
Selection of Suitable Reference Genes for Analysis of Salivary Transcriptome in Non-Syndromic Autistic Male Children

Abstract
:
1. Introduction
2. Results
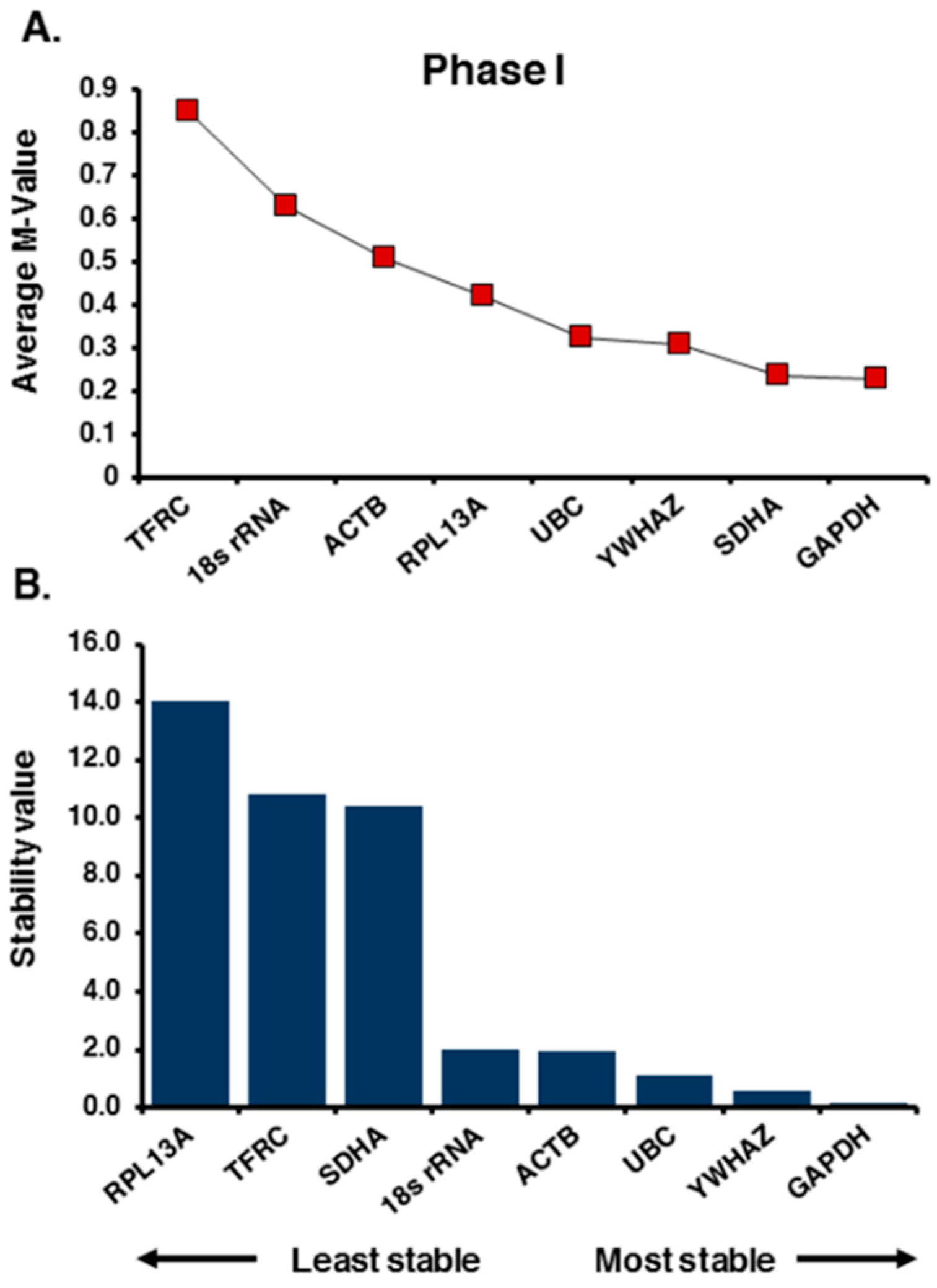
2.1. Selection of Candidate Reference Genes and Initial Screening
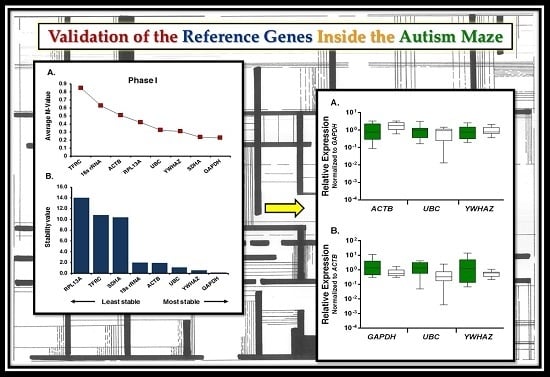
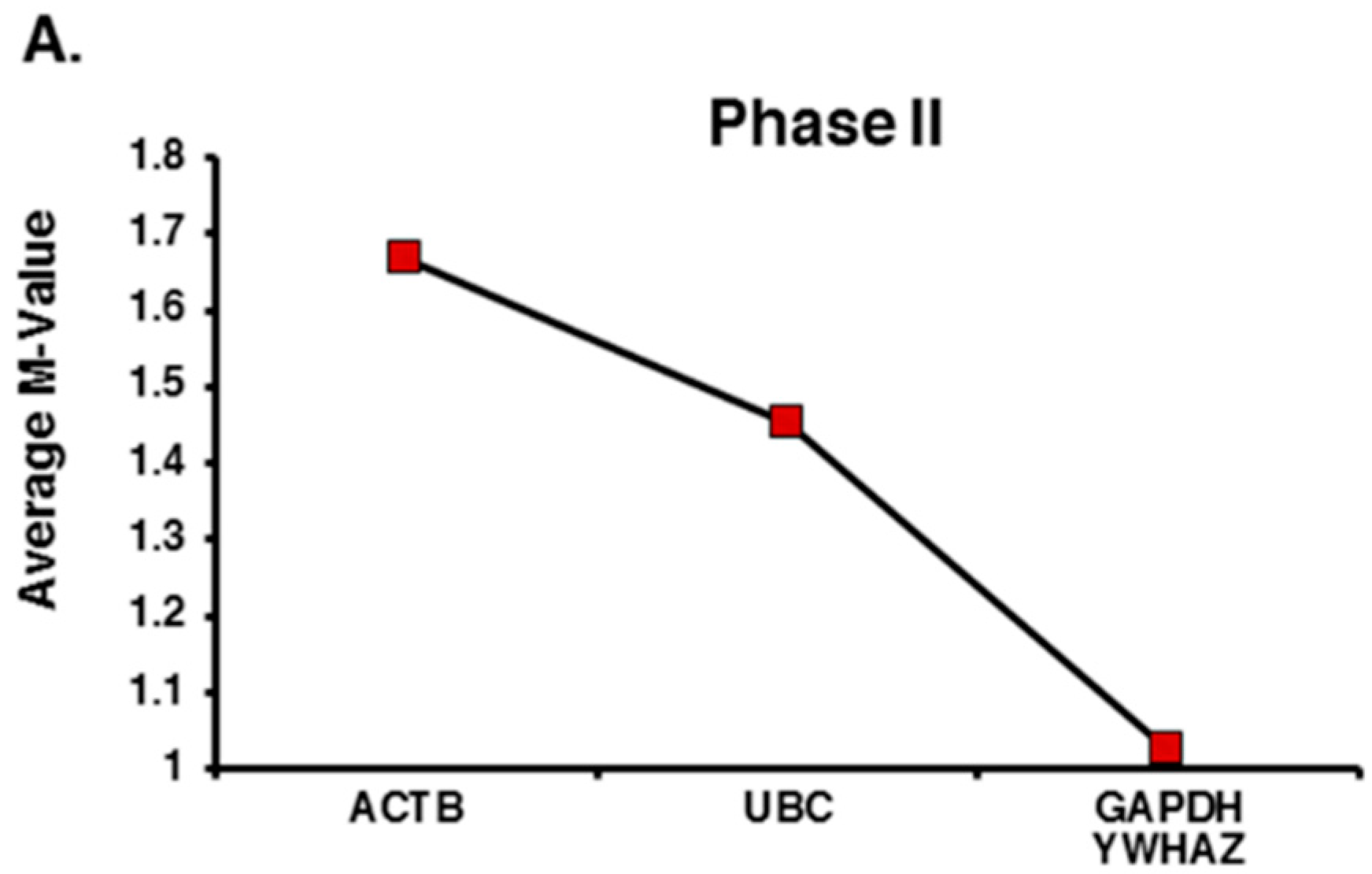
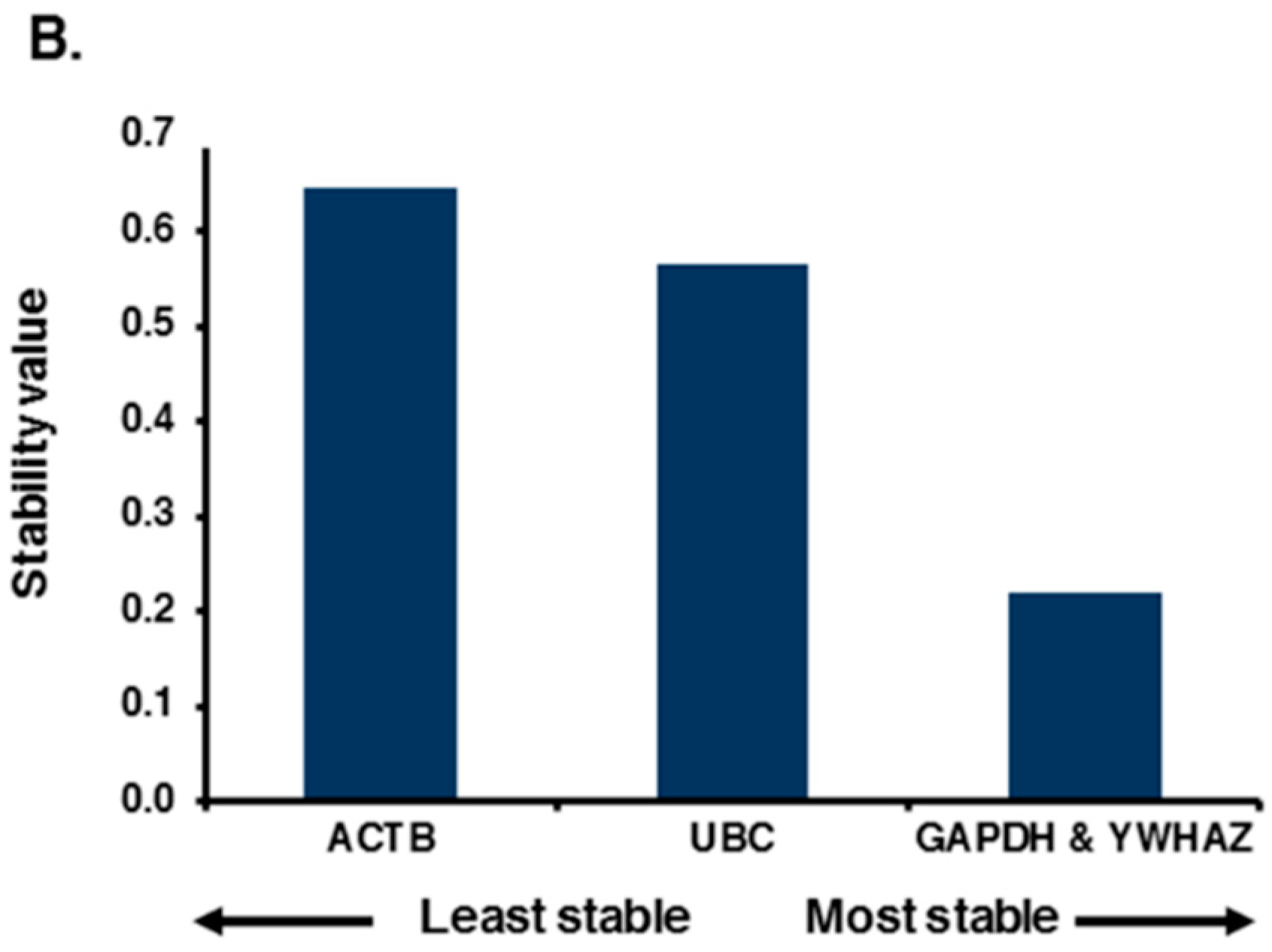
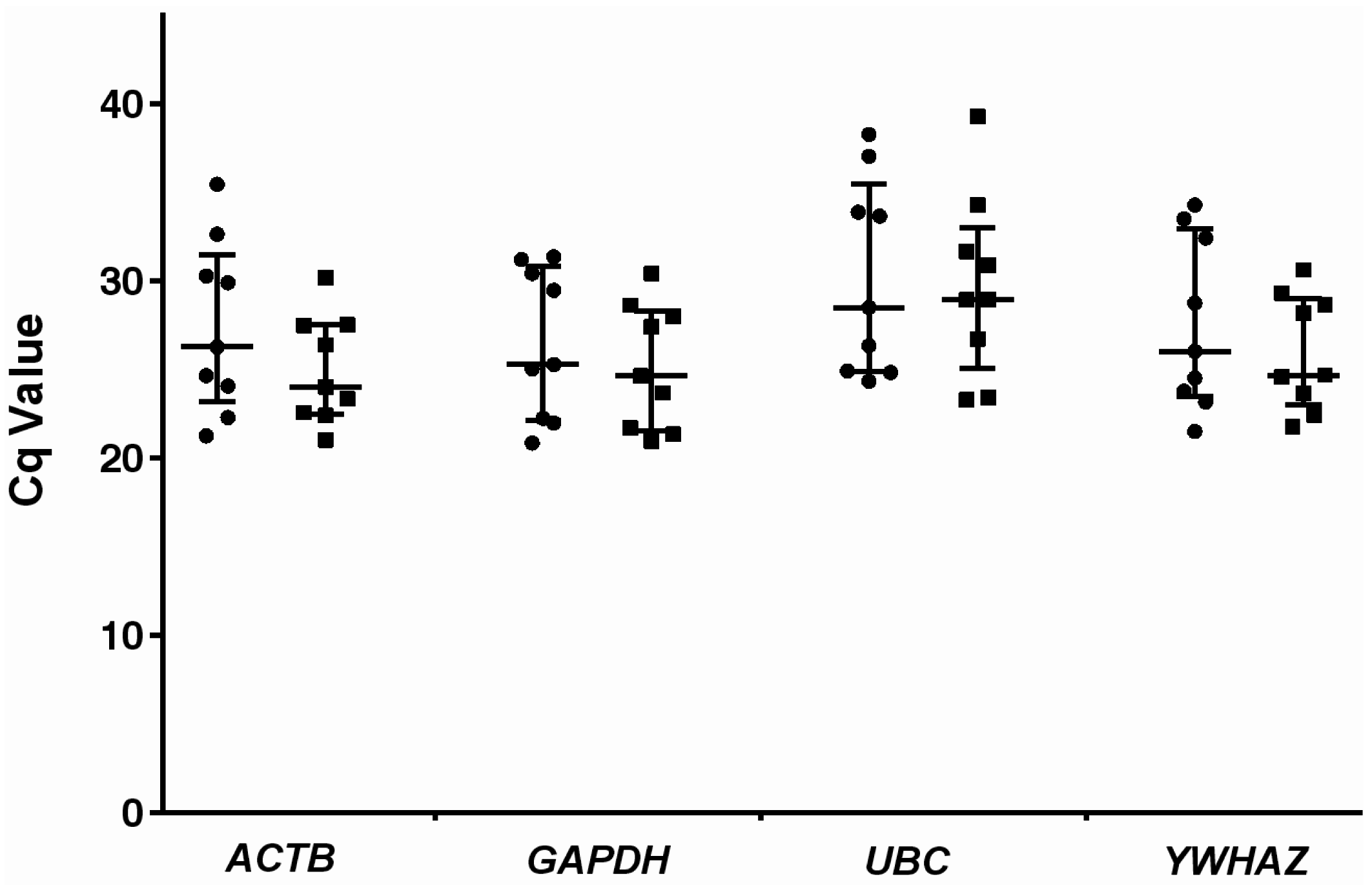
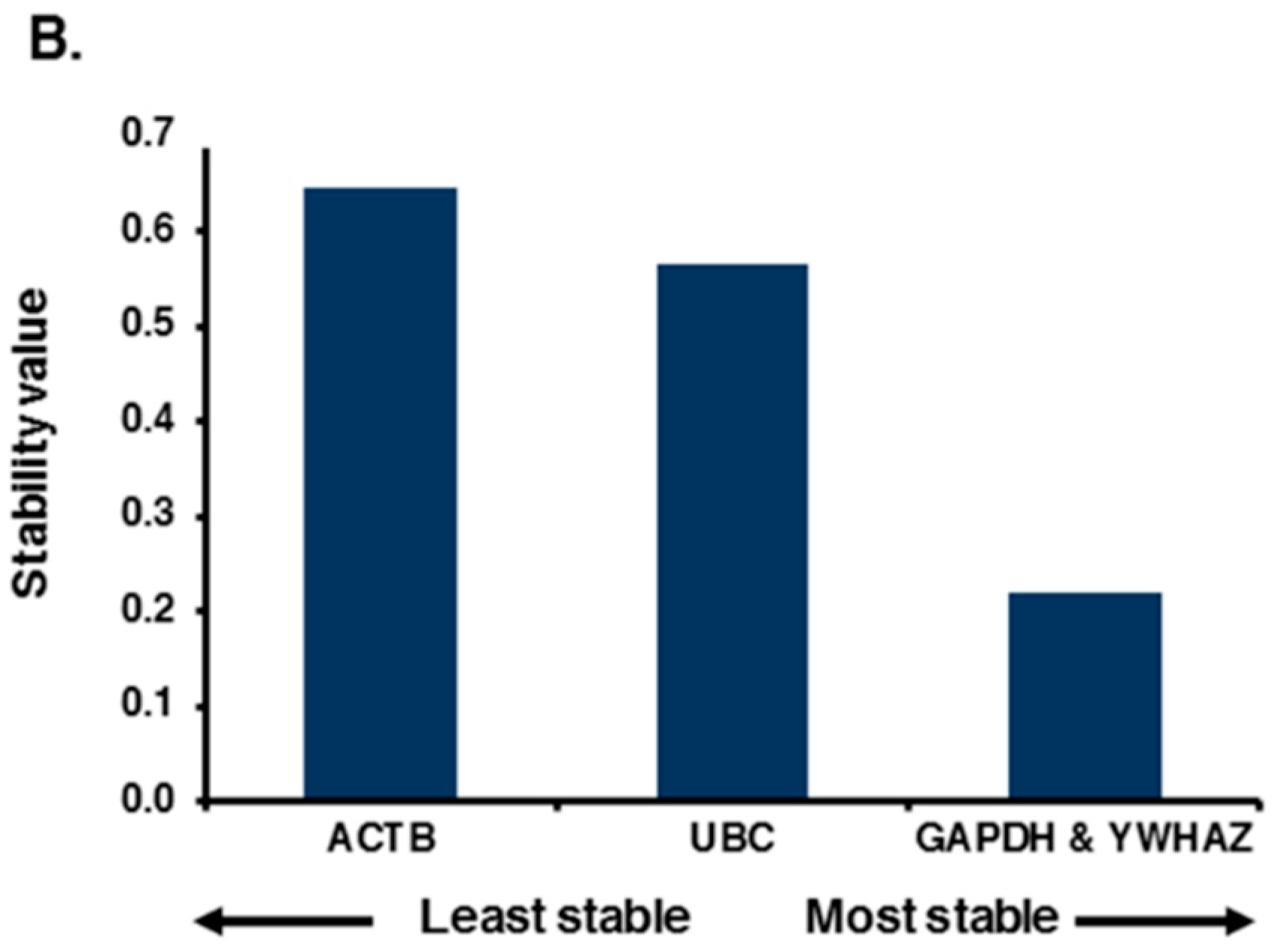
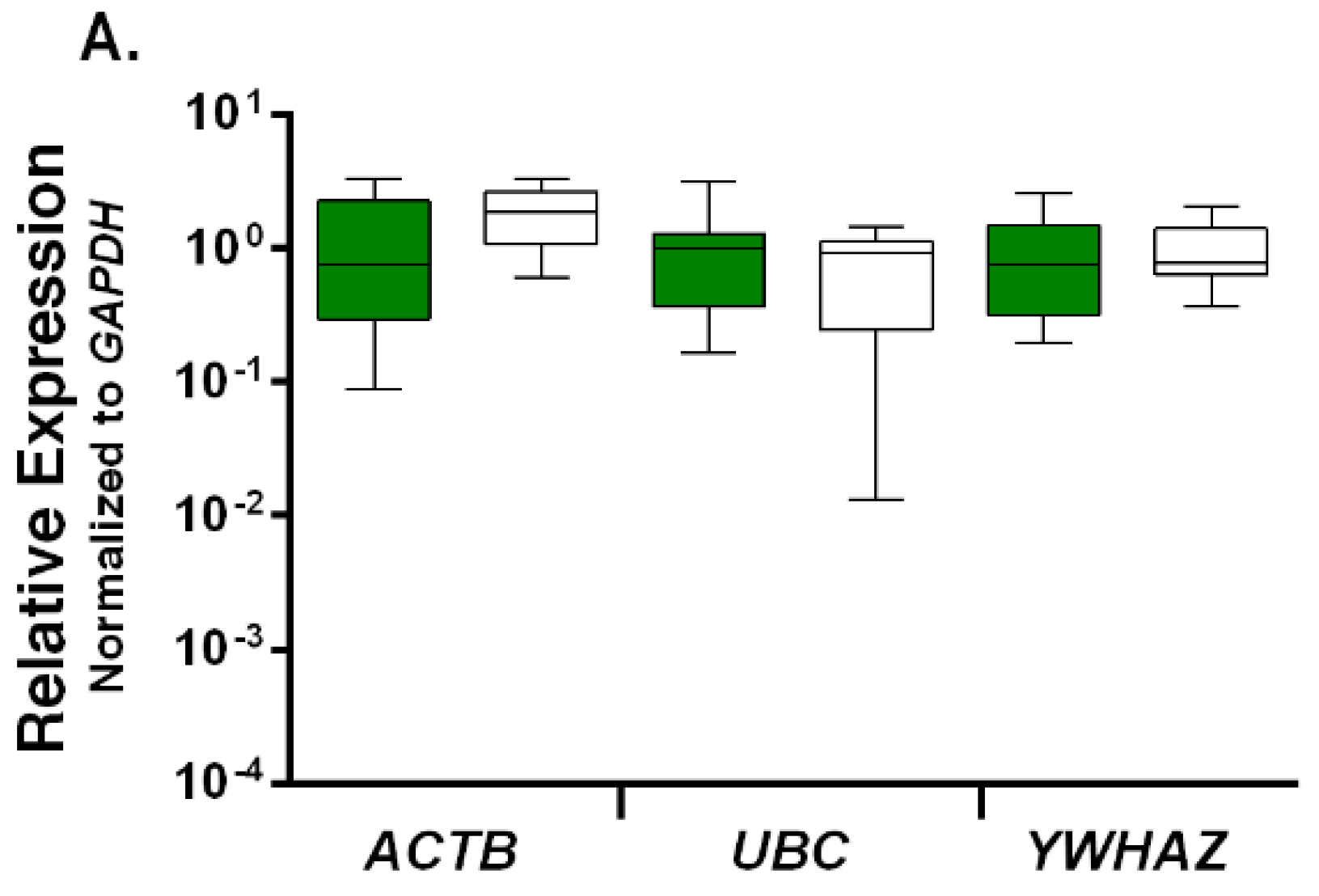
2.2. Expression Profiling and Validation of Candidate Reference Genes
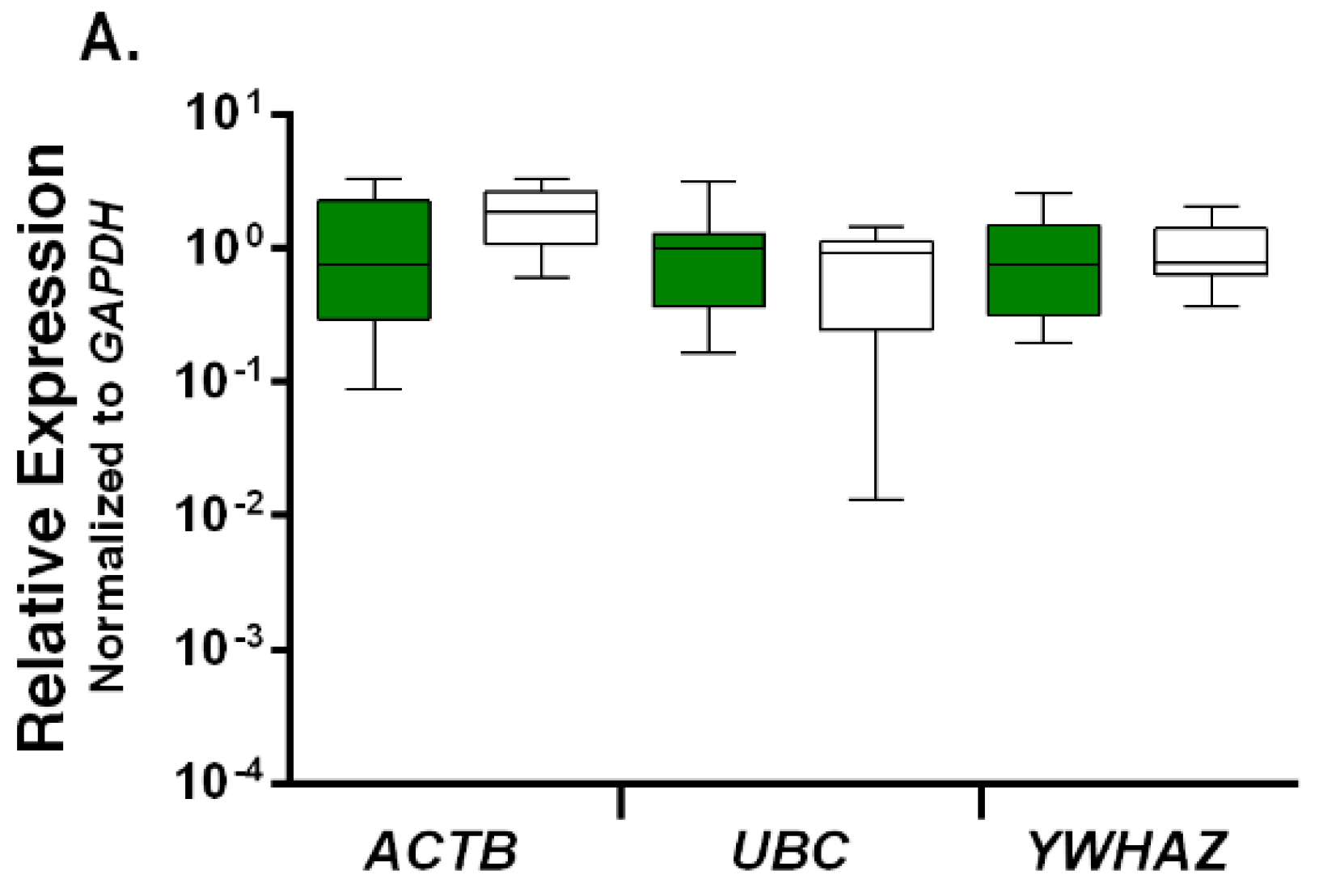
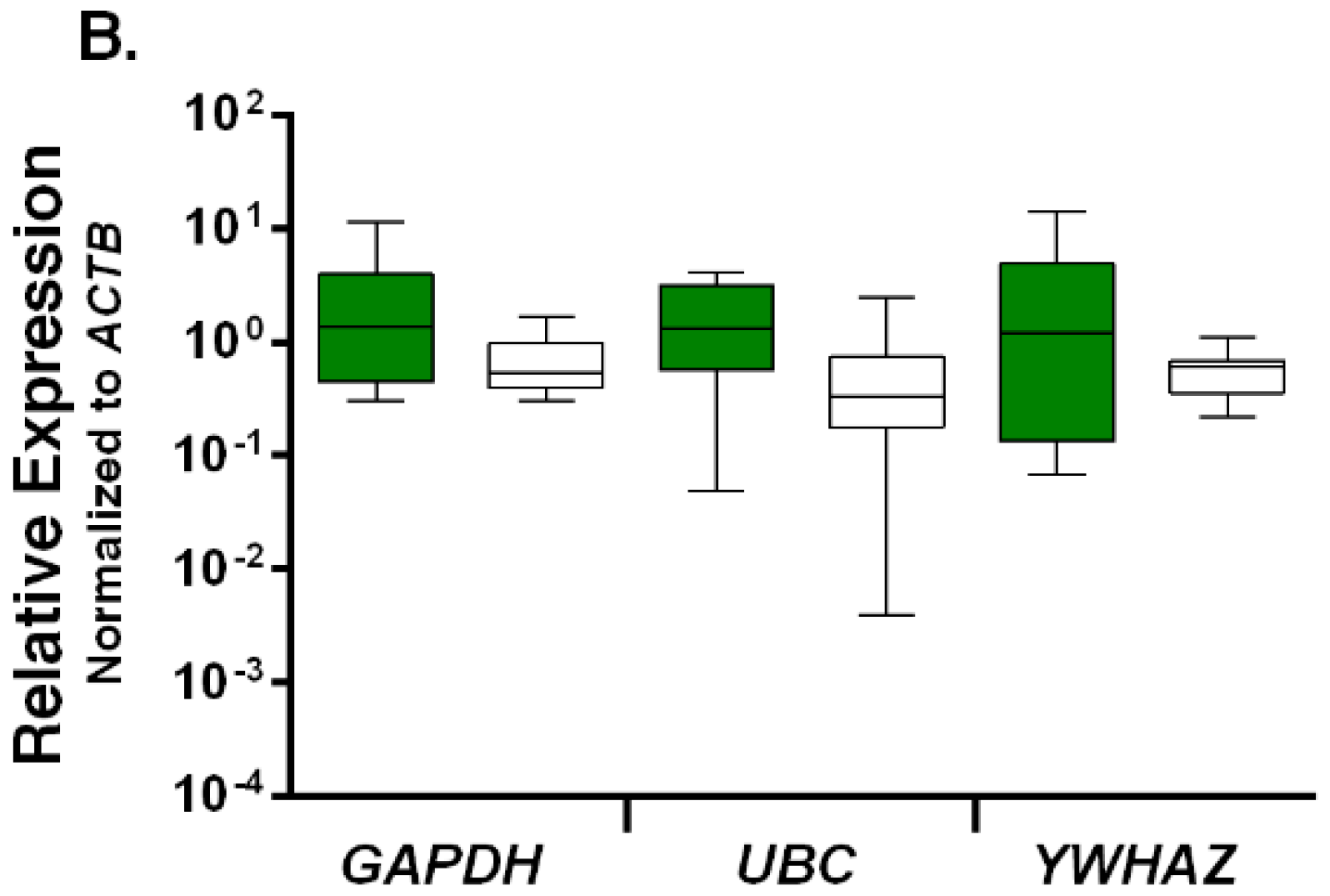
2.3. Relative Expression Levels of Candidate Reference Genes in Autistic vs. Healthy Subjects
3. Discussion
4. Materials and Methods
4.1. Subjects and Sampling Procedure
4.2. Total RNA Preparation and cDNA Synthesis
4.3. Candidate Reference Gene Selection
4.4. Quantitative Real-Time PCR
4.5. Gene Expression Stability Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
References
- Lai, M.C.; Lombardo, M.V.; Baron-Cohen, S. Autism. Lancet 2014, 383, 896–910. [Google Scholar] [CrossRef]
- Lord, C.; Risi, S.; DiLavore, P.S.; Shulman, C.; Thurm, A.; Pickles, A. Autism from 2 to 9 years of age. Arch. Gen. Psychiatry 2006, 63, 694–701. [Google Scholar] [CrossRef] [PubMed]
- Werling, D.M.; Geschwind, D.H. Sex differences in autism spectrum disorders. Curr. Opin. Neurol. 2013, 26, 146–161. [Google Scholar] [CrossRef] [PubMed]
- Betancur, C.; Coleman, M. Etiological Heterogeneity in Autism Spectrum Disorders: Role of Rare Variants. In The Neuroscience of Autism Spectrum Disorders; Joseph, D., Buxbaum, P.R.H., Eds.; Academic Press: Waltham, MA, USA, 2013; pp. 113–144. [Google Scholar]
- Ch’ng, C.; Kwok, W.; Rogic, S.; Pavlidis, P. Meta-analysis of gene expression in autism spectrum disorder. Autism Res. 2015, 8, 593–608. [Google Scholar] [CrossRef] [PubMed]
- Berg, J.M.; Geschwind, D.H. Autism genetics: Searching for specificity and convergence. Genome Biol. 2012, 13, 247–263. [Google Scholar] [CrossRef] [PubMed]
- Morey, J.S.; Ryan, J.C.; van Dolah, F.M. Microarray validation: Factors influencing correlation between oligonucleotide microarrays and real-time PCR. Biol. Proced. Online 2006, 8, 175–193. [Google Scholar] [CrossRef] [PubMed]
- Nolan, T.; Hands, R.E.; Bustin, S.A. Quantification of mrna using real-time RT-PCR. Nat. Protoc. 2006, 1, 1559–1582. [Google Scholar] [CrossRef] [PubMed]
