
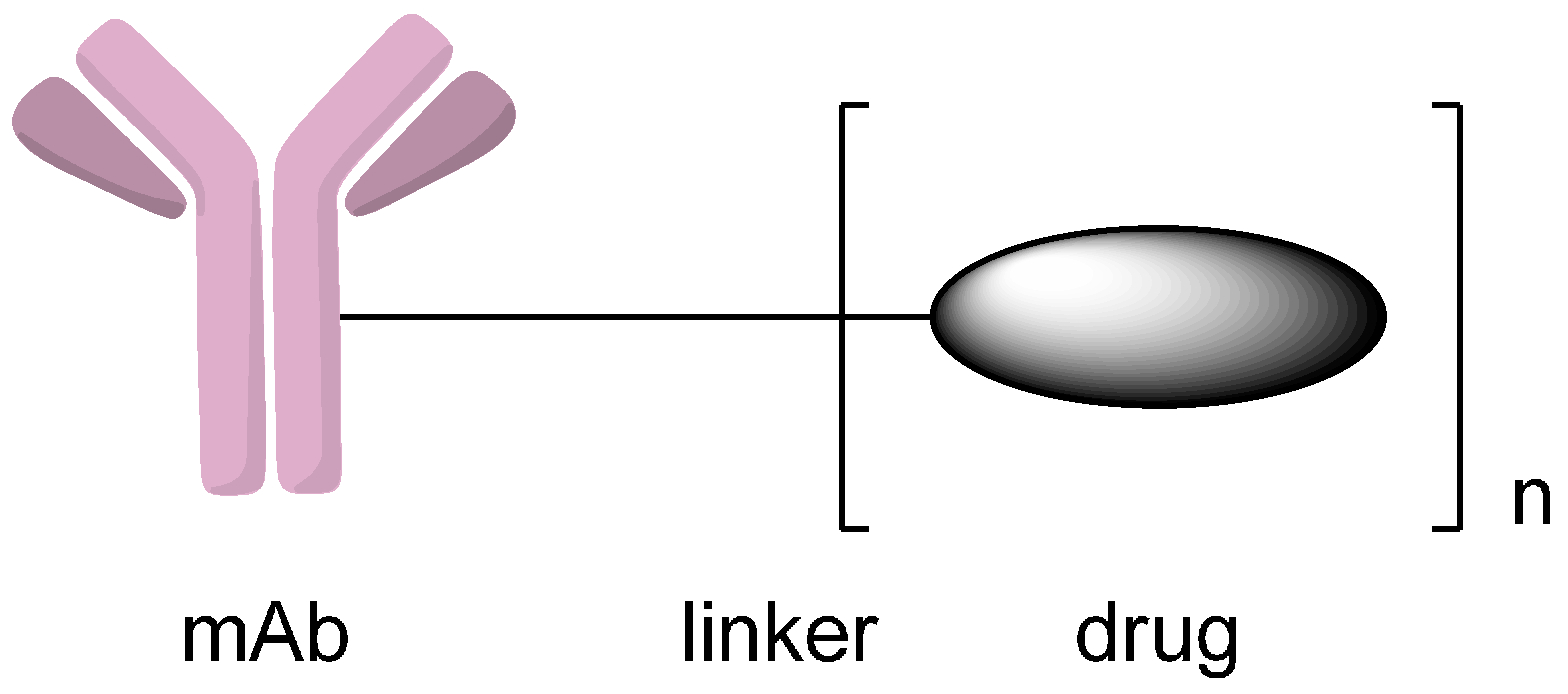
Methods to Design and Synthesize Antibody-Drug Conjugates (ADCs)


Abstract
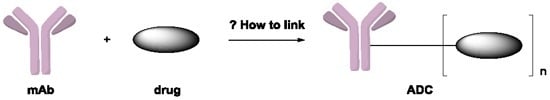
:
1. Introduction
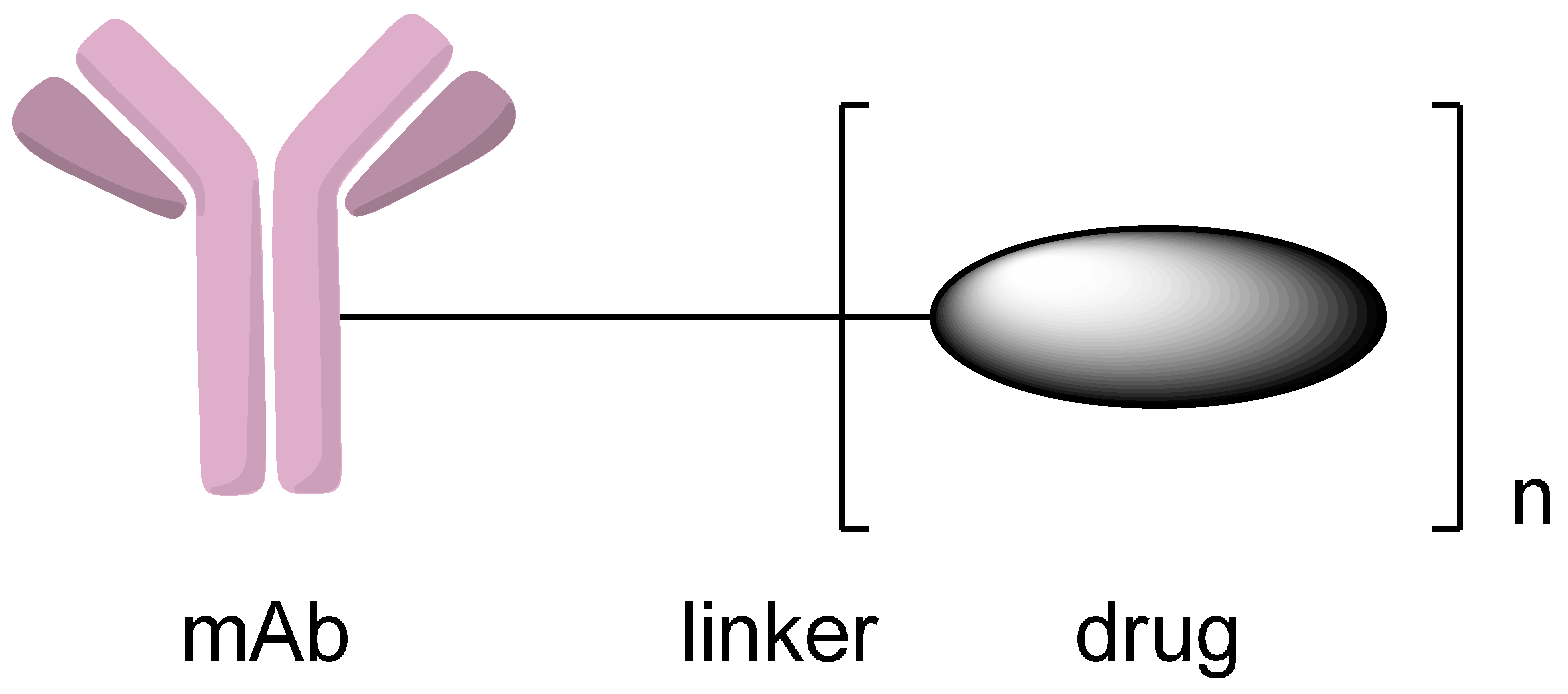
2. Conjugation via Various Functional Groups to Synthesize Antibody-Drug Conjugates (ADCs)
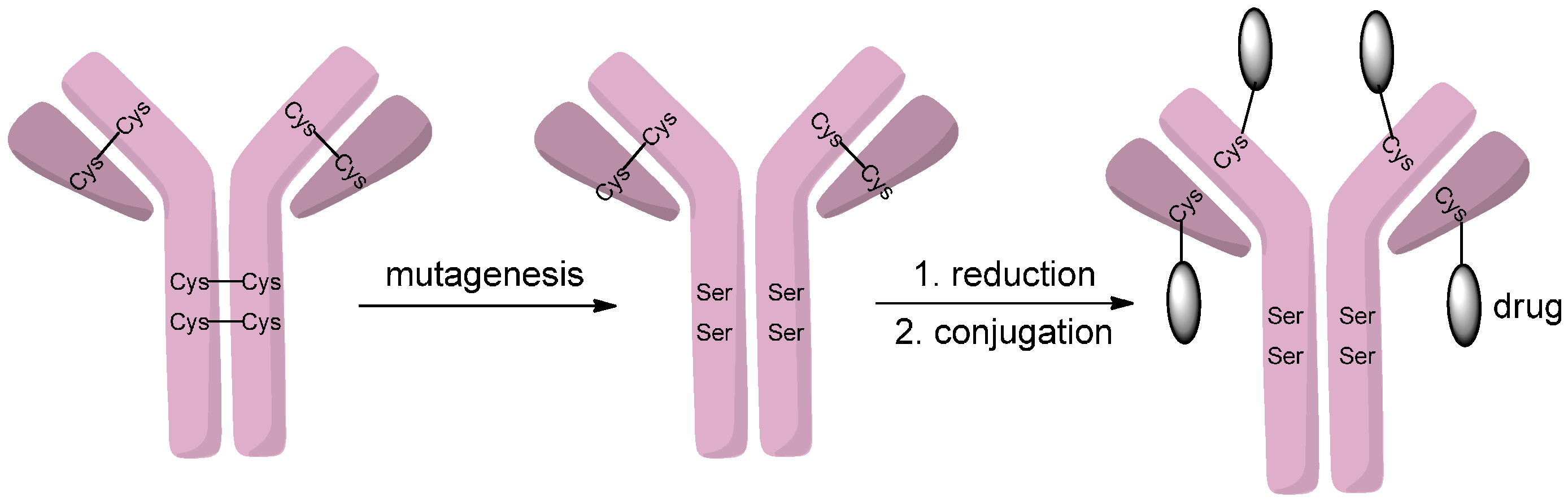
2.1. Conjugation via Thiols
2.1.1. Addition to Maleimides
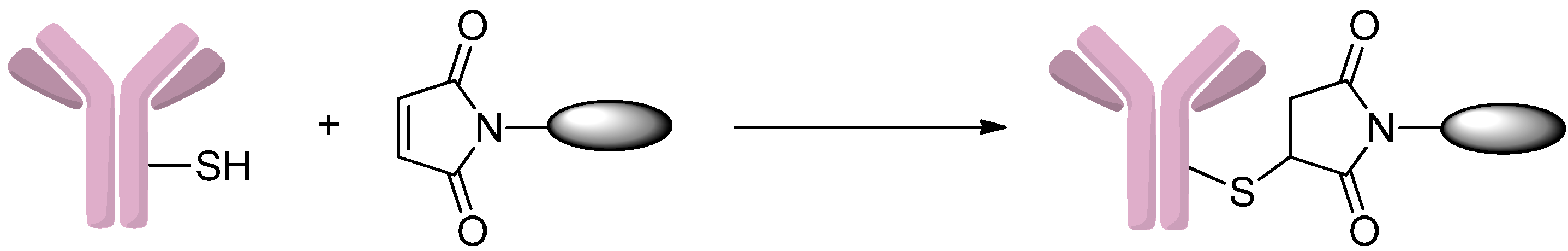
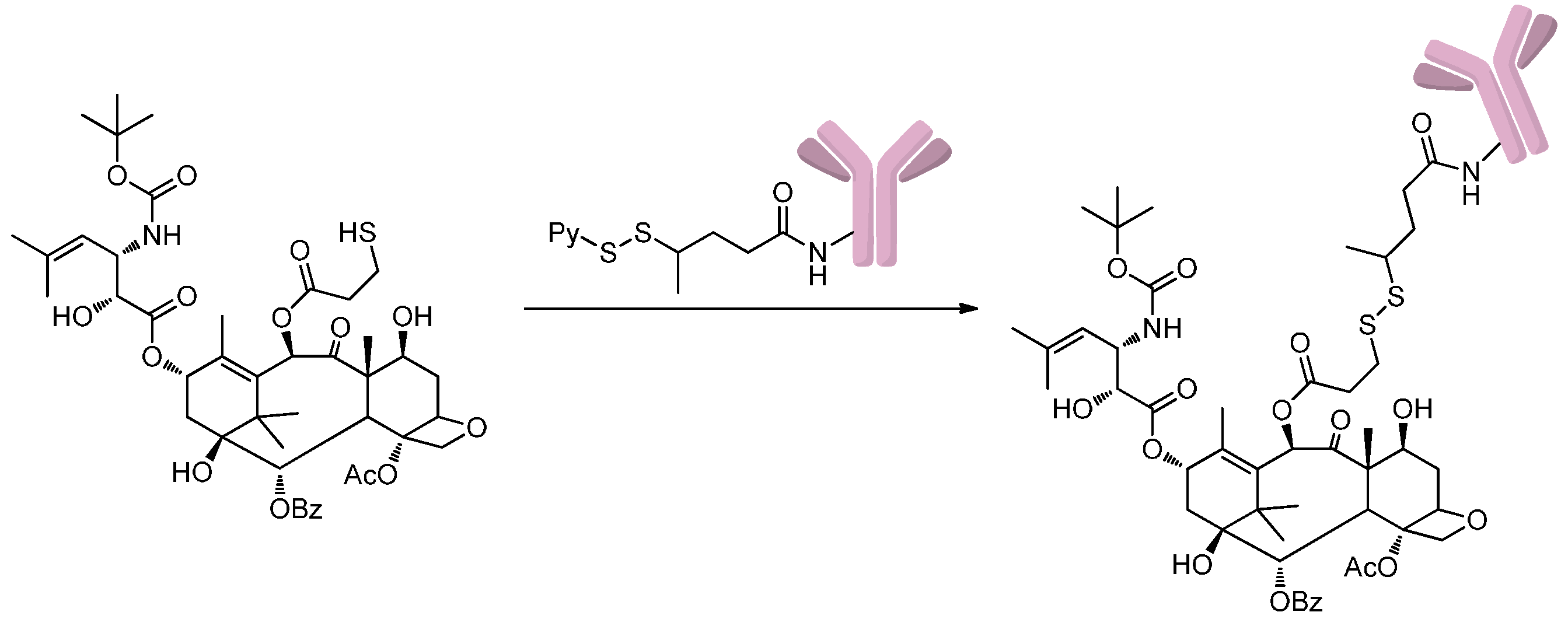
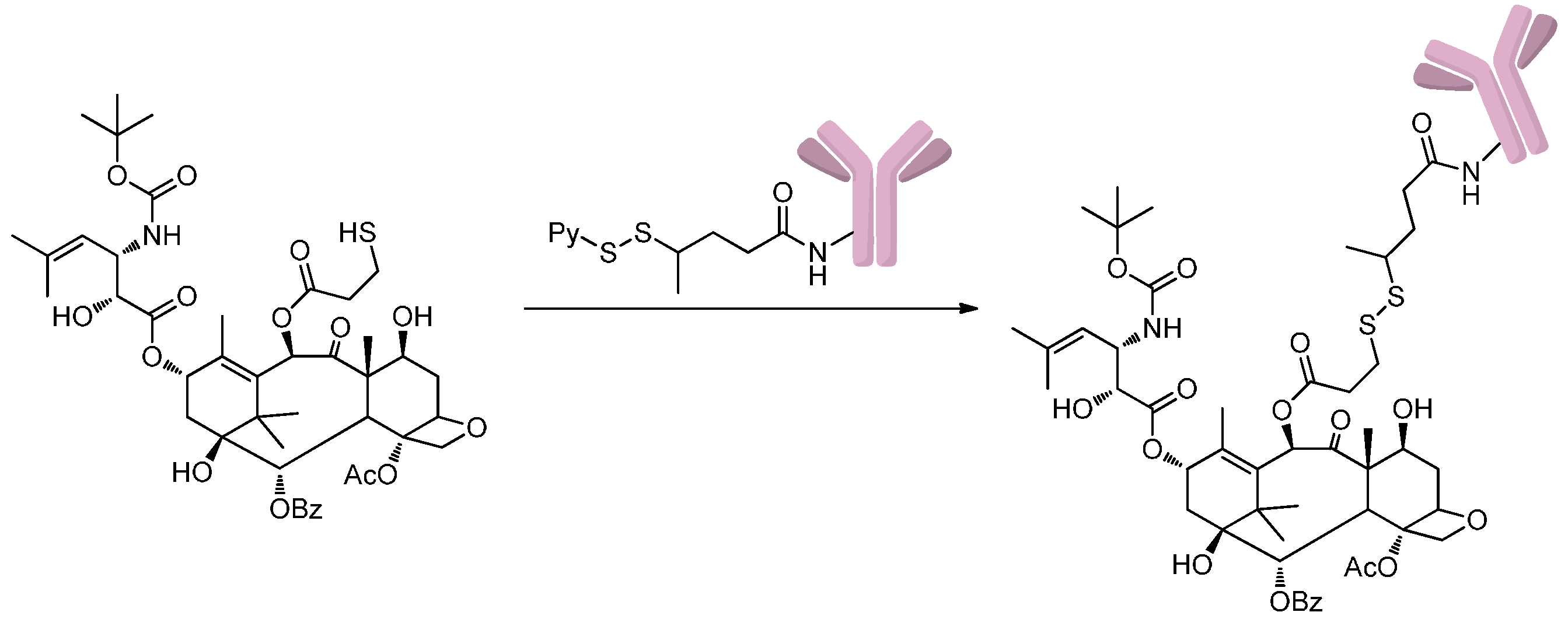
2.1.2. Disulfide-Thiol Exchange
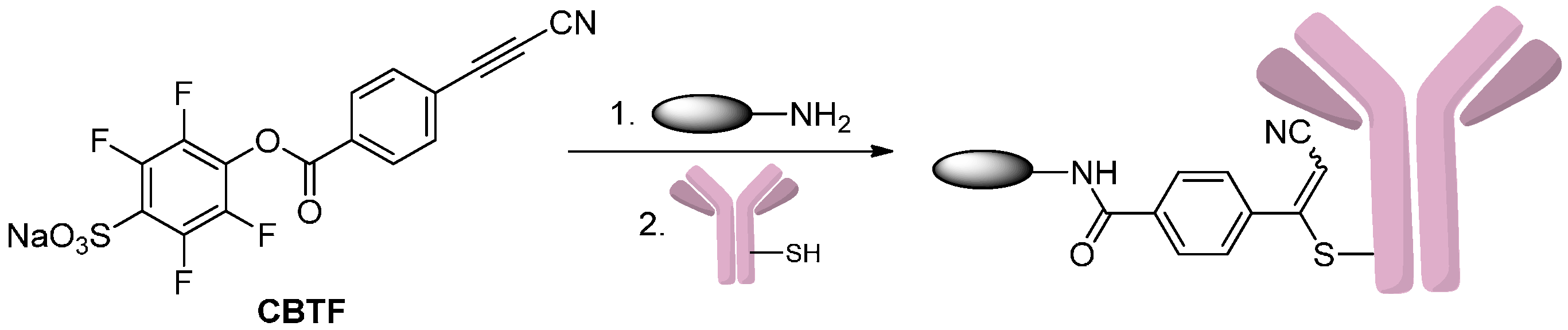
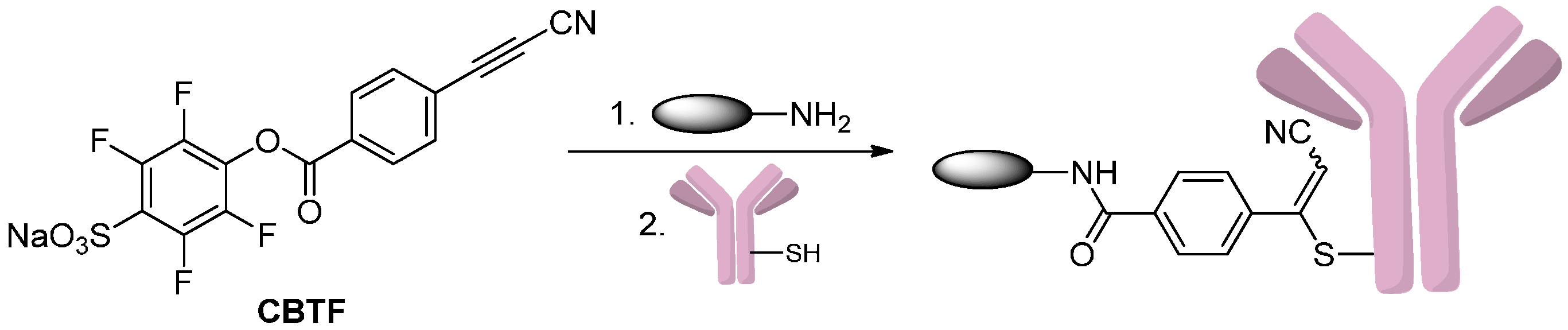
2.1.3. Addition to Alkynes
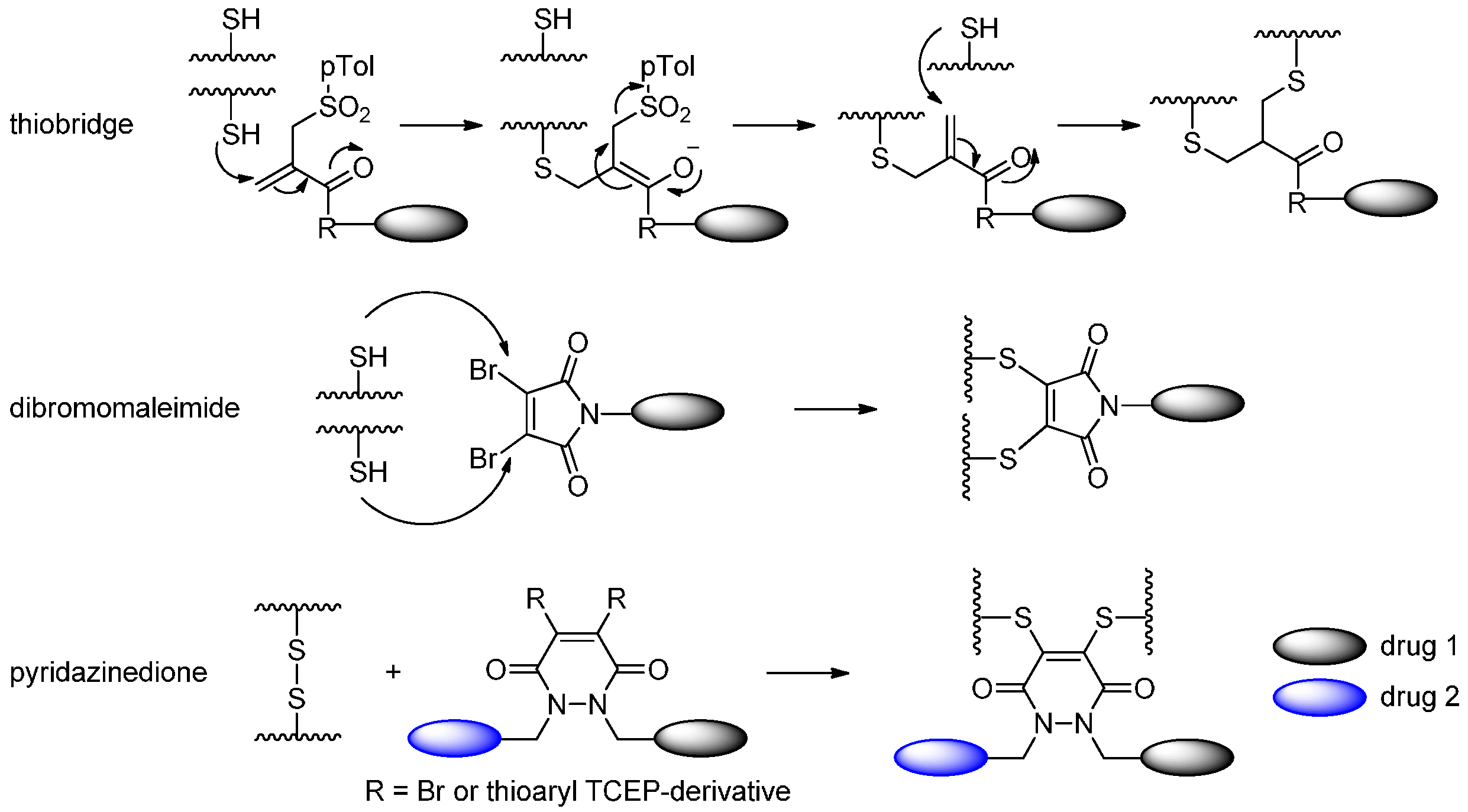
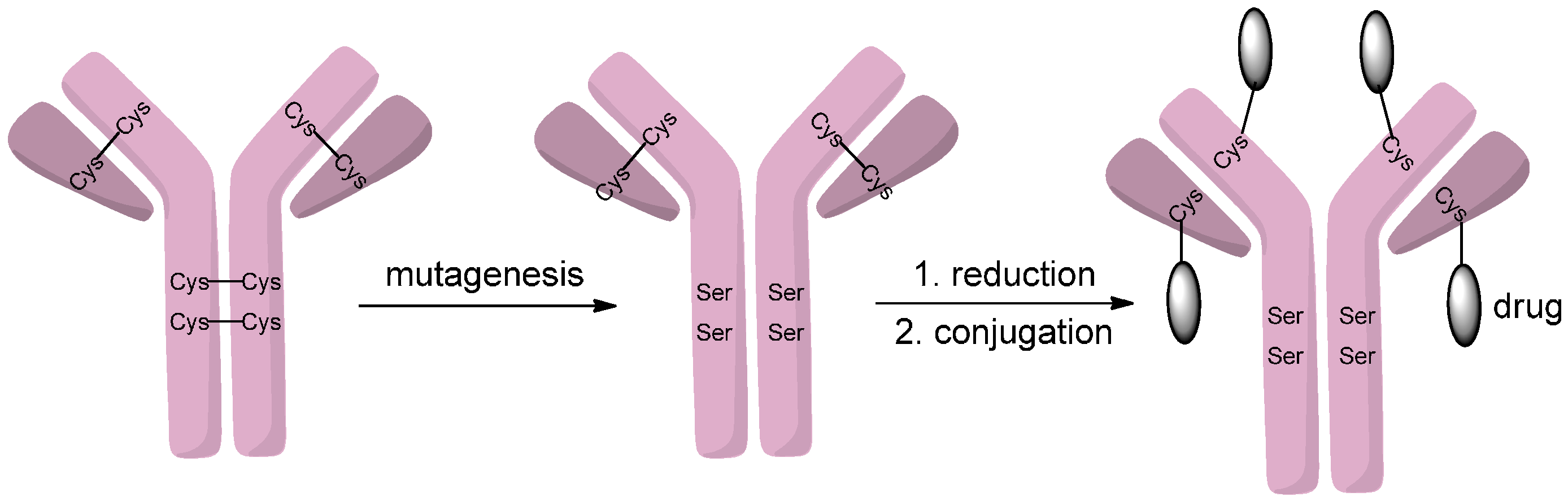
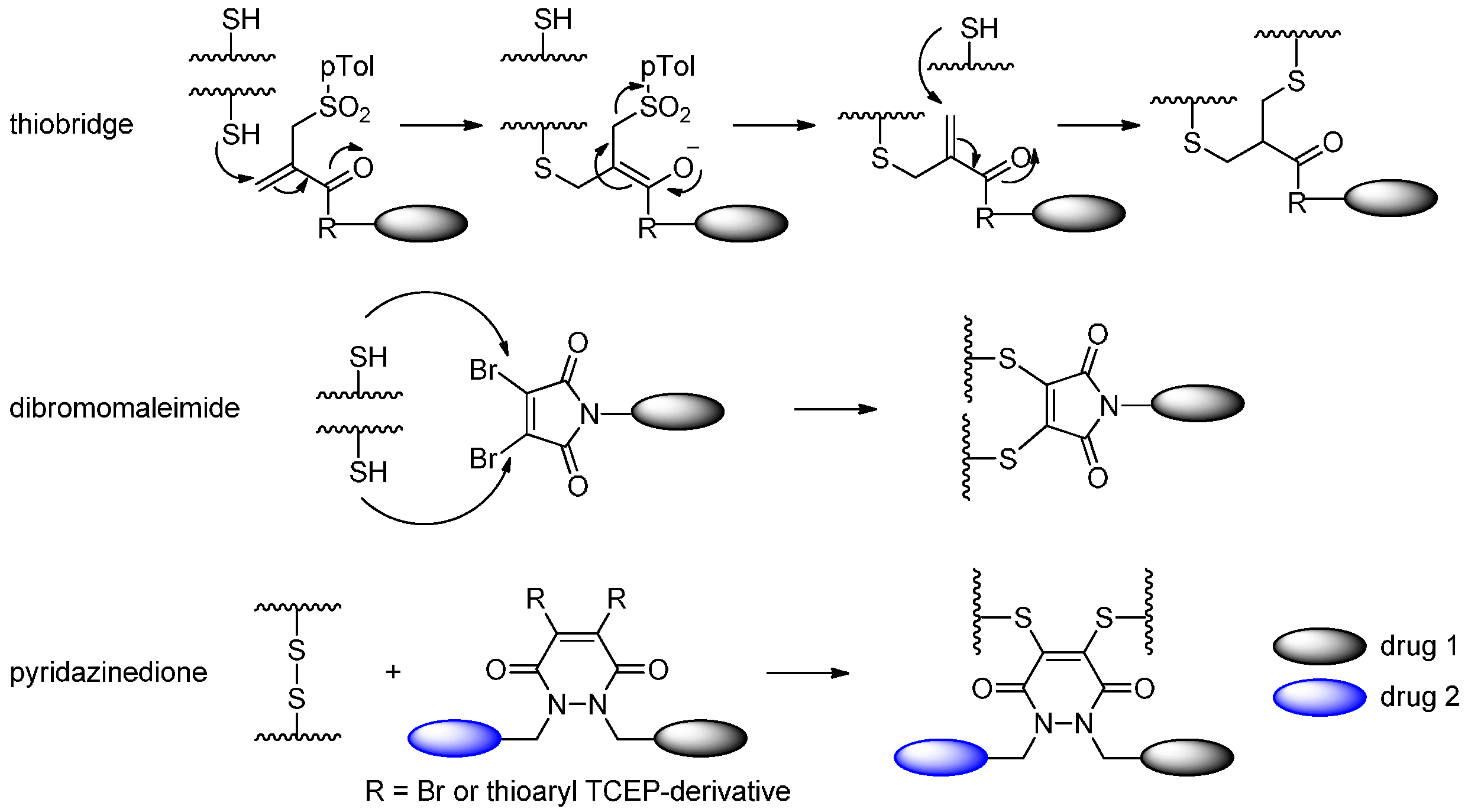
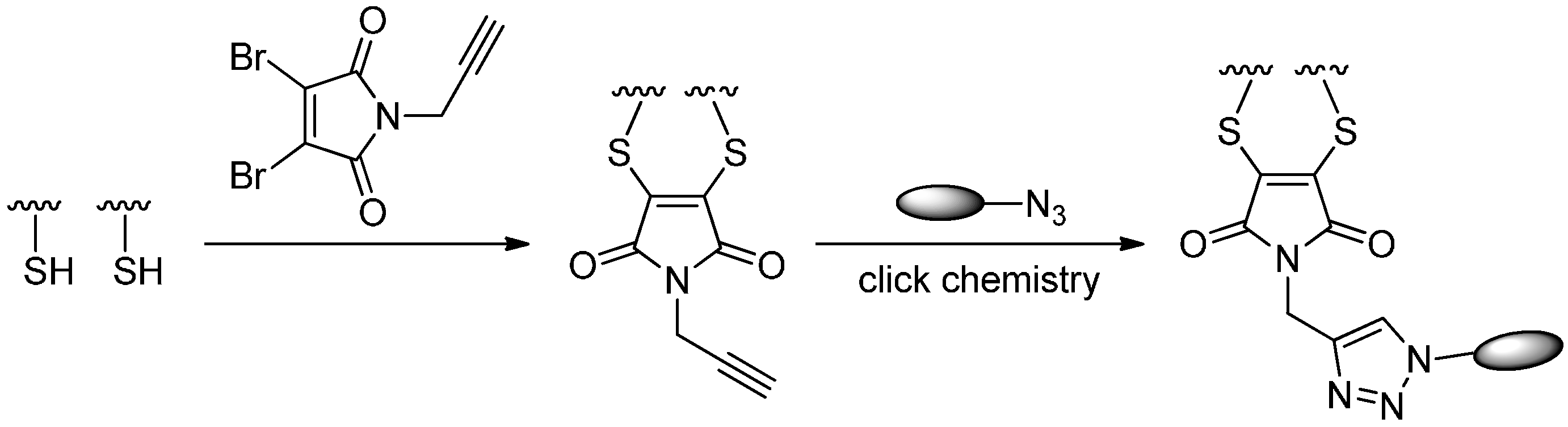
2.1.4. Disulfide Re-Bridging
{kind=link}
{kind=link}
{kind=link}
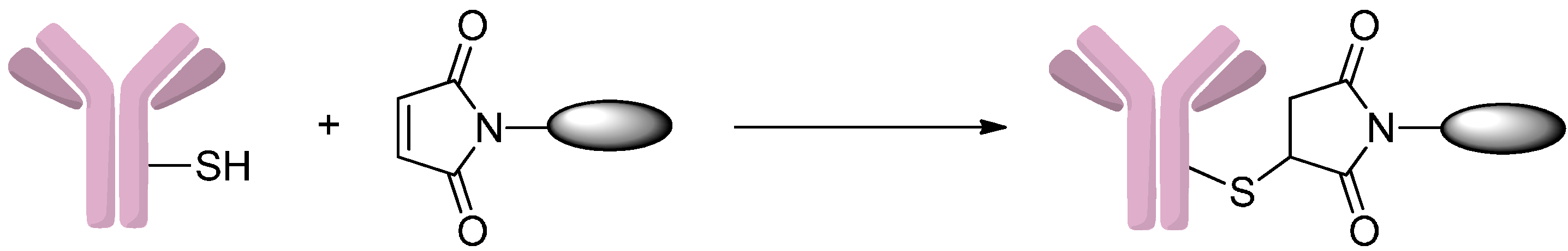{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
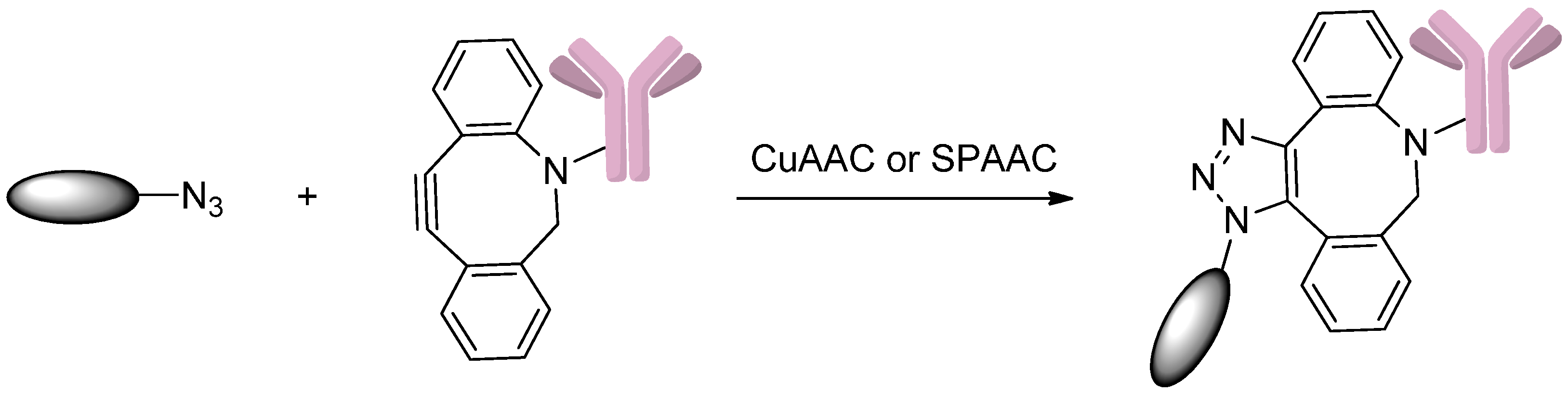{kind=link}
{kind=link}
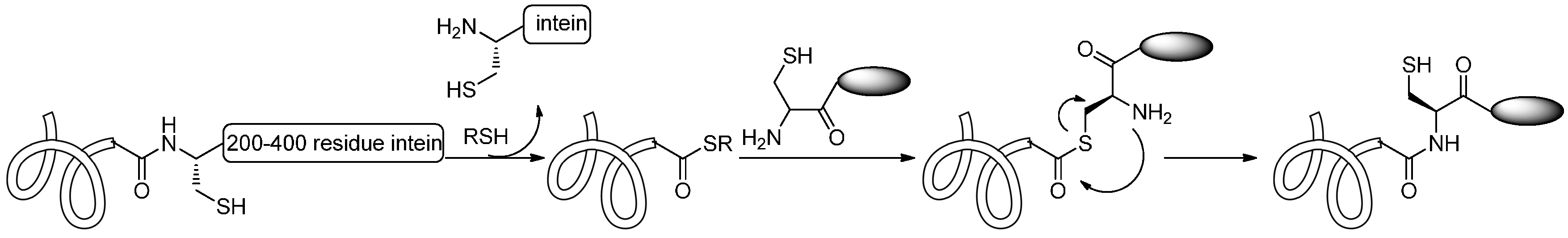
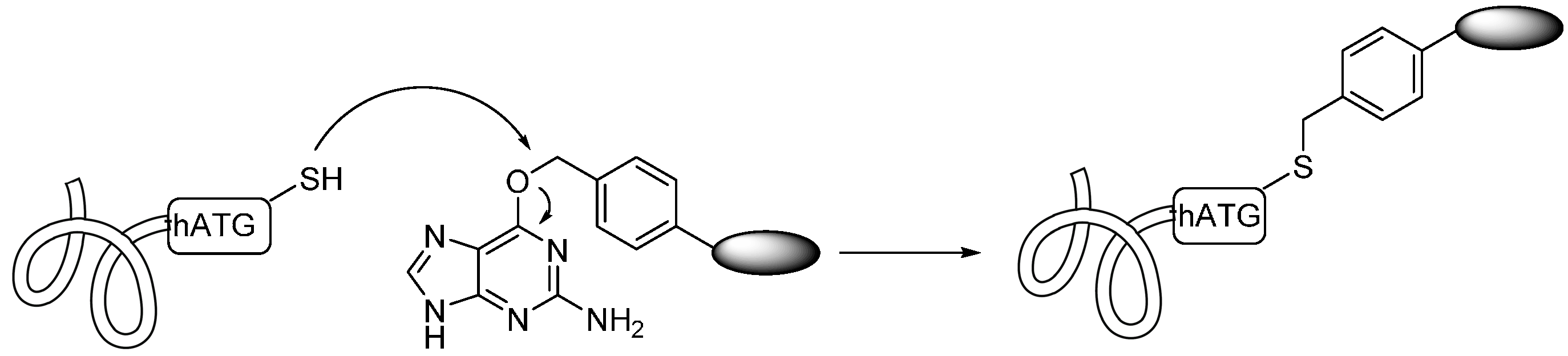
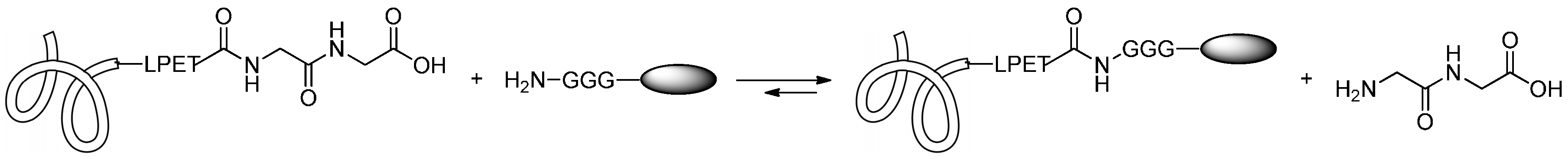
2.1.5. Expressed Protein Ligation (EPL) and Alkylguanine-DNA-Alkyl Transferase (AGT) Reaction
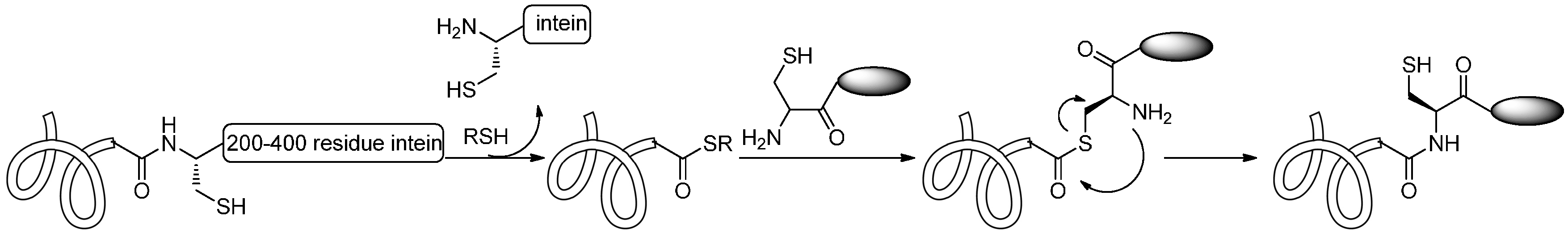
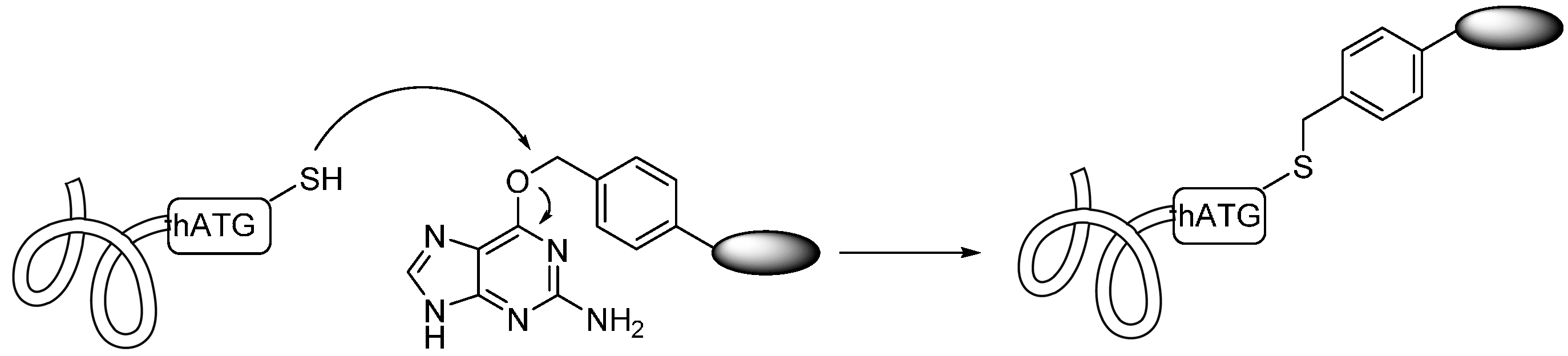
2.2. Conjugation via Amines
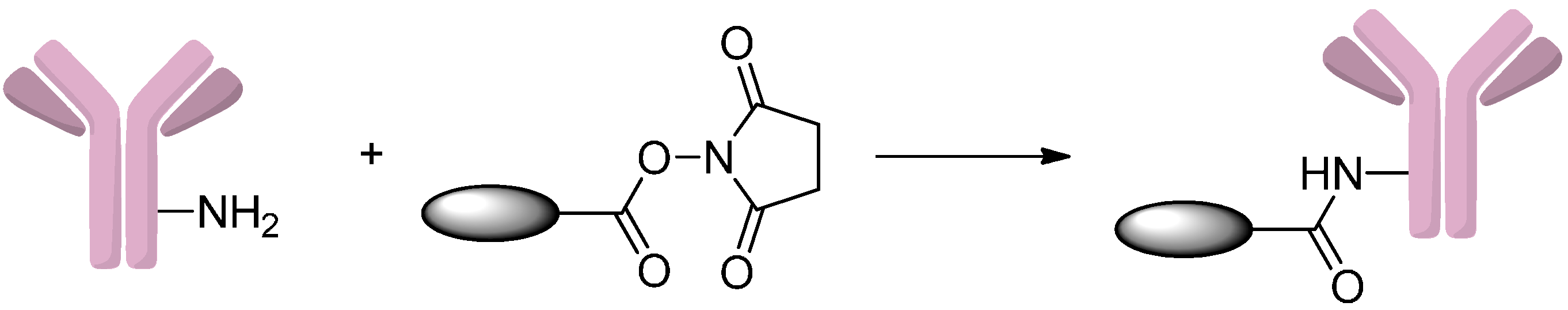
2.2.1. Formation of Amides
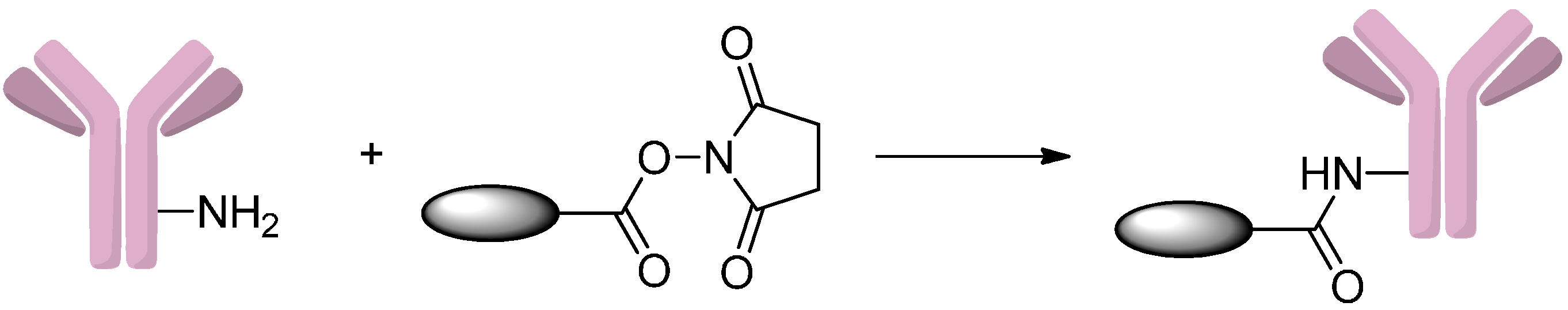
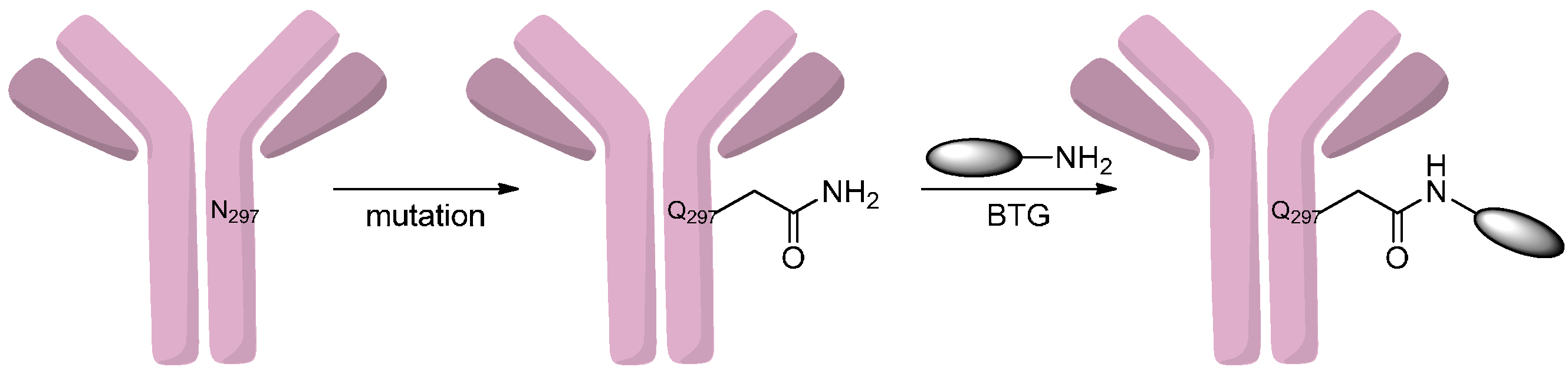
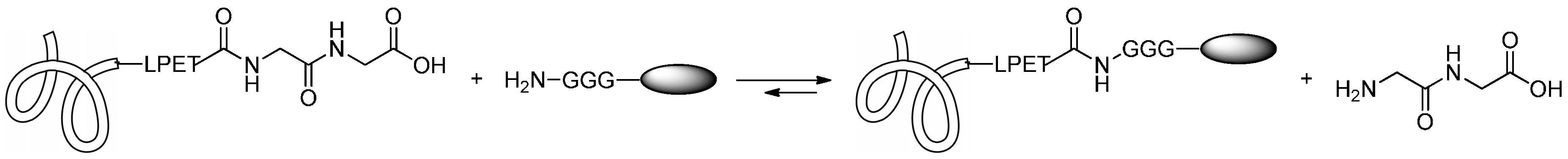
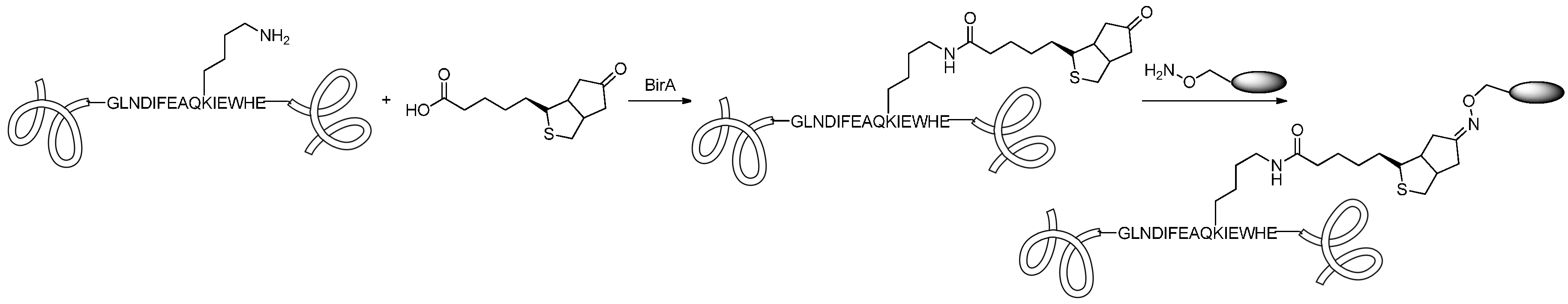
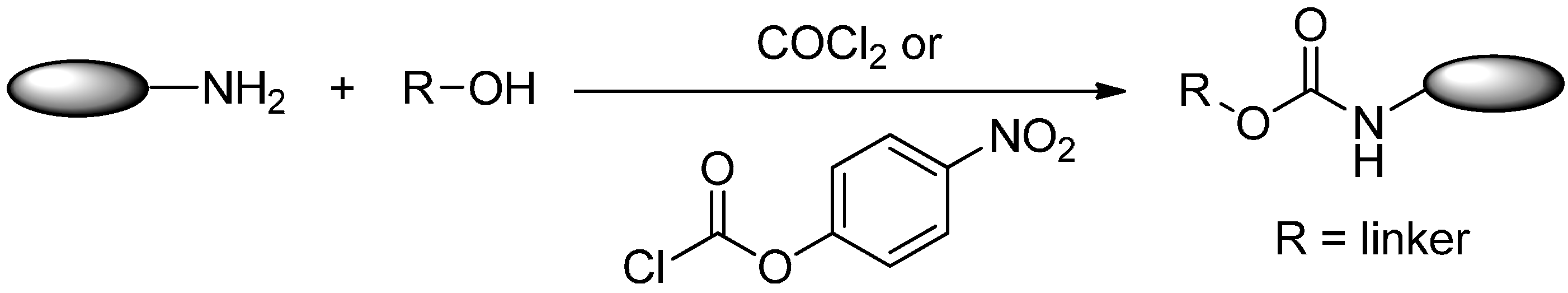
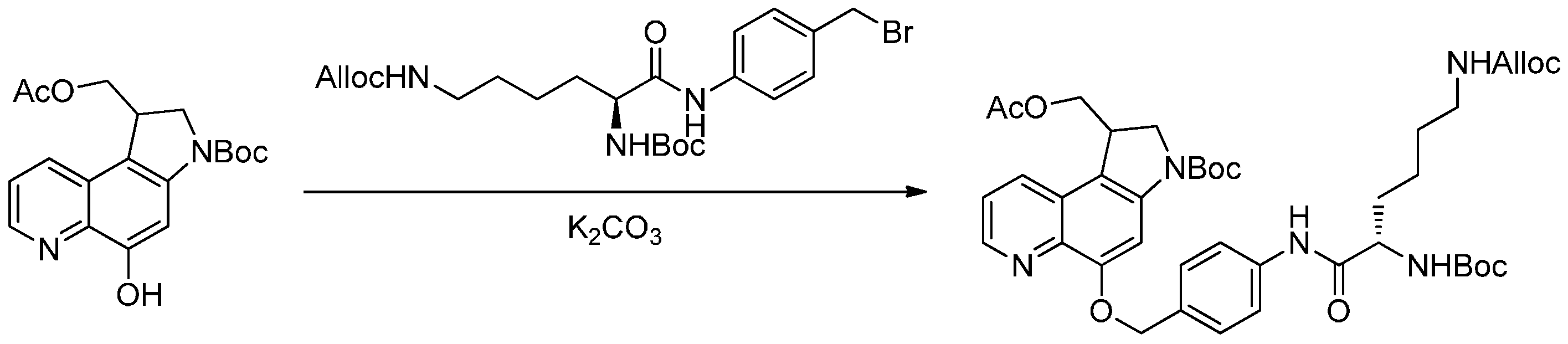
2.2.2. Formation of Carbamates
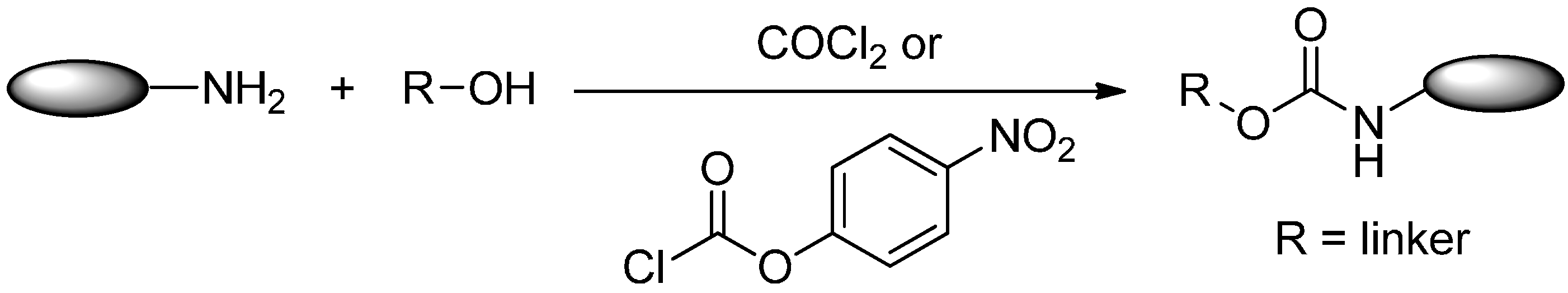
2.3. Conjugation via Alcohols
2.3.1. Formation of Carbonates
2.3.2. Formation of Ether Bonds
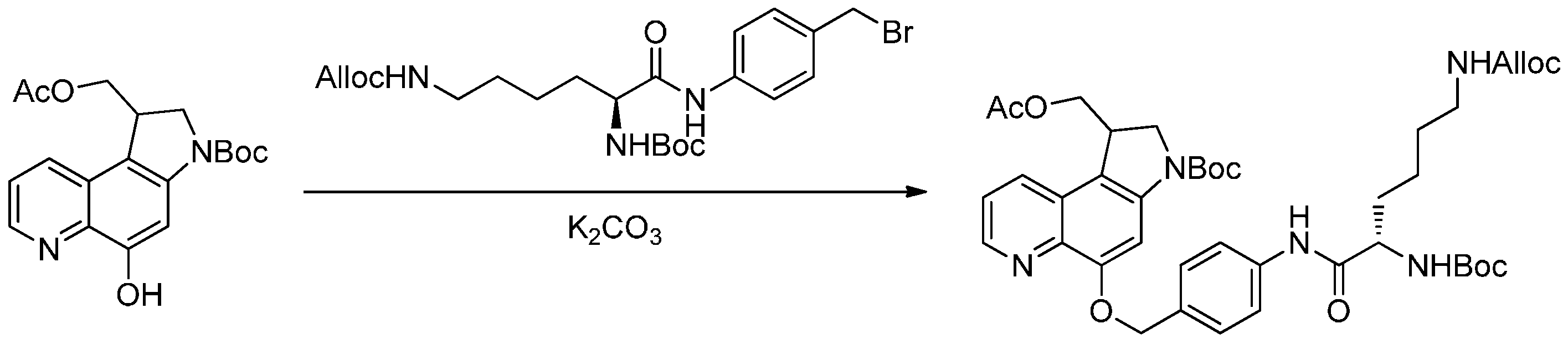
2.3.3. Formation of Ester Bond
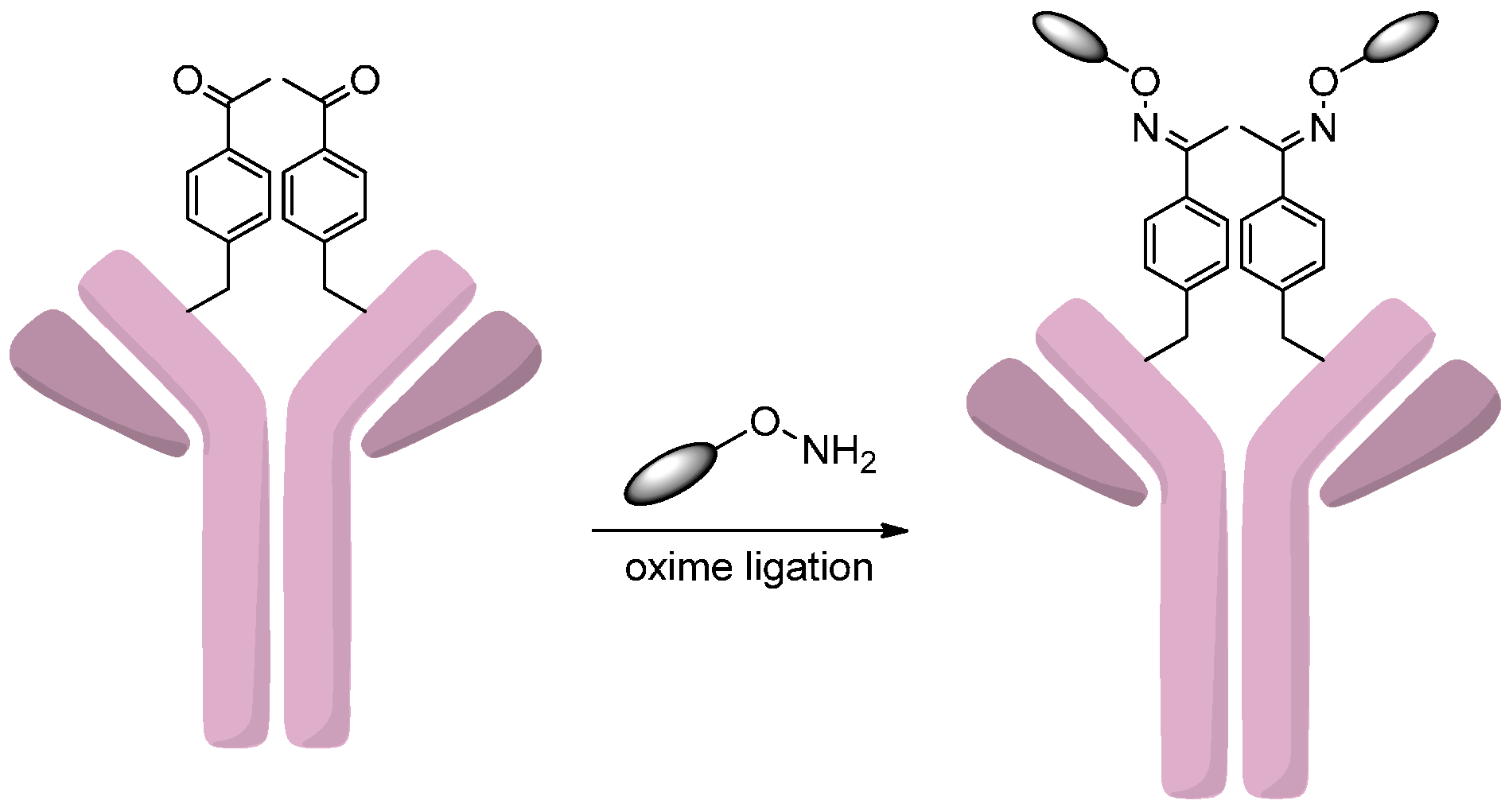
2.4. Conjugation via Aldehydes
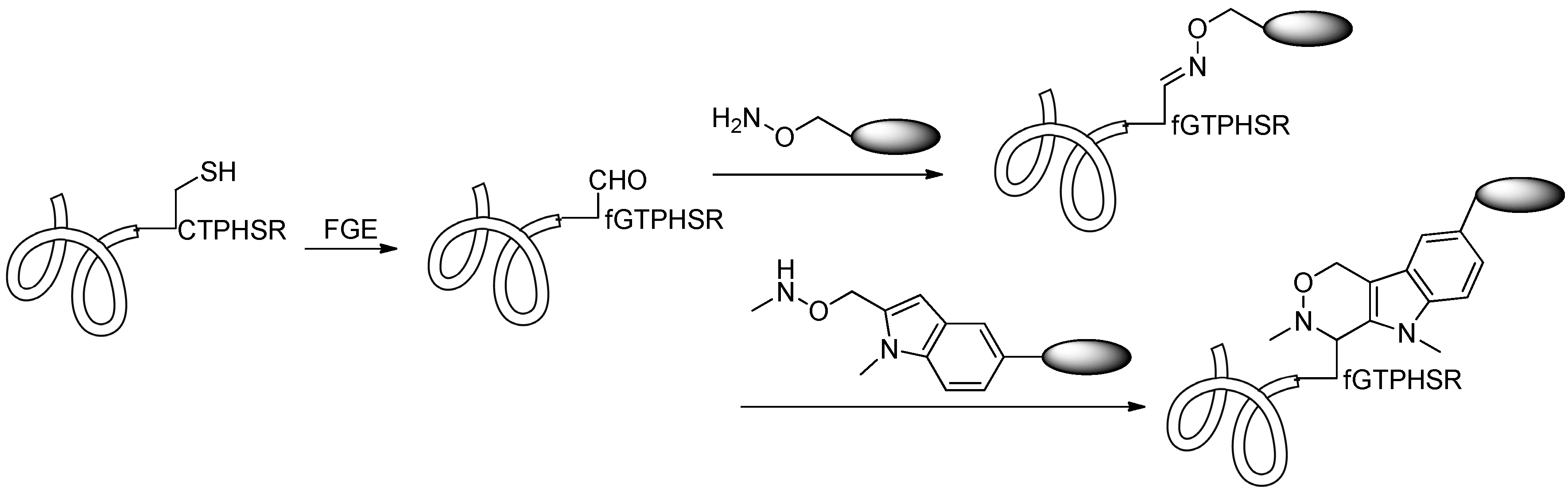
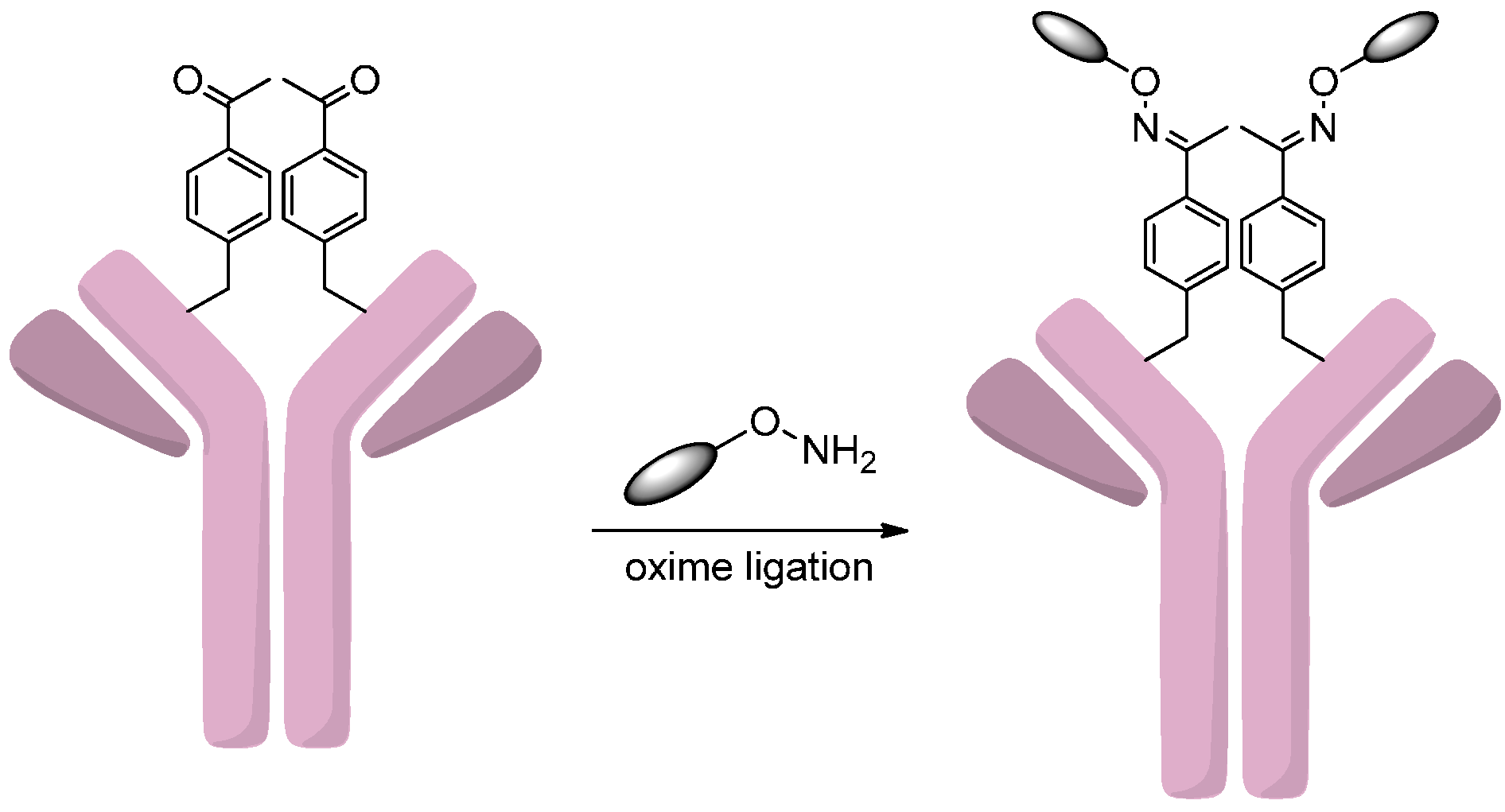
2.4.1. Conjugation via FGE
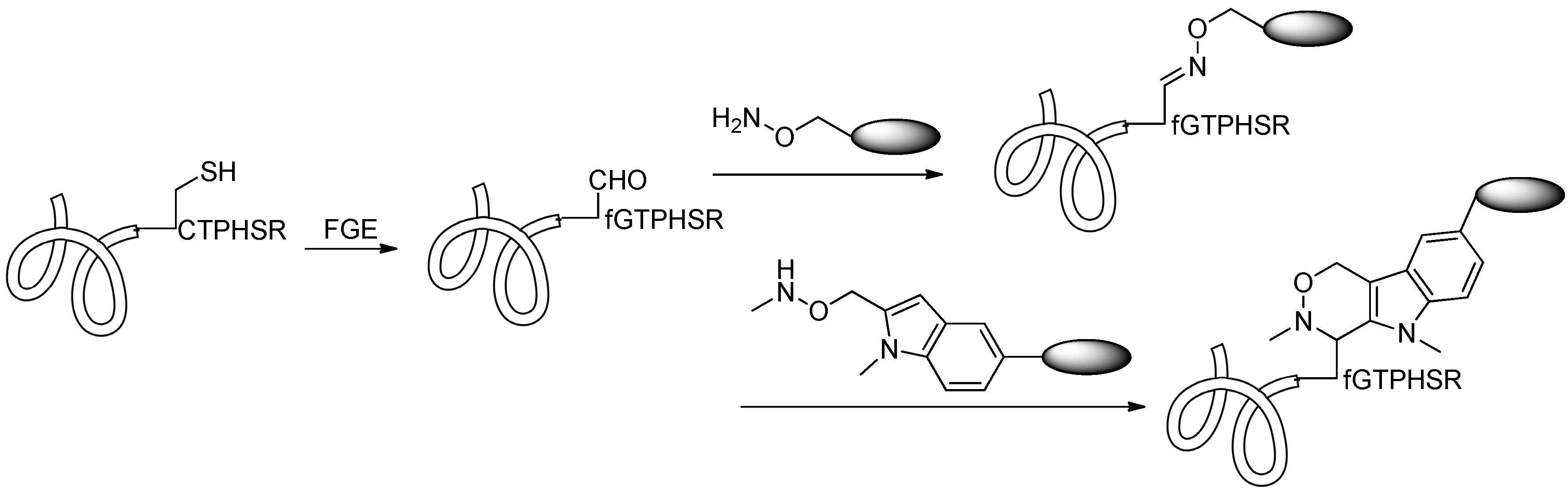
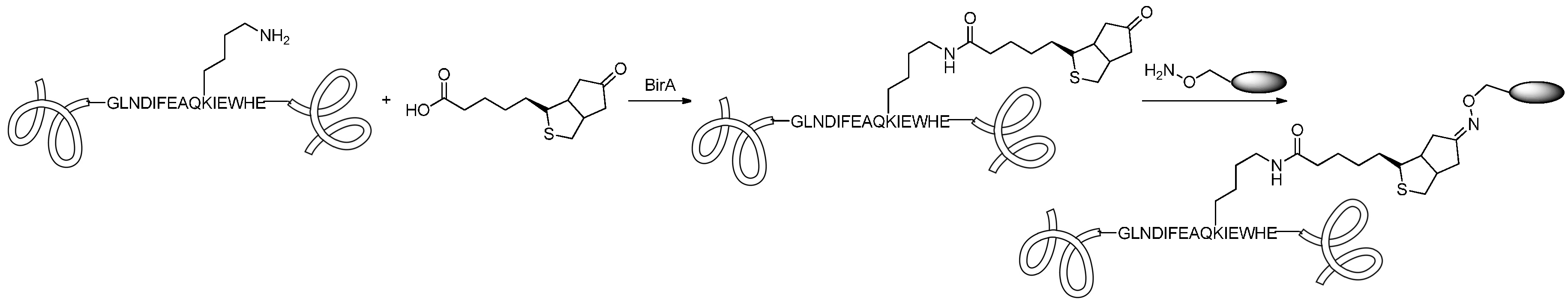
2.4.2. Conjugation via aaRS
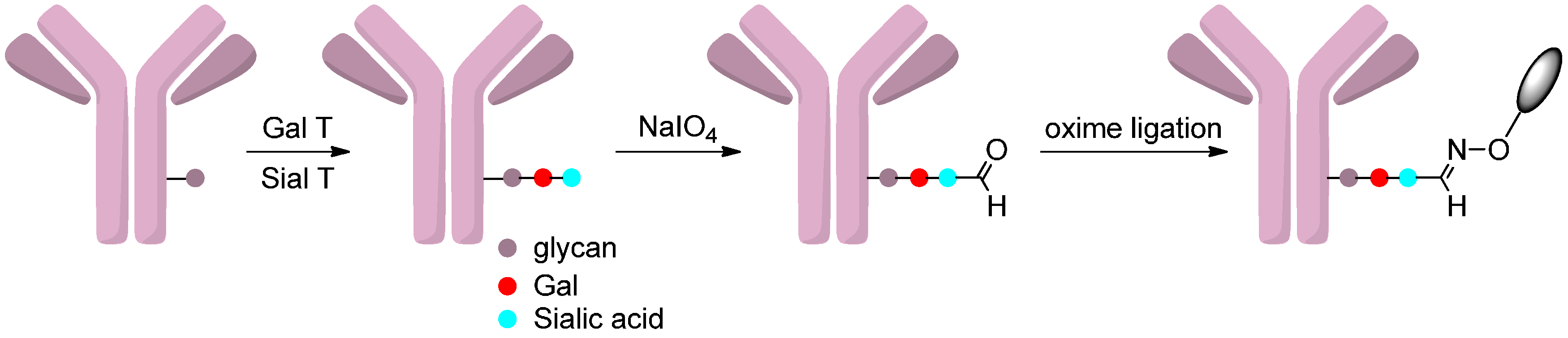
2.4.3. Oxidation of Sialic Acids

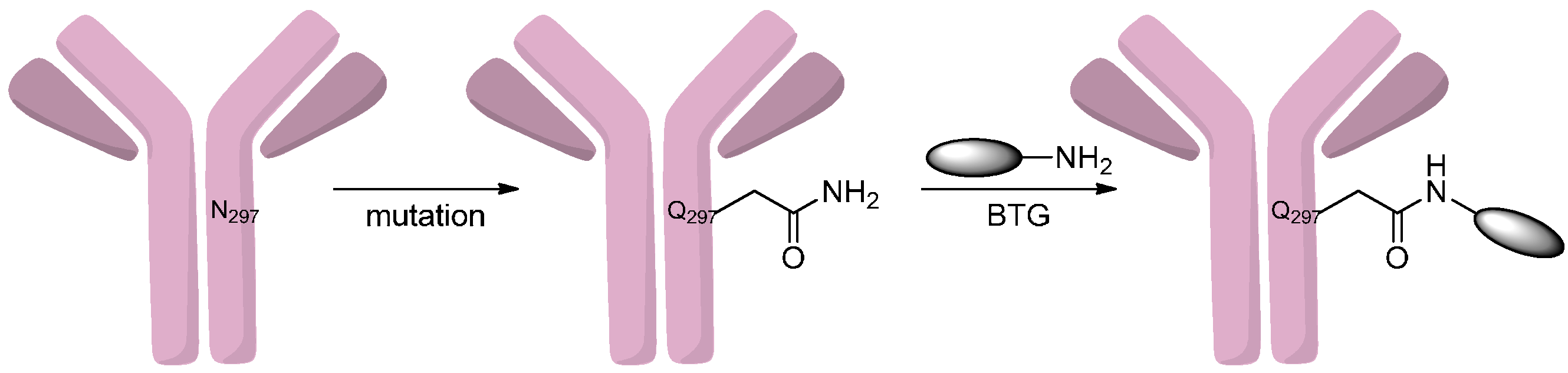
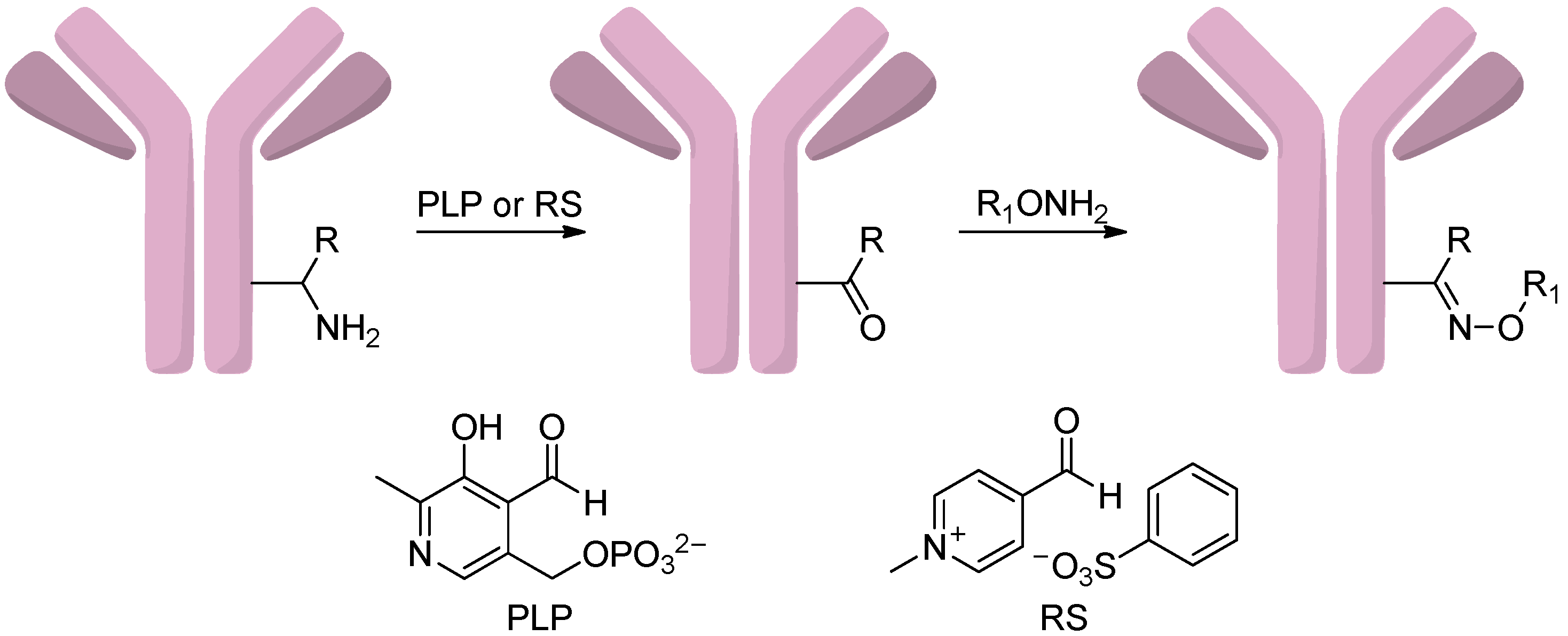
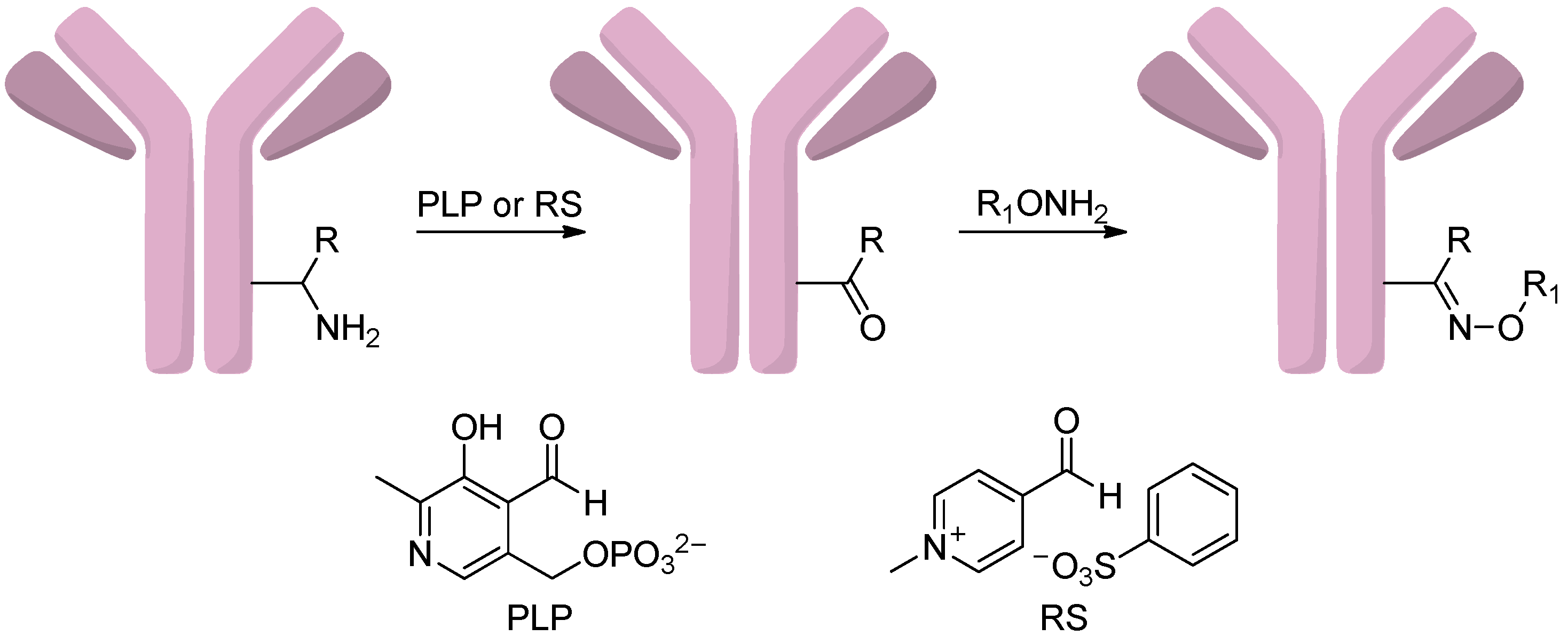
2.4.4. Conjugation via Transamination Reagent
2.5. Conjugation via Azides
2.5.1. Click Reactions with DBCO
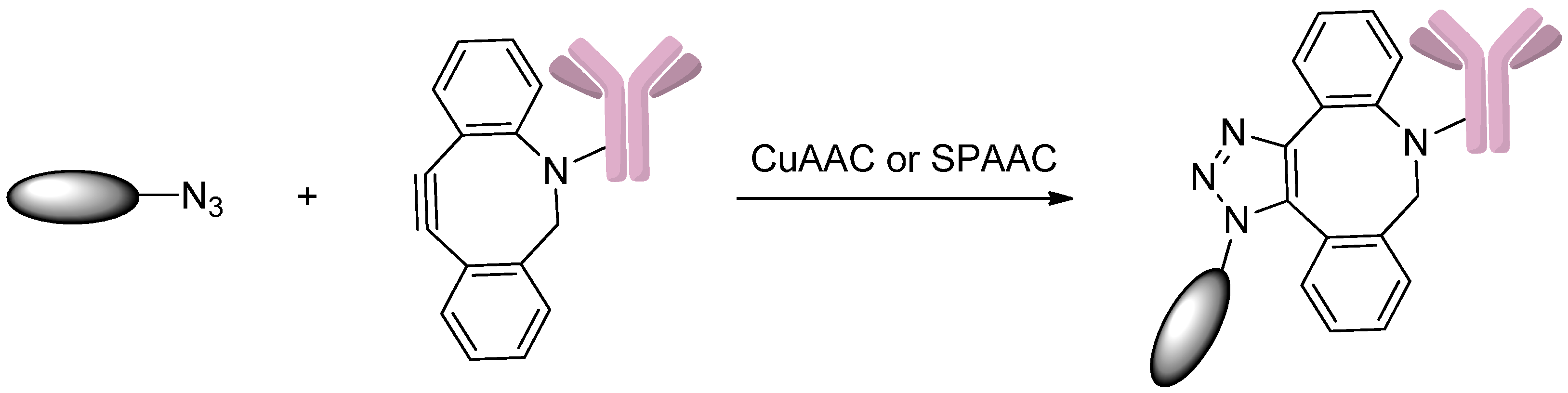
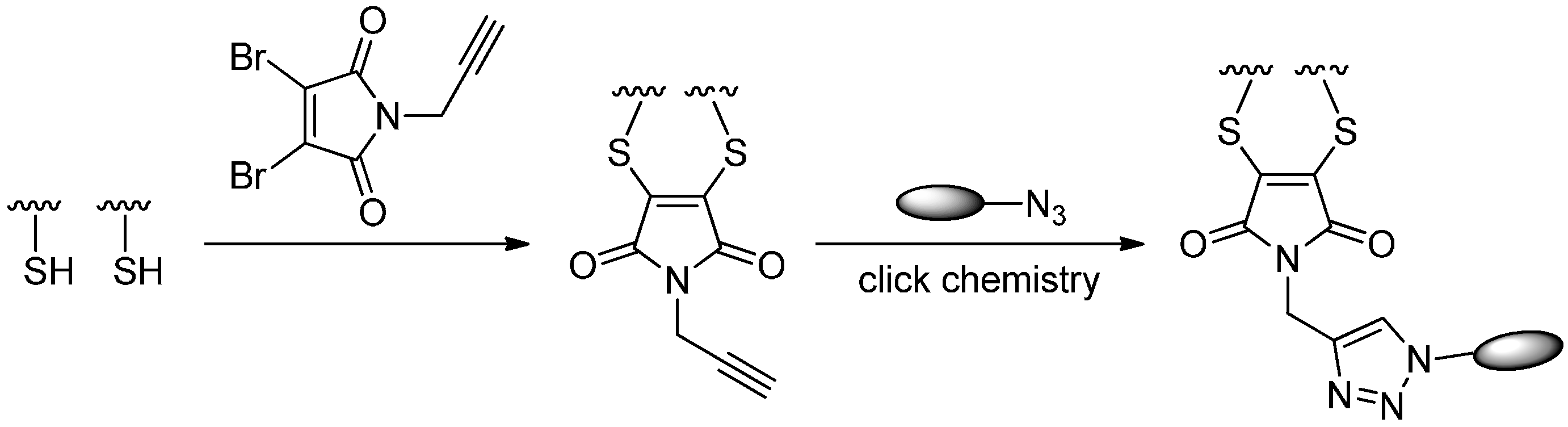
2.5.2. Click Reactions with Terminal Alkynes
3. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lenoir, T. A magic bullet: Research for profit and the growth of knowledge in Germany around 1900. Minerva 1988, 26, 66–88. [Google Scholar] [CrossRef]
- Laurent, D.; Bernhard, S. Antibody-drug conjugates: Linking cytotoxic payloads to monoclonal antibodies. Bioconjug. Chem. 2010, 21, 5–13. [Google Scholar]
- Perez, H.L.; Cardarelli, P.M.; Deshpande, S.; Gangwar, S.; Schroeder, G.M.; Vite, G.D.; Borzilleri, R.M. Antibody-drug conjugates: Current status and future directions. Drug Discov. Today 2014, 19, 869–881. [Google Scholar] [CrossRef] [PubMed]
