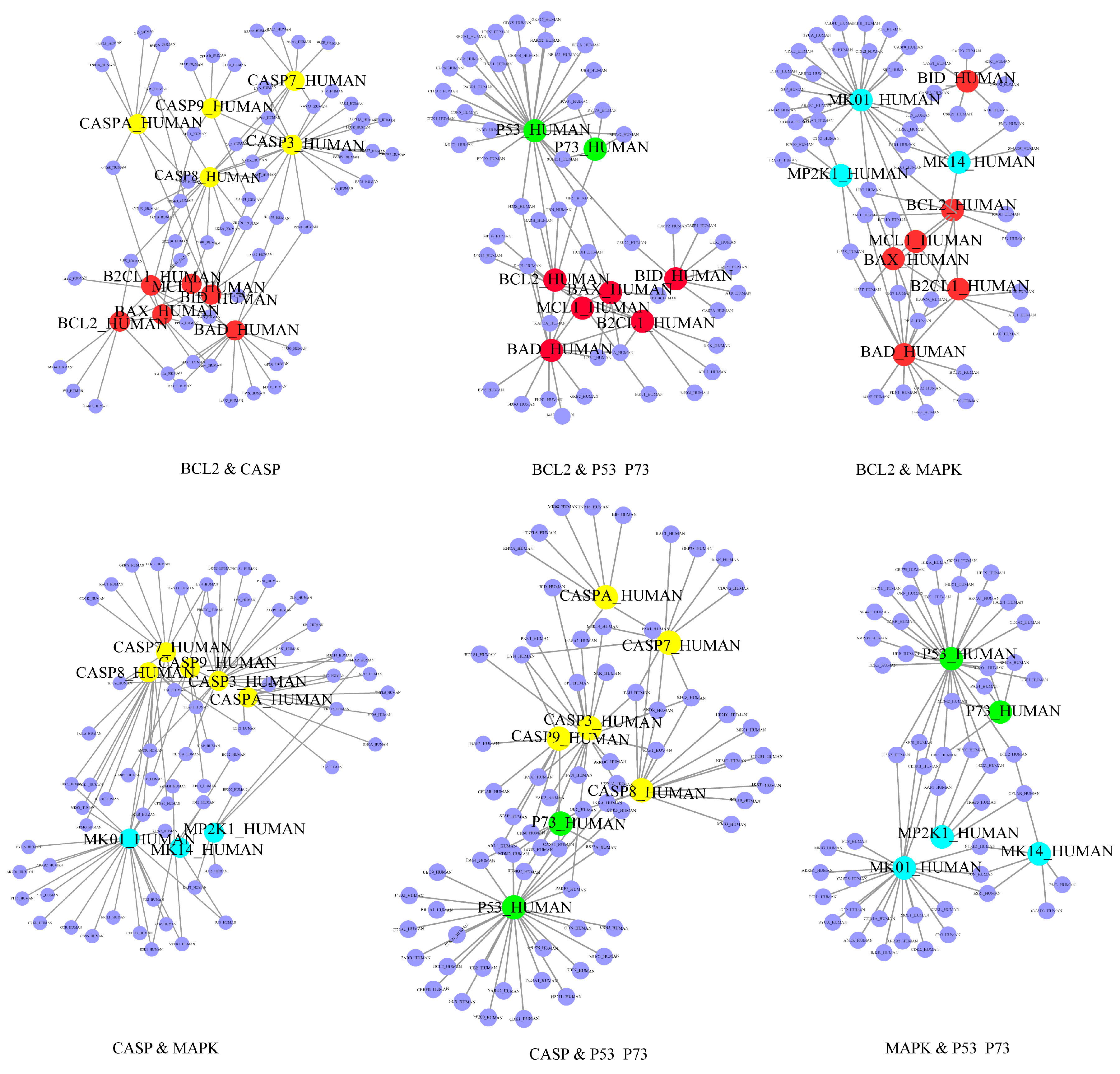
2.2. Identification of the Core Apoptotic Pathways in Human PPI Network
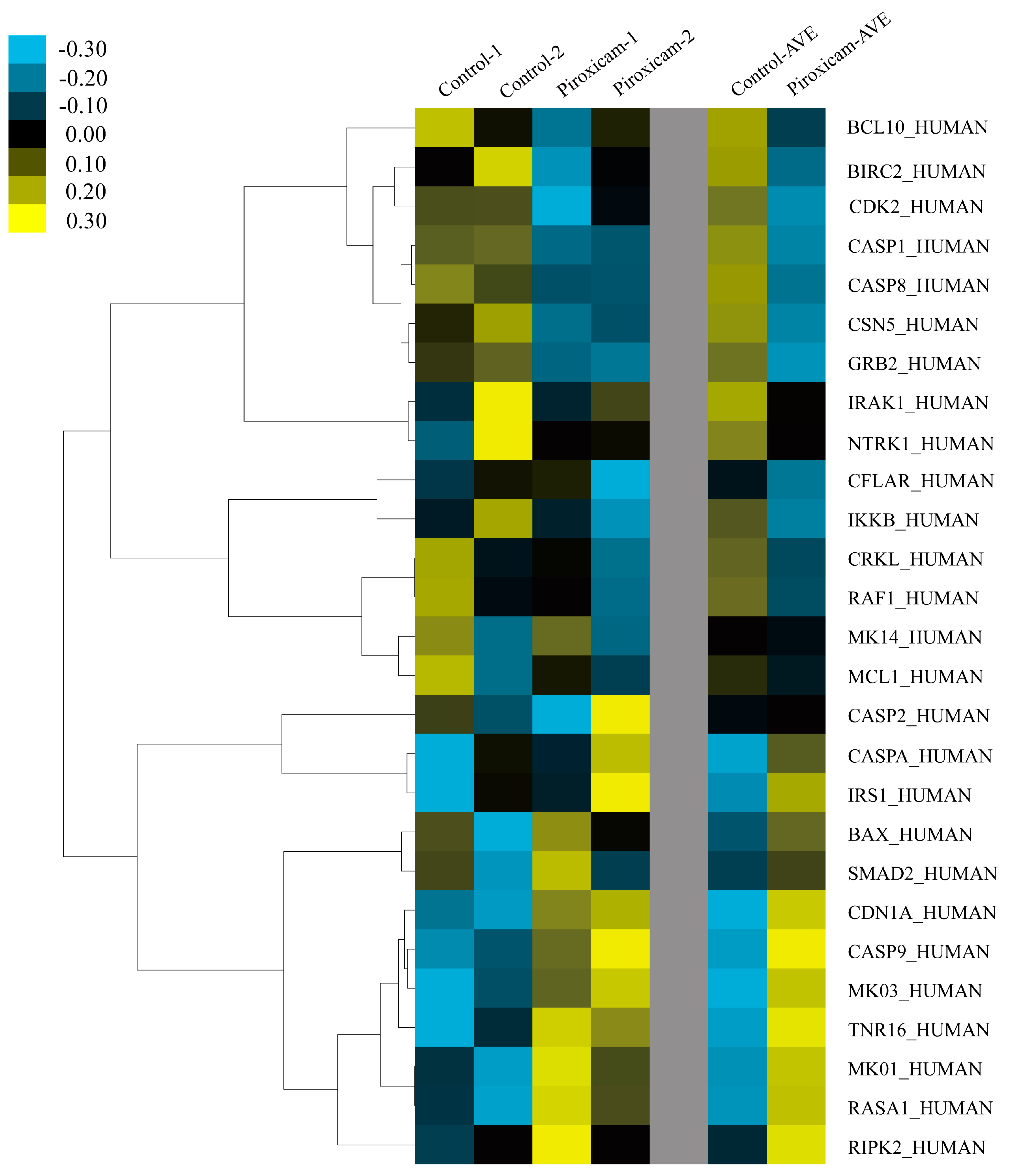
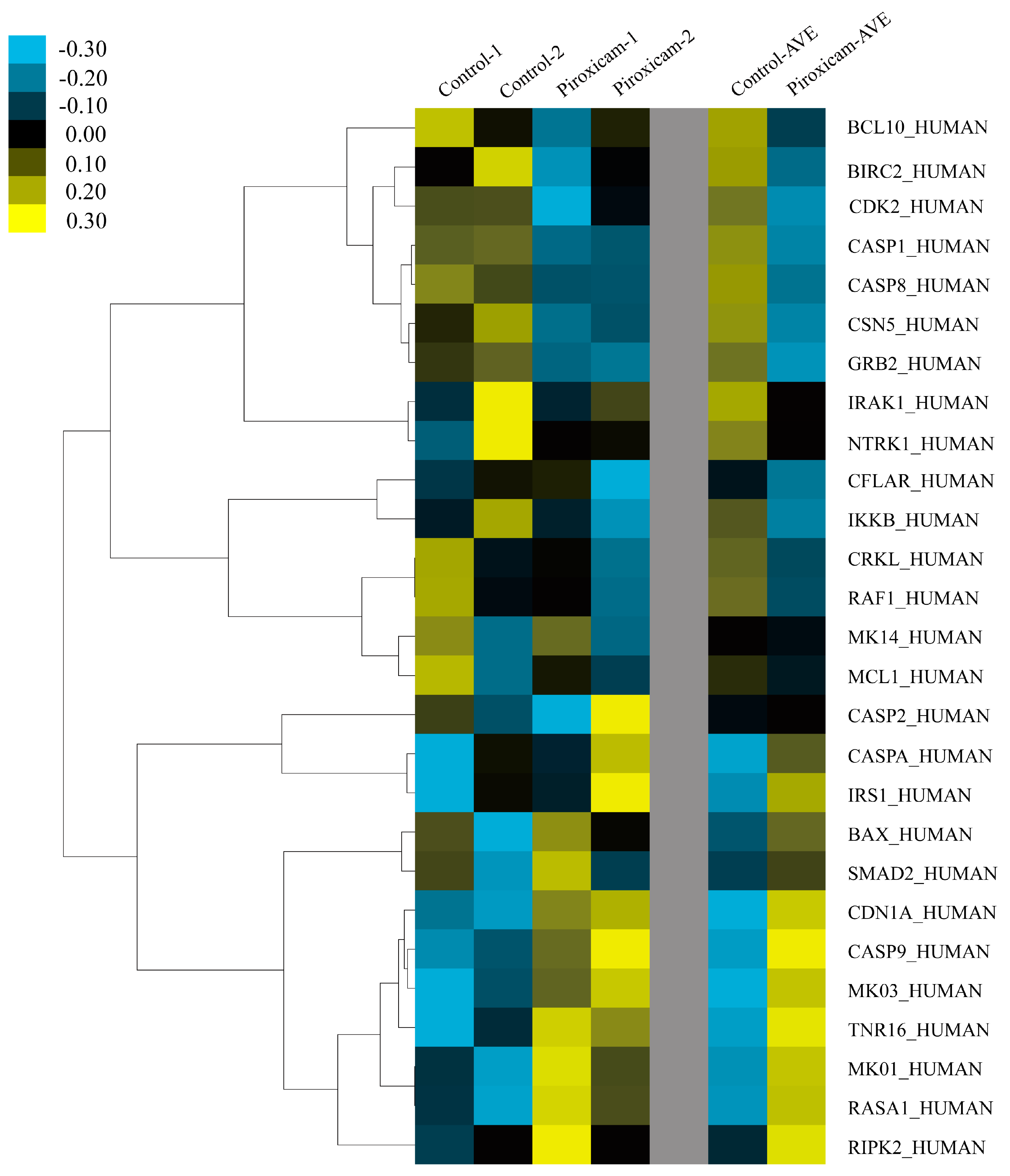
2.3. Identification of Plant Lectin-Induced Novel Apoptotic Pathways in Cancer
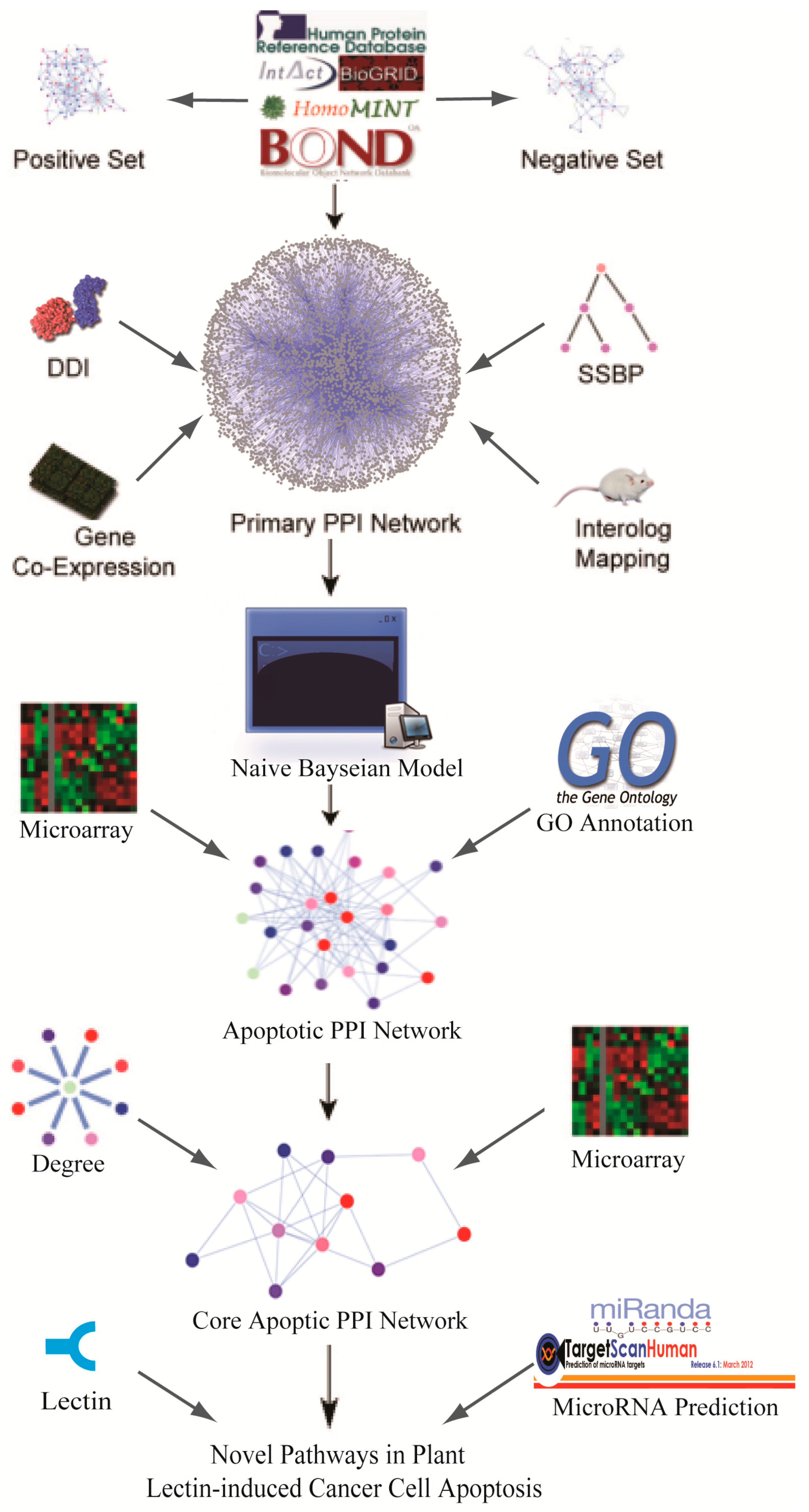
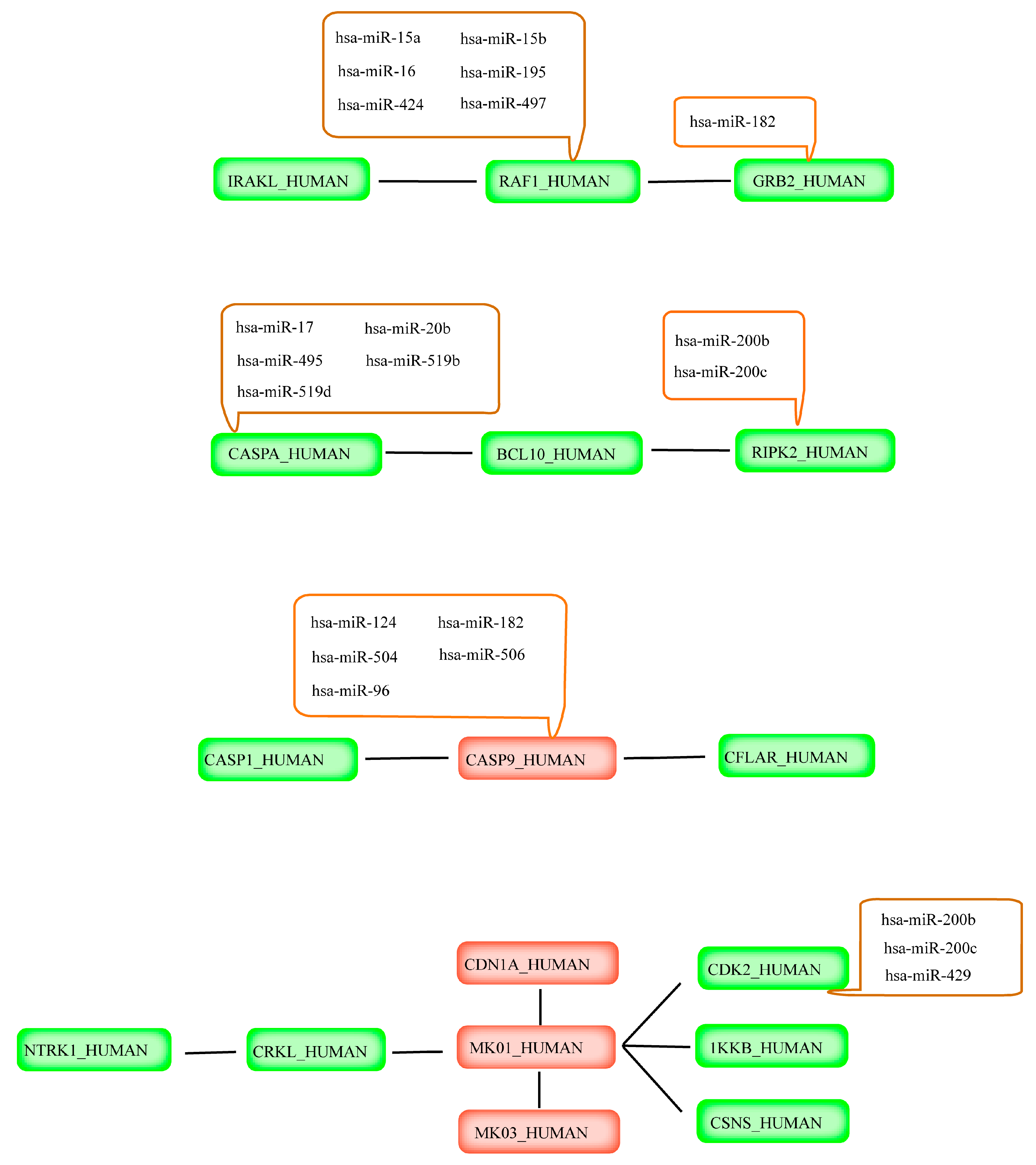
2.4. Prediction of miRNAs Targeting Apoptotic Hub Proteins

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Protein Name | Consensus Results |
|---|---|---|
| CASP9 | Caspase-9 | hsa-miR-124 |
| hsa-miR-182 | ||
| hsa-miR-504 | ||
| hsa-miR-506 | ||
| hsa-miR-96 | ||
| RIPK2 | Receptor-interacting serine/threonine-protein kinase 2 | hsa-miR-200b |
| hsa-miR-200c | ||
| GRB2 | Growth factor receptor-bound protein 2 | hsa-miR-182 |
| CDK2 | Cyclin-dependent kinase 2 | hsa-miR-200b |
| hsa-miR-200c | ||
| hsa-miR-429 | ||
| CASP8 | Caspase-8 | hsa-miR-17 |
| hsa-miR-20b | ||
| hsa-miR-495 | ||
| hsa-miR-519d | ||
| hsa-miR-93 | ||
| RAF1 | Rapidly Accelerated Fibrosarcoma (RAF) proto-oncogene serine/threonine-protein kinase | hsa-miR-15a |
| hsa-miR-15b | ||
| hsa-miR-16 | ||
| hsa-miR-195 | ||
| hsa-miR-424 | ||
| hsa-miR-497 |
2.5. Plant Lectins as Promising Candidate for Drug Development
3. Materials and Methods
3.1. Retrieving of Functional Genomics Data
3.2. Integration of Several Biological Data Sources
3.2.1. Smallest Shared Biological Process
3.2.2. Domain–Domain Interaction
3.2.3. Gene Co-Expression Profiles
3.2.4. Cross-Species Interolog Mapping
3.2.5. Naïve Bayesian Model Analysis
3.2.6. Evaluation and Prediction of Naïve Bayesian Model
3.2.7. Identification of Hub Proteins
3.2.8. Microarray Analyses of Apoptotic Genes
3.2.9. Targeted miRNA Prediction and Classification
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Peumans, W.J.; van Damme, E.J.M.; Barre, A.; Rougé, P. Classification of plant lectins in families of structurally and evolutionary related proteins. Adv. Exp. Med. Biol. 2001, 491, 27–54. [Google Scholar] [PubMed]
