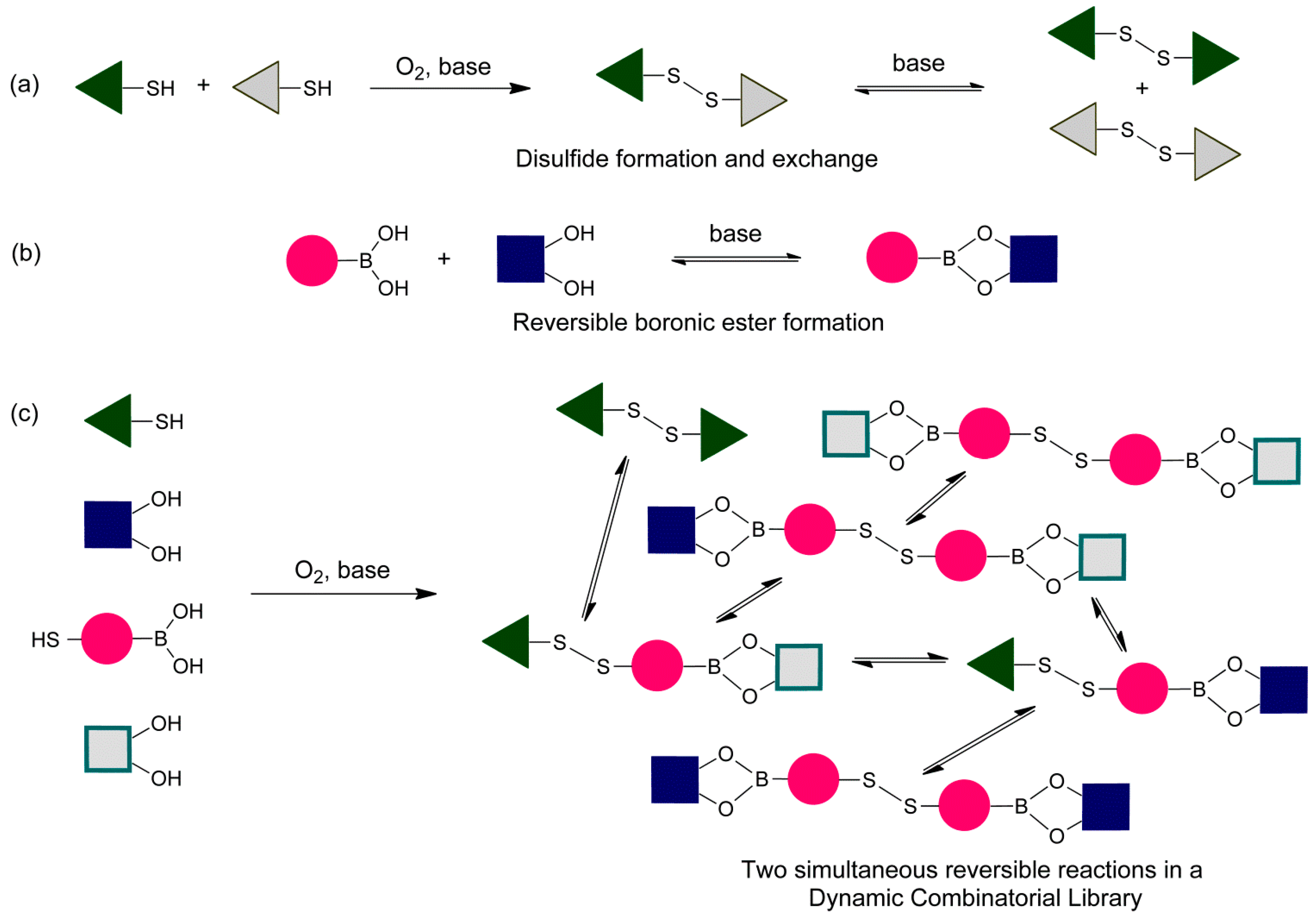
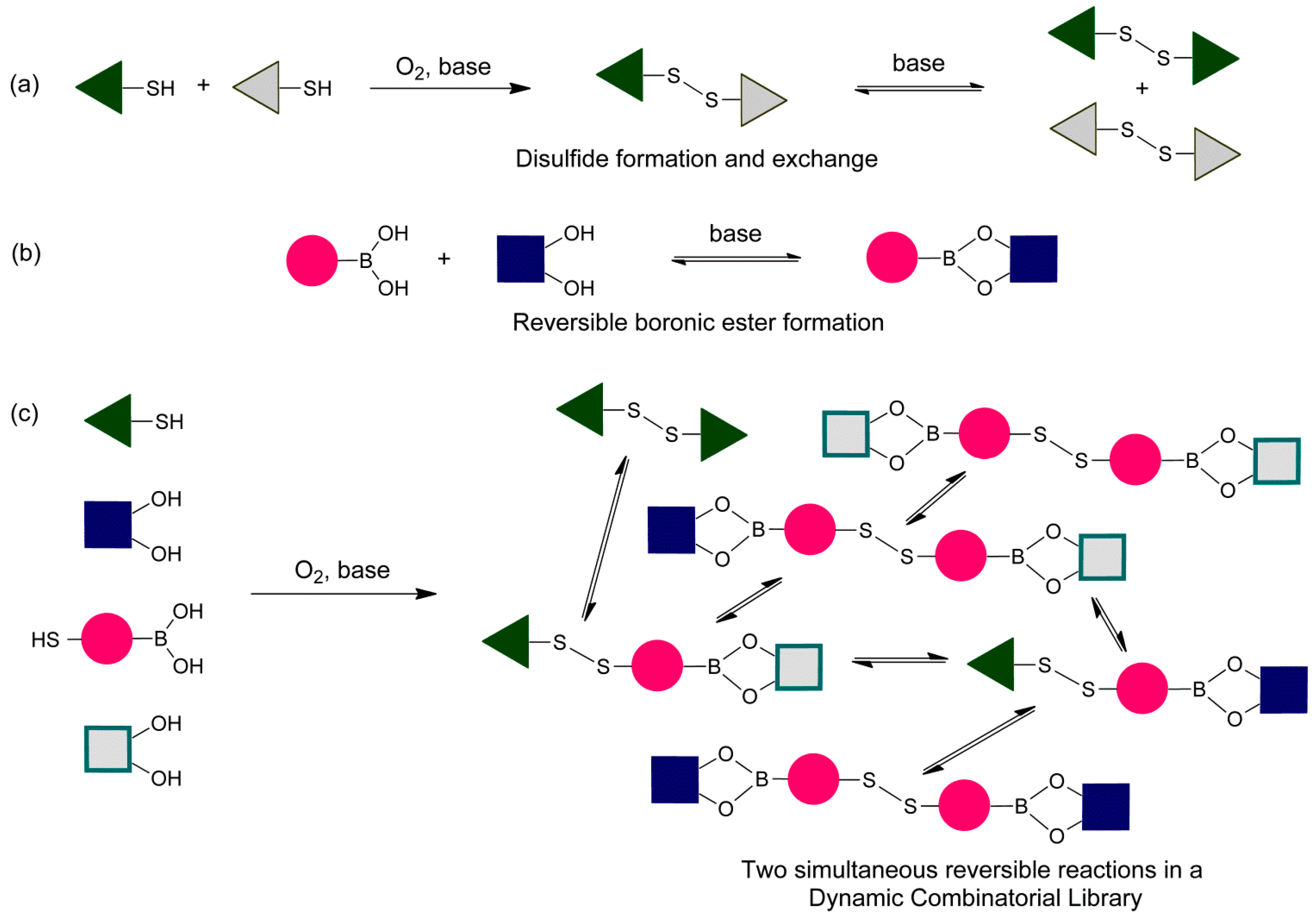
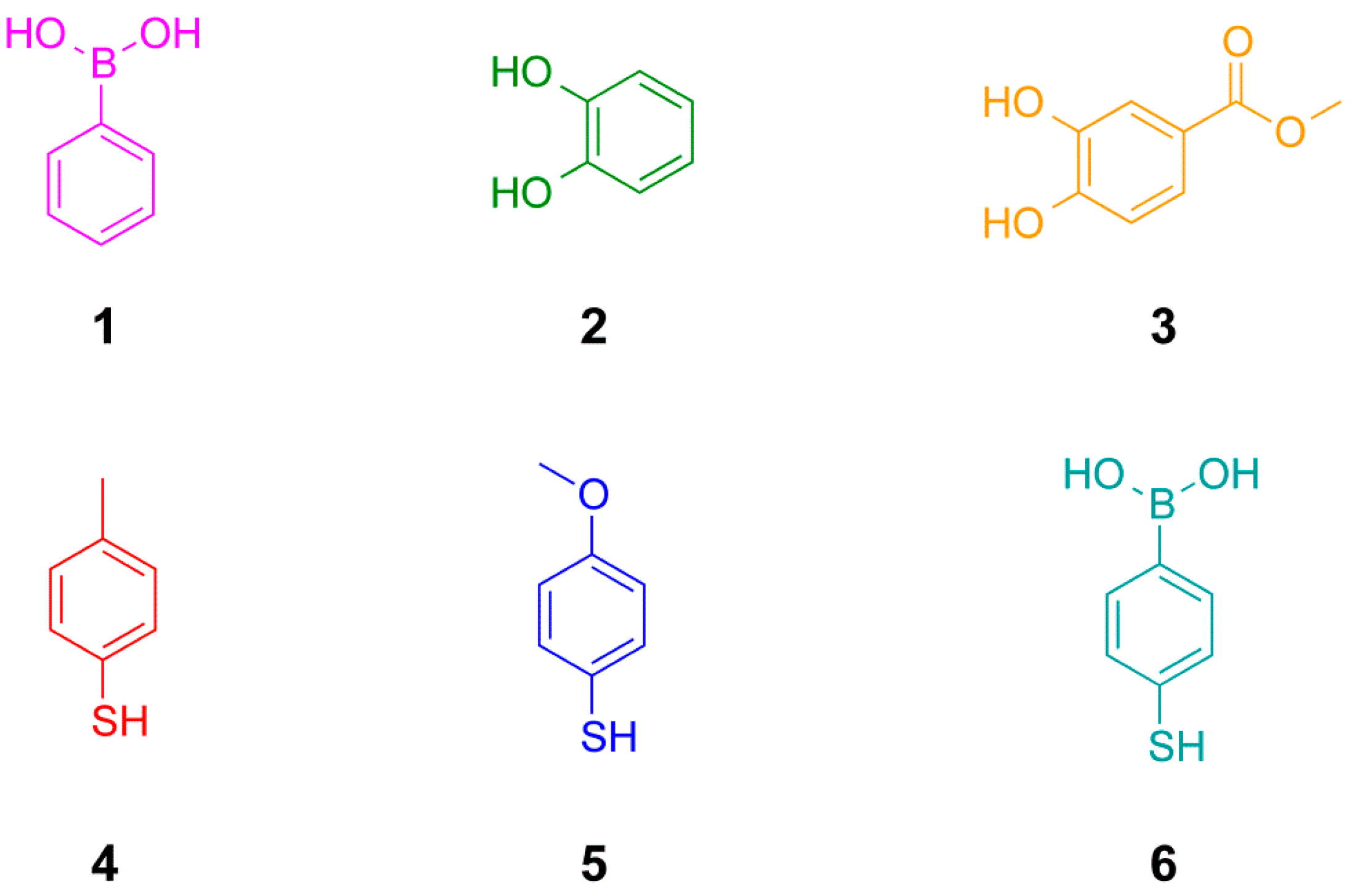
We have chosen to use phenylboronic acids, catechols (1,2-dihydroxy benzenes) and aromatic thiols to develop the conditions for the simultaneous exchange of disulfide and boronic ester building blocks in dynamic combinatorial chemistry (
Figure 2). We first examine the individual reactions and established conditions that ensure reversibility and that the libraries equilibrated under thermodynamic control (
Figure 1). We then determine conditions compatible with both reactions. All experiments have been performed using CDCl
3 as the solvent and a number of different experimental conditions have been examined with regards to other additives, different concentrations and bases.