- Chari, R.V.J.; Miller, M.L.; Widdison, W.C. Antibody-drug conjugates: An emerging concept in cancer therapy. Angew. Chem. Int. Ed. Engl. 2014, 53, 3796–3827. [Google Scholar] [CrossRef] [PubMed]
- Thudium, K.; Bilic, S.; Leipold, D.; Mallet, W.; Kaur, S.; Meibohm, B.; Erickson, H.; Tibbitts, J.; Zhao, H.; Gupta, M. American association of pharmaceutical scientists national biotechnology conference short course: Translational challenges in developing antibody-drug conjugates. MAbs 2013, 5, 5–12. [Google Scholar] [CrossRef]
- Sievers, E.L.; Senter, P.D. Antibody-drug conjugates in cancer therapy. Annu. Rev. Med. 2013, 64, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.; Mao, S.; Strout, P.; Kamal, A. Selecting an optimal antibody for antibody-drug conjugate therapy: Internalization and intracellular localization. Methods Mol. Biol. 2013, 1045, 41–49. [Google Scholar] [PubMed]
- Bander, N.H. Antibody-drug conjugate target selection: Critical factors. Methods Mol. Biol. 2013, 1045, 29–40. [Google Scholar] [PubMed]
- Gerber, H.-P.; Koehn, F.E.; Abraham, R.T. The antibody-drug conjugate: An enabling modality for natural product-based cancer therapeutics. Nat. Prod. Rep. 2013, 30, 625–639. [Google Scholar] [CrossRef] [PubMed]
- Chudasama, V.; Maruani, A.; Caddick, S. Recent advances in the construction of antibody–drug conjugates. Nat. Chem. 2016, 8, 114–119. [Google Scholar] [CrossRef]
- Diamantis, N.; Banerji, U. Antibody-drug conjugates—An emerging class of cancer treatment. Br. J. Cancer 2016. [Google Scholar] [CrossRef] [PubMed]
- McAuley, A.; Jacob, J.; Kolvenbach, C.G.; Westland, K.; Lee, H.J.; Brych, S.R.; Rehder, D.; Kleemann, G.R.; Brems, D.N.; Matsumura, M. Contributions of a disulfide bond to the structure, stability, and dimerization of human IgG1 antibody CH3 domain. Protein Sci. 2008, 17, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.M.C.; Beam, K.S.; Cerveny, C.G.; Hamblett, K.J.; Blackmore, R.S.; Torgov, M.Y.; Handley, F.G.M.; Ihle, N.C.; Senter, P.D.; Alley, S.C. Reduction–alkylation strategies for the modification of specific monoclonal antibody disulfides. Bioconjug. Chem. 2005, 16, 1282–1290. [Google Scholar] [CrossRef] [PubMed]
- Willner, D.; Trail, P.A.; Hofstead, S.J.; King, H.D.; Lasch, S.J.; Braslawsky, G.R.; Greenfield, R.S.; Kaneko, T.; Firestone, R.A. (6-Maleimidocaproyl) hydrazone of doxorubicin-a new derivative for the preparation of immunoconjugates of doxorubicin. Bioconjug. Chem. 1993, 4, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Doronina, S.O.; Toki, B.E.; Torgov, M.Y.; Mendelsohn, B.A.; Cerveny, C.G.; Chace, D.F.; DeBlanc, R.L.; Gearing, R.P.; Bovee, T.D.; Siegall, C.B.; et al. Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nat. Biotechnol. 2003, 21, 778–784. [Google Scholar] [CrossRef]
- Francisco, J.A.; Cerveny, C.G.; Meyer, D.L.; Mixan, B.J.; Klussman, K.; Chace, D.F.; Rejniak, S.X.; Gordon, K.A.; DeBlanc, R.; Toki, B.E.; et al. cAC10-vcMMAE, an anti-CD30-monomethyl auristatin E conjugate with potent and selective antitumor activity. Blood 2003, 102, 1458–1465. [Google Scholar] [CrossRef] [PubMed]