- Van Damme, E.J.M.; Sachiko, N.T.; Smith, D.F.; Maté, O.; Winter, H.C.; Pierre, R.; Goldstein, I.J.; Hanqing, M.; Junko, K.; Rapha, C.; et al. Phylogenetic and specificity studies of two-domain GNA-related lectins: Generation of multispecificity through domain duplication and divergent evolution. Biochem. J. 2007, 404, 51–61. [Google Scholar] [CrossRef] [PubMed]
- De Mejia, E.G.; Traliece Bradford, B.S.; Hasler, C. The anticarcinogenic potential of soybean lectin and lunasin. Nutr. Rev. 2003, 61, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Meyer, A.; Rypniewski, W.M. Structure of mistletoe lectin I from Viscum album in complex with the phytohormone zeatin. Biochim. Biophys. Acta 2008, 1784, 1590–1595. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R. Means to an End: Apoptosis and Other Cell Death Mechanisms; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2011. [Google Scholar]
- Ouyang, L.; Shi, Z.; Zhao, S.; Wang, F.T.; Zhou, T.T.; Liu, B.; Bao, J.K. Programmed cell death pathways in cancer: A review of apoptosis, autophagy and programmed necrosis. Cell Prolif. 2012, 45, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Faheina-Martins, G.V.; da Silveira, A.L.; Cavalcanti, B.C.; Ramos, M.V.; Moraes, M.O.; Pessoa, C.; Araújo, D.A.M. Antiproliferative effects of lectins from Canavalia ensiformis and Canavalia brasiliensis in human leukemia cell lines. Toxicol. In Vitro 2012, 26, 1161–1169. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.L.; Xin, W.; Bao, J.K.; Bo, L. MicroRNA-modulated autophagic signaling networks in cancer. Int. J. Biochem. Cell Biol. 2012, 44, 733–736. [Google Scholar] [CrossRef] [PubMed]
- Lima, R.T.; Busacca, S.; Almeida, G.M.; Gaudino, G.; Fennell, D.A.; Vasconcelos, M.H. MicroRNA regulation of core apoptosis pathways in cancer. Eur. J. Cancer 2011, 47, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Li, L.N.; Zhang, H.D.; Zhi, R.; Yuan, S.J. Down-regulation of some miRNAs by degrading their precursors contributes to anti-cancer effect of mistletoe lectin-I. Br. J. Pharmacol. 