- Huggett, J.; Dheda, K.; Bustin, S.; Zumla, A. Real-time RT-PCR normalisation; strategies and considerations. Genes Immun. 2005, 6, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Shen, Y. An old method facing a new challenge: Re-visiting housekeeping proteins as internal reference control for neuroscience research. Life Sci. 2013, 92, 747–751. [Google Scholar] [CrossRef] [PubMed]
- Purcell, A.E.; Jeon, O.H.; Zimmerman, A.W.; Blue, M.E.; Pevsner, J. Postmortem brain abnormalities of the glutamate neurotransmitter system in autism. Neurology 2001, 57, 1618–1628. [Google Scholar] [CrossRef] [PubMed]
- Fatemi, S.H.; Stary, J.M.; Halt, A.R.; Realmuto, G.R. Dysregulation of reelin and Bcl-2 proteins in autistic cerebellum. J. Autism Dev. Disord. 2001, 31, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Araghi-Niknam, M.; Fatemi, S.H. Levels of Bcl-2 and p53 are altered in superior frontal and cerebellar cortices of autistic subjects. Cell. Mol. Neurobiol. 2003, 23, 945–952. [Google Scholar] [CrossRef] [PubMed]
- Samaco, R.C.; Nagarajan, R.P.; Braunschweig, D.; LaSalle, J.M. Multiple pathways regulate MeCP2 expression in normal brain development and exhibit defects in autism-spectrum disorders. Hum. Mol. Genet. 2004, 13, 629–639. [Google Scholar] [CrossRef] [PubMed]
- Hu, V.W.; Frank, B.C.; Heine, S.; Lee, N.H.; Quackenbush, J. Gene expression profiling of lymphoblastoid cell lines from monozygotic twins discordant in severity of autism reveals differential regulation of neurologically relevant genes. BMC Genom. 2006, 7, 118–136. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, Y.; Martin, C.L.; Vazquez-Lopez, A.; Spence, S.J.; Alvarez-Retuerto, A.I.; Sigman, M.; Steindler, C.; Pellegrini, S.; Schanen, N.C.; Warren, S.T. Genome-wide expression profiling of lymphoblastoid cell lines distinguishes different forms of autism and reveals shared pathways. Hum. Mol. Genet. 2007, 16, 1682–1698. [Google Scholar] [CrossRef] [PubMed]
- Garbett, K.; Ebert, P.J.; Mitchell, A.; Lintas, C.; Manzi, B.; Mirnics, K.; Persico, A.M. Immune transcriptome alterations in the temporal cortex of subjects with autism. Neurobiol. Dis. 2008, 30, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Gregg, J.P.; Lit, L.; Baron, C.A.; Hertz-Picciotto, I.; Walker, W.; Davis, R.A.; Croen, L.A.; Ozonoff, S.; Hansen, R.; Pessah, I.N. Gene expression changes in children with autism. Genomics 2008, 91, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Enstrom, A.M.; Lit, L.; Onore, C.E.; Gregg, J.P.; Hansen, R.L.; Pessah, I.N.; Hertz-Picciotto, I.; van de Water, J.A.; Sharp, F.R.; Ashwood, P. Altered gene expression and function of peripheral blood natural killer cells in children with autism. Brain Behav. Immun. 2009, 23, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Hu, V.W.; Sarachana, T.; Kim, K.S.; Nguyen, A.; Kulkarni, S.; Steinberg, M.E.; Luu, T.; Lai, Y.; Lee, N.H. Gene expression profiling differentiates autism case–controls and phenotypic variants of autism spectrum disorders: Evidence for circadian rhythm dysfunction in severe autism. Autism Res. 2009, 2, 78–97. [Google Scholar] [CrossRef] [PubMed]