- Hamblett, K.J.; Senter, P.D.; Chace, D.F.; Sun, M.M.C.; Lenox, J.; Cerveny, C.G.; Kissler, K.M.; Bernhardt, S.X.; Kopcha, A.K.; Zabinski, R.F.; et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin. Cancer Res. 2004, 10, 7063–7070. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, C.F.; Turcott, E.; Westendorf, L.; Webster, J.B.; Alley, S.C.; Kim, K.; Andreyka, J.; Stone, I.; Hamblett, K.J.; Francisco, J.A.; et al. Engineered antibody-drug conjugates with defined sites and stoichiometries of drug attachment. Prot. Eng. Des. Sel. 2006, 19, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Andersen, D.C.; Reilly, D.E. Production technologies for monoclonal antibodies and their fragments. Curr. Opin. Biotechnol. 2004, 15, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, D.D.; Tankersly, D.L.; Lundblad, J.L. A new preparation of modified immune serum globulin (human) suitable for intravenous administration. I. Standardization of the reduction and alkylation reaction. Vox. Sang. 1981, 40, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Chumsae, C.; Gaza-Bulseco, G.; Hurkmans, K.; Radziejewski, C.H. Ranking the susceptibility of disulfide bonds in human IgG1 antibodies by reduction, differential alkylation, and LC-MS analysis. Anal. Chem. 2010, 82, 5219–5226. [Google Scholar] [CrossRef]
- Nwe, K.; Milenic, D.E.; Ray, G.L.; Kim, Y.-S.; Brechbiel, M.W. Preparation of cystamine core dendrimer and antibody−dendrimer conjugates for MRI angiography. Mol. Pharm. 2012, 9, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Nanaware-Kharade, N.; Gonzalez, G.A.; Lay, J.O.; Hendrickson, H.P.; Peterson, E.C. Therapeutic anti-methamphetamine antibody fragment-nanoparticle conjugates: Synthesis and in vitro characterization. Bioconjug. Chem. 2012, 23, 1864–1872. [Google Scholar] [CrossRef] [PubMed]
- Hermanson, G.T. Bioconjugate Techniques, 2nd ed.; Academic Press: New York, NY, USA, 2008; pp. 1041–1132. [Google Scholar]
- Gauthier, M.A.; Klok, H.-A. Peptide/protein-polymer conjugates: Synthetic strategies and design concepts. Chem. Commun. 2008, 21, 2591–2611. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.-Q.; Xu, K.; Liu, L.; Raab, H.; Bhakta, S.; Kenrick, M.; Parsons-Reponte, K.L.; Tien, J.; Yu, S.-F.; Mai, E.; et al. Conjugation site modulates the in vivo stability and therapeutic activity of antibody-drug conjugates. Nat. Biotechnol. 2012, 30, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Nunes, J.P.M.; Morais, M.; Vassileva, V.; Robinson, E.; Rajkumar, V.S.; Smith, M.E.B.; Pedley, R.B.; Caddick, S.; Baker, J.R.; Chudasama, V. Functional native disulfide bridging enables delivery of a potent, stable and targeted antibody−drug conjugate (ADC). Chem. Commun. 2015, 51, 10624–10627. [Google Scholar] [CrossRef] [PubMed]
- Lyon, R.P.; Setter, J.R.; Bovee, T.D.; Doronina, S.O.; Hunter, J.H.; Anderson, M.E.; Balasubramanian, C.L.; Duniho, S.M.; Leiske, C.I.; Li, F.; et al. Self-hydrolyzing maleimides improve the stability and pharmacological properties of antibody-drug conjugates. Nat. Biotechnol. 2014, 32, 1059–1062. [Google Scholar] [CrossRef] [PubMed]
- Tumey, L.N.; Charati, M.; He, T.; Sousa, E.; Ma, D.; Han, X.; Clark, T.; Casavant, J.; Loganzo, F.; Barletta, F.; et al. Mild method for succinimide hydrolysis on ADCs: Impact on ADC potency, stability, exposure, and efficacy. Bioconjug. Chem. 2014, 25, 1871–1880. [Google Scholar] [CrossRef] [PubMed]