2010, 162, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.L.; Zhao, X.; Xu, H.L.; Wen, X.; Wang, S.Y.; Liu, B.; Bao, J.K.; Wei, Y.Q. Identification of microRNA-regulated autophagic pathways in plant lectin-induced cancer cell death. Cell Prolif. 2012, 45, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Xu, H.L.; Zhao, X.; Wen, X.; Wang, F.T.; Wang, S.Y.; Fu, L.L.; Liu, B.; Bao, J.K. Network-based identification of novel connections among apoptotic signaling pathways in cancer. Appl. Biochem. Biotechnol. 2012, 167, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, D.R.; Tomlins, S.A.; Varambally, S.; Mahavisno, V.; Barrette, T.; Kalyana-Sundaram, S.; Ghosh, D.; Pandey, A.; Chinnaiyan, A.M. Probabilistic model of the human protein–protein interaction network. Nat. Biotechnol. 2005, 23, 951–959. [Google Scholar] [CrossRef] [PubMed]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. In a smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Šali, A.; Blundell, T.L. Comparative protein modeling by satisfaction of spatial restraints. J. Mol. Biol. 1995, 1, 270–277. [Google Scholar]
- Agarwal, S.; Deane, C.M.; Porter, M.A.; Jones, N.S. Approximate solution of Schön’s balance equations for the thermoluminescence and the thermally stimulated conductivity of inorganic photoconducting crystals. J. Immunol. 1966, 18, 307–316. [Google Scholar]
- Arico, S.; Petiot, A.; Bauvy, C.; Dubbelhuis, P.F.; Meijer, A.J.; Codogno, P.; Ogier-Denis, E. The tumor suppresor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway. J. Biol. Chem. 2001, 276, 35243–35246. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Ge, H. Modularity and Dynamics of Cellular Networks. PLoS Comput. Biol. 2006. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Zhao, X.; Chen, L. Identifying responsive functional modules from protein-protein interaction network. PLoS Comput. Biol. 2009, 27, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Deepak, A.; Nicole, J.; Gundara, J.S.; Jingting, Z.; Gill, A.J.; Leigh, D.; Robinson, B.G.; Sidhu, S.B. MicroRNA profiling of sporadic and hereditary medullary thyroid cancer identifies predictors of nodal metastasis, prognosis, and potential therapeutic targets. Clin. Cancer Res. 2011, 17, 4772–4781. [Google Scholar]