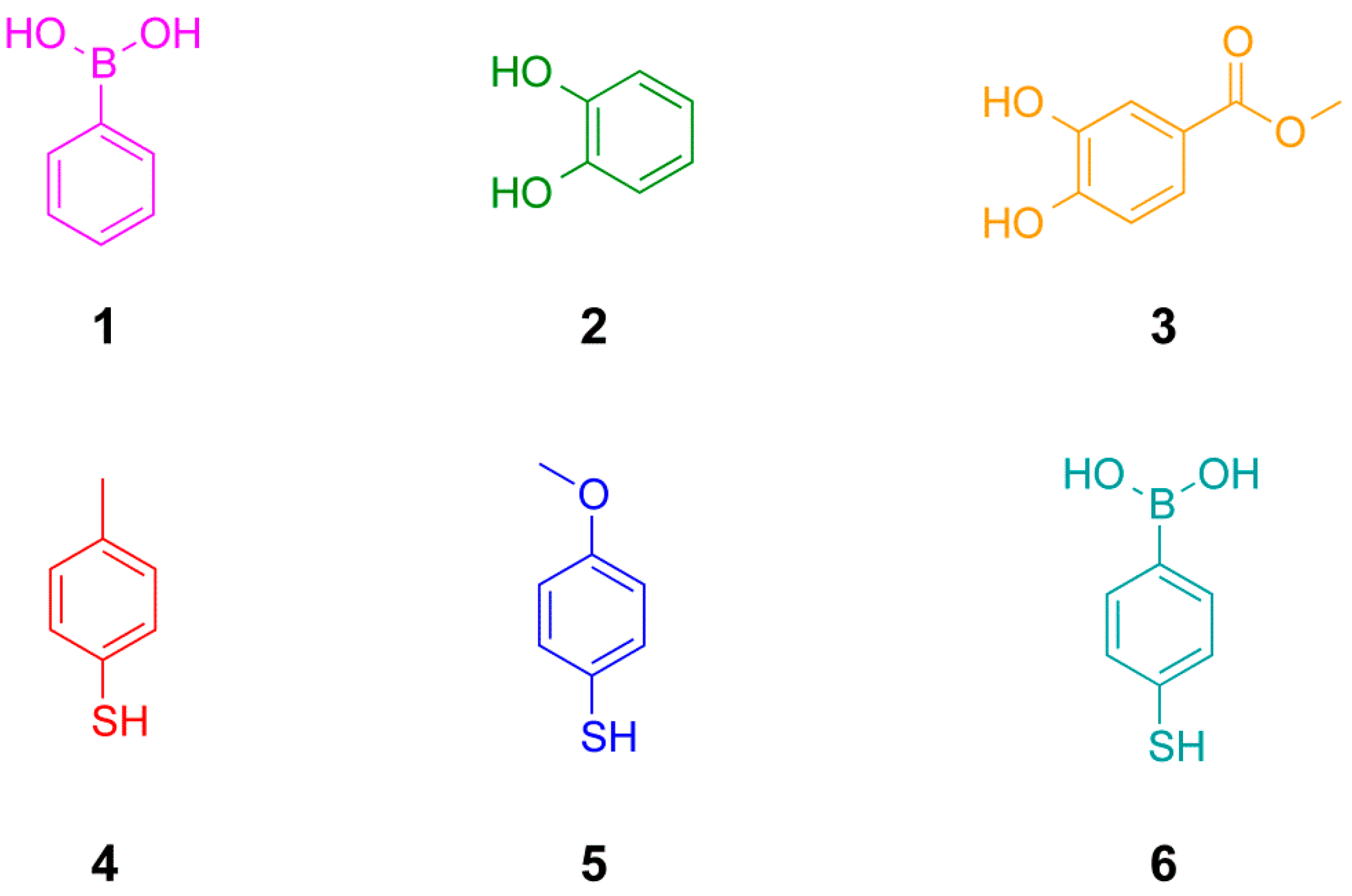
Figure 2.
Structures of building blocks used in this study including phenylboronic acids, catechols (1,2-dihydroxy benzenes) and arylthiols.
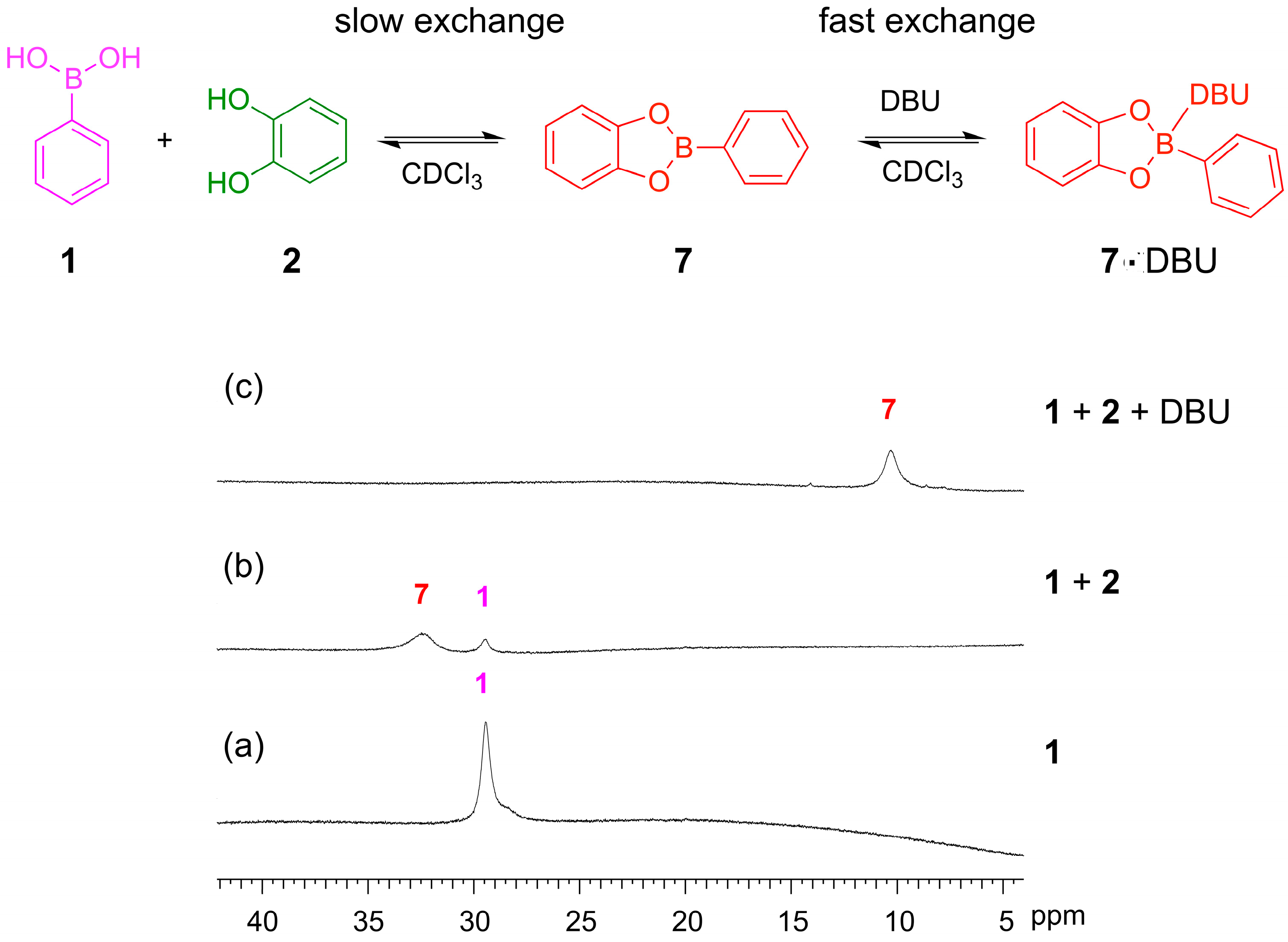
2.1. Boronic Transesterification in CDCl3 Using Et3N as the Base
For the boronic ester exchange, conditions similar to those developed by Kubo
et al. were examined [
27], where phenylboronic acid and a catechol form the boronic acid ester. When recording the
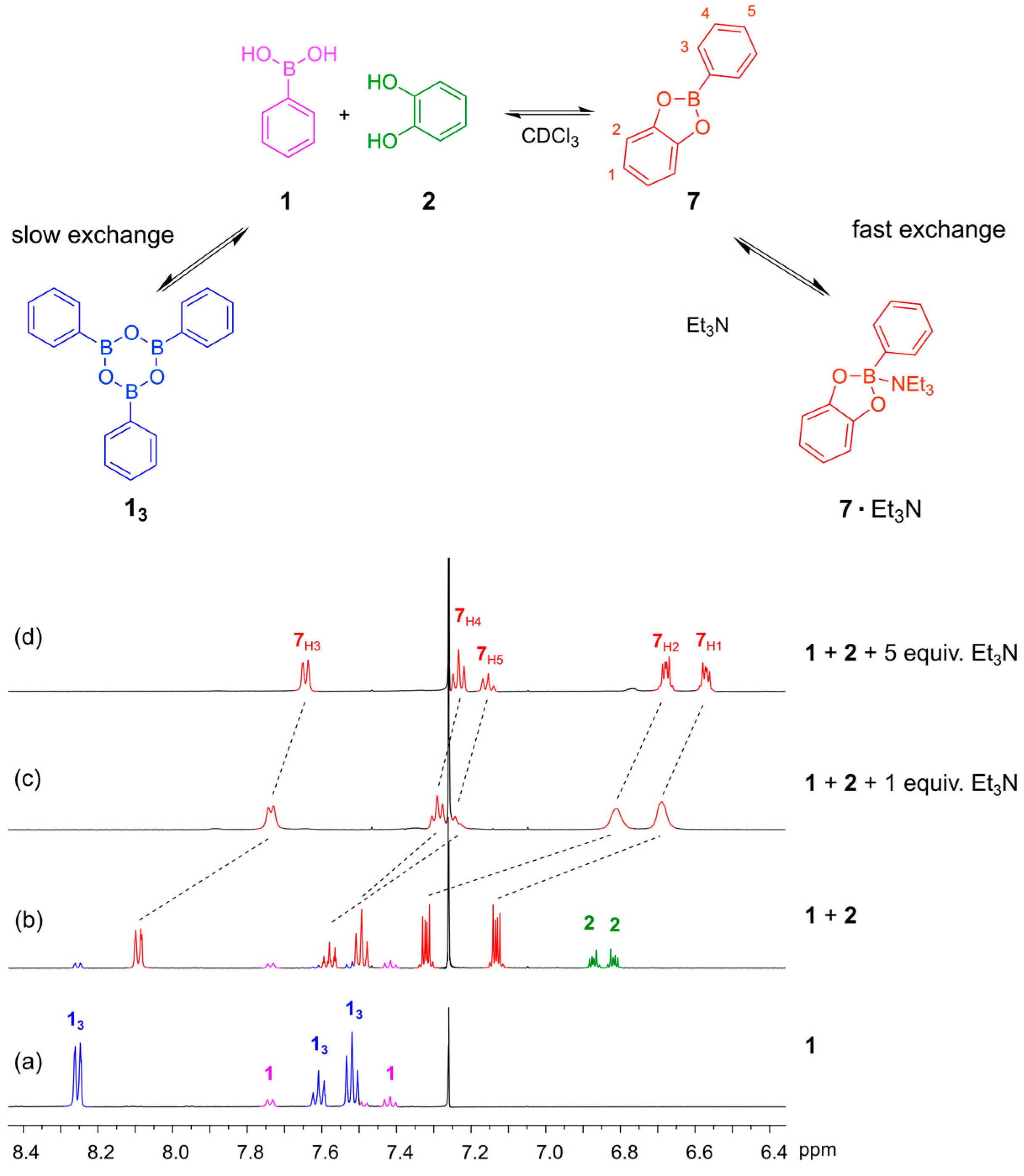
1H-NMR spectra of the boronic acid
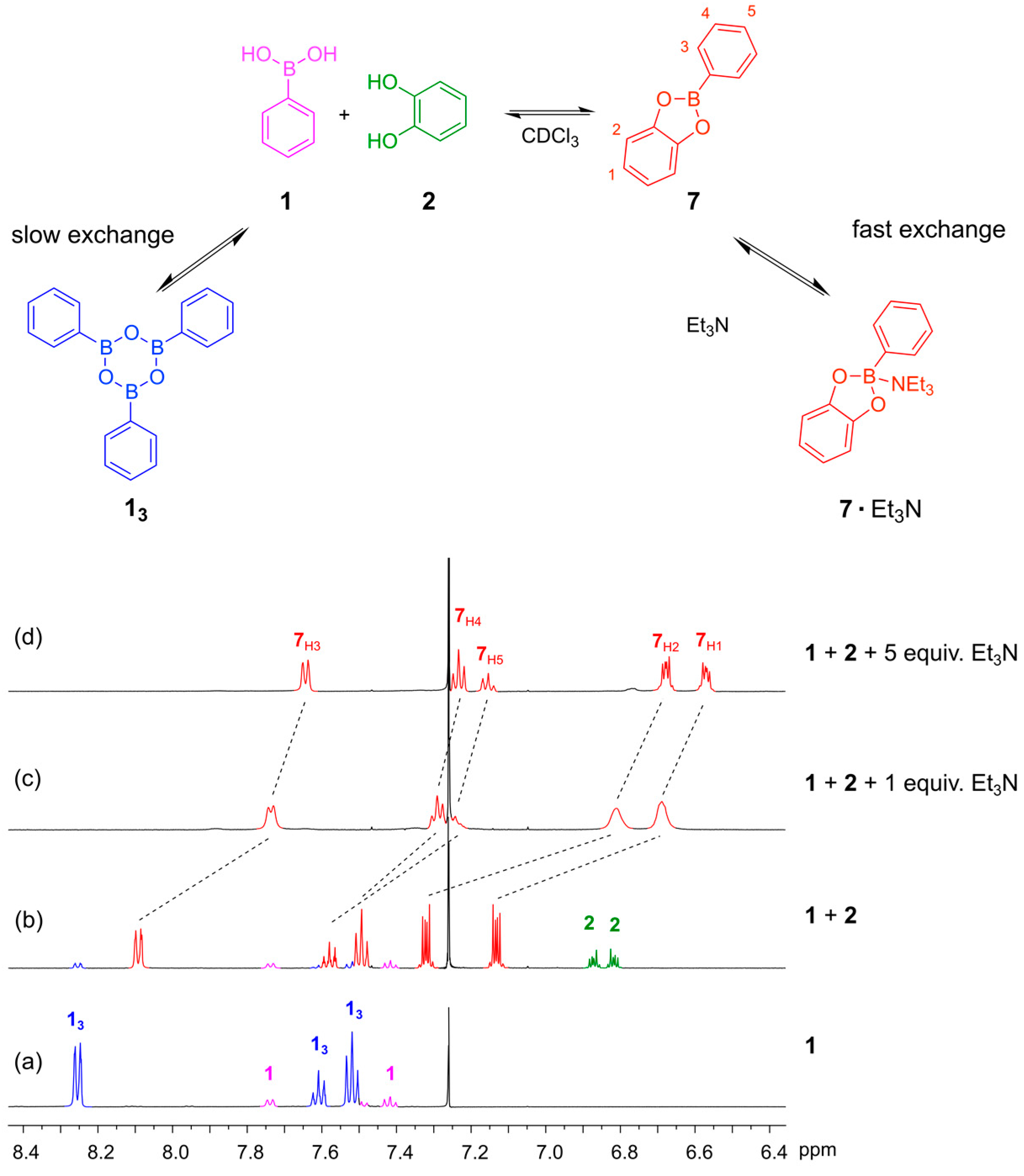
1 before adding the catechol, we observed both the phenylboronic acid and the corresponding boroxine trimer (
13) (
Figure 3a). Triarylboraxines and boronic esters exist in equilibrium with each other, but the equilibrium is completely shifted towards the boronic acid ester in the presence of one equivalent of catechol and a suitable base. We studied the formation of the boronic ester of phenylboronic acid
1 and catechol
2 at 5 mM concentration. This reaction was fast and reached equilibrium within 10 min in CDCl
3. In the absence of base, the boronic ester
7 is formed, but unreacted boronic acid and catechol are both still present, as seen in the
1H-NMR spectrum (
Figure 3b). We proceeded to test how much base was required to push the equilibrium over to the ester. We first examined the use of Et
3N, which has previously been used to promote disulfide exchange in organic solvent, and also tested DMAP (
N,
N’-4-dimethylamino pyridine) and pyridine, which gave similar results. When only one equivalent of Et
3N was added, the ester
7 was formed but the
1H-NMR spectrum showed broad signals, suggesting that the exchange of the free and Et
3N bound
7 occurs at an intermediate rate on the NMR timescale (
Figure 3c). Upon addition of five equivalents of Et