- Ojima, I.; Geng, X.; Wu, X.; Qu, C.; Borella, C.P.; Xie, H.; Wilhelm, S.D.; Leece, B.A.; Bartle, L.M.; Goldmacher, V.S.; et al. Tumor-specific novel taxoid-monoclonal antibody conjugates. J. Med. Chem. 2002, 45, 5620–5623. [Google Scholar] [CrossRef] [PubMed]
- Widdison, W.C.; Wilhelm, S.D.; Cavanagh, E.E.; Whiteman, K.R.; Leece, B.A.; Kovtun, Y.; Goldmacher, V.S.; Xie, H.; Steeves, R.M.; Lutz, R.J.; et al. Semisynthetic maytansine analogues for the targeted treatment of cancer. J. Med. Chem. 2006, 49, 4392–4408. [Google Scholar] [CrossRef] [PubMed]
- Kolodych, S.; Koniev, O.; Baatarkhuu, Z.; Bonnefoy, J.-Y.; Debaene, F.; Cianférani, S.; Dorsselaer, A.V.; Wagner, A. CBTF: New amine-to-thiol coupling reagent for preparation of antibody conjugates with increased plasma stability. Bioconjug. Chem. 2015, 26, 197–200. [Google Scholar] [CrossRef] [PubMed]
- Cal, P.M.; Bernardes, G.J.; Gois, P.M. Cysteine-selective reactions for antibody conjugation. Angew. Chem. Int. Ed. Engl. 2014, 53, 10585–10587. [Google Scholar] [CrossRef] [PubMed]
- Kline, T.; Steiner, A.R.; Penta, K.; Sato, A.K.; Hallam, T.J.; Yin, G. Methods to make homogenous antibody drug conjugates. Pharm. Res. 2015, 32, 3480–3493. [Google Scholar] [CrossRef] [PubMed]
- Badescu, G.; Bryant, P.; Bird, M.; Henseleit, K.; Swierkosz, J.; Parekh, V.; Tommasi, R.; Pawlisz, E.; Jurlewicz, K.; Farys, M.; et al. Bridging disulfides for stable and defined antibody drug conjugates. Bioconjug. Chem. 2014, 25, 1124–1136. [Google Scholar] [CrossRef] [PubMed]
- Bryant, P.; Pabst, M.; Badescu, G.; Bird, M.; McDowell, W.; Jamieson, E.; Swierkosz, J.; Jurlewicz, K.; Tommasi, R.; Henseleit, K.; et al. In vitro and in vivo evaluation of cysteine rebridged trastuzumab-MMAE antibody drug conjugates with defined drug-to-antibody ratios. Mol. Pharm. 2015, 12, 1872–1879. [Google Scholar] [CrossRef] [PubMed]
- Behrens, C.R.; Ha, E.H.; Chinn, L.L.; Bowers, S.; Probst, G.; Fitch-Bruhns, M.; Monteon, J.; Valdiosera, A.; Bermudez, A.; Liao-Chan, S.; et al. Antibody−drug conjugates (ADCs) derived from interchain cysteine cross-linking demonstrate improved homogeneity and other pharmacological properties over conventional heterogeneous ADCs. Mol. Pharm. 2015, 12, 3986–3998. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.T.W.; Maruani, A.; Baker, J.R.; Caddick, S.; Chudasama, V. Next-generation disulfide stapling: Reduction and functional re-bridging all in one. Chem. Sci. 2016, 7, 799–802. [Google Scholar] [CrossRef]
- Maruani, A.; Savoie, H.; Bryden, F.; Caddick, S.; Boyle, R.; Chudasama, V. Site-selective multi-porphyrin attachment enables the formation of a next-generation antibody-based photodynamic therapeutic. Chem. Commun. 2015, 51, 15304–15307. [Google Scholar] [CrossRef] [PubMed]
- Maruani, A.; Smith, M.E.B.; Miranda, E.; Chester, K.A.; Chudasama, V.; Caddick, S. A plug-and-play approach to antibody-based therapeutics via a chemoselective dual click strategy. Nat. Commun. 2015, 6, 6645. [Google Scholar] [CrossRef] [PubMed]
