
Gardenamide A Protects RGC-5 Cells from H2O2-Induced Oxidative Stress Insults by Activating PI3K/Akt/eNOS Signaling Pathway

Abstract
:
1. Introduction
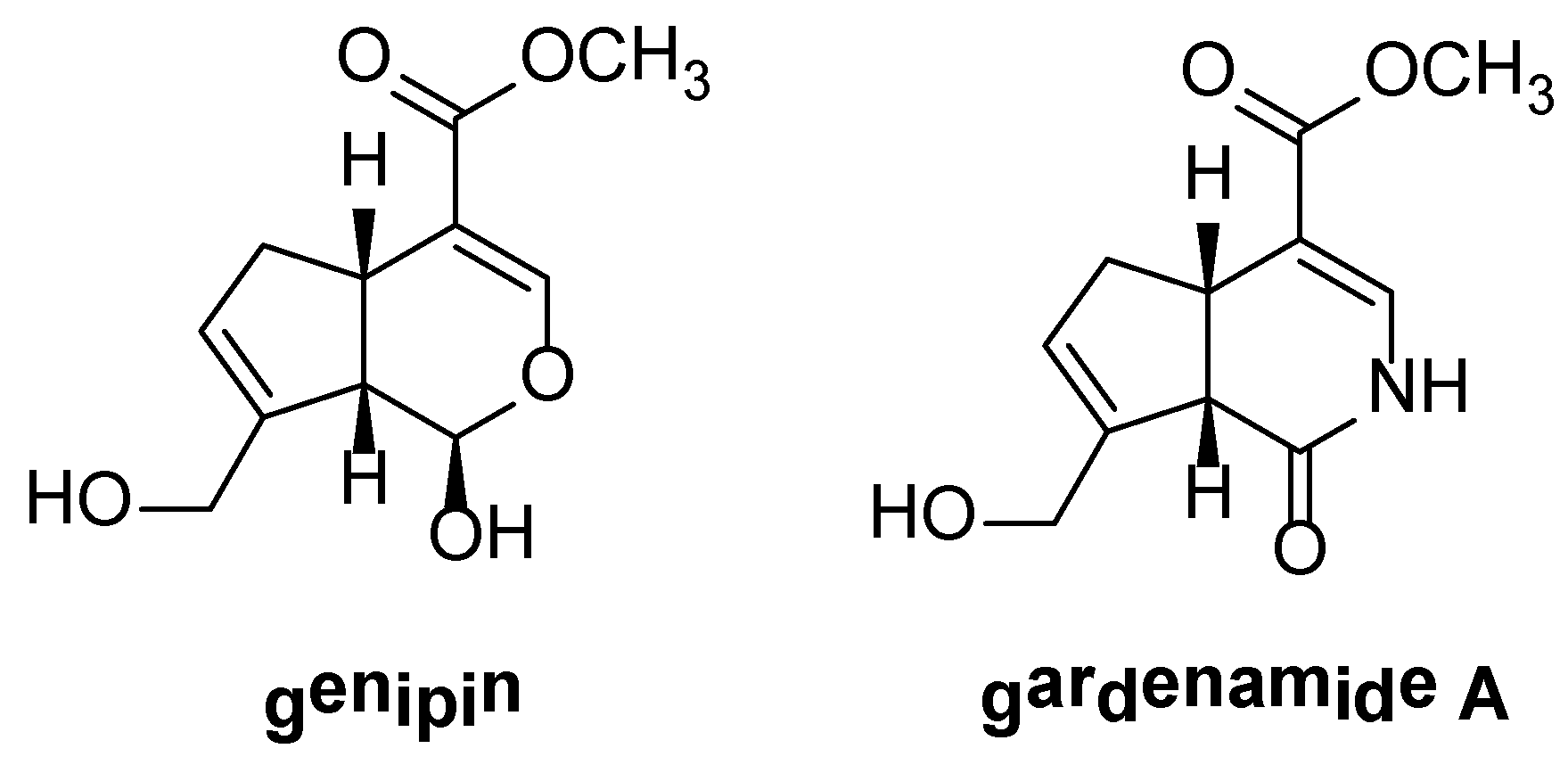
2. Results and Discussion
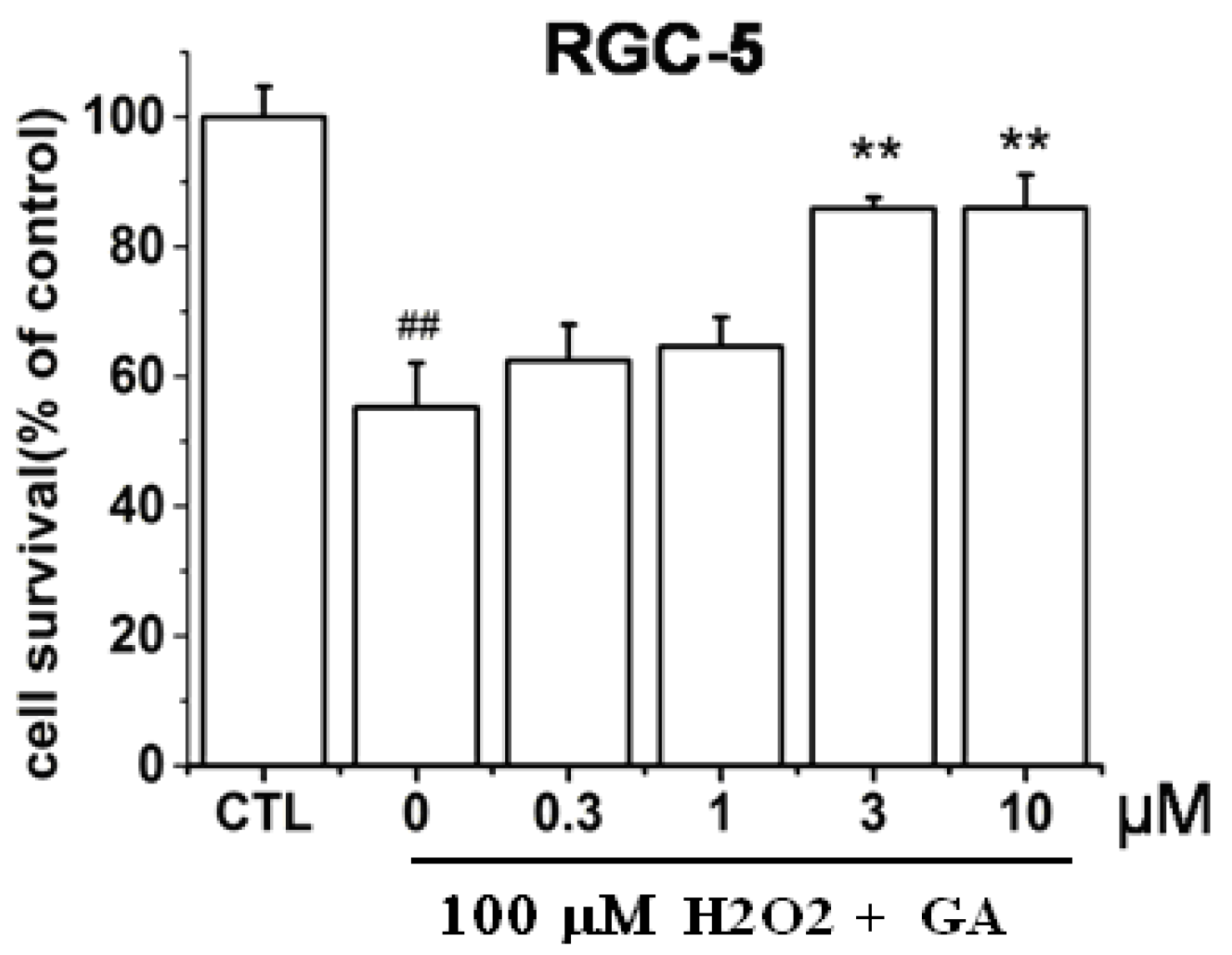
2.1. GA Dose-Dependently Protected RGC-5 Cells from H2O2-Induced Insults

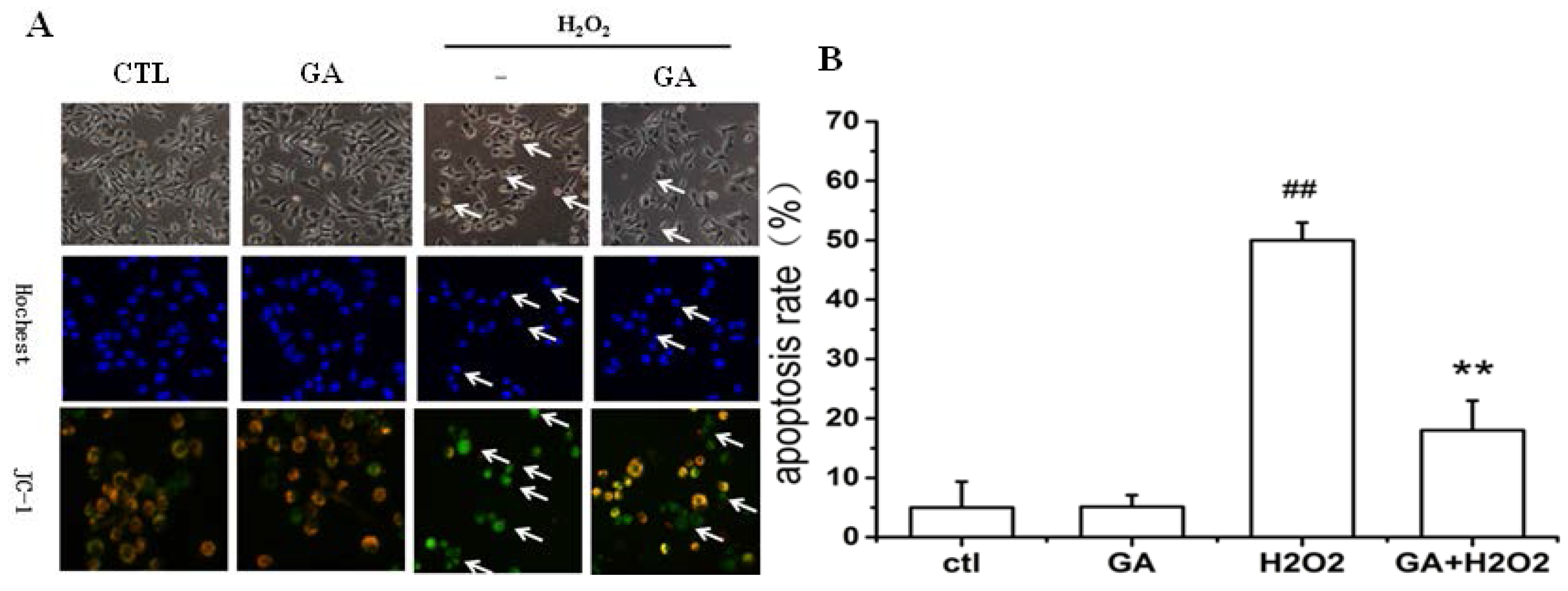
2.2. GA Protected RGC-5 Cells against Apoptosis Induced by H2O2
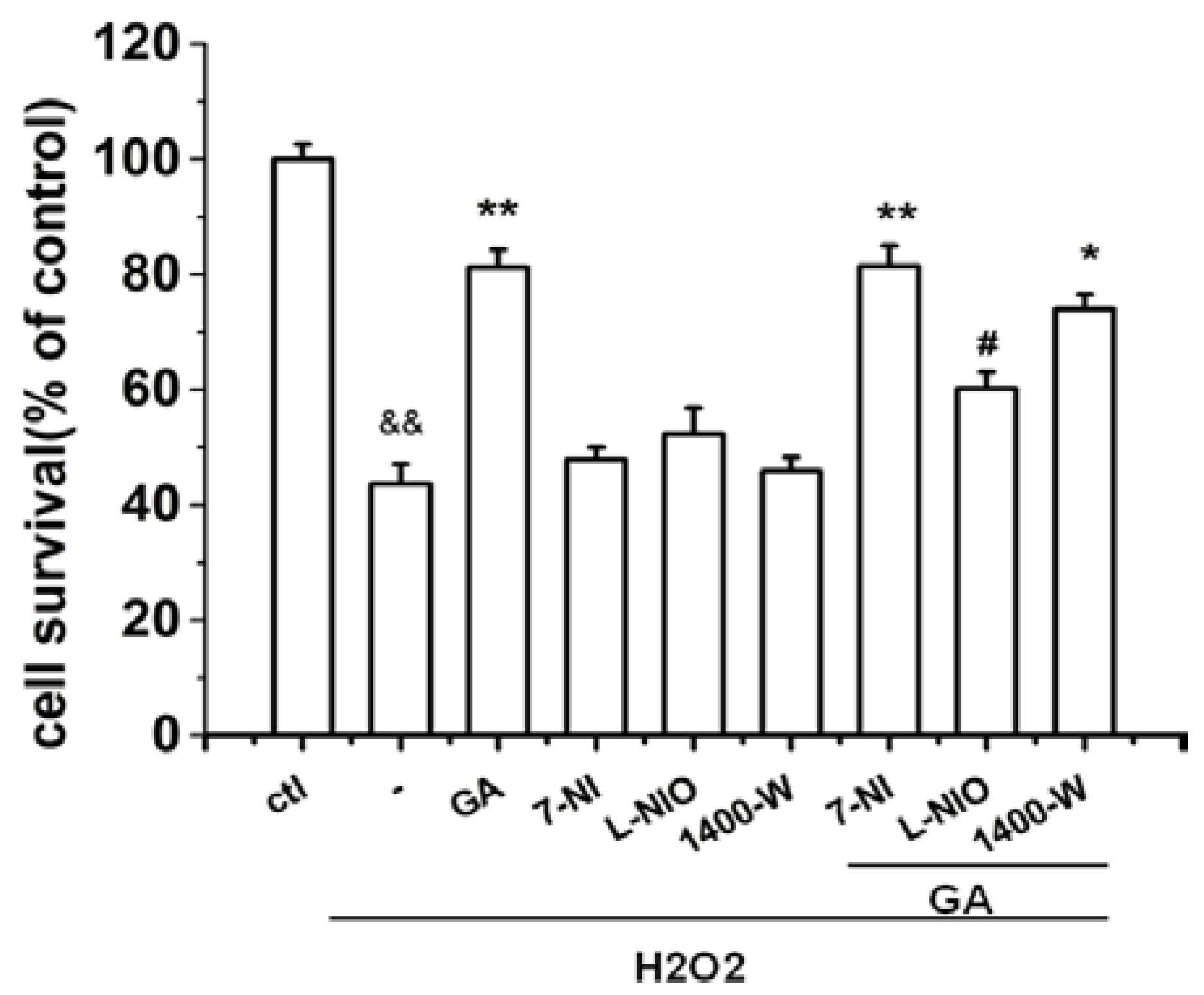
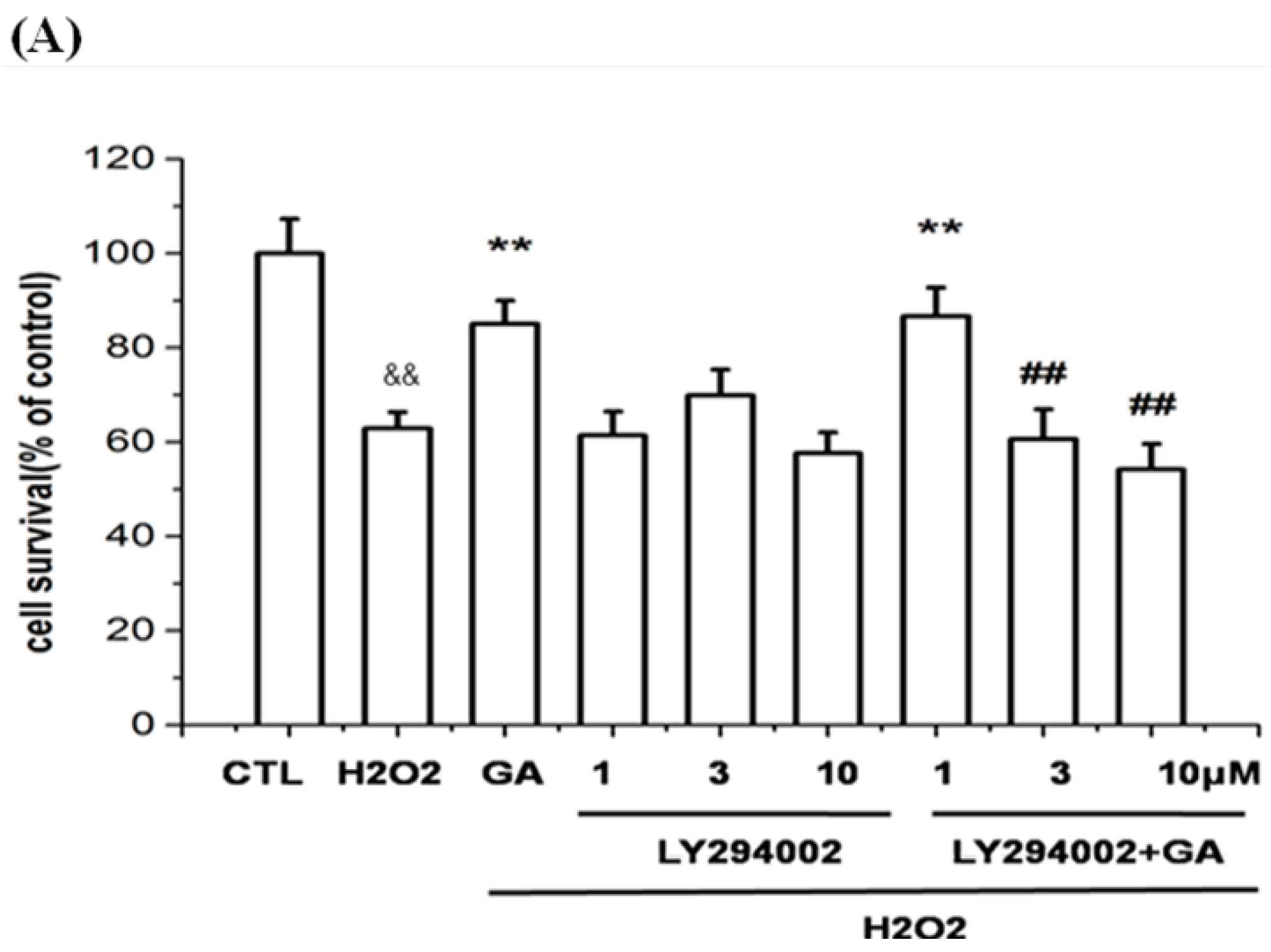
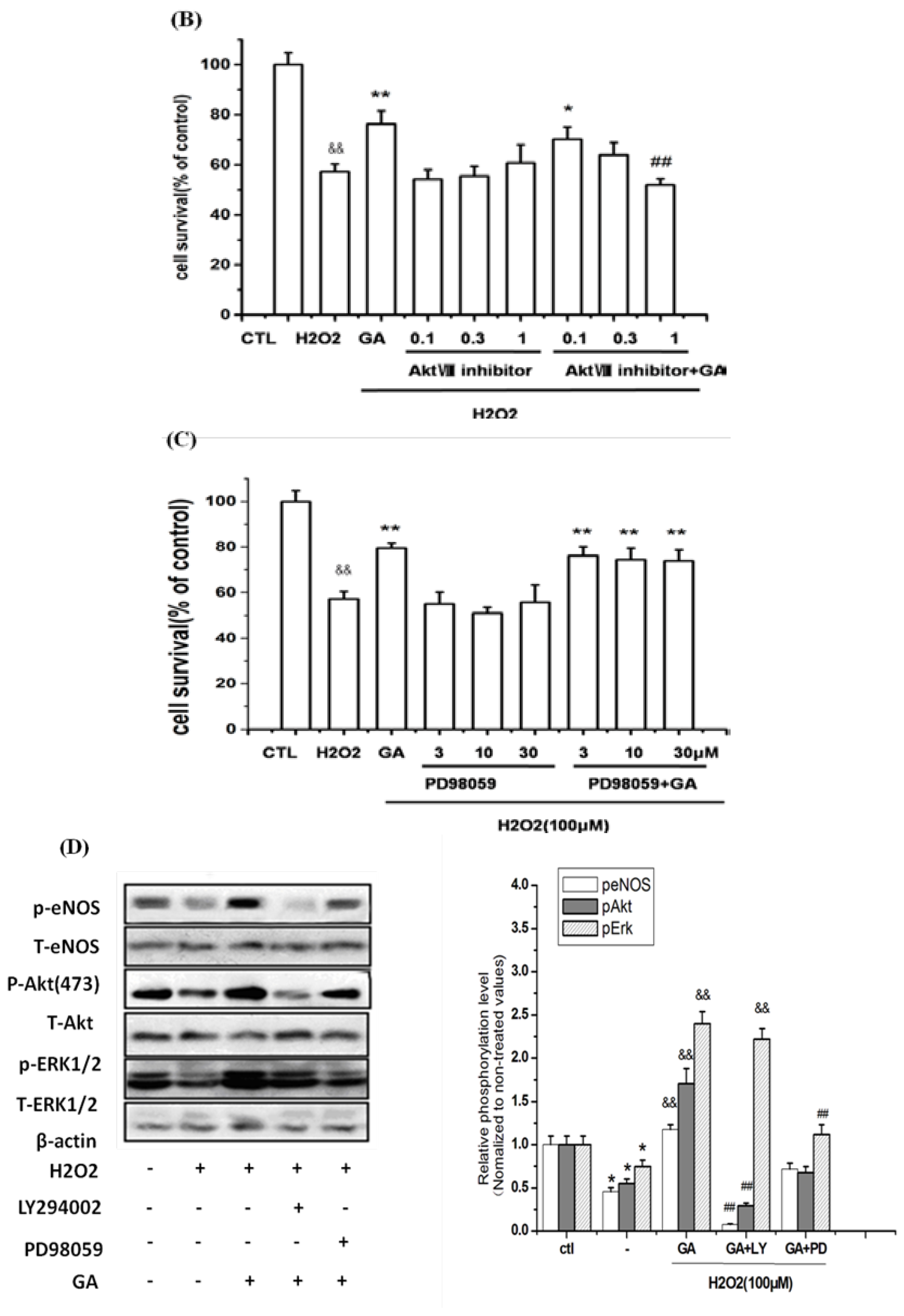
2.3. The Protective Effect of GA on RGC-5 Cells Was Mediated by Endothelial Nitrioxide Synthase (eNOS)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | tNOS (U/mL) | cNOS (U/mL) | eNOS (U/mL) | iNOS (U/mL) |
|---|---|---|---|---|
| ctrl | 8.23 ± 0.43 | 6.42 ± 0.64 | 3.32 ± 0.35 | 1.81 ± 0.33 |
| GA | 9.52 ± 0.34 | 8.36 ± 0.52 && | 4.26 ± 0.22 && | 1.16 ± 0.22 |
| H2O2 | 5.45 ± 0.22 ** | 1.82 ± 0.19 ** | 1.03 ± 0.17 ** | 3.63 ± 0.42 ** |
| H2O2 + GA | 7.51 ± 0.58 ## | 6.20 ± 0.28 ## | 3.14 ± 0.23 ## | 1.31 ± 0.21 ## |
2.4. GA Time- and Dose-Dependently Stimulated the Phosphorylation of ERK1/2 and Akt in RGC-5 Cells
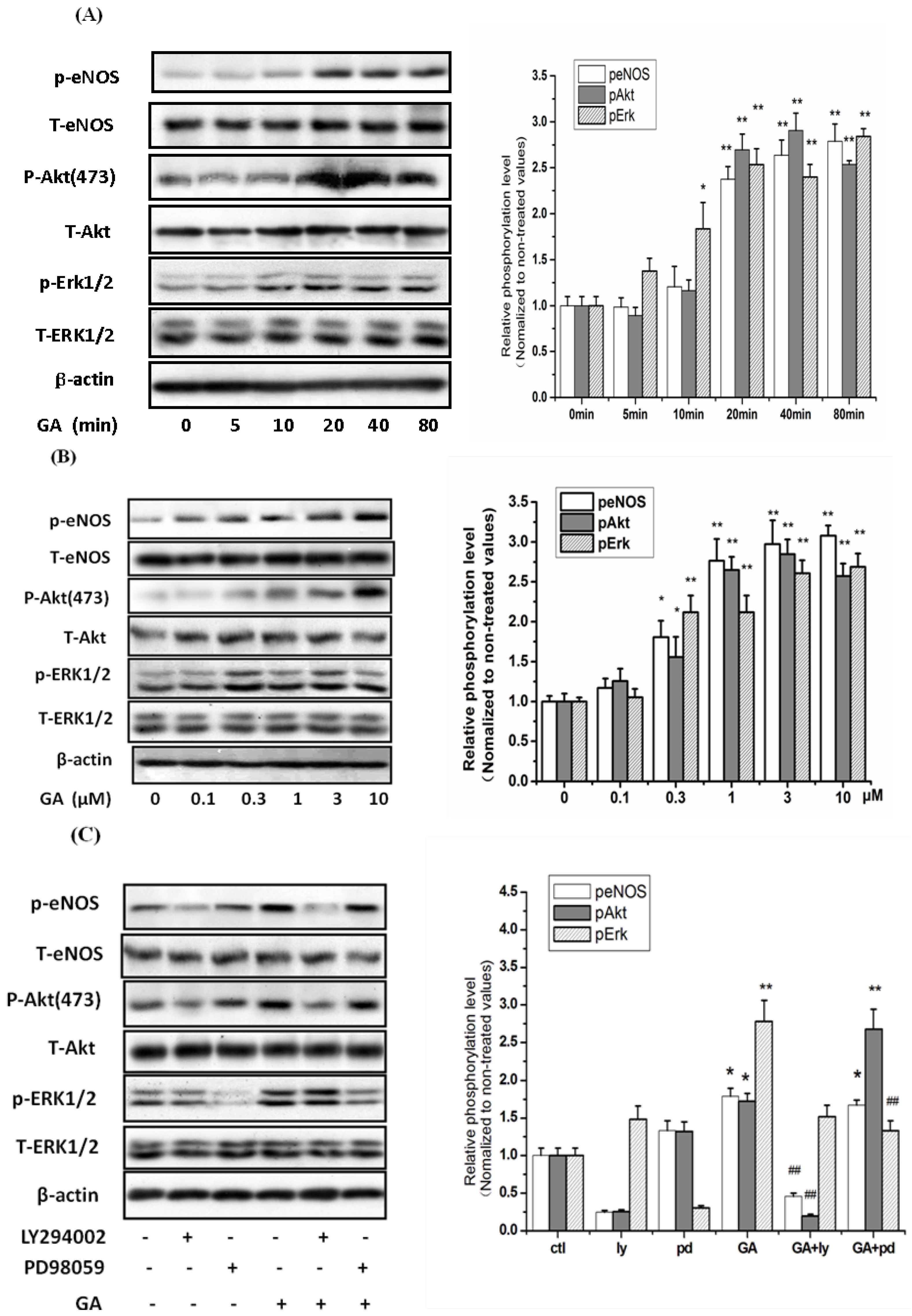
2.5. Neuroprotective Action of GA against H2O2-Induced Impairments to RGC-5 Cells Was Mediated by the Activation of the PI3K/Akt Signaling Pathway
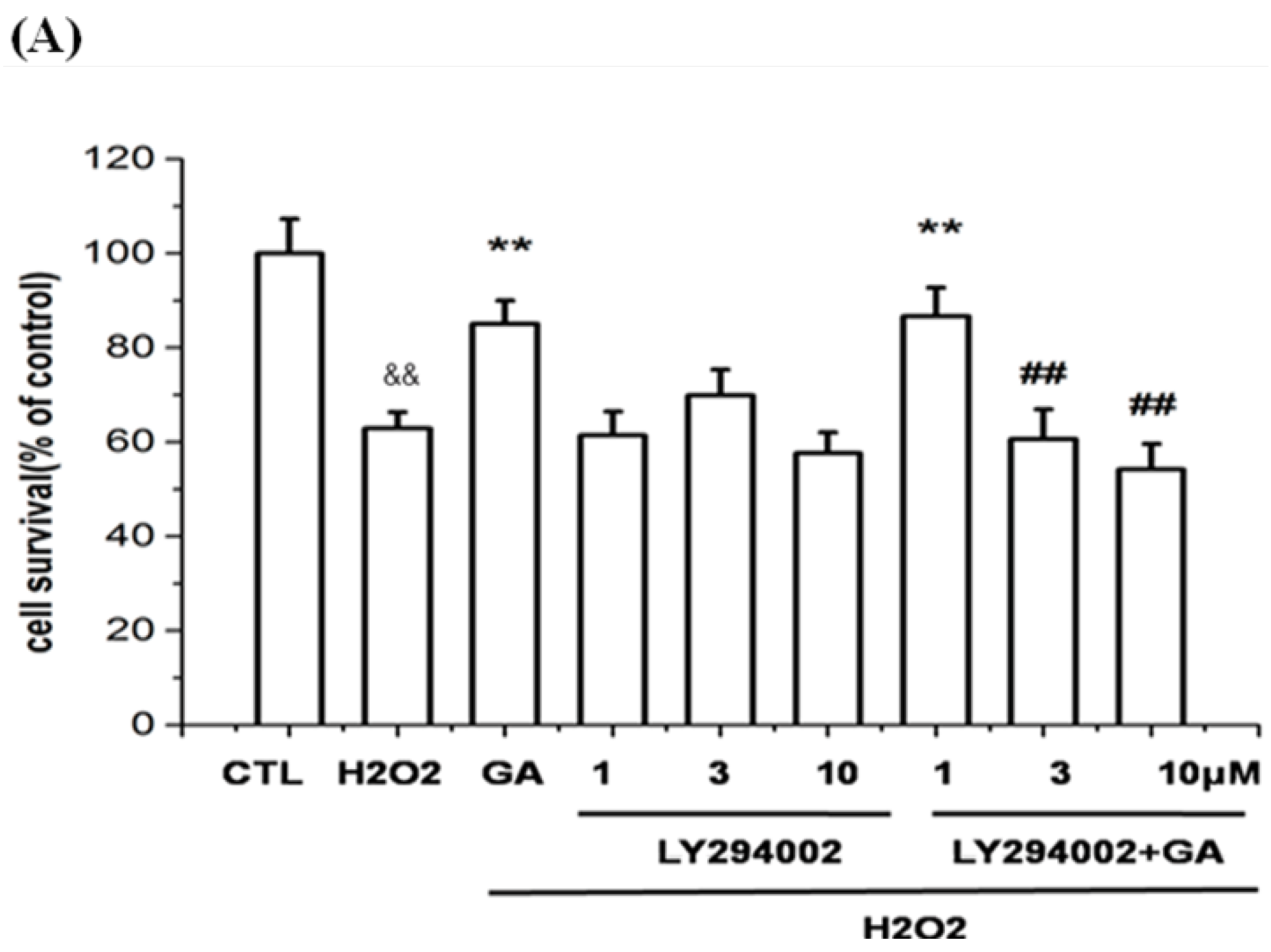
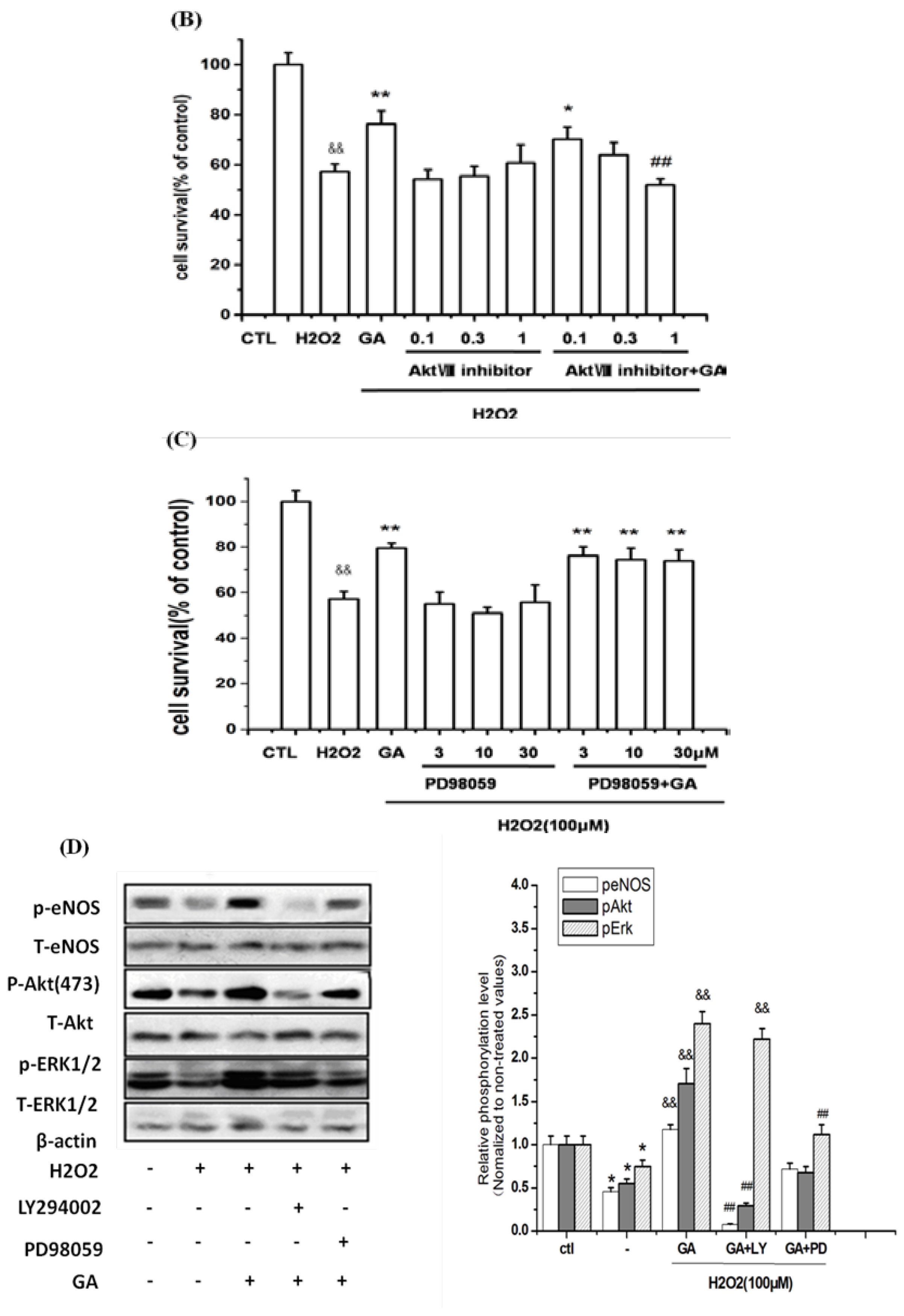
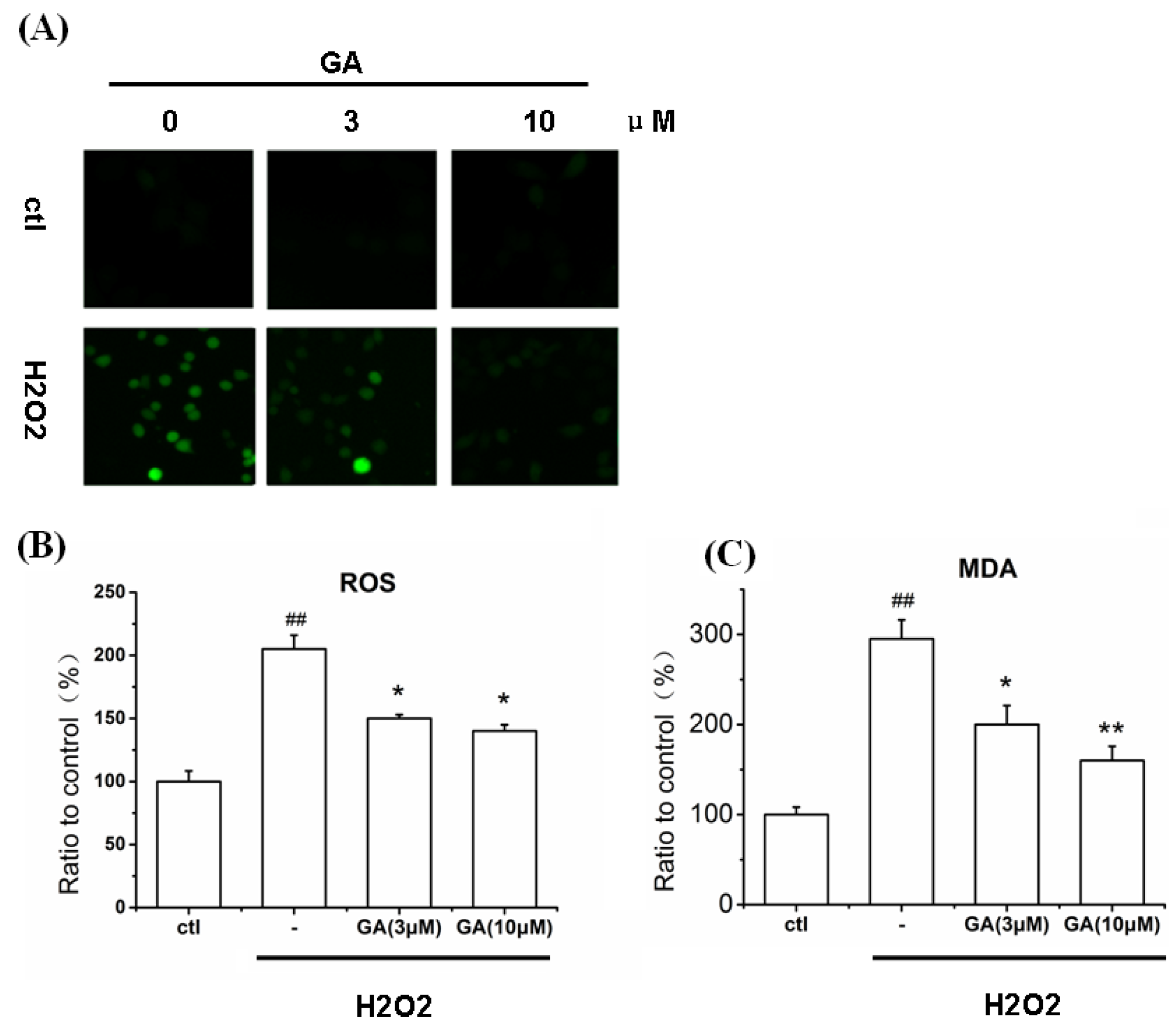
2.6. GA Blocked ROS Production and Lipid Peroxidation Induced by H2O2 in RGC-5 Cells

2.7. Discussion
2.7.1. GA Decreased ROS and MDA Levels in RGC-5 Cells Induced by H2O2
2.7.2. GA Promoted Survival of RGC-5 by Activating eNOS