3N, nice well-defined signals were observed in the NMR spectrum, which is important considering that the objective in dynamic combinatorial chemistry is to generate complex mixtures of several compounds (
Figure 3d).
Figure 3.
(Top) The equilibriums involved when mixing phenylboronic acid 1 with catechol 2 in CDCl3 in the presence and absence of Et3N; (Bottom) 1H-NMR spectra (500 MHz, CDCl3, 300 K): (a) Phenyl boronic acid 1 (10 mM). The spectrum shows that 1 and the corresponding trimeric boroxine 13 are both present under these conditions; (b) The reaction mixture after 24 h when 1 and 2 (5 mM each) are mixed in the absence of base. The spectrum shows incomplete conversion to the boronic acid ester; (c) The reaction mixture after 24 h when 1, 2 (4 mM each) and Et3N (4 mM) are combined in CDCl3. The spectrum shows broad feature indicating that the rate of exchange of the 7·Et3N complex is comparable with the NMR chemical shift timescale; (d) The reaction mixture after 24 h when 1 (4 mM), 2 (4 mM) and Et3N (20 mM) are combined in CDCl3. The spectrum shows full conversion to the amine-bound boronic ester 7·Et3N.
Figure 3.
(Top) The equilibriums involved when mixing phenylboronic acid 1 with catechol 2 in CDCl3 in the presence and absence of Et3N; (Bottom) 1H-NMR spectra (500 MHz, CDCl3, 300 K): (a) Phenyl boronic acid 1 (10 mM). The spectrum shows that 1 and the corresponding trimeric boroxine 13 are both present under these conditions; (b) The reaction mixture after 24 h when 1 and 2 (5 mM each) are mixed in the absence of base. The spectrum shows incomplete conversion to the boronic acid ester; (c) The reaction mixture after 24 h when 1, 2 (4 mM each) and Et3N (4 mM) are combined in CDCl3. The spectrum shows broad feature indicating that the rate of exchange of the 7·Et3N complex is comparable with the NMR chemical shift timescale; (d) The reaction mixture after 24 h when 1 (4 mM), 2 (4 mM) and Et3N (20 mM) are combined in CDCl3. The spectrum shows full conversion to the amine-bound boronic ester 7·Et3N.