- Hu, V.W.; Nguyen, A.; Kim, K.S.; Steinberg, M.E.; Sarachana, T.; Scully, M.A.; Soldin, S.J.; Luu, T.; Lee, N.H. Gene expression profiling of lymphoblasts from autistic and nonaffected sib pairs: Altered pathways in neuronal development and steroid biosynthesis. PLoS ONE 2009, 4, e5775. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, A.; Li, X.; Wen, G.; Tauqeer, Z.; Brown, W.; Malik, M. Cathepsin D and apoptosis related proteins are elevated in the brain of autistic subjects. Neuroscience 2010, 165, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Malik, M.; Sheikh, A.M.; Wen, G.; Spivack, W.; Brown, W.T.; Li, X. Expression of inflammatory cytokines, Bcl-2 and cathepsin D are altered in lymphoblasts of autistic subjects. Immunobiology 2011, 216, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Kuwano, Y.; Kamio, Y.; Kawai, T.; Katsuura, S.; Inada, N.; Takaki, A.; Rokutan, K. Autism-associated gene expression in peripheral leucocytes commonly observed between subjects with autism and healthy women having autistic children. PLoS ONE 2011, 6, e24723. [Google Scholar] [CrossRef] [PubMed]
- Ghahramani Seno, M.M.; Hu, P.; Gwadry, F.G.; Pinto, D.; Marshall, C.R.; Casallo, G.; Scherer, S.W. Gene and miRNA expression profiles in autism spectrum disorders. Brain Res. 2011, 1380, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Sanders, S.J.; Tian, Y.; Voineagu, I.; Huang, N.; Chu, S.H.; Klei, L.; Cai, C.; Ou, J.; Lowe, J.K. Genome-wide transcriptome profiling reveals the functional impact of rare de novo and recurrent CNVs in autism spectrum disorders. Am. J. Hum. Genet. 2012, 91, 38–55. [Google Scholar] [CrossRef] [PubMed]
- Chow, M.L.; Pramparo, T.; Winn, M.E.; Barnes, C.C.; Li, H.-R.; Weiss, L.; Fan, J.-B.; Murray, S.; April, C.; Belinson, H. Age-dependent brain gene expression and copy number anomalies in autism suggest distinct pathological processes at young versus mature ages. PLoS Genet. 2012, 8, e1002592. [Google Scholar] [CrossRef] [PubMed]
- Kong, S.W.; Collins, C.D.; Shimizu-Motohashi, Y.; Holm, I.A.; Campbell, M.G.; Lee, I.-H.; Brewster, S.J.; Hanson, E.; Harris, H.K.; Lowe, K.R. Characteristics and predictive value of blood transcriptome signature in males with autism spectrum disorders. PLoS ONE 2012, 7, e49475. [Google Scholar] [CrossRef] [PubMed]