- Juillerat, A.; Gronemeyer, T.; Keppler, A.; Gendreizig, S.; Pick, H.; Vogel, H.; Johnsson, K. Directed evolution of O6-alkylguanine-DNA alkyltransferase for efficient labeling of fusion proteins with small molecules in vivo. Chem. Biol. 2003, 10, 313–317. [Google Scholar] [CrossRef]
- Cohen, R.; Vugts, D.J.; Visser, G.W.M.; Walsum, M.S.-V.; Bolijn, M.; Spiga, M.; Lazzari, P.; Shankar, S.; Sani, M.; Zanda, M.; et al. Development of novel ADCs: Conjugation of tubulysin analogues to trastuzumab monitored by dual radiolabeling. Cancer Res. 2014, 74, 5700–5710. [Google Scholar] [CrossRef] [PubMed]
- Mueller, B.M.; Wrasidlo, W.A.; Reisfeld, R.A. Determination of the number of e-amino groups available for conjugation of effect or molecules to monoclonal antibodies. Hybridoma 1988, 7, 453–456. [Google Scholar] [CrossRef] [PubMed]
- Wakankar, A.A.; Feeney, M.B.; Rivera, J.; Chen, Y.; Kim, M.; Sharma, V.K.; Wang, Y.J. Physicochemical stability of the antibody-drug conjugate Trastuzumab-DM1: Changes due to modification and conjugation processes. Bioconjug. Chem. 2010, 21, 1588–1595. [Google Scholar] [CrossRef] [PubMed]
- Siegel, M.M.; Tabei, K.; Kunz, A.; Hollander, I.J.; Hamann, R.R.; Bell, D.H.; Berkenkamp, S.; Hillenkamp, F. Calicheamicin derivatives conjugated to monoclonal antibodies: Determination of loading values and distributions by infrared and UV matrix-assisted laser desorption/ionization mass spectrometry and electrospray ionization mass spectrometry. Anal. Chem. 1997, 69, 2716–2726. [Google Scholar] [CrossRef]
- Adamczyk, M.; Gebler, J.; Shreder, K.; Wu, J. Region-selective labeling of antibodies as determined by electrospray ionization-mass spectrometry (ESI-MS). Bioconjug. Chem. 2000, 11, 557–563. [Google Scholar] [CrossRef]
- Hamann, P.R.; Hinman, L.M.; Hollander, I.; Beyer, C.F.; Lindh, D.; Holcomb, R.; Hallett, W.; Tsou, H.R.; Upeslacis, J.; Shochat, D.; et al. Gemtuzumab ozogamicin, a potent and selective anti-CD33 antibody-calicheamicin conjugate for treatment of acute myeloid leukemia. Bioconjug. Chem. 2002, 13, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Zhu, Z.; Chen, W.; Prabakaran, P.; Lin, K.; Dimitrov, D. Conjugates of small molecule drugs with antibodies and other proteins. Biomedicines 2014, 2, 1–13. [Google Scholar] [CrossRef]
- Hong, L.P.T.; Scoble, J.A.; Doughty, L.; Coia, G.; Williams, C.C. Cancer-targeting antibody-drug conjugates: Site-specific conjugation of doxorubicin to anti-EGFR 528 Fab9 through a polyethylene glycol linker. Aust. J. Chem. 2011, 64, 779–789. [Google Scholar] [CrossRef]
- Jeger, S.; Zimmermann, K.; Blanc, A.; Grünberg, J.; Honer, M.; Hunziker, P.; Struthers, H.; Schibli, R. Site-specific and stoichiometric modification of antibodies by bacterial transglutaminase. Angew. Chem. Int. Ed. Engl. 2010, 49, 9995–9997. [Google Scholar] [CrossRef] [PubMed]
- Mazmanian, S.K.; Liu, G.; Ton-That, H.; Schneewind, O. Staphylococcus aureus sortase, an enzyme that anchors surface proteins to the cell wall. Science 1999, 285, 760–763. [Google Scholar] [CrossRef] [PubMed]
- Tsukiji, S.; Nagamune, T. Sortase-mediated ligation: A gift from Gram-positive bacteria to protein engineering. Chem. Biochem. Eur. J. Chem. Biol. 2009, 10, 787–798. [Google Scholar] [CrossRef] [PubMed]