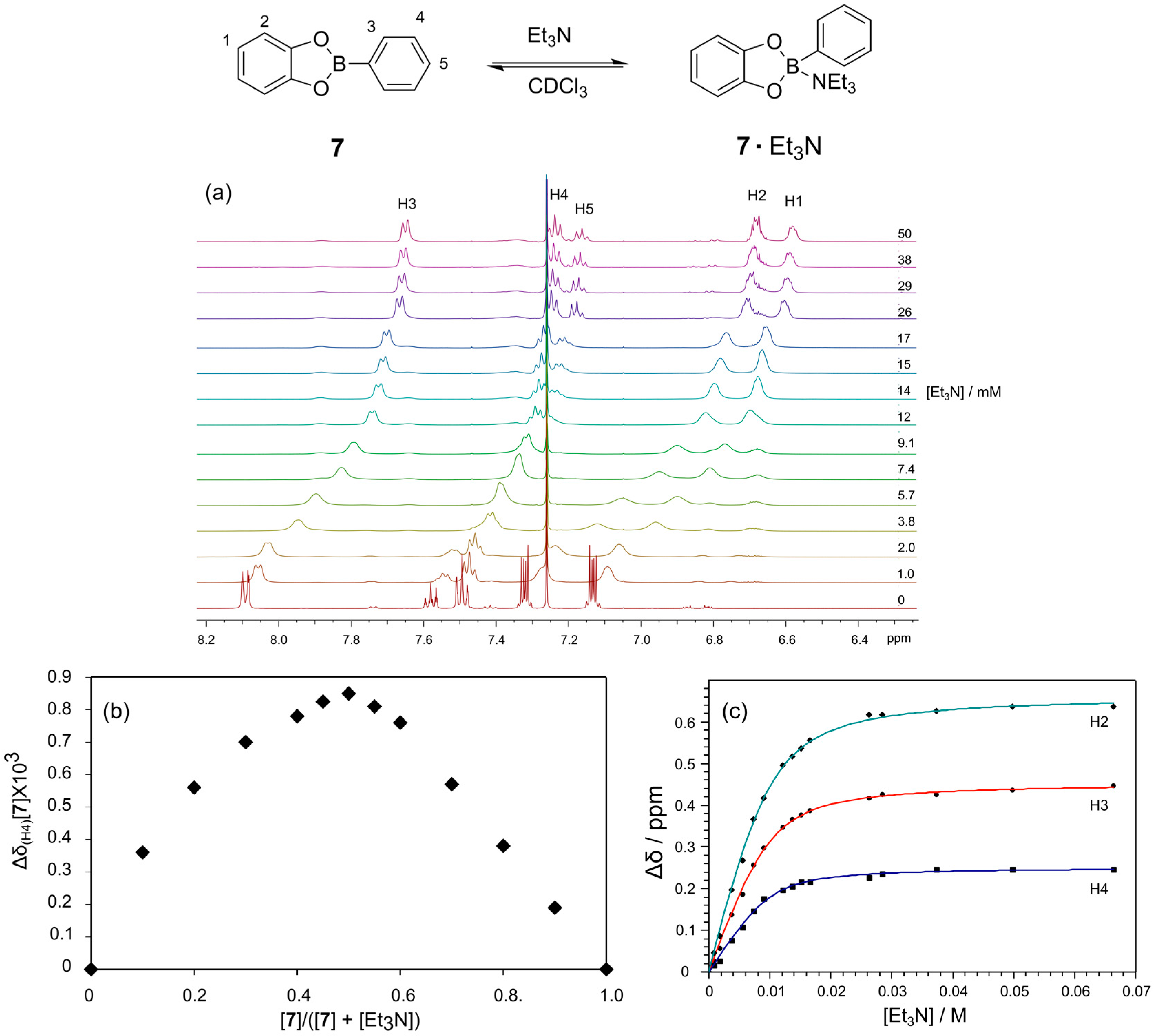
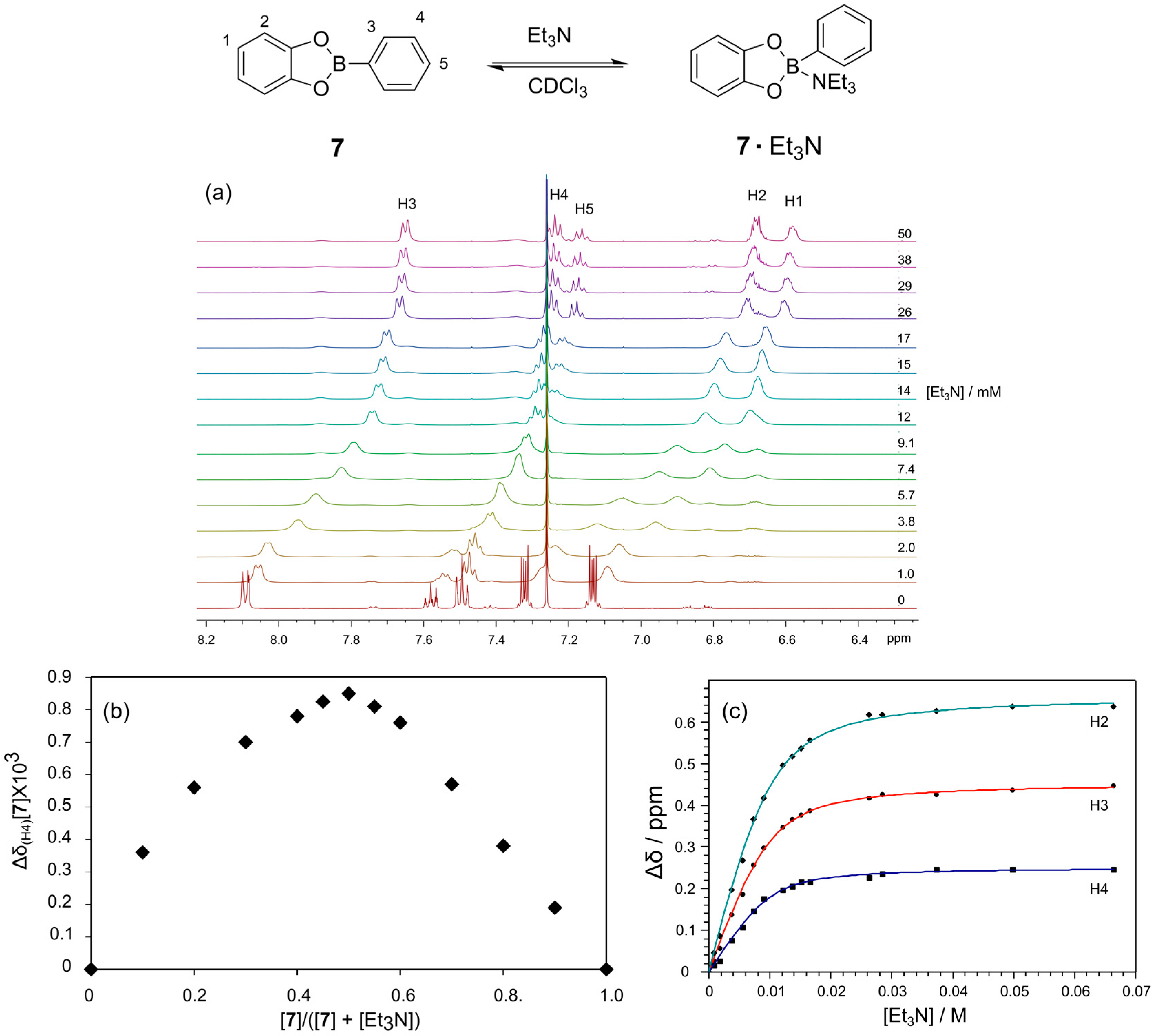
To further understand the influence of the amine base, we studied the interaction of Et
3N with the simple catechol based phenylboronic ester
7 using
1H-NMR spectroscopy (
Figure 4). Upon titration of Et
3N into a solution of
7 (10 mM) in CDCl
3 all the aromatic peaks in the spectrum shifted upfield as a consequence of the binding of Et
3N at the boron to give a tetrahedral geometry (
Figure 4a). These changes in
1H-NMR chemical shift reflect a binding event that is fast on the NMR chemical shift time scale. We have established using a Job plot that Et
3N binds to the boronic ester in a 1:1 fashion; the apex of the Job plot is at 0.5, as seen in
Figure 4b. A plot of the chemical shift change of the meta proton on the boronic acid (H4) as a function of the Et
3N concentration provides a binding isotherm from which a binding constant of 700 M
−1 was determined (
Figure 4c). Fitting of Binding Constants are provided in
Supplementary Material.
Figure 4.
Determination of the binding strength and stoichiometry for the binding of Et3N to phenylboronic ester 7. (a) 1H-NMR spectra (500 MHz, CDCl3, 300 K) of phenylboronic ester 7 (10 mM) in the presence of increasing concentrations of Et3N; (b) Job plot confirming the 1:1 binding stoichiometry for the interaction; (c) Plot of the chemical shift change of H3 of the phenylboronic ester 7 as a function of Et3N concentration and the non-linear fit to determine the binding constant.
Figure 4.
Determination of the binding strength and stoichiometry for the binding of Et3N to phenylboronic ester 7. (a) 1H-NMR spectra (500 MHz, CDCl3, 300 K) of phenylboronic ester 7 (10 mM) in the presence of increasing concentrations of Et3N; (b) Job plot confirming the 1:1 binding stoichiometry for the interaction; (c) Plot of the chemical shift change of H3 of the phenylboronic ester 7 as a function of Et3N concentration and the non-linear fit to determine the binding constant.
2.3. Disulfide Exchange in CDCl3 Using DBU as the Base
To speed up this oxidation reaction, we tried a number of different bases. The more nucleophilic bases DMAP and pyridine were effective at facilitating the exchange of disulfides but the oxidation of thiols was still slow, requiring several days, and was therefore again not practical for DCLs. We reasoned that a stronger base could possibly help resolve this experimental problem. We searched for a base that would: (1) be stronger than Et
3N; (2) be unreactive towards the substrates used; (3) be compatible with the wish to follow the process of the exchange reactions using
1H-NMR spectroscopy; (4) be transparent in the aromatic region of the
1H-NMR spectra; and (5) be simple to handle and non-toxic. With these requirements, we identified the stronger amidine base DBU (1,8-diazabicycloundec-7-ene) as a suitable candidate [
32,
33]. We proceeded to perform a series of experiments in which we determined that a disulfide DCLs could be generated from free thiols in CDCl
3 in the presence of DBU. It was necessary to show that not only oxidation and disulfide formation occurred, but that there was also exchange of building blocks between disulfide library member takes place and that there was thermodynamic control over the DCL.