- Akavia, U.D.; Litvin, O.; Kim, J.; Sanchez-Garcia, F.; Kotliar, D.; Causton, H.C.; Pochanard, P.; Mozes, E.; Garraway, L.A.; Pe’Er, D. An integrated approach to uncover drivers of cancer. Cell 2010, 143, 1005–1017. [Google Scholar] [CrossRef] [PubMed]
- Chu, L.H.; Chen, B.S. Construction of a cancer-perturbed protein. BMC Syst. Biol. 2008, 2, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Pietro, C.D.; Ragusa, M.; Barbagallo, D.; Duro, L.R.; Guglielmino, M.R.; Majorana, A.; Angelica, R.; Scalia, M.; Statello, L.; Salito, L.; et al. The apoptotic machinery as a biological complex system: Analysis of its omics and evolution, identification of candidate genes for fourteen major types of cancer, and experimental validation in CML and neuroblastoma. BMC Med. Genom. 2009, 2. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.L.; Zhou, C.C.; Yao, S.; Yu, J.Y.; Liu, B.; Bao, J.K. Plant lectins: Targeting programmed cell death pathways as antitumor agents. Int. J. Biochem. Cell Biol. 2011, 43, 1442–1449. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; An, N.; Lu, B.M.; Zhou, N.; Yang, S.L.; Zhang, B.; Li, C.Y.; Wang, Z.J.; Wang, F.; Wu, C.F.; et al. Identification of novel kinase inhibitors by targeting a kinase-related apoptotic protein–protein interaction network in HeLa cells. Cell Prolif. 2014, 47, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Strasser, A. The Bcl-2 protein family: Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Schuler, M.; Bossy-Wetzel, E.; Goldstein, J.C.; Fitzgerald, P.; Green, D.R. p53 induces apoptosis by caspase activation through mitochondrial cytochrome c release. J. Biol. Chem. 2000, 275, 7337–7342. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.S. The functional interactions between the p53 and MAPK signaling pathways. Cancer Biol. Ther. 2004, 3, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Sun, R.; Chen, W.P.; Lu, B.M.; Li, X.Y.; Wang, Z.J.; Bao, J.K. A Systematic In Silico Mining of the Mechanistic Implications and Therapeutic Potentials of Estrogen Receptor (ER)-α in Breast Cancer. PLoS ONE 2014, 9, e91894. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Li, C.Y.; Zhao, S.; Yu, Y.; An, N.; Liu, Y.X.; Wu, C.F.; Yue, B.S.; Bao, J.K. A systems biology analysis of autophagy in cancer therapy. Cancer Lett. 2013, 2, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.L.; Wang, Z.J.; Liang, X.M.; Li, X.; Shi, Z.; Zhou, N.; Bao, J.K. In silico identification of novel kinase inhibitors targeting wild-type and T315I mutant ABL1 from FDA-approved drugs. Mol. BioSyst. 2014, 10, 1524–1537. [Google Scholar] [CrossRef] [PubMed]
- Schlatter, R.; Schmich, K.; Vizcarra, I.A.; Scheurich, P.; Sauter, T.; Borner, C.; Ederer, M.; Merfort, I.; Sawodny, O. ON/OFF and Beyond-A Boolean Model of Apoptosis. PLoS Comput. Biol. 2009, 5. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.R.A.; Quaranta, V. Integrative mathematical oncology. Nat. Rev. Cancer 2008, 8, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Zhang, S.; Zhang, L.; Tong, X.; Zhang, J.; Zhang, Y.; Ouyang, L.; Liu, B.; Huang, J. Systems biology network-based discovery of a small molecule activator BL-AD008 targeting AMPK/ZIPK and inducing apoptosis in cervical cancer. Oncotarget 2015, 6, 8071–8088. [Google Scholar] [CrossRef] [PubMed]
- Mishra, G.R.; Suresh, M.; Kumaran, K.; Kannabiran, N.; Suresh, S.; Bala, P.; Shivakumar, K.; Anuradha, N.; Reddy, R.; Raghavan, T.M.; et al. Human protein reference database—2006 update. Nucleic Acids Res. 2006, 34, D411–D414. [Google Scholar] [CrossRef] [PubMed]
- Alfarano, C.; Andrade, C.E.; Anthony, K.; Bahroos, N.; Bajec, M.; Bantoft, K.; Betel, D.; Bobechko, B.; Boutilier, K.; Burgess, E.; et al. The Biomolecular Interaction Network Database and related tools 2005 update. Nucleic Acids Res. 2005, 33, D418–D424. [Google Scholar] [CrossRef] [PubMed]