2.7.3. GA Promoted Survival of RGC-5 by Activation of the PI3K/Akt/eNOS Signaling Pathway
3. Experimental Section
3.1. Chemicals and Reagents
3.2. Cell Culture
3.3. MTT Assay
3.4. Detection of Apoptosis by Hoechst Staining
3.5. Western Blotting
3.6. Mitochondrial Membrane Potential Determination
3.7. Measurement of Intracellular ROS Generation
3.8. Estimation of Lipid Peroxidation
3.9. Determination of NOS Activity
3.10. Data Analysis and Statistics
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Al-Ayadhi, L.Y. Oxidative stress and neurodegenerative disease. Neurosciences 2004, 9, 19–23. [Google Scholar] [PubMed]
- Zawia, N.H.; Lahiri, D.K.; Cardozo-Pelaez, F. Epigenetics, oxidative stress, and Alzheimer disease. Free Radic. Biol. Med. 2009, 46, 1241–1249. [Google Scholar] [CrossRef] [PubMed]
- Beatty, S.; Koh, H.H.; Henson, D.; Boulton, M. The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv. Ophthalmol. 2000, 45, 115–134. [Google Scholar] [CrossRef]
- Cai, J.Y.; Nelson, K.C.; Wu, M.; Sternberg, P.; Jones, D.P. Oxidative damage and protection of the RPE. Prog. Retin. Eye Res. 2000, 19, 205–221. [Google Scholar] [CrossRef]
- Yildirim, Z.; Ucgun, N.I.; Yildirim, F. The role of oxidative stress and antioxidants in the pathogenesis of age-related macular degeneration. Clinics 2011, 66, 743–746. [Google Scholar] [PubMed]
- Ye, J.; Han, Y.; Chen, X.; Xie, J.; Liu, X.; Qiao, S.; Wang, C. L-Carnitine attenuates H2O2-induced neuron apoptosis via inhibition of endoplasmic reticulum stress. Neurochem. Int. 2014, 78, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Wei, T.; Tian, W.; Yan, H.; Shao, G.; Xie, G. Protective Effects of Phillyrin on H2O2-induced Oxidative Stress and Apoptosis in PC12 Cells. Cell. Mol. Neurobiol. 