- Griesi-Oliveira, K.; Moreira Dde, P.; Davis-Wright, N.; Sanders, S.; Mason, C.; Orabona, G.M.; Vadasz, E.; Bertola, D.R.; State, M.W.; Passos-Bueno, M.R. A complex chromosomal rearrangement involving chromosomes 2, 5, and X in autism spectrum disorder. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2012, 159, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Anitha, A.; Nakamura, K.; Thanseem, I.; Yamada, K.; Iwayama, Y.; Toyota, T.; Matsuzaki, H.; Miyachi, T.; Yamada, S.; Tsujii, M.; et al. Brain region-specific altered expression and association of mitochondria-related genes in autism. Mol. Autism 2012, 3, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, M.R.; Rubin, R.A.; Falcone, T.; Ting, A.H.; Natowicz, M.R. Brain transcriptional and epigenetic associations with autism. PLoS ONE 2012, 7, e44736. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Ababon, M.R.; Soliman, M.; Lin, Y.; Brzustowicz, L.M.; Matteson, P.G.; Millonig, J.H. Autism associated gene, engrailed2, and flanking gene levels are altered in post-mortem cerebellum. PLoS ONE 2014, 9, e87208. [Google Scholar] [CrossRef] [PubMed]
- Nardone, S.; Sams, D.S.; Reuveni, E.; Getselter, D.; Oron, O.; Karpuj, M.; Elliott, E. DNA methylation analysis of the autistic brain reveals multiple dysregulated biological pathways. Transl. Psychiatry 2014, 4, e433. [Google Scholar] [CrossRef] [PubMed]
- Pandit, P.; Cooper-White, J.; Punyadeera, C. High-yield RNA-extraction method for saliva. Clin. Chem. 2013, 59, 1118–1122. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Zhou, H.; Reiss, J.K.; Yan, X.; Zhang, L.; Chia, D.; Wong, D.T. Direct saliva transcriptome analysis. Clin. Chem. 2011, 57, 1295–1302. [Google Scholar] [CrossRef] [PubMed]
- Park, N.J.; Li, Y.; Yu, T.; Brinkman, B.M.; Wong, D.T. Characterization of RNA in saliva. Clin. Chem. 2006, 52, 988–994. [Google Scholar] [CrossRef] [PubMed]
- Seugnet, L.; Boero, J.; Gottschalk, L.; Duntley, S.P.; Shaw, P.J. Identification of a biomarker for sleep drive in flies and humans. Proc. Natl. Acad. Sci. USA 2006, 103, 19913–19918. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L. Primer sequence disclosure: A clarification of the MIQE guidelines. Clin. Chem. 2011, 57, 919–921. [Google Scholar] [CrossRef] [PubMed]
- Vandesompele, J.; de Preter, K.; Pattyn, F.; Poppe, B.; van Roy, N.; de Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, 1–12. [Google Scholar] [CrossRef]
- Andersen, C.L.; Jensen, J.L.; Orntoft, T.F. Normalization of real-time quantitative reverse transcription-PCR data: A model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 2004, 64, 5245–5250. [Google Scholar] [CrossRef] [PubMed]