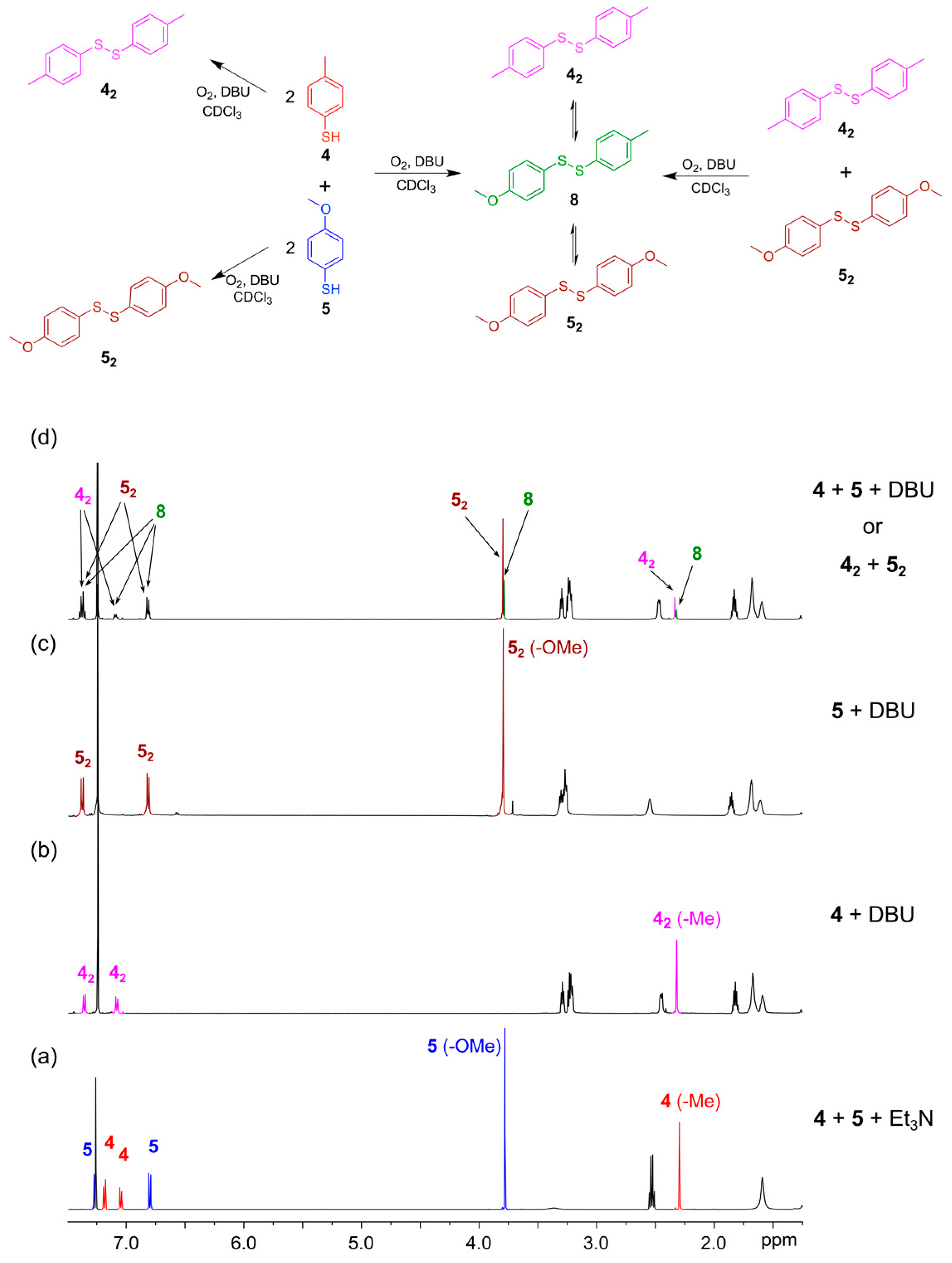
To validate that the disulfide exchange reaction is reversible and that a DCL operates under thermodynamic control, it must be possible to generate that DCL from different starting points. For example, if a DCL can be generated by mixing thiol building blocks, 4-methylphenylthiol (
4) and 4-methoxyphenylthiol (
5), it must also be possible to generate the same library composition by mixing the disulfides
42 and
52 in the presence of a catalytic amount of free thiol. To test this experimentally, we initiated the DCL from these two different starting points. In the first experiment, the DCL was initiated by mixing thiols
4 and
5 (each 4 mM) in CDCl
3 with DBU (20 mM) in a NMR tube. The
1H-NMR spectrum was recorded regularly over three days. In the second experiment, the aryl thiols
4 and
5 (each 8 mM) were individually dissolved in two different NMR tubes with DBU (20 mM) added. After 24 h, the NMR spectrum of each sample was recorded showing the formation of homodimers
42 and
52 (
Figure 5b,c). The two samples were then mixed in equal portions to form a second DCL and the
1H-NMR spectrum of the mixture was recorded regularly during the following three days. After one day, the NMR spectra of the two DCLs showed identical mixtures containing the homodimers,
42 and
52, as well as the heterodimer
8, indicating that exchange of building blocks took place and the libraries had reached equilibrium (
Figure 5d). A GC/MS analysis of the two samples verified the formation of the homo- and hetero-disulfides. Disulfide Exchange is provided in
Supplementary Material.
Figure 5.
Establishing thermodynamic equilibrium with disulfides using DBU as the base in CDCl3. (Top) Reaction scheme illustrating how a DCL containing homo and heterodimers 42, 52 and 8 can be formed either from the building blocks 4 and 5 or by mixing the in situ pre-formed dimers 42 and 52; (Bottom) 1H-NMR spectra (500 MHz, CDCl3, 300 K): (a) The two thiols 4 (4 mM) and 5 (4 mM) after mixing in the presence of Et3N (20 mM) as the base. After 48 h only the thiols are present; (b) Disulfide 42 (4 mM) generated in the presence of DBU (20 mM) in situ; (c) Disulfide 52 (4 mM) generated in the presence of DBU (20 mM) in situ; (d) Equilibrium constitution achieved when either 4 and 5 (4 mM each) were reacted directly or equal volumes of solutions of in situ generated 42 and 52 (5 mM each) are combined in the presence of DBU (20 mM). Note the appearance of the heterodimer 8.
Figure 5.
Establishing thermodynamic equilibrium with disulfides using DBU as the base in CDCl3. (Top) Reaction scheme illustrating how a DCL containing homo and heterodimers 42, 52 and 8 can be formed either from the building blocks 4 and 5 or by mixing the in situ pre-formed dimers 42 and 52; (Bottom) 1H-NMR spectra (500 MHz, CDCl3, 300 K): (a) The two thiols 4 (4 mM) and 5 (4 mM) after mixing in the presence of Et3N (20 mM) as the base. After 48 h only the thiols are present; (b) Disulfide 42 (4 mM) generated in the presence of DBU (20 mM) in situ; (c) Disulfide 52 (4 mM) generated in the presence of DBU (20 mM) in situ; (d) Equilibrium constitution achieved when either 4 and 5 (4 mM each) were reacted directly or equal volumes of solutions of in situ generated 42 and 52 (5 mM each) are combined in the presence of DBU (20 mM). Note the appearance of the heterodimer 8.
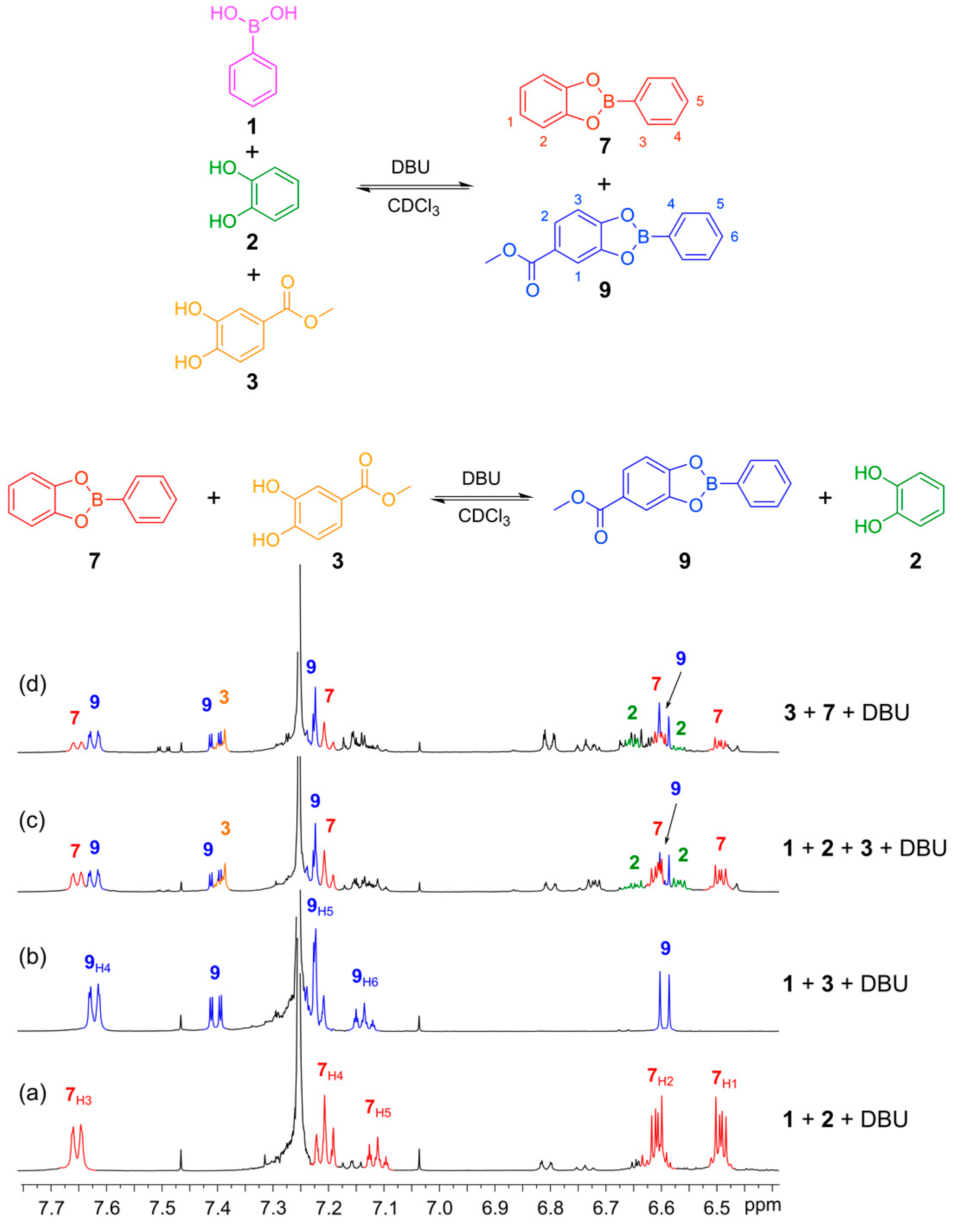
2.4. Boronic Transesterfication and Disulfide Exchange in CDCl3 Using DBU as the Base
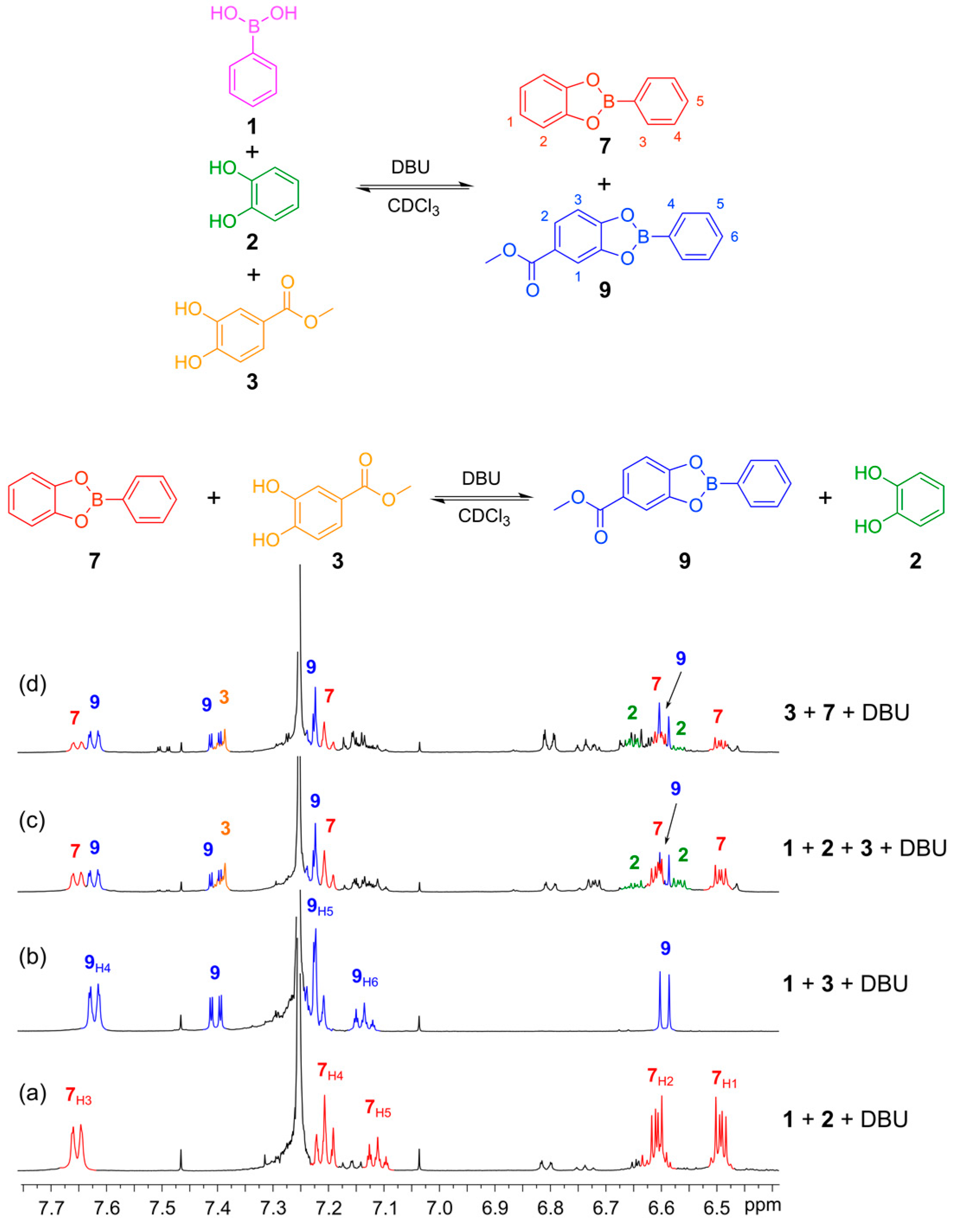
With conditions in place to perform the disulfide exchange under thermodynamic control, we proceeded to verify the reversible nature of the boronic ester transesterification using DBU as the base. First, we verified that DBU mediated the formation of the boronic esters from phenyl boronic acid (
1) with catechol (
2) and methyl 3,4-dihydroxybenzoate (
3). Five equivalents of DBU were utilized and boronic esters
7 and
9 were formed quantitatively giving well-defined
1H-NMR spectra with sharp features recorded after 30 min reaction time (