2014, 34, 1165–1173. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Oxidative stress and neurodegeneration: Where are we now? J. Neurochem. 2006, 97, 1634–1658. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, V.; Cornelius, C.; Dinkova-Kostova, A.T.; Calabrese, E.J.; Mattson, M.P. Cellular stress responses, the hormesis paradigm, and vitagenes: Novel targets for therapeutic intervention in neurodegenerative disorders. Antioxid. Redox Signal. 2010, 11, 1763–1811. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, N.; Ghosh, R.; Mandal, S.C. Antioxidant protection: A promising therapeutic intervention in neurodegenerative disease. Free Radic. Res. 2011, 8, 888–905. [Google Scholar] [CrossRef] [PubMed]
- Machida, K.; Oyama, K.; Ishii, M.; Kakuda, R.; Yaoita, Y.; Kikuchi, M. Studies of the constituents of Gardenia species. II. Terpenoids from Gardeniae Fructus. Chem. Pharm. Bull. 2000, 48, 746–748. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Wang, R.; Huang, Z.; Yang, J.; Yao, X.; Chen, H.; Zheng, W. Synthesis of stable genipin derivatives and studies of their neuroprotective activity in PC12 cells. ChemMedChem 2012, 7, 1661–1668. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.H.; Kar, S.; Quirion, R. Insulin-like growth factor-1-induced phosphorylation of transcription factor FKHRL1 is mediated by phosphatidylinositol 3-kinase/Akt kinase and role of this pathway in insulin-like growth factor-1-induced survival of cultured hippocampal neurons. Mol. Pharmacol. 2002, 62, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.R.; Dudek, H.; Tao, X.; Masters, S.; Fu, H.; Gotoh, Y.; Greenberg, M.E. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997, 91, 231–241. [Google Scholar] [CrossRef]