- Lallemant, B.; Evrard, A.; Combescure, C.; Chapuis, H.; Chambon, G.; Raynal, C.; Reynaud, C.; Sabra, O.; Joubert, D.; Hollande, F.; et al. Reference gene selection for head and neck squamous cell carcinoma gene expression studies. BMC Mol. Biol. 2009, 10, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−∆∆Ct method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W.; Tichopad, A.; Prgomet, C.; Neuvians, T.P. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: Bestkeeper—Excel-based tool using pair-wise correlations. Biotechnol. Lett. 2004, 26, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Keller, S.; Sarchiapone, M.; Zarrilli, F.; Videtic, A.; Ferraro, A.; Carli, V.; Sacchetti, S.; Lembo, F.; Angiolillo, A.; Jovanovic, N.; et al. Increased BDNF promoter methylation in the Wernicke area of suicide subjects. Arch. Gen. Psychiatry 2010, 67, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Arion, D.; Sabatini, M.; Unger, T.; Pastor, J.; Alonso-Nanclares, L.; Ballesteros-Yanez, I.; Garcia Sola, R.; Munoz, A.; Mirnics, K.; DeFelipe, J. Correlation of transcriptome profile with electrical activity in temporal lobe epilepsy. Neurobiol. Dis. 2006, 22, 374–387. [Google Scholar] [CrossRef] [PubMed]
- Arion, D.; Unger, T.; Lewis, D.A.; Levitt, P.; Mirnics, K. Molecular evidence for increased expression of genes related to immune and chaperone function in the prefrontal cortex in schizophrenia. Biol. Psychiatry 2007, 62, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Xiao, H.; Wong, D.T. Salivary biomarkers for clinical applications. Mol. Diagn. Ther. 2009, 13, 245–259. [Google Scholar] [CrossRef] [PubMed]
- Saxena, V.; Yadev, N.; Juneja, V.; Singh, A.; Tiwari, U.; Santha, B. Saliva: A miraculous biofluid for early detection of disease. J. Oral Health Community Dent. 2013, 7, 64–68. [Google Scholar]
- Zhang, L.; Xiao, H.; Karlan, S.; Zhou, H.; Gross, J.; Elashoff, D.; Akin, D.; Yan, X.; Chia, D.; Karlan, B.; et al. Discovery and preclinical validation of salivary transcriptomic and proteomic biomarkers for the non-invasive detection of breast cancer. PLoS ONE 2010, 5, e15573. [Google Scholar] [CrossRef] [PubMed]
- Nohesara, S.; Ghadirivasfi, M.; Mostafavi, S.; Eskandari, M.R.; Ahmadkhaniha, H.; Thiagalingam, S.; Abdolmaleky, H.M. DNA hypomethylation of MB-COMT promoter in the DNA derived from saliva in schizophrenia and bipolar disorder. J. Psychiatr. Res. 2011, 45, 1432–1438. [Google Scholar] [CrossRef] [PubMed]