- Kerrien, S.; Alam-Faruque, Y.; Aranda, B.; Bancarz, I.; Bridge, A.; Derow, C.; Dimmer, E.; Feuermann, M.; Friedrichsen, A.; Huntley, R.; et al. IntAct—Open source resource for molecular interaction data. Nucleic Acids Res. 2007, 35, D561–D565. [Google Scholar] [CrossRef] [PubMed]
- Persico, M.; Ceol, A.; Gavrila, C.; Hoffmann, R.; Florio, A.; Cesareni, G. HomoMINT: An inferred human network based on orthology mapping of protein interactions discovered in model organisms. BMC Bioinform. 2005, 6. [Google Scholar] [CrossRef] [PubMed]
- Winter, A.G.; Wildenhain, J.; Tyers, M. BioGRID REST Service, BiogridPlugin2 and BioGRID WebGraph: New tools for access to interaction data at BioGRID. Bioinformatics 2011, 27, 1043–1044. [Google Scholar] [CrossRef] [PubMed]
- Xenarios, I.; Salwínski, L.; Duan, X.J.; Higney, P.; Kim, S.M.; Eisenberg, D. DIP, the Database of Interacting Proteins: A research tool for studying cellular networks of protein interactions. Nucleic Acids Res. 2002, 30, 303–305. [Google Scholar] [CrossRef] [PubMed]
- Punta, M.; Coggill, P.C.; Eberhardt, R.Y.; Mistry, J.; Tate, J.; Boursnell, C.; Pang, N.; Forslund, K.; Ceric, G.; Clements, J.; et al. The Pfam protein families database. Nucleic Acids Res. 2012, 40, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Nacher, J.; Hayashida, M.; Akutsu, T. Emergence of scale-free distribution in protein–protein interaction networks based on random selection of interacting domain pairs. Biosystems 2009, 95, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.I.; Whitfield, M.L.; Trinklein, N.C.; Myers, R.M.; Brown, P.O.; Botstein, D. Diverse and specific gene expression responses to stresses in cultured human cells. Mol. Biol. Cell 2004, 15, 2361–2374. [Google Scholar] [CrossRef] [PubMed]
- Nan, Z.; Jinchun, Z.; Ling, F.; Bangmin, L.; Zijie, W.; Rong, S.; Chuanfang, W.; Jinku, B. IntApop: A web service for predicting apoptotic protein interactions in humans. Biosystems 2013, 114, 238–244. [Google Scholar]