- Michell, B.J.; Griffiths, J.E.; Mitchelhill, K.I.; Rodriguez-Crespo, I.; Tiganis, T.; Bozinovski, S.; de Montellano, P.R.; Kemp, B.E.; Pearson, R.B. The Akt kinase signals directly to endothelial nitric oxide synthase. Curr. Biol. 1999, 9, 845–848. [Google Scholar] [CrossRef]
- Zheng, W.; Wang, H.; Zeng, Z.; Lin, J.; Little, P.J.; Srivastava, L.K.; Quirion, R. The possible role of the Akt signaling pathway in schizophrenia. Brain Res. 2012, 1470, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Ho, F.M.; Lin, W.W.; Chen, B.C.; Chao, C.M.; Yang, C.R.; Lin, L.Y.; Lai, C.C.; Liu, S.H.; Lian, C.S. High glucose-induced apoptosis in human vascular endothelial cells is mediated through NF-κB and c-Jun NH2-terminal kinase pathway and prevented by PI3K/Akt/eNOS pathway. Cell Signal. 2006, 18, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.X.; Zhou, B.; Chen, Z.; Ren, Q.; Lu, S.H.; Sawamura, T.; Han, Z.C. Oxidized low density lipoprotein impairs endothelial progenitor cells by regulation of endothelial nitric oxide synthase. J. Lipid Res. 2006, 47, 1227–1237. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, R.R.; Clark, A.F.; Daudt, D.; Vishwanatha, J.K.; Yorio, T. A forensic path to RGC-5 cell line identification: Lessons learned. Investig. Ophthalmol. Vis. Sci. 2013, 54, 5712–5719. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Gao, E.; Yue, T.L.; Ohlstein, E.H.; Lopez, B.L.; Christopher, T.A.; Ma, X.L. Nitric oxide mediates the antiapoptotic effect of insulin in myocardial ischemia-reperfusion: The roles of PI3-kinase, Akt, and endothelial nitric oxide synthase phosphorylation. Circulation 2002, 105, 1497–1502. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, M.; Chiba, K. Cyclic GMP-dependent neurite outgrowth by genipin and nerve growth factor in PC12h cells. Eur. J. Pharmacol. 