Figure 6a,b). Next, we tested for reversibility and thermodynamic control over the system by initiating DCLs from different starting points.
Figure 6c shows a DCL formed from the mixing of phenylboronic acid (
1) with catechols
2 and
3 in a 1:1:1 ratio to give a mixture containing the two boronic esters,
7 and
9, as well as excess of catechols
2 and
3.
Figure 6d shows the DCL formed by mixing the
in situ formed boronic ester
7 (seen in spectrum 6a) with methyl 3,4-dihydroxybenzoate (
3). The two spectra (
Figure 6c,d) exhibit similar features, indicating that reversible transesterification proceeds and the mixture is under thermodynamic control. Establishing Equilibrium for Boronic Ester Transesterification is provided in
Supplementary Material.
Figure 6.
Establishing thermodynamic equilibrium with boronic ester transesterification using DBU as the base in CDCl3. (Top) Reaction schemes illustrating how boronic esters 7 and 9 can be formed either from their constituent building blocks 1, 2 and 3 or by reacting the preformed boronic ester 7 with catechol 3 to give an equilibrium mixture containing esters 7 and 9 and catechols 2 and 3; (Bottom) 1H-NMR spectra (500 MHz, CDCl3, 300 K): (a) in situ-generated phenylboronic ester 7 formed from 1 (4 mM) and 2 (4 mM) in the presence of DBU (20 mM) after 30 min; (b) In situ-generated phenylboronic ester 9 formed from 1 (4 mM) and 3 (4 mM) in the presence of DBU (20 mM) after 30 min; (c) DCL formed from 1 (2 mM), 2 (2 mM) and 3 (2 mM); (d) DCL formed from 7 (2 mM) and 3 (2 mM). Note that the spectra in c and d show the same composition. There are catechol decomposition products seen at 6.8 and 7.2 ppm. These are formed because the experiments are performed with a 1:1:1 ration of 1, 2 and 7 so there is an excess of catechol.
Figure 6.
Establishing thermodynamic equilibrium with boronic ester transesterification using DBU as the base in CDCl3. (Top) Reaction schemes illustrating how boronic esters 7 and 9 can be formed either from their constituent building blocks 1, 2 and 3 or by reacting the preformed boronic ester 7 with catechol 3 to give an equilibrium mixture containing esters 7 and 9 and catechols 2 and 3; (Bottom) 1H-NMR spectra (500 MHz, CDCl3, 300 K): (a) in situ-generated phenylboronic ester 7 formed from 1 (4 mM) and 2 (4 mM) in the presence of DBU (20 mM) after 30 min; (b) In situ-generated phenylboronic ester 9 formed from 1 (4 mM) and 3 (4 mM) in the presence of DBU (20 mM) after 30 min; (c) DCL formed from 1 (2 mM), 2 (2 mM) and 3 (2 mM); (d) DCL formed from 7 (2 mM) and 3 (2 mM). Note that the spectra in c and d show the same composition. There are catechol decomposition products seen at 6.8 and 7.2 ppm. These are formed because the experiments are performed with a 1:1:1 ration of 1, 2 and 7 so there is an excess of catechol.
![Ijms 16 21858 g006]()
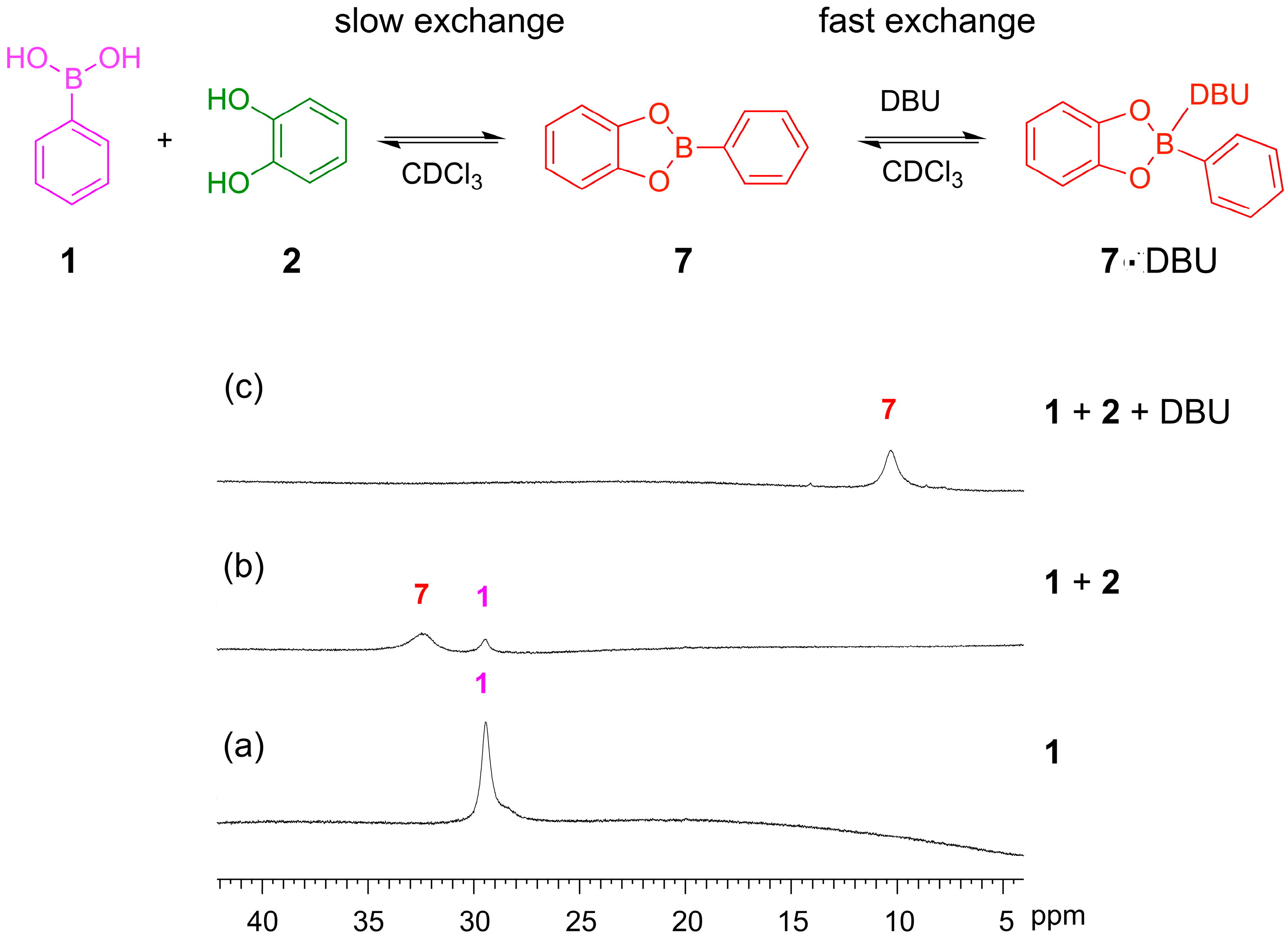
11B-NMR spectroscopy provides a useful tool to monitor the formation of boronic esters and study coordination at the boron center. We used
11B-NMR spectroscopy to observe the formation of boronic ester
7 from its constituent building blocks
1 and
2 and to examine the role of DBU in mediating the reaction. Upon addition of catechol
2 to a solution of phenylboronic acid
1, a new signal is observed at 32 ppm, which can be assigned to the boronic ester
7 while some of boronic acid
1 is still present, illustrating the incomplete formation of the ester in the absence of base (
Figure 7a,b). When five equivalents of DBU were added, the signal for the boronic acid
1 disappears and the signal for the ester
7 increases in size and shifts upfield to 10 ppm (
Figure 7c). DBU binds to the boronic ester
7, stabilizing this product, and thus mediates the complete conversion from the acid to the boronic ester. The upfield shift upon base binding is consistent with a change in hybridization of the boron center from trigonal planar to tetrahedral, as reported previously in the literature [
34]. This result is similar to our previous observation that Et
3N binds to boronic ester
7.