- O’Brien, K.P.; Remm, M.; Sonnhammer, E.L.L. Inparanoid: A comprehensive database of eukaryotic orthologs. Nucleic Acids Res. 2005, 33, D476–D480. [Google Scholar] [CrossRef] [PubMed]
- Zou, M.; Conzen, S.D. A new dynamic Bayesian network (DBN) approach for identifying gene regulatory networks from time course microarray data. Bioinformatics 2005, 21, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Baldi, P.; Brunak, S.Y.; Andersen, C.; Nielsen, H. Assessing the accuracy of prediction algorithms for classification: An Overview. Bioinformatics 2000, 16, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Baldi, A.; Piccolo, M.T.; Boccellino, M.R.; Donizetti, A.; Cardillo, I.; La Porta, R.; Quagliuolo, L.; Spugnini, E.P.; Cordero, F.; Citro, G.; et al. Apoptosis induced by piroxicam plus cisplatin combined treatment is triggered by p21 in mesothelioma. PLoS ONE 2011, 6, e23569. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.W.; Juan, H.F.; Huang, H.C. Characterization of microRNA-regulated protein-protein interaction network. Proteomics 2008, 8, 1975–1979. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.H.; Chin, C.H.; Wu, H.H.; Ho, C.W.; Ko, M.T.; Lin, C.Y. Cyto-Hubba: A Cytoscape plug-in for hub object analysis in network biology. In Proceedings of the 20th International Conference on Genome Informatics, Yokohama, Japan, 14–16 December 2009.
- Ho, S.Y.; Hsieh, C.H.; Chen, H.M.; Huang, H.L. Interpretable gene expression classifier with an accurate and compact fuzzy rule base for microarray data analysis. Biosystems 2006, 85, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Tusher, V.G.; Tibshirani, R.; Chu, G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. USA 2001, 98, 5116–5121. [Google Scholar] [CrossRef] [PubMed]
- De Hoon, M.J.L.; Imoto, S.; Nolan, J.; Miyano, S. Open Source Clustering Software. Bioinformatics 2004, 20, 1453–1454. [Google Scholar] [CrossRef] [PubMed]