2008, 581, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.H.; Kar, S.; Quirion, R. FKHRL1 and its homologs are new targets of nerve growth factor Trk receptor signaling. J. Neurochem. 2002, 80, 1049–1061. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Cawley, N.Y.; Loh, Y.P. Carboxypeptidase E/NFα1: A new neurotrophic factor against oxidative stress-induced apoptotic cell death mediated by ERK and PI3-K/AKT pathways. PLoS ONE 2013, 8, e71578. [Google Scholar] [CrossRef] [PubMed]
- Schafer, M.; Goodenough, S.; Moosmann, B.; Behl, C. Inhibition of glycogen synthase kinase 3 beta is involved in the resistance to oxidative stress in neuronal HT22 cells. Brain Res. 2004, 1005, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Jiang, R.; Zhu, X.; Zhang, X.; Zhan, Z. Genipin inhibits TNF-alpha-induced vascular smooth muscle cell proliferation and migration via induction of HO-1. PLoS ONE 2013, 8, e74826. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.A.; Kanuga, N.; Romero, I.A.; Greenwood, J.; Luthert, P.J.; Cheetham, M.E. Oxidative stress affects the junctional integrity of retinal pigment epithelial cells. Investig. Ophthalmol. Vis. Sci. 2004, 45, 675–684. [Google Scholar] [CrossRef]
- Usui, S.; Oveson, B.C.; Lee, S.Y.; Jo, Y.J.; Yoshida, T.; Miki, A.; Miki, K.; Iwase, T.; Lu, L.; Campochiaro, P.A. NADPH oxidase plays a central role in cone cell death in retinitis pigmentosa. J. Neurochem. 2009, 110, 1028–1037. [Google Scholar] [CrossRef] [PubMed]
- Liang, F.Q.; Godley, B.F. Oxidative stress-induced mitochondrial DNA damage in human retinal pigment epithelial cells: A possible mechanism for RPE aging and age-related macular degeneration. Exp. Eye Res. 2003, 76, 397–403. [Google Scholar] [CrossRef]