- Rahbar, M.H.; Samms-Vaughan, M.; Ma, J.; Bressler, J.; Dickerson, A.S.; Hessabi, M.; Loveland, K.A.; Grove, M.L.; Shakespeare-Pellington, S.; Beecher, C.; et al. Synergic effect of GSTP1 and blood manganese concentrations in autism spectrum disorder. Res. Autism Spectr. Disord. 2015, 18, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Hicks, S.D.; Ignacio, C.; Gentile, K.; Middleton, F.A. Salivary mirna profiles identify children with autism spectrum disorder, correlate with adaptive behavior, and implicate asd candidate genes involved in neurodevelopment. BMC Pediatr. 2016, 16, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Farrell, J.J.; Zhou, H.; Elashoff, D.; Akin, D.; Park, N.H.; Chia, D.; Wong, D.T. Salivary transcriptomic biomarkers for detection of resectable pancreatic cancer. Gastroenterology 2010, 138, 941–947. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.R.; Kuo, H.C.; Huang, H.C.; Huang, L.T.; Tain, Y.L.; Chen, C.C.; Liang, C.D.; Sheen, J.M.; Lin, I.C.; Wu, C.C.; et al. Glyceraldehyde-3-phosphate dehydrogenase is a reliable internal control in western blot analysis of leukocyte subpopulations from children. Anal. Biochem. 2011, 413, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Sochivko, D.; Beck, H.; Marechal, D.; Wiestler, O.D.; Becker, A.J. Activity-induced expression of common reference genes in individual cns neurons. Lab. Investig. 2001, 81, 913–916. [Google Scholar] [CrossRef] [PubMed]
- Silberberg, G.; Baruch, K.; Navon, R. Detection of stable reference genes for real-time PCR analysis in schizophrenia and bipolar disorder. Anal. Biochem. 2009, 391, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Penna, I.; Vella, S.; Gigoni, A.; Russo, C.; Cancedda, R.; Pagano, A. Selection of candidate housekeeping genes for normalization in human postmortem brain samples. Int. J. Mol. Sci. 2011, 12, 5461–5470. [Google Scholar] [CrossRef] [PubMed]
- Steinhausen, H.C.; Erdin, A. Abnormal psychosocial situations and ICD-10 diagnoses in children and adolescents attending a psychiatric service. J. Child Psychol. Psychiatry 1992, 33, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.; Pickles, A.; McLennan, J.; Rutter, M.; Bregman, J.; Folstein, S.; Fombonne, E.; Leboyer, M.; Minshew, N. Diagnosing autism: Analyses of data from the autism diagnostic interview. J. Autism Dev. Disord. 1997, 27, 501–517. [Google Scholar] [CrossRef] [PubMed]
- Goodman, R. The strengths and difficulties questionnaire: A research note. J. Child. Psychol. Psychiatry 1997, 38, 581–586. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | RNA/Protein Source | Reference Gene(s) | Age (Year) | N | Sex |
|---|---|---|---|---|---|
| Purcell et al. (2001) [12] | Brain tissue | ACTIN WB | 5–54 | 10 | M/F 1 |