- Saldanha, A.J. Java Treeview-extensible visualization of microarray data. Bioinformatics 2004, 20, 3246–3248. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; An, N.; Zhao, S.; Li, X.; Bao, J.K.; Yue, B.S. In silico analysis of molecular mechanisms of legume lectin-induced apoptosis in cancer cells. Cell Prolif. 2013, 46, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Friedman, R.C. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2011, 19, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Doron, B.; Manda, W.; Aaron, G.; Marks, D.S.; Chris, S. The microRNA.org resource: Targets and expression. Nucleic Acids Res. 2008, 36, D149–D153. [Google Scholar]
- Maragkakis, M.; Alexiou, P.; Papadopoulos, G.L.; Reczko, M.; Dalamagas, T.; Giannopoulos, G.; Goumas, G.; Koukis, E.; Kourtis, K.; Simossis, V.A.; et al. Accurate microRNA target prediction correlates with protein repression levels. BMC Bioinform. 2009, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, Z.; Sun, R.; Yu, T.; Liu, R.; Cheng, L.-J.; Bao, J.-K.; Zou, L.; Tang, Y. Identification of Novel Pathways in Plant Lectin-Induced Cancer Cell Apoptosis. Int. J. Mol. Sci. 2016, 17, 228. https://doi.org/10.3390/ijms17020228
Shi Z, Sun R, Yu T, Liu R, Cheng L-J, Bao J-K, Zou L, Tang Y. Identification of Novel Pathways in Plant Lectin-Induced Cancer Cell Apoptosis. International Journal of Molecular Sciences. 2016; 17(2):228. https://doi.org/10.3390/ijms17020228
Chicago/Turabian StyleShi, Zheng, Rong Sun, Tian Yu, Rong Liu, Li-Jia Cheng, Jin-Ku Bao, Liang Zou, and Yong Tang. 2016. "Identification of Novel Pathways in Plant Lectin-Induced Cancer Cell Apoptosis" International Journal of Molecular Sciences 17, no. 2: 228. https://doi.org/10.3390/ijms17020228
APA StyleShi, Z., Sun, R., Yu, T., Liu, R., Cheng, L.-J., Bao, J.-K., Zou, L., & Tang, Y. (2016). Identification of Novel Pathways in Plant Lectin-Induced Cancer Cell Apoptosis. International Journal of Molecular Sciences, 17(2), 228. https://doi.org/10.3390/ijms17020228