- Paradies, G.; Petrosillo, G.; Paradies, V.; Ruggiero, F.M. Mitochondrial dysfunction in brain aging: Role of oxidative stress and cardiolipin. Neurochem. Int. 2011, 58, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Kim, Y.; Chung, J. Mitochondrial dysfunction and Parkinson’s disease genes: Insights from Drosophila. Dis. Model. Mech. 2009, 2, 336–340. [Google Scholar] [CrossRef] [PubMed]
- Morais, V.A.; de Strooper, B. Mitochondria dysfunction and neurodegenerative disorders: Cause or Consequence. J. Alzheimers Dis. 2010, 20, S255–S263. [Google Scholar] [PubMed]
- Xie, W.; Wan, O.W.; Chung, K.K. New insights into the role of mitochondrial dysfunction and protein aggregation in Parkinson’s disease. Biochim. Biophys. Acta 2010, 1802, 935–941. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Yang, J.; Liao, S.; Xiao, G.; Luo, J.; Zhang, L.; Little, P.J.; Chen, H.; Zheng, W. Stereoselective Reduction of 1-O-Isopropyloxygenipin Enhances Its Neuroprotective Activity in Neuronal Cells from Apoptosis Induced by Sodium Nitroprusside. ChemMedChem 2014, 9, 1397–1403. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Yamazaki, M.; Chiba, K.; Sawanishi, H. Characteristic properties of genipin as an activator in neuronal nitric oxide synthase. J. Health Sci. 2007, 53, 730–733. [Google Scholar] [CrossRef]
- Koriyama, Y.; Takagi, Y.; Chiba, K.; Yamazaki, M.; Arai, K.; Matsukawa, T.; Suzuki, K.; Sugitani, H.; Kagechika, H.; Kato, S. Long-acting genipin derivative protects retinal ganglion cells from oxidative stress models in vitro and in vivo through the Nrf2/antioxidant response element signaling pathway. J. Neurochem. 2011, 119, 1232–1242. [Google Scholar] [CrossRef] [PubMed]