| Cerebellum | GAPDH RP | ||||
| Fatemi et al. (2001) [13] | Brain tissue | ACTIN WB | 23.5 ± 4.8 | 5 | M |
| Cerebellum | |||||
| Araghi-Niknam et al. (2003) [14] | Brain tissue | ACTIN WB | 23.8 ± 4.9 | 5 | M |
| Cerebellum, Frontal Cortex | |||||
| Samaco et al. (2004) [15] | Brain tissue | TFRC and ACTIN IF | 2–32 | 13 | M/F 8 |
| Frontal Cortex | GAPDH, H1, and TFRC FISH | ||||
| Hu et al. (2006) [16] | LCL | 18s rRNA qP | 6–16 | 10 | M |
| Nishimura et al. (2007 [17]) | LCL | HPRT1 qP | AGRE | 42 | M |
| Garbett et al. (2008) [18] | Brain tissue | ACTB MA−qP | 4–30 | 6 | M/F 2 |
| Sup. temporal gyrus | |||||
| Gregg et al. (2008) [19] | Whole blood | DDR1, RPL37A, and | CHARGE | 35 | M/F 6 |
| SMARC2 MA−qP | |||||
| Enstrom et al. (2009) [20] | PBL, Natural killer cells | DDR1, RPL37A, and SMARC2 MA−qP | 2.3–5.6 | 52 | M |
| Hu et al. (2009a) [21] | LCL | MDH1, ARF1, ACSL5 MA−qP | 12.3 ± 3.7 | 116 | M |
| Hu et al. (2009b) [22] | LCL | 18s rRNA MA−qP | 7.8 ± 3.4 | 20 | NA |
| Sheikh et al. (2010) [23] | Brain tissue | ACTIN WB | 8.9 ± 3.2 | 9 | M/F 4 |
| Frontal Cortex | |||||
| Malik et al. (2011) [24] | PBL | ACTIN WB, NB | 8.4 ± 0.3 | 6 | NA |
| Kuwano et al. (2011) [25] | Whole blood | GAPDH and HDAC1 MA−qP | 26.7 ± 5.5 | 21 | M/F 4 |
| Ghahramani Seno et al. (2011) [26] | LCL | TMEM32 qP TUBB WB | 2–18 | 20 | M/F 7 |
| Luo et al. (2012) [27] | LCL | GAPDH MA−qP | SSC | 42 | NA |
| Chow et al. (2012) [28] | Brain tissue | ACTB, TBP, and | 2–56 | 20 | M |
| Prefrontal Cortex | RPL13A MA−qP | ||||
| Kong et al. (2012) [29] | Whole blood | GAPDH MA−qP | 8.2 ± 3.0 | 170 | M |
| Griesi-Oliveira et al. (2012) [30] | Dental pulp stem cells | GAPDH, HPRT1, SDHA, and HMBS qP | 10 | 1 | F |
| Anitha et al. (2012) [31] | Brain tissue | GAPDH WB | 8–29 | 8 | M/F 2 |
| Anterior cingulated gyrus, Motor Cortex, thalamus | B2M, HPRT1, RPL13A, GAPDH, and ACTB qP | ||||
| Ginsberg et al. (2012) [32] | Brain tissue | GAPDH qP | 2–60 | 9 | M/F |
| Occipital and cerebellar hemispheric cortices | |||||
| Choi et al. (2014) [33] | Brain Tissue | GAPDH qP | 4.5–82 | 29 | M/F 8 |
| Cerebellum | |||||
| Nardone et al. (2014) [34] | Brain Tissue | GAPDH, HPRT1, POLR2α, and SDHA qP | 18–51 | 13 | M/F 2 |
| Anterior cingulated gyrus | |||||
| Prefrontal Cortex |
| Gene Name | Function | Gene Symbol | mRNA Accession No. | Amplicon 2 Length (bp) |
|---|---|---|---|---|
| 18S ribosomal RNA | Ribosomal RNA Subunit | 18s rRNA | M10098 1 | 99 * |
| β-actin | Cytoskeletal structural protein | ACTB | NM_001101 | 94 |
| Glyceraldehyde-3-phosphate dehydrogenase | Glycolytic enzyme | GAPDH | NM_002046 | 142 * |