- Madej, M.P.; Coia, G.; Williams, C.C.; Caine, J.M.; Pearce, L.A.; Attwood, R.; Bartone, N.A.; Dolezal, O.; Nisbet, R.M.; Nuttall, S.D.; et al. Engineering of an anti-epidermal growth factor receptor antibody to single chain format and labeling by Sortase A-mediated protein ligation. Biotechnol. Bioeng. 2012, 109, 1461–1470. [Google Scholar] [CrossRef] [PubMed]
- Ta, H.T.; Peter, K.; Hagemeyer, C.E. Enzymatic antibody tagging: Toward a universal biocompatible targeting tool. Trends Cardiovasc. Med. 2012, 22, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Williamson, D.J.; Fascione, M.A.; Webb, M.E.; Turnbull, W.B. Efficient N-terminal labeling of proteins by use of sortase. Angew. Chem. 2012, 124, 9511–9514. [Google Scholar] [CrossRef]
- Chen, I.; Howarth, M.; Lin, W.; Ting, A.Y. Site-specific labeling of cell surface proteins with biophysical probes using biotin ligase. Nat. Methods 2005, 2, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, S.C.; Torgov, M.Y.; Andreyka, J.B.; Boddington, L.; Cerveny, C.G.; Denny, W.A.; Gordon, K.A.; Gustin, D.; Haugen, J.; Toni Kline, T.; et al. Design, synthesis, and in vitro evaluation of dipeptide-based antibody minor groove binder conjugates. J. Med. Chem. 2005, 48, 1344–1358. [Google Scholar] [CrossRef] [PubMed]
- Burke, P.J.; Senter, P.D.; Meyer, D.W.; Miyamoto, J.B.; Anderson, M.; Toki, B.E.; Manikumar, G.; Wani, M.C.; Kroll, D.J.; Jeffrey, S.C. Design, synthesis, and biological evaluation of antibody-drug conjugates comprised of potent camptothecin analogues. Bioconjug. Chem. 2009, 20, 1242–1250. [Google Scholar] [CrossRef] [PubMed]
- Dubowchik, G.M.; Firestone, R.A.; Padilla, L.; Willner, D.; Hofstead, S.J.; Mosure, K.; Knipe, J.O.; Lasch, S.J.; Trail, P.A. Cathepsin B-labile dipeptide linkers for lysosomal release of doxorubicin from internalizing immunoconjugates: Model studies of enzymatic drug release and antigen-specific in vitro anticancer activity. Bioconjug. Chem. 2002, 13, 855–869. [Google Scholar] [CrossRef] [PubMed]
- King, H.D.; Dubowchik, G.M.; Mastalerz, H.; Willner, D.; Hofstead, S.J.; Firestone, R.A.; Lasch, S.J.; Trail, P.A. Monoclonal antibody conjugates of doxorubicin prepared with branched peptide linkers: Inhibition of aggregation by methoxytriethyleneglycol chains. J. Med. Chem. 2002, 45, 4336–4343. [Google Scholar] [CrossRef] [PubMed]
- Walker, M.A.; Dubowchik, G.M.; Hofstead, S.J.; Trail, P.A.; Firestone, R.A. Synthesis of an immunoconjugate of camptothecin. Bioorg. Med. Chem. Lett. 2002, 12, 217–219. [Google Scholar] [CrossRef]
- Elgersma, R.C.; Coumans, R.G.E.; Huijbregts, T.; Menge, W.M.P.B.; Joosten, J.A.F.; Spijker, H.J.; de Groot, F.M.H.; van der Lee, M.M.C.; Ubink, R.; van den Dobbelsteen, D.J. Design, synthesis, and evaluation of linker-duocarmycin payloads: Toward selection of HER2-targeting antibody−drug conjugate SYD985. Mol. Pharm. 2015, 12, 1813–1835. [Google Scholar] [CrossRef]
- Moon, S.-J.; Govindan, S.V.; Cardillo, T.M.; D’Souza, C.A.; Hansen, H.J.; Goldenberg, D.M. Antibody conjugates of 7-ethyl-10-hydroxycamptothecin (SN-38) for targeted cancer chemotherapy. J. Med. Chem. 2008, 51, 6916–6926. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Rubio, B.; Sapra, P.; Wu, D.; Reddy, P.; Sai, P.; Martinez, A.; Gao, Y.; Lozanguiez, Y.; Longley, C.; et al. Novel prodrugs of SN-38 using multiarm poly(ethylene glycol) linkers. Bioconjug. Chem. 2008, 19, 849–859. [Google Scholar] [CrossRef] [PubMed]