Figure 7.
11B NMR. (160 MHz, CDCl3, 300 K) illustrating boronic ester formation and binding of DBU: (a) Phenylboronic acid 1 (10 mM); (b) Phenylboronic acid 1 (3.3 mM) and catechol 2 (3.3 mM) recorded after 24 h and showing the incomplete formation of the phenylboronic ester 7 with residual phenylboronic acid 1; (c) 1 (3.3 mM), 2 (3.3 mM) and DBU (16.7 mM) recorded after 24 h and showing the formation of the tetrahedral boronate complex with DBU.
Figure 7.
11B NMR. (160 MHz, CDCl3, 300 K) illustrating boronic ester formation and binding of DBU: (a) Phenylboronic acid 1 (10 mM); (b) Phenylboronic acid 1 (3.3 mM) and catechol 2 (3.3 mM) recorded after 24 h and showing the incomplete formation of the phenylboronic ester 7 with residual phenylboronic acid 1; (c) 1 (3.3 mM), 2 (3.3 mM) and DBU (16.7 mM) recorded after 24 h and showing the formation of the tetrahedral boronate complex with DBU.
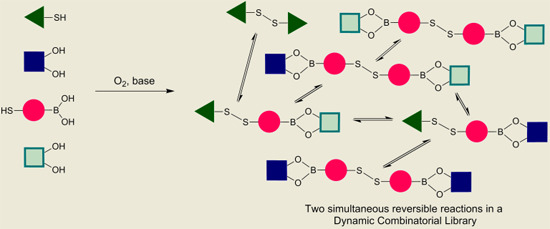
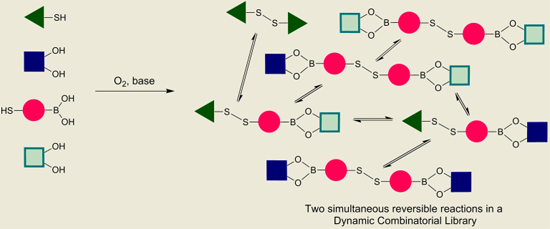
With proof of both reactions equilibrating under thermodynamic control using identical conditions (CDCl
3, 5 equivalents DBU), we continued to test whether the two reversible reactions function simultaneously. This was confirmed according to the experiment outlined in
Figure 8 and
Figure 9. In both experiments, we use the bifunctional building block
6, which contains both a boronic acid functionality and a thiol functionality.
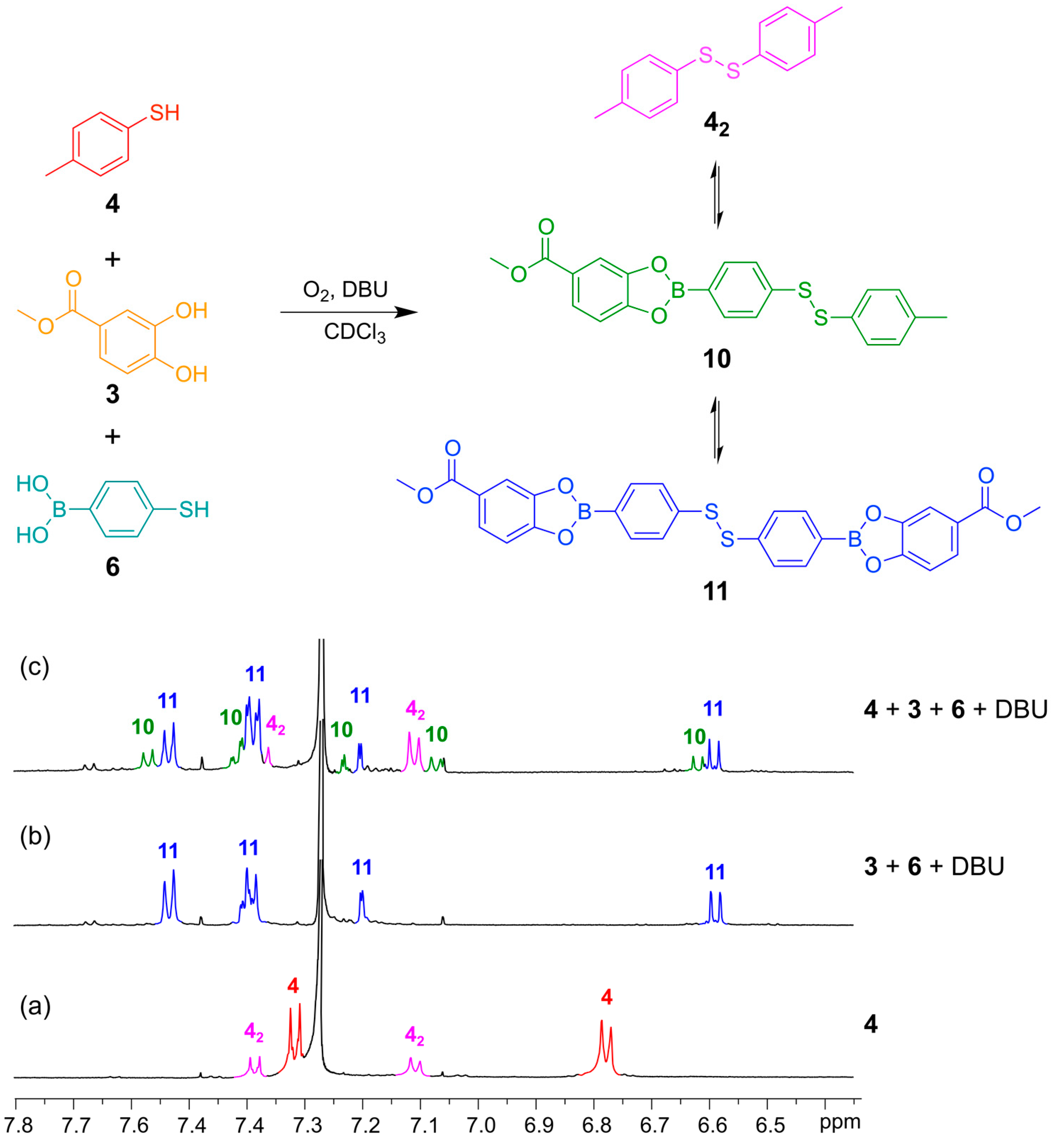
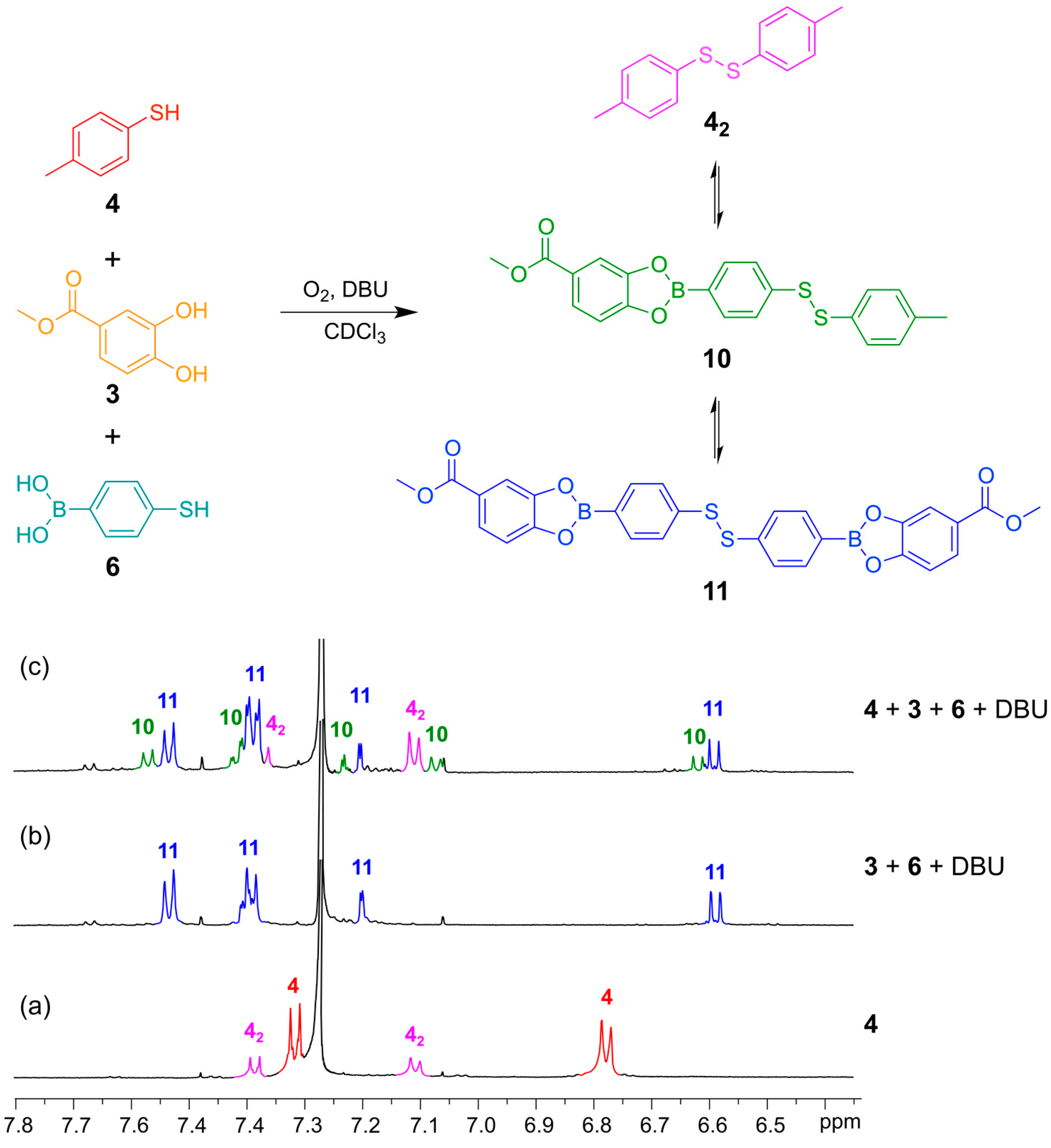
In
Figure 8 is shown
1H-NMR spectra that illustrate how the bifunctional building block
6 can engage in DCLs with both another thiol (4-methylphenylthiol (
4)) and a catechol (methyl 3,4-dihydroxybenzoate (
3)). A DCL containing the symmetrical homo-disulfides,
42 and
11, and the hetero-disulfide
10 formed within 24 h and the composition of this DCL was independent of the order of addition of the building blocks (
Figure 8c).