- Koriyama, Y.; Chiba, K.; Yamazaki, M.; Suzuki, H.; Muramoto, K.; Kato, S. Neuritogenic activity of a genipin derivative in retinal ganglion cells is mediated by retinoic acid receptor beta expression through nitric oxide/S-nitrosylation signaling. J. Neurochem. 2010, 115, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Li, W.M.; Pi, R.B.; Chan, H.H.N.; Fu, H.J.; Lee, N.T.K.; Tsang, H.W.; Pu, Y.M.; Chang, D.C.; Li, C.Y.; Luo, J.L.; et al. Novel dimeric acetylcholinesterase inhibitor bis(7)-tacrine, but not donepezil, prevents glutamate-induced neuronal apoptosis by blocking n-methyl-d-aspartate receptors. J. Biol. Chem. 2005, 280, 18179–18188. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.J.; Nie, A.F.; Zhang, Z.J.; Zhang, Z.G.; Du, L.; Li, X.Y.; Ning, G. SIRT1 represses estrogen-signaling, ligand-independent ERα-mediated transcription, and cell proliferation in estrogen-responsive breast cells. J. Endocrinol. 2013, 216, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Chen, H.; Qiao, B.; Luo, L.; Ma, H.; Li, H.; Jiang, J.; Niu, D.; Yin, Z. Opposing effects of ERK and p38 MAP kinases on HeLa cell apoptosis induced by dipyrithione. Mol. Cells 2007, 23, 30–38. [Google Scholar] [PubMed]
- Han, B.; Wei, W.; Hua, F.; Cao, T.; Dong, H.; Yang, T.; Yang, Y.; Pan, H.; Xu, C. Requirement for ERK activity in sodium selenite-induced apoptosis of acute promyelocytic leukemia-derived NB4 cells. J. Biochem. Mol. Biol. 2007, 40, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, M.; Chiba, K.; Mohri, T. Fundamental role of nitric oxide in neuritogenesis of PC12h cells. Br. J. Pharmacol. 2005, 146, 662–669. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, M.; Chiba, K.; Mohri, T.; Hatanaka, H. Genipin exhibits neurotrophic effects through a common signaling pathway in nitric oxide synthase-expressing cells. Eur. J. Pharmacol. 2004, 488, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.H.; Quirion, R. Glutamate acting on N-methyl-d-aspartate receptors attenuates insulin-like growth factor-1 receptor tyrosine phosphorylation and its survival signaling properties in rat hippocampal neurons. J. Biol. Chem. 2009, 284, 855–861. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, R.; Peng, L.; Zhao, J.; Zhang, L.; Guo, C.; Zheng, W.; Chen, H. Gardenamide A Protects RGC-5 Cells from H2O2-Induced Oxidative Stress Insults by Activating PI3K/Akt/eNOS Signaling Pathway. Int. J. Mol. Sci. 2015, 16, 22350-22367. https://doi.org/10.3390/ijms160922350
Wang R, Peng L, Zhao J, Zhang L, Guo C, Zheng W, Chen H. Gardenamide A Protects RGC-5 Cells from H2O2-Induced Oxidative Stress Insults by Activating PI3K/Akt/eNOS Signaling Pathway. International Journal of Molecular Sciences. 2015; 16(9):22350-22367. https://doi.org/10.3390/ijms160922350
Chicago/Turabian StyleWang, Rikang, Lizhi Peng, Jiaqiang Zhao, Laitao Zhang, Cuiping Guo, Wenhua Zheng, and Heru Chen. 2015. "Gardenamide A Protects RGC-5 Cells from H2O2-Induced Oxidative Stress Insults by Activating PI3K/Akt/eNOS Signaling Pathway" International Journal of Molecular Sciences 16, no. 9: 22350-22367. https://doi.org/10.3390/ijms160922350
APA StyleWang, R., Peng, L., Zhao, J., Zhang, L., Guo, C., Zheng, W., & Chen, H. (2015). Gardenamide A Protects RGC-5 Cells from H2O2-Induced Oxidative Stress Insults by Activating PI3K/Akt/eNOS Signaling Pathway. International Journal of Molecular Sciences, 16(9), 22350-22367. https://doi.org/10.3390/ijms160922350