- Toki, B.E.; Cerveny, C.G.; Wahl, A.F.; Senter, P.D. Protease mediated fragmentation of p-amidobenzyl ethers: A new strategy for the activation of anticancer prodrugs. J. Org. Chem. 2002, 67, 1866–1872. [Google Scholar] [CrossRef]
- Denny, W.A. DNA minor groove alkylating agents. Curr. Med. Chem. 2001, 8, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Quiles, S.; Raisch, K.P.; Sanford, L.L.; Bonner, J.A.; Safavy, A. Synthesis and preliminary biological evaluation of high-drug-load paclitaxel-antibody conjugates for tumor-targeted chemotherapy. J. Med. Chem. 2010, 53, 586–594. [Google Scholar] [CrossRef] [PubMed]
- Drake, P.M.; Albers, A.E.; Baker, J.; Banas, S.; Barfield, R.M.; Bhat, A.S.; de Hart, G.W.; Garofalo, A.W.; Holder, P.; Jones, L.C.; et al. Aldehyde tag coupled with HIPS chemistry enables the production of ADCs conjugated site-specifically to different antibody regions with distinct in vivo efficacy and PK outcomes. Bioconjug. Chem. 2014, 25, 1331–1341. [Google Scholar] [CrossRef]
- Rabuka, D.; Rush, J.S.; de Hart, G.W.; Wu, P.; Bertozzi, C.R. Site-specific chemical protein conjugation using genetically encoded aldehyde tags. Nat. Protoc. 2012, 7, 1052–1067. [Google Scholar] [CrossRef]
- Agarwal, P.; Kudirka, R.; Albers, A.E.; Barfield, R.M.; de Hart, G.W.; Drake, P.M.; Jones, L.C.; Rabuka, D. Hydrazino-Pictet-Spengler ligation as a biocompatible method for the generation of stable protein conjugates. Bioconjug. Chem. 2013, 24, 846–851. [Google Scholar] [CrossRef]
- Lemke, E.A. The exploding genetic code. ChemBioChem 2014, 15, 1691–1694. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.H.; Axup, J.Y.; Schultz, P.G. Protein conjugation with genetically encoded unnatural amino acids. Curr. Opin. Chem. Biol. 2013, 17, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Hutchins, B.M.; Kazane, S.A.; Staflin, K.; Forsyth, J.S.; Felding-Habermann, B.; Smider, V.V.; Schultz, P.G. Selective formation of covalent protein heterodimers with an unnatural amino acid. Chem. Biol. 2011, 18, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Axup, J.Y.; Bajjuri, K.M.; Ritland, M.; Hutchins, B.M.; Kim, C.H.; Kazane, S.A.; Halder, R.; Forsyth, J.S.; Santidrian, A.F.; Stafin, K.; et al. Synthesis of site-specific antibody–drug conjugates using unnatural amino acids. Proc. Natl. Acad. Sci. USA 2012, 109, 16101–16106. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Stefano, J.E.; Manning, C.; Kyazike, J.; Chen, B.; Gianolio, D.A.; Park, A.; Busch, M.; Bird, J.; Zheng, X.; et al. Site-specific antibody–drug conjugation through glycoengineering. Bioconjug. Chem. 2014, 25, 510–520. [Google Scholar] [CrossRef]
- Witus, L.S.; Francis, M. Site-Specific protein bioconjugation via a pyridoxal 5′-phosphate-mediated N-terminal transamination reaction. Curr. Protoc. Chem. Biol. 2010, 2, 125–134. [Google Scholar] [PubMed]
- Kalia, J.; Raines, R.T. Hydrolytic stability of hydrazones and oximes. Angew. Chem. Int. Ed. 2008, 47, 7523–7526. [Google Scholar] [CrossRef] [PubMed]