| Ribosomal protein L13a | Structural component of large subunit of Ribosome | RPL13A | NM_012423 | 223 * |
| Succinate dehydrogenase complex subunit A, flavoprotein | Electron transporter in the Krebs cycle | SDHA | NM_004168 | 154 * |
| Transferrin receptor | Cellular iron uptake | TFRC | NM_003234 | 134 |
| Ubiquitin C | Protein degradation | UBC | NM_021009 | 192 * |
| Tyrosine 3 monooxygenase activation protein, zeta polypeptide | Signal transduction | YWHAZ | NM_003406 | 150 * |
| Gene Symbol | Control | Autism | Overall SD | ||||
|---|---|---|---|---|---|---|---|
| Mean | SD | N | Mean | SD | N | ||
| 18s rRNA | 16.06 | 1.78 | 2 | 14.58 | 0.66 | 2 | 1.04 |
| ACTB | 25.87 | 0.38 | 2 | 29.18 | 1.77 | 2 | 2.34 |
| GAPDH | 26.27 | 0.29 | 2 | 27.01 | 0.44 | 2 | 0.52 |
| RPL13A | 31.09 | 0.89 | 2 | 32.72 | NA | 1 | 1.15 |
| SDHA | 33.73 | NA | 1 | 28.93 | 0.32 | 2 | 3.40 |
| TFRC | 31.14 | 1.73 | 2 | 33.22 | 1.58 | 2 | 1.47 |
| UBC | 30.22 | 0.62 | 2 | 32.23 | 0.59 | 2 | 1.42 |
| YWHAZ | 27.54 | 0.61 | 2 | 28.07 | 1.11 | 2 | 0.37 |
| Gene Symbol | Slope | R2 | Efficiency (%) |
|---|---|---|---|
| ACTB | −3.400 | 0.981 | 96.8 |
| GAPDH | −3.303 | 0.989 | 100.9 |
| UBC | −3.747 | 0.974 | 84.8 |
| YWHAZ | −3.398 | 0.984 | 97.2 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panahi, Y.; Salasar Moghaddam, F.; Ghasemi, Z.; Hadi Jafari, M.; Shervin Badv, R.; Eskandari, M.R.; Pedram, M. Selection of Suitable Reference Genes for Analysis of Salivary Transcriptome in Non-Syndromic Autistic Male Children. Int. J. Mol. Sci. 2016, 17, 1711. https://doi.org/10.3390/ijms17101711
Panahi Y, Salasar Moghaddam F, Ghasemi Z, Hadi Jafari M, Shervin Badv R, Eskandari MR, Pedram M. Selection of Suitable Reference Genes for Analysis of Salivary Transcriptome in Non-Syndromic Autistic Male Children. International Journal of Molecular Sciences. 2016; 17(10):1711. https://doi.org/10.3390/ijms17101711
Chicago/Turabian StylePanahi, Yasin, Fahimeh Salasar Moghaddam, Zahra Ghasemi, Mandana Hadi Jafari, Reza Shervin Badv, Mohamad Reza Eskandari, and Mehrdad Pedram. 2016. "Selection of Suitable Reference Genes for Analysis of Salivary Transcriptome in Non-Syndromic Autistic Male Children" International Journal of Molecular Sciences 17, no. 10: 1711. https://doi.org/10.3390/ijms17101711
APA StylePanahi, Y., Salasar Moghaddam, F., Ghasemi, Z., Hadi Jafari, M., Shervin Badv, R., Eskandari, M. R., & Pedram, M. (2016). Selection of Suitable Reference Genes for Analysis of Salivary Transcriptome in Non-Syndromic Autistic Male Children. International Journal of Molecular Sciences, 17(10), 1711. https://doi.org/10.3390/ijms17101711