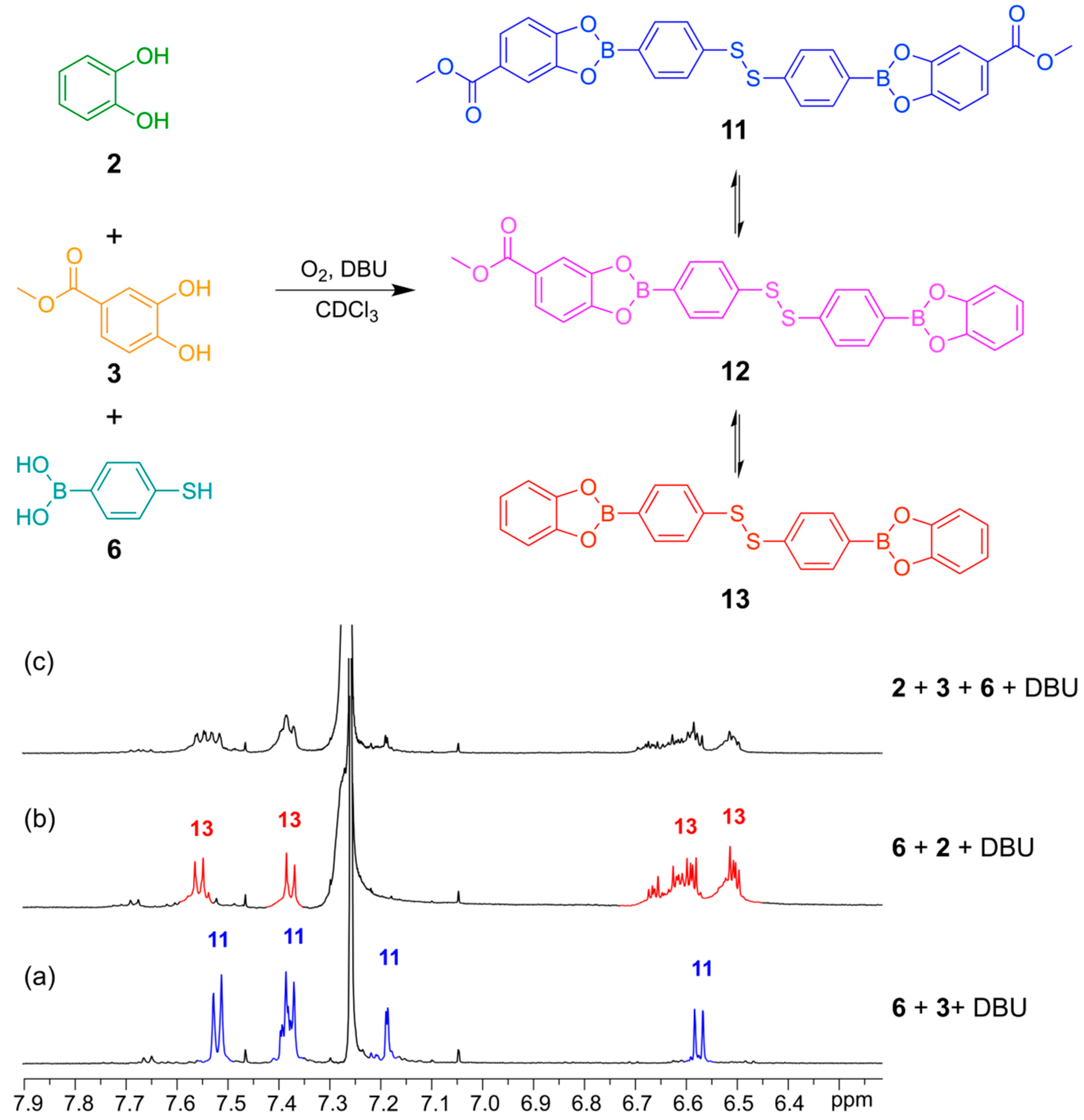
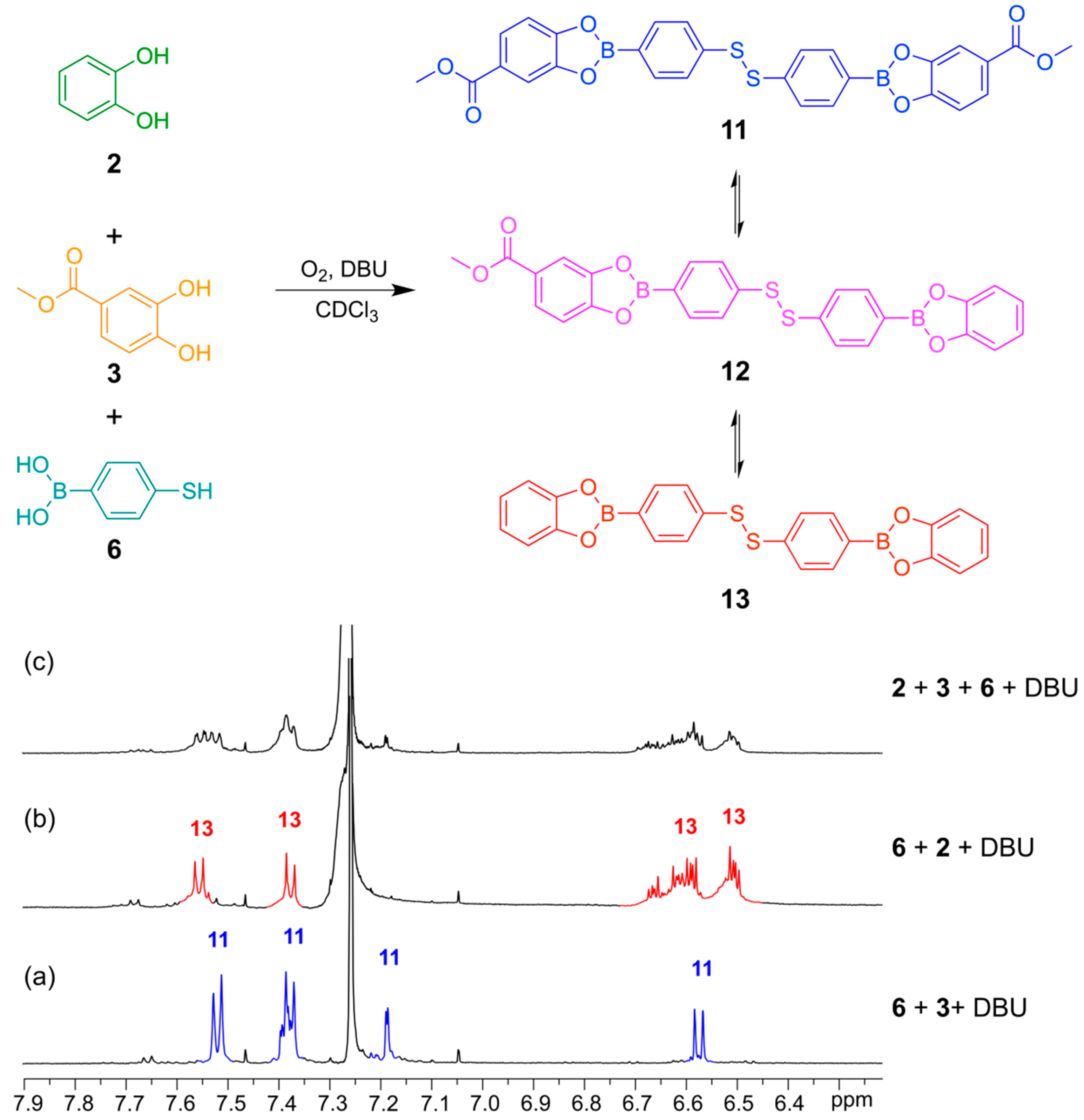
In
Figure 9 is illustrated how two different catechols (
2 and
3) combine with thiol-functionalized phenyl boronic acid
6 to gives the three possible linear disulfide oligomers
11,
12 and
13. Once again, the same distribution of library members in the DCL was obtained after 24 h reaction time regardless of the order of addition of the building blocks.
Figure 8.
(Top) Simultaneous disulfide exchange and boronic ester transesterification combining two different thiol building blocks; (Bottom) 1H-NMR spectra (500 MHz, CDCl3, 300 K): (a) Thiol 4 (contains minor amounts of the disulfide 42); (b) Bifunctional building block 6 (4 mM) reacted with catechol 3 (4 mM) in the presence of DBU (20 mM); (c) Bifunctional building block 6 (2 mM) reacted with catechol 3 (2 mM) and thiol 4 (2 mM) in the presence of DBU (20 mM). All spectra recorded after 24 h reaction time.
Figure 8.
(Top) Simultaneous disulfide exchange and boronic ester transesterification combining two different thiol building blocks; (Bottom) 1H-NMR spectra (500 MHz, CDCl3, 300 K): (a) Thiol 4 (contains minor amounts of the disulfide 42); (b) Bifunctional building block 6 (4 mM) reacted with catechol 3 (4 mM) in the presence of DBU (20 mM); (c) Bifunctional building block 6 (2 mM) reacted with catechol 3 (2 mM) and thiol 4 (2 mM) in the presence of DBU (20 mM). All spectra recorded after 24 h reaction time.
Figure 9.
(Top) Simultaneous disulfide exchange and boronic ester transesterification combining two different diol building blocks; (Bottom) 1H-NMR spectra (500 MHz, CDCl3, 300 K): (a) Bifunctional building block 6 (4 mM) reacted with catechol 3 (4 mM) in the presence of DBU (20 mM); (b) Bifunctional building block 6 (4 mM) reacted with catechol 2 (4 mM) in the presence of DBU (50 Mm); (c) Bifunctional building block 6 (4 mM) reacted with catechol 2 (2 mM) and catechol 3 (2 mM) in the presence of DBU (20 mM). All spectra recorded after 24 h reaction time.
Figure 9.
(Top) Simultaneous disulfide exchange and boronic ester transesterification combining two different diol building blocks; (Bottom) 1H-NMR spectra (500 MHz, CDCl3, 300 K): (a) Bifunctional building block 6 (4 mM) reacted with catechol 3 (4 mM) in the presence of DBU (20 mM); (b) Bifunctional building block 6 (4 mM) reacted with catechol 2 (4 mM) in the presence of DBU (50 Mm); (c) Bifunctional building block 6 (4 mM) reacted with catechol 2 (2 mM) and catechol 3 (2 mM) in the presence of DBU (20 mM). All spectra recorded after 24 h reaction time.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}