- Buckley, T.F.; Rapoport, H. Mild and simple biomimetic conversion of amines to carbonyl compounds. J. Am. Chem. Soc. 1982, 104, 4446–4450. [Google Scholar] [CrossRef]
- Witus, L.S.; Netirojjanakul, C.; Palla, K.S.; Muehl, E.M.; Weng, C.-H.; Iavarone, A.T.; Francis, M.B. Site-Specific Protein Transamination Using N-Methylpyridinium-4-Carboxaldehyde. J. Am. Chem. Soc. 2013, 135, 17223–17229. [Google Scholar] [CrossRef] [PubMed]
- Dirksen, A.; Madsen, M.; Iacono, G.D.; Matin, M.J.; Bacica, M.; Stanković, N.; Callans, S.; Bhat, A. Parallel Synthesis and screening of peptide conjugates. Bioconjug. Chem. 2014, 25, 1052–1060. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Gui, J.; Pan, C.-M.; Albone, E.; Cheng, X.; Suh, E.M.; Grasso, L.; Ishihara, Y.; Baran, P.S. Bioconjugation by native chemical tagging of C–H bonds. J. Am. Chem. Soc. 2013, 135, 12994–12997. [Google Scholar] [CrossRef] [PubMed]
- Griffin, M.; Casadio, R.; Bergamini, C.M. Transglutaminases: Nature’s biological glues. Biochem. J. 2002, 396, 377–396. [Google Scholar] [CrossRef] [PubMed]
- Dennler, P.; Chiotellis, A.; Fischer, E.; Brégeon, D.; Belmant, C.; Gauthier, L.; Lhospice, F.; Romagne, F.; Schibli, R. Transglutaminase-based chemo-enzymatic conjugation approach yields homogeneous antibody–drug conjugates. Bioconjug. Chem. 2014, 25, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Strop, P. Versatility of microbial transglutaminase. Bioconjug. Chem. 2014, 25, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Strop, P.; Liu, S.H.; Dorywalska, M.; Delaria, K.; Dushin, R.G.; Tran, T.T.; Ho, W.H.; Farias, S.; Casas, M.G.; Abdiche, Y.; et al. Location matters: Site of conjugation modulates stability and pharmacokinetics of antibody drug conjugates. Chem. Biol. 2013, 20, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, E.S.; Heibeck, T.H.; Gill, A.; Li, X.; Murray, C.J.; Madlansacay, M.R.; Tran, C.; Uter, N.T.; Yin, G.; Rivers, P.J. Production of site-specific antibody–drug conjugates using optimized non-natural amino acids in a cell-free expression system. Bioconjug. Chem. 2014, 25, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Bryden, F.; Maruani, A.; Savoie, H.; Chudasama, V.; Smith, M.E.B.; Caddick, S.; Boyle, R.W. Regioselective and stoichiometrically controlled conjugation of photodynamic sensitizers to a HER2 targeting antibody fragment. Bioconjug. Chem. 2014, 25, 611–617. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yao, H.; Jiang, F.; Lu, A.; Zhang, G. Methods to Design and Synthesize Antibody-Drug Conjugates (ADCs). Int. J. Mol. Sci. 2016, 17, 194. https://doi.org/10.3390/ijms17020194
Yao H, Jiang F, Lu A, Zhang G. Methods to Design and Synthesize Antibody-Drug Conjugates (ADCs). International Journal of Molecular Sciences. 2016; 17(2):194. https://doi.org/10.3390/ijms17020194
Chicago/Turabian StyleYao, Houzong, Feng Jiang, Aiping Lu, and Ge Zhang. 2016. "Methods to Design and Synthesize Antibody-Drug Conjugates (ADCs)" International Journal of Molecular Sciences 17, no. 2: 194. https://doi.org/10.3390/ijms17020194
APA StyleYao, H., Jiang, F., Lu, A., & Zhang, G. (2016). Methods to Design and Synthesize Antibody-Drug Conjugates (ADCs). International Journal of Molecular Sciences, 17(2), 194. https://doi.org/10.3390/ijms17020194